
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescent solar concentrators with outstanding optical properties by employment of D–A–D quinoxaline fluorophores†
Costanza
Papucci
abc,
Rima
Charaf
d,
Carmen
Coppola
 ce,
Adalgisa
Sinicropi
ace,
Mariangela
di Donato
af,
Maria
Taddei
af,
Paolo
Foggi
fgh,
Antonella
Battisti
i,
Bastiaan
de Jong
cj,
Lorenzo
Zani
a,
Alessandro
Mordini
ab,
Andrea
Pucci
*ad,
Massimo
Calamante
*ab and
Gianna
Reginato
*a
ce,
Adalgisa
Sinicropi
ace,
Mariangela
di Donato
af,
Maria
Taddei
af,
Paolo
Foggi
fgh,
Antonella
Battisti
i,
Bastiaan
de Jong
cj,
Lorenzo
Zani
a,
Alessandro
Mordini
ab,
Andrea
Pucci
*ad,
Massimo
Calamante
*ab and
Gianna
Reginato
*a
aInstitute of Chemistry of Organometallic Compounds (CNR-ICCOM), Via Madonna del Piano 10, 50019 Sesto Fiorentino, Italy
bDepartment of Chemistry “U. Schiff”, University of Florence, Via della Lastruccia 13, 50019 Sesto Fiorentino, Italy
cDepartment of Biotechnology, Chemistry and Pharmacy, University of Siena, Via A. Moro 2, 53100 Siena, Italy
dDepartment of Chemistry and Industrial Chemistry, University of Pisa, Via G. Moruzzi 13, 56124 Pisa, Italy
eCSGI, Consorzio per lo Sviluppo dei Sistemi a Grande Interfase, 50019 Sesto Fiorentino, Italy
fLENS, European Laboratory for Non-Linear Spectroscopy, via N. Carrara 1, 50019 Sesto Fiorentino, Italy
gDepartment of Chemistry, Biology and Biotechnology, University of Perugia, via Elce di Sotto 8, 06123 Perugia, Italy
hINRIM (Istituto Nazionale di Scienza Metrologica), Strada delle Cacce 91, 10135 Torino, Italy
iNanoscience Institute, CNR and NEST, Scuola Normale Superiore, P.zza S. Silvestro 12, 56127 Pisa, Italy
jCicci Research s.r.l., Via Giordania 227, 58100 Grosseto, Italy
First published on 11th October 2021
Abstract
Luminescent solar concentrators (LSCs) are devices designed to efficiently collect both direct and diffuse solar radiation and concentrate it on photovoltaic cells to foster their use in building-integrated photovoltaics (BIPV). The optimization of LSC performances involves the adjustment of both the fluorophore and the guest polymer matrix. On this account, we investigated a series of high quantum yield, donor–acceptor–donor (D–A–D) photostable fluorophores (DQ1–5), presenting a central quinoxalinic acceptor core, not previously employed in LSCs, and triarylamines or phenothiazine as donor groups. The molecules were also decorated with alkyl chains on the central core and/or the donor groups, to explore their compatibility with the poly(methyl methacrylate) (PMMA) and poly(cyclohexyl methacrylate) (PCMA) matrices utilized in this study. The PMMA and PCMA films (25 μm thick), containing 0.2–2.2 wt% of DQ1–5, absorbed in the 370–550 nm range and presented emission maxima at 550–600 nm, with fluorescence quantum yields higher than 40% even at the highest doping contents. Notably, the DQ1/PMMA thin-films showed enhanced phase compatibility and excellent quantum yields, i.e., >95%. Accordingly, they were designed to obtain 25 cm2 area LSCs with remarkable internal (ηint) and external (ηext) photon efficiencies of 42.9% and 6.2%, respectively, higher than those observed from state-of-the-art devices based on the Lumogen Red 305 (LR305) as the reference fluorophore. Overall, these were the best results ever achieved in our laboratory for thin-film LSCs built with organic fluorescent emitters.
1. Introduction
In the past few decades, the need to reduce CO2 emissions generated by human activities has prompted the scientific community to study new and efficient methods to utilize renewable energy sources, such as sunlight, for power generation, aiming to progressively replace the use of fossil fuels.1 Among the currently available technologies, silicon photovoltaics (Si-PVs) have been the subject of continuous development efforts, which ultimately led to the reduction in the production prices of the modules at an actual electricity cost of about 0.2 $ per Watt peak (Wp)2 without detrimenting performances.Nevertheless, Si-PV panels provide the maximum efficiency only under direct light irradiation and require an efficient dissipation of the excess heat due to the unconverted energy. Luminescent solar concentrators (LSCs) rely on a technology studied since the 1970s3,4 and were developed with the aim of obtaining large-area, semi-transparent and cheap photovoltaic devices capable of concentrating both direct and diffuse solar radiation on small solar cells at their edges. Specifically, they consist of a panel of a common plastic material (e.g., poly(methyl methacrylate), PMMA) containing a fluorescent dopant capable of absorbing direct and indirect sunlight and emitting it at longer wavelengths. Commonly used fluorescent compounds can be quantum dots, perovskites, rare-earth complexes and organic molecules.5 The emitted radiation is mainly concentrated, via total internal reflection, at the edge of the panel (where the solar cells are placed) thanks to the different refractive indexes of air and the plastic material. This, together with the aesthetic characteristics of LSCs (color and shape tunability), supports their use in building-integrated photovoltaics (BIPVs).6
In order to obtain high-performance LSC devices, a careful study of the materials used for their assembly must be performed, concerning both the selection of the fluorophore and the plastic material in which it is dispersed.5 Regarding the fluorophore, the most critical parameters influencing the final properties of the device are its fluorescence quantum yield (Φf) and Stokes shift (SS) which, together with the match between its emission spectrum and the electronic band-gap of the PV cell, govern the efficiency of the solar collector.
These properties, particularly Φf, can depend on the polarity of the medium (solution or polymer) in which the fluorophore is dispersed. For the polymer matrix, the main factor to be considered is its ability to disperse large amounts of fluorophores to promote solar harvesting, without activating fluorescence quenching phenomena due to aggregation7,8 (aggregation caused quenching – ACQ). Furthermore, a good matrix might be able to affect the stability of the fluorophore excited state and thus enhance its fluorescence quantum yield. Clearly, a smart selection of the polymer, specifically tailored to fit the fluorophore properties, is an important factor for improving the device performance.
Among the different kinds of fluorophores applicable in LSCs, small organic molecules with a donor–acceptor type (D–A) structure, whose excitation determines the formation of a charge-transfer state with a significant dipole moment,9 appear particularly interesting. We recently investigated the properties of a series of organic fluorophores with donor–acceptor–donor (D–A–D) structures, presenting a benzo[1,2-d:4,5-d′]bisthiazole acceptor core, dispersed in three methacrylic polymer matrices of different polarity.10 Notably, we found that by decreasing the polarity of the matrix by passing from PMMA to poly(cyclohexyl methacrylate) (PCMA, Fig. 1), the Φf of the molecules increased due to the decreased stabilization of the charge-transfer excited state, resulting in a superior optical efficiency of the corresponding LSCs.
 | ||
| Fig. 1 Chemical structure of fluorophores based on 2,3-diphenyl-quinoxaline (DQ1-3) or 2,3-dihexyl-quinoxaline (DQ4-5) acceptor cores and of the used polymer matrices. | ||
To further explore the interactions between organic emitters and polymer matrices of varying polarity and assess their effects on the performances of the corresponding LSCs, we decided to investigate a different series of luminophores, still presenting the same D–A–D architecture. We focused on quinoxaline, a molecular scaffold which is quite easy to functionalize and is characterized by good light harvesting ability, high Φf and large SS.11–18
Owing to their photophysical properties, quinoxalines are good candidates for application in LSCs but, although they have been extensively applied in organic optoelectronics,19 their use in such devices has not yet been reported. Therefore, starting from a previously reported compound, DQ1,20,21 a series of symmetrical derivatives were designed and synthesized by modifying both the donor groups at the 5,8 positions and the substituents at the 2,3 positions (Fig. 1).
