
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pressure-induced Jahn–Teller switch in the homoleptic hybrid perovskite [(CH3)2NH2]Cu(HCOO)3: orbital reordering by unconventional degrees of freedom†
Rebecca
Scatena
 *a,
Michał
Andrzejewski
b,
Roger D.
Johnson
c and
Piero
Macchi
d
*a,
Michał
Andrzejewski
b,
Roger D.
Johnson
c and
Piero
Macchi
d
aDepartment of Physics, Clarendon Laboratory, University of Oxford, Parks Road, Oxford OX1 3PU, UK. E-mail: rebecca.scatena@physics.ox.ac.uk
bDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, Bern CH-3012, Switzerland
cDepartment of Physics and Astronomy, University College London, Gower Street, London WC1 6BT, UK
dDepartment of Chemistry, Materials, and Chemical Engineering, Polytechnic of Milan, Via Mancinelli 7, Milan 20131, Italy
First published on 9th June 2021
Abstract
Through in situ, high-pressure X-ray diffraction experiments we have shown that the homoleptic perovskite-like coordination polymer [(CH3)2NH2]Cu(HCOO)3 undergoes a pressure-induced orbital reordering phase transition above 5.20 GPa. This transition is distinct from previously reported Jahn–Teller switching in coordination polymers, which required at least two different ligands that crystallize in a reverse spectrochemical series. We show that the orbital reordering phase transition in [(CH3)2NH2]Cu(HCOO)3 is instead primarily driven by unconventional octahedral tilts and shifts in the framework, and/or a reconfiguration of A-site cation ordering. These structural instabilities are unique to the coordination polymer perovskites, and may form the basis for undiscovered orbital reorientation phenomena in this broad family of materials.
Introduction
Transition metal perovskites of general chemical formula ABX3 are a compositionally rich family of materials in which a strong interplay between charge, orbital and magnetic degrees of freedom is manifest. In some cases, this interplay leads to technologically significant materials properties such as superconductivity and colossal magneto-resistance.1–3 Orbital ordering in particular affords an efficient mechanism for coupling the transition metal's electronic degrees of freedom to the crystal lattice via the Jahn–Teller (JT) effect,4 and hence is key to our understanding, development, and exploitation of structure–property relationships in these materials. For example, in the case of magnetic order the nature and strength of magnetic superexchange interactions between metal ions is regulated by the ordered spatial orientation of d-orbitals,5–7 and in the case of charge ordering, the local electronic configuration of cations is directly coupled to orbital occupation and the respective orbital order. Hence, the ability to tune and control orbital order may provide a handle on multiple interacting electronic degrees of freedom. Given the strong coupling between orbital order and the crystal structure it is natural to attempt control of orbital order by applied hydrostatic pressure or strain–with hydrostatic pressure being advantageous with regards to mechanical stability and its applicability to polycrystalline ceramics that are commonplace in technology. In this context, the role of strain and pressure in tuning JT physics has been discussed in theory.8,9A canonical example of structure–property relationships established through orbital ordering is found in the fully inorganic transition metal perovskite KCuF3. In this compound the orbital ordering scheme can be appreciated by the alternating position of JT axes of the Cu2+ ions, which directly determines the A-type magnetic structure of ferromagnetic (FM) ab-plane layers coupled antiferromagnetically (AFM) along the c-axis,1 as predicted by the Goodenough–Kanamori–Anderson (GKA) rules.5–7 Neutron diffraction experiments performed on KCuF3 under high pressure showed that in the limit of quasi-hydrostatic compression from ∼0 to 8 GPa, all Cu–F bond lengths progressively shorten.10 However, the JT elongated Cu–F bonds are compressed by almost 0.18(1) Å, whereas the equatorial Cu–F bonds only by 0.04(1) Å. This anisotropic response of the Cu–F bonds to hydrostatic compression leads to a considerable suppression of the JT distortion, possibly restoring the degeneracy between eg orbitals at higher pressure. Whilst this experiment clearly demonstrated tuning of the orbital state in KCuF3, it did not demonstrate any dramatic reconfiguration of the orbital order.
In this article, we have identified a homoleptic coordination polymer structural analogue of KCuF3; dimethylammonium copper formate, [(CH3)2NH2]Cu(HCOO)3 (albeit with additional conventional octahedral tilts and rotations). Here, the A site K+ ions are replaced by dimethylammonium counter cations [(CH3)2NH2]+, and the X site F− ions are replaced by formate anions (HCOO)−. Both substitutions are homovalent, hence preserving the Jahn–Teller active 2+ oxidation state of copper at the B sites. At ambient pressure [(CH3)2NH2]Cu(HCOO)3 adopts a monoclinic I2/a structure (here denoted α-phase), with B site CuII cations bridged by formate anions in anti–anti fashion along all three directions, forming centrosymmetric axially elongated CuO6 octahedra arranged in a conventional (a−a−c−) tilt pattern (in Glazer notation).11 The axial (JT elongated) bonds alternate between subsequent CuII ions along the a + b and a − b crystallographic directions, whereas only equatorial bonds are pointing along c (Fig. 1). Therefore, at ambient pressure [(CH3)2NH2]Cu(HCOO)3 has the same orbital order as found in KCuF3, and consequently the two systems support the same A-type magnetic structure at low temperature.12 However, coordination polymers that adopt a perovskite-like framework have additional structural degrees of freedom associated with A-site ordering and unconventional octahedral tilts and columnar shifts.13 Based on these key differences, one might anticipate significantly different behaviour under applied hydrostatic pressure.
 | ||
| Fig. 1 Crystal structure of the α-phase at ambient pressure and γ-phase at 9.1 GPa. The equatorial planes are drawn in blue and red. JT axis are shown as yellow sticks. | ||
Experimental
High-pressure crystallography
Single crystals of [(CH3)2NH2]Cu(HCOO)3 were loaded in a Merrill–Bassett diamond-anvil cell (DAC)14 (Fig. S1 ESI†). A mixture of MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH (4:1 volume ratio) was used as pressure-transmitting medium.15 The pressure inside the DAC was calibrated using a ruby fluorescence method.16,17 The experiment was reaped on two crystals at room temperature. Single-crystal diffraction experiments for loading A were carried out in house on an Oxford Diffraction SuperNova area-detector diffractometer employing mirror optics, monochromated, and Al filtered18 microsource Mo Kα radiation (λ = 0.71073 Å). Single-crystal diffraction experiments for loading B were performed at the X04SA Material Science beamline of Swiss Light Source (Paul Scherrer Institute, Switzerland) with a Pilatus 6M detector (λ = 0.49647 Å).19 The crystal was gradually compressed up to about 9 GPa. Data collection and reduction were performed by using CrysAlisPro software.20 As the hydrostatic limit of the mixture was reached, the pressure inside the DAC was decreased to 7 GPa. Two pressure points, before and after the transition, were remeasured using synchrotron radiation to determine more accurate atomic positions, at 4.75 and 8.3 GPa, respectively. Crystal structures were solved by using SHELXT21 and the spherical independent atom model (IAM) was refined with SHELXL,22 as implemented in Olex2 interface.23 Selected crystallographic data and refinement details are reported in Tables S1 and S2 (ESI†).
:EtOH (4:1 volume ratio) was used as pressure-transmitting medium.15 The pressure inside the DAC was calibrated using a ruby fluorescence method.16,17 The experiment was reaped on two crystals at room temperature. Single-crystal diffraction experiments for loading A were carried out in house on an Oxford Diffraction SuperNova area-detector diffractometer employing mirror optics, monochromated, and Al filtered18 microsource Mo Kα radiation (λ = 0.71073 Å). Single-crystal diffraction experiments for loading B were performed at the X04SA Material Science beamline of Swiss Light Source (Paul Scherrer Institute, Switzerland) with a Pilatus 6M detector (λ = 0.49647 Å).19 The crystal was gradually compressed up to about 9 GPa. Data collection and reduction were performed by using CrysAlisPro software.20 As the hydrostatic limit of the mixture was reached, the pressure inside the DAC was decreased to 7 GPa. Two pressure points, before and after the transition, were remeasured using synchrotron radiation to determine more accurate atomic positions, at 4.75 and 8.3 GPa, respectively. Crystal structures were solved by using SHELXT21 and the spherical independent atom model (IAM) was refined with SHELXL,22 as implemented in Olex2 interface.23 Selected crystallographic data and refinement details are reported in Tables S1 and S2 (ESI†).