The optical properties of the new molecules were investigated in solution and after dispersion in the PMMA and PCMA matrices to determine their absorption and emission features as well as their Φf. Furthermore, transient absorption spectroscopy (TAS) and fluorescence lifetime studies were also carried out to monitor the excited state evolution and correlate the device characteristics with the dynamic optical properties of the fluorophores. Due to the outstanding performances of these emitters also when dispersed into polymers, LSCs presented excellent optical properties, even superior to those found using the commercial reference fluorophore Lumogen Red 305 (LR305).5,8,10
2. Experimental
2.1 General remarks
All commercially available compounds were purchased from Merck KgaA, Fluorochem Ltd and T.C.I. Co. Ltd, and were used without further purification unless stated otherwise. Anhydrous toluene was obtained after drying with a PureSolv Micro apparatus (Inert). Organometallic reactions were carried out under dry nitrogen using Schlenk techniques. Solvent degassing was carried out according to the “freeze–pump–thaw” method. Reactions were monitored by TLC on Kieselgel 60 F254 (Merck) aluminium sheets and the products were visualized by exposing the plate to UV light or by staining it with KMnO4 solution. Flash column chromatography was performed using a Merck Kieselgel 60 (300–400 mesh) as the stationary phase. 1H-NMR spectra were recorded at 200–400 MHz, and 13C-NMR spectra were recorded at 50.3–100.6 MHz, using Varian Gemini/Mercury/INOVA series instruments. Chemical shifts (δ) are reported in parts per million (ppm) and are referenced to the residual solvent peak (CDCl3, δ = 7.26 ppm for 1H-NMR and δ = 77.0 ppm for 13C-NMR; CD2Cl2, δ = 5.32 ppm for 1H-NMR and δ = 53.84 ppm for 13C-NMR), while coupling constants (J) are reported in Hz. ESI-MS spectra were obtained by direct injection of the sample solution using a Thermo Scientific LCQ-FLEET instrument, while HRMS spectra were measured using a Thermo Scientific LTQ Orbitrap (FT-MS) instrument (carried out at the Interdepartmental Centre for Mass Spectrometry of the University of Florence, CISM); main peaks for both are reported in the form m/z.2.2 Spectroscopic analysis in solution
UV-vis absorption spectra were recorded with a Shimadzu 2600 series spectrometer, and fluorescence spectra were recorded with a JASCO FP-8300 spectrofluorometer, irradiating the sample at the wavelength corresponding to maximum absorption in the UV-vis spectrum. Absolute fluorescence quantum yields (Φf) in toluene solution were determined at room temperature using an integration sphere.2.3 LSC film preparation
Polymer thin films containing the selected fluorophore were prepared by pouring 1.5 mL of toluene (Sigma-Aldrich, HPLC Plus, ≥99.9%) solution containing about 60 mg of the polymer and different concentrations (0.2–2.2 wt%) of the fluorophore on a 50 × 50 × 3 mm optically pure glass substrate (Edmund Optics Ltd BOROFLOAT window 50 × 50 TS). The amount of fluorophore was taken from a mother solution at a concentration of 1.5 mg mL−1. The glass slides were cleaned with chloroform and immersed in 6M HCl for at least 12 h and then dried according to previously reported procedures.22–27 Fluorophore-doped polymer thin films with a thickness of 25 ± 5 μm (Starrett micrometer) were obtained after complete evaporation of toluene at room temperature under a saturated atmosphere of chloroform (Merck, ACS reagent, ≥99.8%). After LSC characterization, the polymer films were carefully detached from the glass surface by immersing the LSC in water, stored in a desiccator and then analysed by means of absorption and emission spectroscopies.2.4 Spectroscopic analysis of polymer films
UV-vis absorption spectroscopy on polymer films was performed at room temperature by using an Agilent Cary 5000 spectrophotometer. Fluorescence spectra on polymer films were recorded at room temperature with a Horiba Jobin-Yvon Fluorolog®-3 spectrofluorometer equipped with a 450 W xenon arc lamp and double-grating excitation and single-grating emission monochromators. Fluorescence quantum yields (Φf) in the solid state were determined using a 152-mm-diameter “Quanta-φ” integrating sphere, coated with Spectralon® and following the procedures reported earlier.22,28,29 Epifluorescence microscopy was accomplished using a LED epifluorescence microscope (Schaefer South-East Europe S.r.l., Rovigo, Italy) equipped with a LED blue and green 5W light source and a DeltaPix Invenio 2EIII microscope camera (DeltaPix, Smorum, Denmark).2.5 Transient absorption spectroscopy and fluorescence lifetime imaging (FLIM)
Ultrafast transient absorption spectra of all samples have been recorded on a system consisting of a home-built Ti:sapphire laser oscillator and regenerative amplifier system (Amplitude Pulsar) which produced 80 femtoseconds pulses at 810 nm with an average output power of 450–500 mW. Excitation pulses at 400 nm were obtained by the second harmonic generation of the fundamental laser output in a 2 mm-thick β-barium borate (BBO) crystal. For all measurements, the pump beam polarization was set at the magic angle with respect to the probe beam by rotating a λ/2 plate to exclude rotational contributions to the transient signal.30,31 The excitation powers were on the order of 50–100 nJ. The probe pulses were generated by focusing a portion of the 800 nm radiation beam on a 3 mm-thick CaF2 window. The time delay between the pump and probe pulses was introduced by sending the portion of 800 nm light used for probe generation through a motorised stage. After passing through the sample, the white light probe was sent to a flat field monochromator coupled to a home-made CCD detector. For samples in solution, measurements were performed in a quartz cell (2 mm thick) mounted on a movable stage in order to refresh the solution and avoid undesired photochemical degradation of the sample. In the case of samples dispersed in PMMA, the polymeric films were directly mounted on the same motorised stage used for liquid samples. Analysis of the transient data was performed applying singular value decomposition (SVD)32 and global analysis,33 using GLOTARAN software.34 Global analysis allows the simultaneous fit of all the measured wavelengths with a combination of exponential decay functions and retrieves the kinetic constants describing the dynamic evolution of the system and the corresponding spectral component, called evolution associated difference spectra (EADS). A linear decay kinetic scheme was employed for data analysis.Fluorescence lifetime imaging (FLIM) was carried out on PMMA films doped with fluorophores DQ1–5 (1.0 wt%) using a inverted confocal microscope Leica TCS SP5 (Leica Microsystems, Wetzlar, GE) coupled with an external pulsed diode laser (λex = 405 nm) and a TCSPC acquisition card (PicoHarp 300, PicoQuant, Berlin, GE) connected to internal spectral detectors. The laser repetition rate was set to 20 or 40 Hz. The images (256 × 256 pixels) were usually acquired with a scan speed of 400 Hz (lines per second). The pinhole aperture was adjusted depending on the fluorescence intensity and films were imaged under a 40×/NA 0.60 dry objective (Leica Microsystems). Fluorescence emission was monitored in the 480 and 620 nm range using the built-in acousto-optical beam splitter detection system of the microscope. Acquisitions were stopped after the collection of about 100–200 photons per pixel, at a photon counting rate of about 200–300 kHz. The decay curves were obtained from the binned lifetime images and fitted using PicoQuant SymPhoTime software.
2.6 Determination of LSC optical properties
Internal (ηint) and external (ηext) photon efficiencies were evaluated in agreement with recent protocols35 (see the ESI† for additional discussion and pictures). All the measurements were performed using a commercially available system (Arkeo – Cicci research s.r.l.) containing a CMOS-based spectrometer with a symmetrical Czerny–Turner optical bench connected to an integrating sphere. A fiber-based tunable LED source was used to excite the center of the LSC device with a circular spot of 2 mm in diameter and at a distance of 0.5 mm. The platform includes a tunable LED source composed of 10 monochromatic diodes (from 360 to 960 nm) and 2 white diodes (warm and cold) used to match AM 1.5G (Fig. S22 and S23, ESI†). The use of LED source allowed the irradiation stability with time. An integrating sphere of 5 cm of diameter and 1 cm of aperture is placed along the edge of the glass plate, such that the aperture of the sphere is fully covered by the glass and one corner coincides with the edge of the aperture hole. The integrating sphere was moved along the side of the LSC until all the slab edges had been scanned. The spectrally resolved edge output photon count was collected from the CMOS-based spectrometer and calibrated into optical power (W) and then in irradiance. Aimed at limiting reflections of unabsorbed light, an absorbing matte black background was placed in contact with the LSC rear side. The fiber was kept close and perpendicular to the centre of the LSC front surface to minimize the diverge of the excitation beam and to avoid the direct illumination of the integrating sphere.36 A series of 3–5 measurements were repeated in order to align the integration sphere to collect the maximum single-edge output power. For the auto-absorption experiments, the Arkeo platform included a motorised translation stage and was equipped with a 405 nm, 1 mW laser.2.7 Synthesis of DQs
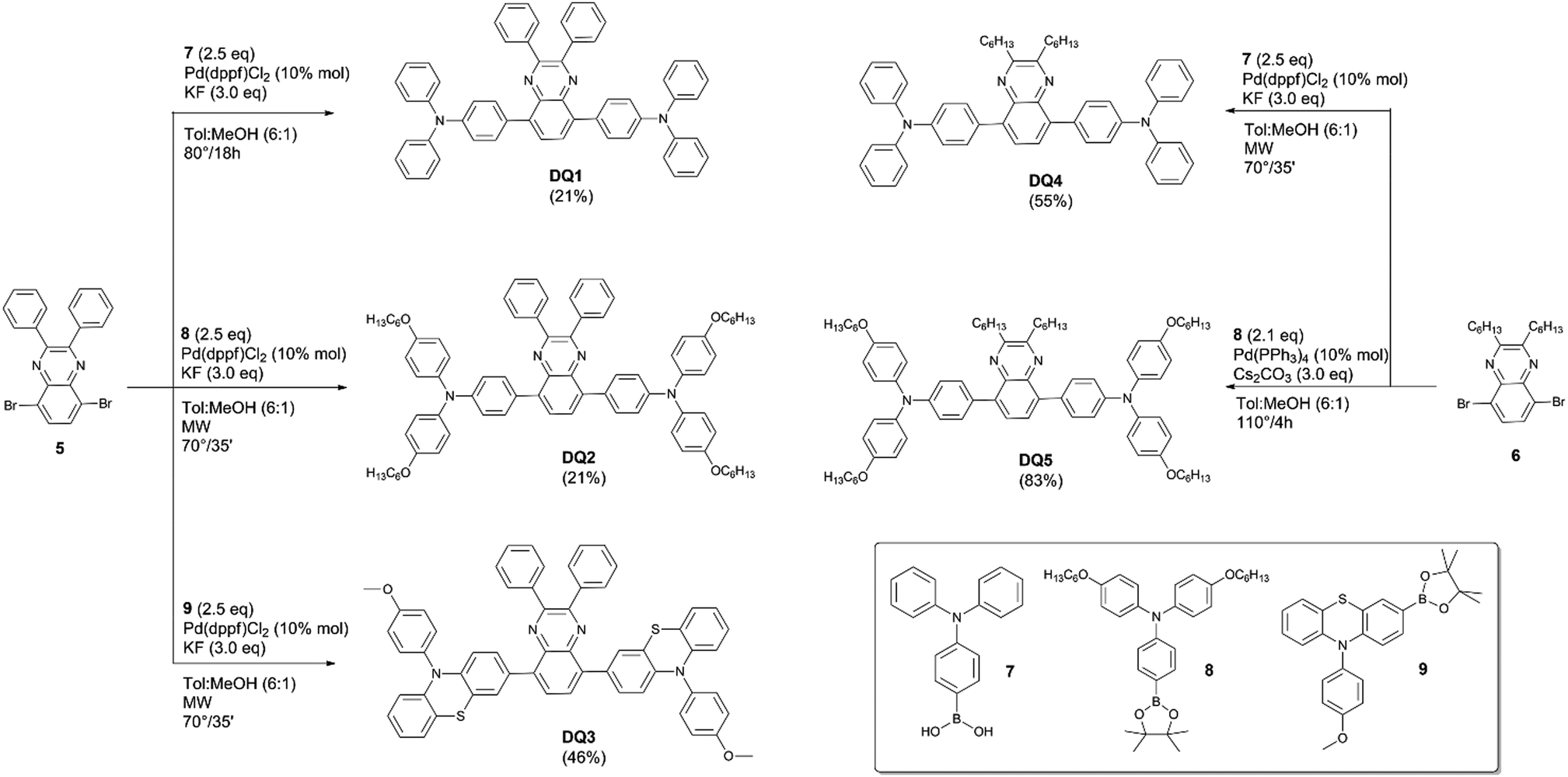
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (4 mL). KF (39 mg, 0.68 mmol, 3.0 eq.) and boronic acid 7 (164 mg, 0.57 mmol, 2.5 eq.) were added to the mixture. The solution was heated at 80 °C for 18 h. The mixture was then cooled at room temperature and water (5 mL) was added. The mixture was extracted with EtOAc (3 × 4 mL). The organic phase was washed with water and brine and then dried on anhydrous Na2SO4. Purification by flash column chromatography (SiO2, petroleum ether/DCM 3:1) gave product DQ1 as an orange solid (37 mg, 0.048 mmol, 21%). 1H-NMR (400 MHz, CDCl3): δ = 7.89 (s, 2H), 7.80 (d, J = 8.4 Hz, 4H), 7.63 (d, J = 7.6 Hz, 4H), 7.27–7.38 (m, 14H), 7.20–7.27 (m, 12H), 7.07 (dd, J = 7.6, 6.4 Hz, 4H) ppm. 13C-NMR (100 MHz, CDCl3): δ = 151.0, 147.7, 147.3, 139.1, 138.6, 138.4, 132.1, 131.7, 130.1, 129.4, 129.3, 128.8, 128.2, 124.7, 123.0, 122.7 ppm. HRMS: m/z calculated for C56H41N4: 769.3326. Found: 769.3315 [M + H]+.
:1 (4 mL). KF (39 mg, 0.68 mmol, 3.0 eq.) and boronic acid 7 (164 mg, 0.57 mmol, 2.5 eq.) were added to the mixture. The solution was heated at 80 °C for 18 h. The mixture was then cooled at room temperature and water (5 mL) was added. The mixture was extracted with EtOAc (3 × 4 mL). The organic phase was washed with water and brine and then dried on anhydrous Na2SO4. Purification by flash column chromatography (SiO2, petroleum ether/DCM 3:1) gave product DQ1 as an orange solid (37 mg, 0.048 mmol, 21%). 1H-NMR (400 MHz, CDCl3): δ = 7.89 (s, 2H), 7.80 (d, J = 8.4 Hz, 4H), 7.63 (d, J = 7.6 Hz, 4H), 7.27–7.38 (m, 14H), 7.20–7.27 (m, 12H), 7.07 (dd, J = 7.6, 6.4 Hz, 4H) ppm. 13C-NMR (100 MHz, CDCl3): δ = 151.0, 147.7, 147.3, 139.1, 138.6, 138.4, 132.1, 131.7, 130.1, 129.4, 129.3, 128.8, 128.2, 124.7, 123.0, 122.7 ppm. HRMS: m/z calculated for C56H41N4: 769.3326. Found: 769.3315 [M + H]+.
General procedure for microwave-activated Suzuki–Miyaura cross coupling
In a microwave tube, Pd(dppf)Cl2 (10 mol%) was added to a solution of appropriate dibromoquinoxaline (1.0 eq.) in toluene. Meanwhile, in a Schlenk tube, under a nitrogen atmosphere, boronic ester/acid (2.5 eq.) and KF (3.0 eq.) were dissolved in MeOH. After complete dissolution, the latter solution was added into the MW tube. The mixture was stirred for 30′ at room temperature and then heated under MW irradiation at 70 °C for 35′. The mixture was cooled at room temperature and the solvent was removed under a vacuum.:1 → 3:1) obtaining DQ2 as an orange solid (28 mg, 0.024 mmol, 21%). 1H-NMR (400 MHz, CDCl3): δ = 7.86 (s, 2H), 7.69 (d, J = 9.0 Hz, 4H), 7.58 (d, J = 7.4 Hz, 4H), 7.37–7.29 (m, 6H), 7.15 (d, J = 9.0 Hz, 8H), 7.04 (d, J = 11.9 Hz, 4H), 6.88 (d, J = 9.1 Hz, 8H), 3.96 (t, J = 6.4 Hz, 8H), 1.83–1.74 (m, 8H), 1.52–1.44 (m, 8H), 1.40–1.33 (m, 16H), 0.93 (t, J = 6.6 Hz, 12H) ppm. 13C-NMR (100 MHz, CDCl3): δ = 155.8, 151.1, 148.4, 140.4, 139.2, 138.3, 131.3, 129.9, 129.8, 129.15, 128.6, 128.1, 127.0, 118.9, 115.2, 68.3, 31.6, 29.3, 25.7, 22.6, 13.8 ppm. HRMS: m/z calculated for C80H89O4N4: 1169.6878. Found: 1169.6864 [M + H]+.
:1 → 2:1) obtaining DQ3 as a yellow solid (93 mg, 0.10 mmol, 46%). 1H-NMR (400 MHz, CDCl3): δ = 7.74 (s, 2H), 7.59 (dd, J = 6.0, 3.6 Hz, 6H), 7.36 (d, J = 6.0 Hz, 4H), 7.33–7.29 (m, 8H), 7.15 (dd, J = 7.6, 3.2 Hz, 4H), 7.03 (dd, J = 5.6, 1.6 Hz, 2H), 6.82 (ddd, J = 9.2, 4.0, 1.2 Hz, 4H), 6.30 (d, J = 8.8 Hz, 2H), 6.19 (dd, J = 4.0, 2.0 Hz, 2H), 3.92 (s, 6H) ppm. 13C-NMR (100 MHz, CDCl3): δ = 159.2, 151.1, 144.4, 144.0, 138.3, 137.5, 133.2, 132.3, 132.2, 130.1, 129.1, 128.8, 128.7, 128.2, 126.8, 126.6, 122.3, 119.5, 118.9, 115.9, 115.6, 115.2, 55.5 ppm. HRMS: m/z calculated for C58H57N4: 889.2665. Found: 889.2652 [M + H]+.
:1 → 2:1 → EtOAc) obtaining DQ4 as a yellow solid (67 mg, 0.08 mmol, 55%). 1H-NMR (400 MHz, CDCl3): δ = 7.77 (s, 2H), 7.72 (d, J = 8.4 Hz, 4H), 7.29 (dd, J = 15.9, 8.0 Hz, 8H), 7.24–7.15 (m, 12H), 7.04 (m, 4H), 2.97 (t, J = 7.6 Hz, 4H), 1.91–1.81 (m, 4H), 1.46–1.37 (m, 4H), 1.36–1.21 (m, 6H), 0.86 (t, J = 6.0 Hz, 6H) ppm. 13C-NMR (100 MHz, CDCl3): δ = 155.1, 148.0, 147.1, 138.8, 138.3, 132.9, 131.8, 129.4, 128.5, 124.7, 123.0, 34.9, 32.0, 29.3, 27.3, 22.8, 14.2 ppm. HRMS: m/z calculated for C56H57N4: 785.4577. Found: 785.4564 [M + H]+.
:1 → 1:1) obtaining DQ5 as an orange solid (108 mg, 0.091 mmol, 83%). 1H-NMR (400 MHz, CDCl3): δ = 7.72 (s, 2H), 7.65 (d, J = 8.4 Hz, 4H), 7.13 (d, J = 9.2 Hz, 8H), 7.03 (d, J = 8.4 Hz, 4H), 6.84 (d, J = 9.0 Hz, 8H), 3.94 (t, J = 1.2 Hz, 8H), 2.96 (t, J = 1.2 Hz, 4H), 1.74–1.90 (m, 14H), 1.51–1.43 (m, 10H), 1.37–1.33 (m, 21H), 0.95–0.89 (m, 14H), 0.88–0.82 (m, 7H) ppm. 13C-NMR (100 MHz, CDCl3): δ = 155.4, 154.7, 147.9, 140.8, 138.7, 138.0, 131.4, 130.6, 128.1, 126.7, 119.6, 115.2, 68.2, 34.7, 31.8, 31.6, 29.3, 29.2, 27.3, 25.8, 22.6, 14.1, 14.0 ppm. HRMS: m/z calculated for C80H105O4N4: 1184.8058. Found: 1184.8042 [M + H]+.
3. Results and Discussions
3.1 Synthesis of chromophores DQ1–5
Compounds DQ1–5 were rationally designed to modulate their relevant electronic and physico-chemical properties. By inserting different electron-donating groups at the 5,8 positions of the quinoxaline core, the HOMO–LUMO energy difference could be tuned, thus effectively modulating their absorption spectra.38 In addition, the nature of the substituents at the 2,3 positions can affect the electron-accepting capacity of the quinoxaline unit, stabilizing the LUMO orbital.21 Finally, the presence of alkyl chains on the acceptor and donor groups, other than modifying the electronic and optical properties, can also alter the dispersibility of the fluorophores in the polymer matrix, controlling the potential formation of non-emissive aggregates. Compounds DQ1–5 (Fig. 1) were synthesized by means of an easy and flexible three-step route, making use of well-known Suzuki–Miyaura cross-coupling reactions as key steps. The two acceptor cores were prepared following a common strategy, shown in Scheme 1. Diamine 2 was synthesized in good yield by the reduction of commercially available 4,7-dibromo-2,1,3-benzothiadiazole (1) using a large excess of NaBH4 in EtOH.17 Compound 2 was used immediately, without purification, for the subsequent step involving a cyclization reaction using diketones 3 or 4, under slightly different conditions, to obtain quinoxalines 5 and 6, respectively. The latter were employed in the Suzuki–Miyaura cross-coupling reactions, whose conditions were individually optimized to enhance the yield of each product (Scheme 2). | ||
| Scheme 1 Synthesis of the quinoxaline cores. | ||
 | ||
| Scheme 2 Synthetic procedures for the preparation of DQ fluorophores. | ||
In particular, compounds DQ2–4 were synthesized by reacting quinoxalines 5 or 6 with the corresponding boronic acid/ester (7–9) in the presence of Pd(dppf)Cl2 as the catalyst and KF as the base under microwave (MW) heating. Compared to conventional heating, MW irradiation allowed shortening the reaction time (approx. 35 minutes at 70 °C) and reducing the number of side-products. A mixture of toluene and MeOH (6/1) was identified as the best medium to absorb microwaves efficiently. However, the MW irradiation conditions were surprisingly ineffective in the synthesis of DQ1 and DQ5. So, DQ1 was obtained under similar conditions to DQ2–4 (Scheme 2), but applying conventional heating for 18 h, while DQ5 was prepared using a synthetic method reported by Kang et al. for a different compound.20 This last reaction was run using Pd(PPh3)4 and Cs2CO3 as the catalyst and base, respectively, at a temperature of 100 °C for 4 hours.
3.2 Spectroscopic characterization in solution
| Compound | λ abs (nm) | ε × 104 (M−1 cm−1) | λ emi (nm) | SS (nm [eV]) | Φ f (%) |
|---|---|---|---|---|---|
| a Absolute quantum yield of solutions. | |||||
| DQ1 | 433 | 3.1 | 538 | 105 [0.56] | 74 |
| DQ2 | 457 | 3.3 | 567 | 110 [0.53] | 66 |
| DQ3 | 452 | 1.2 | 601 | 149 [0.68] | 46 |
| DQ4 | 398 | 1.2 | 512 | 117 [0.71] | 44 |
| DQ5 | 416 | 2.0 | 529 | 112 [0.62] | 67 |
 | ||
| Fig. 2 Normalized absorption spectra (solid lines) and normalized fluorescence emission spectra (dash dot lines) of DQ1–5 in toluene (0.01 mM) with λexc at 433, 457, 452, 398 and 416 nm, respectively. | ||
In addition, all the absorption spectra featured broad peaks, maintaining a constant shape at different concentrations (see Fig. S2–S6, ESI†).
The ground-state frontier molecular orbitals of the DQ series were calculated using Gaussian 16,40 Revision C.01 suite of programs, at the B3LYP41,42/6-31G* level of theory in toluene.
Solvent effects were included using the polarizable continuum model (PCM).43 The inspection of the frontier molecular orbitals (see Fig. S1, ESI†) suggests that in all cases, the light absorption in the visible region could involve an internal charge-transfer (ICT) mechanism, as already observed in DQ1 and similar molecules.21,44 This is in agreement with the spectroscopic results.
The fluorescence spectra showed an intense emission band between 512 and 600 nm (Fig. 2, dash dot lines), relatively well-matched with the external quantum efficiency of a Si-based PV module. No vibronic structure was observed, and the constant shape of the emission band at different concentrations proved the absence of dye aggregation in toluene for all DQs in the explored concentration range (Fig. S2–S6, ESI†). Furthermore, in all cases, large Stokes shifts (0.53–0.71 eV, corresponding to >100 nm) with limited overlap between the absorption and emission spectra were observed, leading to minimal re-absorption phenomena (Table 1).
 | ||
| Fig. 3 Transient absorption spectra (a–c) and evolution associated difference spectra (EADS) (d–f) of DQ1–3 (from left to right) in toluene solutions. | ||
 | ||
| Fig. 4 Transient absorption spectra (a and b) and evolution associated difference spectra (EADS) (c and d) of DQ4-5 (from left to right) in toluene solutions. | ||
2,3-Diphenyl-substituted compounds DQ1–3 showed a broad positive excited state absorption (ESA) band, covering almost all the probed spectral interval (Fig. 3a–c). DQ1 and DQ2 showed very similar spectra, indicating a minor influence of the alkyl chains on the excited state dynamics, with a small negative band centred at about 440 nm, corresponding to the ground-state bleaching, and a more intense broad positive one, featuring a double-peaked structure because of an evident overlap with the negative stimulated emission (SE) band. In the corresponding EADS panels (Fig. 3d and e), the first component (black line) of both samples presents a broad positive band peaked at 600 nm, living about 2 ps in case of DQ1 and 700 fs for DQ2.
In the following evolution (red lines in Fig. 3d and e), the ESA assumes a double-peaked shape, because of the increase in the positive signal in the 450–500 nm region and the superposition between the broad positive absorption band and the stimulated emission band (peaked at about 530 nm for DQ1 and 550 nm for DQ2). This spectral evolution can be assigned to a fast electronic relaxation of the molecule, as evidenced by a large red-shift of the stimulated emission signal.
According to the theoretical analysis (see Fig. S1, ESI†), the lowest excited state has a charge-transfer character. The positive band above 700 nm noticed in the second EADS could be interpreted in terms of the simultaneous localization of the negative charge on the quinoxaline unit and the positive charge on the triphenylamine donor, as evidenced in previous studies.45,46 After 14.4 ps for DQ1 (7.3 ps for DQ2), the red component evolves towards the blue EADS.
Although the spectral shape of the third EADS remains mostly unaffected, the stimulated emission signal partially recovers and red-shifts at about 30 nm reflecting a dynamic Stokes-shift due to solvent-induced excited state relaxation. On the same timescale, the band peaked at 600 nm narrows and slightly blue-shifts, as the effect of vibrational cooling occurring in the excited state. The narrowing at 600 nm is particularly evident for DQ2. Finally, the signal intensity slightly decreases on the following 448 ps timescale for DQ1 (540 ps for DQ2). For both samples, the final spectral component (green EADS) lives beyond the time interval of the measurement (1.5 ns).
The transient spectrum of DQ3 appears slightly different as compared to the previously described samples, reflecting the presence of the phenothiazine donor group (Fig. 3c and f), but the excited evolution can be interpreted in a similar way. Also in this case, after a fast electronic relaxation occurring in about 500 fs, a positive band peaked at about 500 nm is observed to increase, together with a broad absorption extending between 600 and >750 nm, and presenting a minimum at about 570 nm, due to the superposition with the stimulated emission band. The band peaked at 500 nm may reflect the localization of positive charge on the phenothiazine ring, while the signal in the red part of the spectrum (>600 nm) can be assigned to the localization of the negative charge on the quinoxaline core, thus confirming the formation of a charge-transfer state also in this case. The spectral dynamics observed on the 5 ps timescale reflects a dynamic Stokes shift of the emission band. The intensity of the overall transient spectrum slightly decreases on the following 212 ps timescale and the final spectral component (green line, Fig. 3f) once again lives longer than the investigated timescale.
The transient absorption spectra and EADS of 2,3-di-n-hexylquinoxalines DQ4 and DQ5 are reported in Fig. 4. For both samples, a negative band, peaked at about 500 nm, is assigned as the SE signal, and a broad positive ESA band is noticed in the spectra. Opposite to the previous series of compounds, here the spectral evolution observed within the investigated timescale is quite limited. Inspection of the EADS shows a progressive red-shift of the SE band, ascribed to a solvent-induced excited state relaxation, occurring within 10 ps for both samples (evolution from the black to the red component and from the red to the blue one, Fig. 4c and d). The intensity of the ESA band partially decreases in about 325 ps for DQ4 (about 430 ps for DQ5). Already in the second spectral component, a positive band at about 600 nm can be recognized, possibly related to the partial localization of the positive charge on the triphenylamine groups and therefore signalling the emergence of a charge-transfer state upon photoexcitation. Also for these samples, the final spectral component lives longer than the investigated timescale. Overall, the transient absorption spectroscopy measurements of all the investigated compounds confirm the charge-transfer characteristic of the lowest energy excited state, furthermore indicating that no intermediate dark states are involved in its decay, suggesting that its main relaxation channel is of radiative nature, a very beneficial feature for compounds to be employed as LSC emitters.
3.3 Spectroscopic characterization in polymer matrices
DQ luminophores were characterized in polymer matrices by dispersing them in transparent and totally amorphous PMMA and PCMA, obtaining films with a 25 μm ± 5 μm thickness, at different concentrations (0.2–2.2 wt%). All molecules visually showed good dispersion in the polymer matrices, which appeared homogenous and with negligible macroscopic phase separation at the film surface even at the highest fluorophore content (Fig. 5). | ||
| Fig. 5 PMMA and PCMA films doped with DQ1–5 at 1.4 wt% concentration under visible light illumination. | ||
 | ||
| Fig. 6 Normalized absorption spectra (solid lines) and normalized fluorescence emission spectra (dash dot lines) of DQ1–5 in PMMA (1.0 wt%) with λexc at 430, 454, 456, 400 and 415 nm, respectively. UV-vis absorption and emission spectra as a function of fluorophore concentration are reported in the ESI† (Fig. S7–S11). | ||
| Compound | PMMA | PCMA | ||||
|---|---|---|---|---|---|---|
| λ abs (nm) | λ emi (nm) | SS (nm [eV]) | λ abs (nm) | λ emi (nm) | SS (nm [eV]) | |
| DQ1 | 430 | 546 | 116 [0.61] | 432 | 538 | 106 [0.56] |
| DQ2 | 447 | 580 | 133 [0.64] | 459 | 569 | 110 [0.52] |
| DQ3 | 446 | 583 | 137 [0.65] | 449 | 566 | 117 [0.57] |
| DQ4 | 406 | 513 | 107 [0.64] | 404 | 493 | 89 [0.55] |
| DQ5 | 414 | 553 | 139 [0.75] | 419 | 519 | 100 [0.57] |
This is confirmed by the inspection of epifluorescence microscopy images, revealing the presence of only a few microsized fluorophore aggregates with an average size of about 15 μm (Fig. 7), characterized by brilliant emission. Such behaviour appears very promising in obtaining solar collectors with improved optical performances.
 | ||
| Fig. 7 Fluorescent microscopy images of 1.4 wt% DQ1–5/PMMA films. Scale bar = 100 μm. | ||
Conversely, DQ2/PMMA films showed Φf values of 30–40% within the entire range of concentrations (Fig. S8, ESI†), lower than those recorded in solutions. No beneficial effect was exerted by the polymer matrix in this case, which can be attributed to the emersion of a large fraction of phase-separated microscopic fluorophore aggregates (Fig. 7). We can hypothesize that, in the initial toluene solution used for film preparation, DQ2 molecules were well-dispersed. Upon the evaporation of the solvent, their local concentration increased, and thanks to the interaction of their long alkyl chains, they started to form small aggregates, which became progressively entrapped in the polymer matrix. This caused the size of the aggregates to increase, leading to the observed microscopic phase separation (Fig. 7).
The phase dispersion in PMMA also influenced the quantum efficiency of DQ3/PMMA and DQ4/PMMA films, since the values of 56% and 80% measured at the lowest fluorophore content (0.2 wt%) gradually collapsed with the increasing content to 33% and 58%, respectively (Fig. S9 and S10, ESI†). In these cases, the lower fluorophore compatibility with the PMMA matrix progressively gave rise to the emersion of micro-scale aggregates that, in turn, triggered the activation of the typical ACQ effect (Fig. 7).52,53
This phenomenon appeared much less severe in the case of DQ5/PMMA films, whose fluorescence quantum yield remained constant at 50–60% in the entire range of investigated concentrations (Fig. S11, ESI†).
 | ||
| Fig. 8 Normalized absorption spectra (solid lines) and normalized emission spectra (dash dot lines) of DQ1–5 in PCMA (1.0 wt%) with λexc at 430, 459, 447, 400, and 419 nm, respectively. UV-vis absorption and emission spectra as a function of fluorophore concentration are reported in the ESI† (Fig. S12–S16). | ||
The quantum yields of DQ1–3 fluorophores appeared generally maintained or increased in the PCMA matrix compared to PMMA. Accordingly, DQ1/PCMA films confirmed high values of Φf of around 96–98% (Fig. S12, ESI†), whereas a striking increase from 40% to 71% and from 33% to 53% was recorded moving from PMMA to PCMA for DQ2 (Fig. S13, ESI†) and DQ3 (Fig. S14, ESI†), respectively, at the highest fluorophore content. This behaviour suggests that a less polar environment increases the phase compatibility between the fluorophores and the polymer matrix, as indicated by the images obtained by fluorescence microscopy of compound DQ2 in PCMA films (Fig. 9, left). Clearly, a reduced number of smaller DQ2 aggregates were present in PCMA with respect to the more phase-separated DQ2/PMMA system (Fig. 7).
 | ||
| Fig. 9 Fluorescent microscopy images of a 1.4 wt% DQ2,5/PCMA film. Scale bar = 100 μm. | ||
Moving to DQ4 and DQ5 (Fig. S15 and S16, ESI†), the introduction of hexyl chains into their acceptor core resulted in a less evident quenching of fluorescence for concentrations higher than 1.4 wt% and in a less pronounced bathochromic shift of the emission maximum. Nevertheless, the alkyl characteristic of DQ4 and DQ5 affected divergently the Φf variations with the fluorophore content. While DQ4 showed a considerable increase of Φf with concentrations (i.e., from 65% at 1.0 wt% to 90% at 1.8 wt%), the quantum yield of DQ5 declined drastically from 80% at 0.2 wt% to around 53% at 1.8 wt%, i.e., comparable to data gathered from PMMA films at the same content. Φf declining in DQ5/PCMA films was attributed to the formation of phase-separated aggregates triggering the formation of micro-cracks over the surface of the PCMA film as revealed by fluorescence microscopy (Fig. 9, right). Such hypothesis seems to be confirmed by the emission spectra of DQ5/PCMA (Fig. S16, ESI†), where a second emission at a longer wavelength appeared for high concentrations in agreement with the formation of emissive aggregates. Overall, the introduction of alkyl chains into the backbone of the fluorophores seems to promote better dispersion and compatibility with the less polar PCMA matrix, as especially evident in the case of compounds DQ2 and DQ4. Notwithstanding this general observation, DQ5, featuring the highest number of alkyl chains of the series, diverges from such behaviour, requiring a more refined explanation.54,55 Indeed, it must be considered that DQ5, despite the flexibility of the n-hexyloxy substituents, is the largest molecule among those examined, clearly larger than DQ1, but also than DQ4. Since the relative motions of the polymer film constituents are restricted, unlike what happens in solutions, it is possible that increasing the concentration of DQ5 above a certain limit prevented the polymer chains to come sufficiently close due to steric reasons, decreasing the mechanical strength of the films. This caused the appearance of the observed micro-cracks, accompanied by a progressive phase-segregation of the emitter molecules, which was supported by the above-mentioned change in shape of the emission spectra (Fig. S16, ESI†).
Overall, DQ1 displayed the highest quantum yields in both polymer matrices. Notably, compared to the other compounds studied in this work, DQ1 does not present either any flexible alkyl substituent on its backbone or phenothiazine substituents, which can assume different conformations due to the bending of the tricyclic system. Therefore, compared to the other compounds, it should be less prone to the non-radiative energy losses resulting from vibrational relaxation of the excited state. This is supported by its relatively low SS value, as can be seen in Table 1, indicative of a lower degree of structural reorganization in the excited state. Finally, from the TAS studies reported in Fig. 3 and 4, it can also be seen that the initial spectral evolution for DQ1, assigned to a fast electronic relaxation of the molecule, appears slower than that of the other emitters seem to indicate similar rate constants for all compounds. The variation of Φf values with concentrations for all compounds both in the PMMA and PCMA matrices is reported in Fig. 10. All the collected results possibly suggest that even PCMA could experience compatibility issues at the highest fluorophore content of 1.8–2 wt%, which often requires maximizing the solar harvesting features of the LSC collectors. An accessible solution to overcome such an issue would be using copolymers made from methyl methacrylate and cyclohexyl methacrylate to take advantage of both the film stability induced by the former and the effective fluorophore dispersion allowed by the latter. Research efforts aimed at developing these new aspects are currently being pursued in our labs.
 | ||
| Fig. 10 Φ f values (%) of the PMMA (left) and PCMA (right) films tested at different fluorophore concentrations. | ||
The dynamic Stokes shift observed in solution is barely observed in the matrix, because of the lower vibrational freedom and more constrained geometry experienced by the molecules in such an environment. The intensity of the absorption signal decayed bi-exponentially in all DQ samples, with the excited state showing a relatively long lifetime of >1.5 ns. Overall, these measurements show that the excited state relaxation is dominated by the radiative channel also in the polymer, and that the variation in Φf observed when moving from the solution to the solid phase can mainly be associated with both the more restricted conformation adopted by the different molecules in the polymer and the degree of dispersibility of the various compounds in the matrix. To demonstrate the effect of the matrix, which can also induce a decreased emission efficiency in the case of suboptimal compatibility, Fig. 11 reports the comparison of the kinetic traces measured at the maximum of the excited state absorption band (640 nm) for DQ5 in toluene and in the two polymer matrices, PMMA and PCMA (1.0 wt%). It is evident that the fast decay component has a higher weight in the polymers compared to the solution, which affects the overall Φf observed in the different environments. This is most probably due to the lower conformational freedom experienced by the molecules in the polymer matrix, which reduces the degrees of structural rearrangement in the excited state, as also evidenced by the reduced dynamic Stokes shift of the stimulated emission band (see Fig. S17 and S18, ESI†). To complete the characterization of the films, fluorescence lifetime measurements have also been performed for all compounds dispersed in PMMA at 1.0 wt% concentration. The fluorescence decay rates are similar for all compounds, with lifetimes in the 4–8 ns time range, quite typical of organic fluorophores. Fluorescence lifetimes are slightly shorter for DQ4 and DQ5 if compared to the other molecules. We found that the best fitting results (with a χ2 value of about 1) were obtained for a bi-exponential decay: this could reflect either the presence of small inhomogeneities in the chemical environment surrounding the fluorophore molecules or the formation of oligomers with slightly different decay rates. The fluorescence decay curves and lifetimes are reported in the ESI† (Fig. S19 and Table S1).

3.4 LSC performances
Before determining the photon efficiencies of the prepared thin-film LSCs, auto-absorption in the active film was analysed. The measurements were performed by irradiating the surface of the selected devices with a laser (λexc = 405 nm, 1 mW cm−2) and the edge-emitted power was recorded as a function of the optical pathlength distance between the excitation spot and the collecting edge (Fig. S21, ESI†). LSCs based on LR305, DQ1 and DQ4 were evaluated. LR305 was selected to be as the state-of-the-art fluorophore in LSC, while DQ1 and DQ4 as fluorophores with top and intermediate fluorescence quantum efficiencies, as reported in Figure 10. Fluorophore content was selected in terms of the maximum absolute Φf, i.e., 1.4 wt% for DQ1 and DQ4. As far as LR305 is concerned, the concentration of 1.0 wt% was selected in terms of the maximum LSC performance as determined in a previous study.8Notably, as the optical pathlength distance increased, the edge-emitted power was found to progressively decrease in all cases (Fig. S21a, ESI†). This phenomenon appeared more evident for the LSC based on LR305, possibly due to the lowest SS of 36 nm27 compared to 106 nm for DQ1 and 89 nm for DQ4. Moreover, by comparing the collected emission spectra, an evident red-shift of about 20 nm in the emission maximum was observed in the case of LR305, thus supporting emission losses by the occurrence of reabsorption phenomena (Fig. S21b, ESI†).
The optical performances of LSC based on LR305, DQ1 and DQ4 were then evaluated in terms of the internal and external photon efficiency (ηint and ηext, respectively) (Table 3). ηint and ηext were obtained according to the recently published protocols35 and calculated from eqn (S1) and (S2) (ESI†). In detail, ηint represents the ratio of photons collected at the LSC edges to the number of absorbed photons by the embedded fluorophores, and it is a key parameter to determine all lightguide losses. ηext is the ratio of photons collected at the LSC edges to the number of total incident photons, and it also provides information about the solar harvesting features of the device.
| Chromophore | Matrix | Concentration (wt%) | Φ f (%) | η int (%) | η ext (%) |
|---|---|---|---|---|---|
| LR305 | PMMA | 1.0 | 59.3 | 16.6 | 4.7 |
| DQ1 | PMMA | 1.4 | 96.7 | 42.9 | 6.2 |
| DQ4 | PMMA | 1.4 | 64.4 | 27.9 | 3.6 |
It is worth noting that for both the ηint and ηext parameters, DQ1 was the best fluorophore in providing the highest LSC efficiencies with values of 42.9% and 6.2%, respectively. Surprisingly, the ηint of the LSC based on LR305 resulted the lowest (16.6%). According to the ηint determination, the broadest absorption band of LR305 adversely affects this parameter in largest extent.
Conversely, ηext shows a different trend of performances, i.e., with DQ1 as the best fluorophore followed by the state-of-the-art LR305. DQ4, notwithstanding the higher but similar Φf values (64.4 against 59.3%), possibly suffers from inferior solar harvesting characteristics with respect to the perylene bisimide chromophore.
Moreover, the photon efficiencies obtained here appear to be higher than those reported in a recent study.56 Overall, DQ1 turns out to be a very promising fluorophore for employment in LSCs.
However, for LSC practical applications, DQ1/PMMA stability must be assessed through performance measurements over time. Notably, DQ1/PMMA films did not show any noticeable variation in their visible appearance following their preparation, and with optical performances maintained over months. Also, the thermal stability of the fluorophore was evaluated through thermogravimetric analysis showing that the samples maintained at least 95% of their weight up to 300 °C (Fig. S25, ESI†). The photostability of DQ1/PMMA LSCs was then preliminarily investigated by continuously irradiating a 0.25 cm2 spot of the film at 430 nm with a 450 W Xe arc lamp under aerobic conditions. The film samples were placed in the spectrofluorometer chamber and fixed in the solid-state holder, thus receiving irradiances of about 50 μW cm−2. Notably, DQ1 retained 97.8% of its emission after two hours of continuous excitation compared to the 98.8% of LR305 (Fig. S26, ESI†), thus suggesting a potentially sufficient photostability. Further experiments will be carried out in order to definitely assess this behaviour.
4. Conclusions
In conclusion, we have reported the preparation of thin-film LSCs having optical properties superior to those of the state-of-the-art solar collectors. This result was obtained by replacing the reference fluorophore LR305 in PMMA and PCMA films with a series of new high quantum yield, donor–acceptor–donor (D–A–D) photostable fluorophores (DQ1–5) characterized by a central quinoxalinic acceptor core and triarylamines or phenothiazine as donor groups. Thanks to their structures, all fluorophores presented excited states characterized by a high degree of intramolecular charge transfer, which was confirmed by both DFT computational analysis and transient absorption spectroscopy studies. Notably, they displayed intense emissions between 550 and 600 nm with Stokes shifts at about 80–120 nm that helped in minimizing the auto-absorption effects when dispersed in polymers at high content. Furthermore, decoration with long alkyl chains at different parts of the molecular backbone allowed tuning their dispersibility in polymer films. It is worth noting that DQ1 in PMMA provided outstanding LSC ηint and ηext values of 42.9 and 6.2%, respectively. This result was addressed to the effective combination between high phase compatibility and excellent quantum yield, which for DQ1 reached remarkable values of >95% in both PMMA and PCMA matrices.In view of these performances, our findings provide new advances for the widespread distribution of the LSC/PV system in the integrated photovoltaics, which is the backbone of the zero-energy building European target.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank “Regione Toscana” (COLOURS project, POR FESR 2014-2020, grant no. 3553.04032020.158000411) and “Fondazione Cassa di Risparmio di Firenze” (SUNNYSIDE project, grant no. 2020.1408) for financial support. A. S. and C. C. acknowledge MIUR Grant – Department of Excellence 2018–2022. Computational resources were provided by hpc@dbcf (http://molsys.dbcf.unisi.it/hpc). MDD and MT acknowledge European Union's Horizon 2020 research and innovation program under grant agreement no. 871124 Laserlab-Europe. Dr Giulia Tuci (CNR-ICCOM) and Mr Francesco Ventura are kindly acknowledged for their help in TGA experiments and fluorescence microscopy, respectively. The Mass Spectrometry Center (CISM) of the University of Florence is kindly acknowledged for HRMS analyses.Notes and references
- N. Armaroli and V. Balzani, Chem. – Eur. J., 2016, 22, 32–57 CrossRef CAS PubMed.
- G. M. Wilson, M. Al-Jassim, W. K. Metzger, S. W. Glunz, P. Verlinden, G. Xiong, L. M. Mansfield, B. J. Stanbery, K. Zhu, Y. Yan, J. J. Berry, A. J. Ptak, F. Dimroth, B. M. Kayes, A. C. Tamboli, R. Peibst, K. Catchpole, M. O. Reese, C. S. Klinga, P. Denholm, M. Morjaria, M. G. Deceglie, J. M. Freeman, M. A. Mikofski, D. C. Jordan, G. TamizhMani and D. B. Sulas-Kern, J. Phys. D: Appl. Phys., 2020, 53, 493001 CrossRef CAS.
- J. S. Batchelder, A. H. Zewai and T. Cole, Appl. Opt., 1979, 18, 3090 CrossRef CAS PubMed.
- W. H. Weber and J. Lambe, Appl. Opt., 1976, 15, 2299 CrossRef CAS PubMed.
- J. Roncali, Adv. Energy Mater., 2020, 10, 2001907 CrossRef CAS.
- F. Meinardi, F. Bruni and S. Brovelli, Nat. Rev. Mater., 2017, 2, 17072 CrossRef CAS.
- G. Griffini, Front. Mater., 2019, 6, 29 CrossRef.
- F. J. Ostos, G. Iasilli, M. Carlotti and A. Pucci, Polymers, 2020, 12, 2898 CrossRef CAS PubMed.
- T. M. Clarke, K. C. Gordon, W. M. Kwok, D. L. Phillips and D. L. Officer, J. Phys. Chem. A, 2006, 110, 7696–7702 CrossRef CAS PubMed.
- C. Papucci, A. Dessì, C. Coppola, A. Sinicropi, G. Santi, M. di Donato, M. Taddei, P. Foggi, L. Zani, G. Reginato, A. Pucci, M. Calamante and A. Mordini, Dyes Pigm., 2021, 188, 109207 CrossRef CAS.
- H. J. Song, D. H. Kim, E. J. Lee and D. K. Moon, J. Mater. Chem. A, 2013, 1, 6010–6020 RSC.
- B. Li, M. Zhao and F. Zhang, ACS Mater. Lett., 2020, 2, 905–917 CrossRef CAS.
- P. Y. Gu, J. Zhang, G. Long, Z. Wang and Q. Zhang, J. Mater. Chem. C, 2016, 4, 3809–3814 RSC.
- L. Lyu, R. Su, S. Y. Al-Qaradawi, K. A. Al-Saad and A. El-Shafei, Dyes Pigm., 2019, 171, 107683 CrossRef CAS.
- W. Liu, Z. Liu, J. Yan, L. Wang, H. Xu, H. Wang and B. Zhao, Dyes Pigm., 2021, 191, 109305 CrossRef CAS.
- D. Gedefaw, M. Prosa, M. Bolognesi, M. Seri and M. R. Andersson, Adv. Energy Mater., 2017, 7, 1700575 CrossRef.
- M. L. Jiang, J. Wen, Z. Chen, W. Tsai, T. Lin, T. J. Chow and Y. J. Chang, ChemSusChem, 2019, 12, 3654–3665 CrossRef CAS PubMed.
- B. D. Lindner, Y. Zhang, S. Höfle, N. Berger, C. Teusch, M. Jesper, K. I. Hardcastle, X. Qian, U. Lemmer, A. Colsmann, U. H. F. Bunz and M. Hamburger, J. Mater. Chem. C, 2013, 1, 5718–5724 RSC.
- H. Zhang, Y. Wu, W. Zhang, E. Li, C. Shen, H. Jiang, H. Tian and W. H. Zhu, Chem. Sci., 2018, 9, 5919–5928 RSC.
- H. J. Son, W. S. Han, D. H. Yoo, K. T. Min, S. N. Kwon, J. Ko and S. O. Kang, J. Org. Chem., 2009, 74, 3175–3178 CrossRef CAS PubMed.
- Y. Chen, Y. Ling, L. Ding, C. Xiang and G. Zhou, J. Mater. Chem. C, 2016, 4, 8496–8505 RSC.
- T. A. Geervliet, I. Gavrila, G. Iasilli, F. Picchioni and A. Pucci, Chem. – Asian J., 2019, 14, 877–883 CrossRef CAS PubMed.
- R. Mori, G. Iasilli, M. Lessi, A. B. Muñoz-García, M. Pavone, F. Bellina and A. Pucci, Polym. Chem., 2018, 9, 1168–1177 RSC.
- J. Lucarelli, M. Lessi, C. Manzini, P. Minei, F. Bellina and A. Pucci, Dyes Pigm., 2016, 135, 154–162 CrossRef CAS.
- G. Iasilli, R. Francischello, P. Lova, S. Silvano, A. Surace, G. Pesce, M. Alloisio, M. Patrini, M. Shimizu, D. Comoretto and A. Pucci, Mater. Chem. Front., 2019, 3, 429–436 RSC.
- F. De Nisi, R. Francischello, A. Battisti, A. Panniello, E. Fanizza, M. Striccoli, X. Gu, N. L. C. Leung, B. Z. Tang and A. Pucci, Mater. Chem. Front., 2017, 1, 1406–1412 RSC.
- M. Carlotti, E. Fanizza, A. Panniello and A. Pucci, Sol. Energy, 2015, 119, 452–460 CrossRef CAS.
- G. Albano, T. Colli, T. Biver, L. A. Aronica and A. Pucci, Dyes Pigm., 2020, 178, 108368 CrossRef CAS.
- G. Albano, T. Colli, L. Nucci, R. Charaf, T. Biver, A. Pucci and L. A. Aronica, Dyes Pigm., 2020, 174, 108100 CrossRef CAS.
- P. L. Gentili, M. Mugnai, L. Bussotti, R. Righini, P. Foggi, S. Cicchi, G. Ghini, S. Viviani and A. Brandi, J. Photochem. Photobiol., A, 2007, 187, 209–221 CrossRef CAS.
- M. Di Donato, A. Iagatti, A. Lapini, P. Foggi, S. Cicchi, L. Lascialfari, S. Fedeli, S. Caprasecca and B. Mennucci, J. Phys. Chem. C, 2014, 118, 23476–23486 CrossRef CAS.
- E. R. Henry, Biophys. J., 1997, 72, 652–673 CrossRef CAS PubMed.
- I. H. M. Van Stokkum, D. S. Larsen and R. Van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- M. G. Debije, R. C. Evans and G. Griffini, Energy Environ. Sci., 2021, 14, 293–301 RSC.
- C. Yang, D. Liu and R. R. Lunt, Joule, 2019, 3, 2871–2876 CrossRef.
- B. Mátravölgyi, T. Hergert, A. Thurner, B. Varga, N. Sangiorgi, R. Bendoni, L. Zani, G. Reginato, M. Calamante, A. Sinicropi, A. Sanson, F. Faigl and A. Mordini, Eur. J. Org. Chem., 2017, 1843–1854 CrossRef.
- G. Qian, B. Dai, M. Luo, D. Yu, J. Zhan, Z. Zhang, M. Dongge and Z. Y. Wang, Chem. Mater., 2008, 20, 6208–6216 CrossRef CAS.
- A. Dessì, M. Calamante, A. Mordini, M. Peruzzini, A. Sinicropi, R. Basosi, F. Fabrizi De Biani, M. Taddei, D. Colonna, A. Di Carlo, G. Reginato and L. Zani, RSC Adv., 2015, 5, 32657–32668 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, GAUSSIAN16: Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- X. Lu, S. Fan, J. Wu, X. Jia, Z.-S. Wang and G. Zhou, J. Org. Chem., 2014, 79, 6480–6489 CrossRef CAS PubMed.
- K. Lušpai, A. Staško, V. Lukeš, D. Dvoranová, Z. Barbieriková, M. Bella, V. Milata, P. Rapta and V. Brezová, J. Solid State Electrochem., 2015, 19, 113–122 CrossRef.
- K. Y. Chiu, T. X. Su, J. H. Li, T. H. Lin, G. S. Liou and S. H. Cheng, J. Electroanal. Chem., 2005, 575, 95–101 CrossRef CAS.
- F. Donati, A. Pucci, C. Cappelli, B. Mennucci and G. Ruggeri, J. Phys. Chem. B, 2008, 112, 3668–3679 CrossRef CAS PubMed.
- J. M. Drake, M. L. Lesiecki, J. Sansregret and W. R. L. Thomas, Appl. Opt., 1982, 21, 2945 CrossRef CAS PubMed.
- M. G. Debije and P. P. C. Verbunt, Adv. Energy Mater., 2012, 2, 12–35 CrossRef CAS.
- J. Donovalová, M. Cigáň, H. Stankovičová, J. Gašpar, M. Danko, A. Gáplovský and P. Hrdlovič, Molecules, 2012, 17, 3259–3276 CrossRef PubMed.
- M. Kaholek and P. Hrdlovič, J. Photochem. Photobiol., A, 1997, 108, 283–288 CrossRef.
- F. Gianfaldoni, F. De Nisi, G. Iasilli, A. Panniello, E. Fanizza, M. Striccoli, D. Ryuse, M. Shimizu, T. Biver and A. Pucci, RSC Adv., 2017, 7, 37302–37309 RSC.
- C. Papucci, T. A. Geervliet, D. Franchi, O. Bettucci, A. Mordini, G. Reginato, F. Picchioni, A. Pucci, M. Calamante and L. Zani, Eur. J. Org. Chem., 2018, 2657–2666 CrossRef CAS.
- A. Pucci, N. Tirelli, G. Ruggeri and F. Ciardelli, Macromol. Chem. Phys., 2005, 206, 102–111 CrossRef CAS.
- N. Tirelli, S. Amabile, C. Cellai, A. Pucci, L. Regoli, G. Ruggeri and F. Ciardelli, Macromolecules, 2001, 34, 2129–2137 CrossRef CAS.
- F. Corsini, A. Nitti, E. Tatsi, G. Mattioli, C. Botta, D. Pasini and G. Griffini, Adv. Opt. Mater., 2021, 9, 2100182 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc02923a |
| This journal is © The Royal Society of Chemistry 2021 |