Mode decomposition analysis
The tetragonal P4/mmm [a = 6.2514 Å, c = 5.7444 Å] hypothetical high-symmetry parent cell was used to derive the irreducible representation constituting the low-symmetry structures (Table S3 ESI†). A structural CIF file of the parent cell with space-group symmetry, cell parameters and the positions of the atoms excluding the hydrogen atoms was imported in ISODISTORT24 to derive, with “method 4”, the mode decomposition of the distorted structures i.e. the monoclinic I2/a α-phase and triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) γ-phase. The basis (1,−1,0)(1,1,0)(0,0,2) was applied to transform between parent and distorted cells, and obtain the lattice parameters of the undistorted supercell: a = 8.84082, b = 8.84082, c = 11.48882, alpha = 90.0, beta = 90.0, gamma = 90.0 used in the definition of the modes. The PCR file produced by ISODISTORT includes the description of the modes through the polarization vectors of the displacive symmetry modes for each atom and the refinable amplitudes of the displacive symmetry modes. The refinement of amplitudes and isotropic thermal parameters against the experimental structure factor was carried out using Fullprof.25 The refined mode amplitudes were converted into absolute displacements in Å through normalization factor and the polarization vector direction (Tables S4 and S5 ESI†). The irreducible representations derived for the tetragonal to monoclinic and tetragonal to triclinic distortions are shown, respectively, in Fig. S5 and S6 (ESI†).
γ-phase. The basis (1,−1,0)(1,1,0)(0,0,2) was applied to transform between parent and distorted cells, and obtain the lattice parameters of the undistorted supercell: a = 8.84082, b = 8.84082, c = 11.48882, alpha = 90.0, beta = 90.0, gamma = 90.0 used in the definition of the modes. The PCR file produced by ISODISTORT includes the description of the modes through the polarization vectors of the displacive symmetry modes for each atom and the refinable amplitudes of the displacive symmetry modes. The refinement of amplitudes and isotropic thermal parameters against the experimental structure factor was carried out using Fullprof.25 The refined mode amplitudes were converted into absolute displacements in Å through normalization factor and the polarization vector direction (Tables S4 and S5 ESI†). The irreducible representations derived for the tetragonal to monoclinic and tetragonal to triclinic distortions are shown, respectively, in Fig. S5 and S6 (ESI†).
Results and discussion
We performed high-pressure single-crystal X-ray diffraction measurements using diamond anvil cells loaded with a single crystal sample of [(CH3)2NH2]Cu(HCOO)3 and a mixed MeOH:EtOH pressure transmitting medium. The experiments were carried out in the range 0–9 GPa using both laboratory-based Mo Kα radiation and synchrotron radiation (λ = 0.49647 Å). The α-phase was found to be stable up to 5.20 GPa (Table 1). Throughout this low pressure phase the Cu–O coordinative bonds were found to be the most compressible bonds in the structure, and by 5.20 GPa the axial and equatorial bonds were shortened by 0.207(13) Å and 0.036(8) Å, respectively–consistent with the pressure induced anisotropic octahedral compression found in KCuF3.10
| P/GPa | 0.0001 | 1.0 | 2.23 | 3.40 | 4.75 | 5.20 | 7.15 | 8.3 | 9.1 |
|---|---|---|---|---|---|---|---|---|---|
| SG | I2/a | I2/a | I2/a | I2/a | I2/a | I2/a |
P |
P |
P |
| a/Å | 8.8330(4) | 8.6895(13) | 8.5019(14) | 8.323(3) | 8.2878(9) | 8.086(4) | 7.2638(16) | 7.2432(13) | 7.2332(8) |
| b/Å | 8.7093(4) | 8.5925(10) | 8.5043(10) | 8.4733(14) | 8.4645(6) | 8.466(2) | 8.5726(15) | 8.5444(11) | 8.5452(10) |
| c/Å | 11.4145(5) | 11.3323(4) | 11.2256(4) | 11.1183(7) | 11.0942(3) | 10.9541(9) | 11.2929(17) | 11.2169(19) | 11.085(4) |
| α/° | 90 | 90 | 90 | 90 | 90 | 90 | 92.384(13) | 92.441(13) | 92.908(17) |
| β/° | 96.224(4) | 95.623(7) | 95.055(7) | 94.553(12) | 94.521(4) | 94.215(17) | 101.797(16) | 102.055(15) | 102.278(18) |
| γ/° | 90 | 90 | 90 | 90 | 90 | 90 | 91.352(16) | 91.298(13) | 91.268(10) |
| V/Å3 | 872.93(7) | 842.05(16) | 808.48(17) | 781.7(3) | 775.86(10) | 747.8(4) | 687.4(2) | 677.92(19) | 668.2(3) |
Analysis of diffraction data collected at 7.15 GPa showed a dramatic departure of the α and γ angles from 90° and a breaking of the I-centering translational symmetry through emergence of reflection intensities with h + k + l = 2n + 1 where n is an integer (Fig. S2 ESI†). Together, these observations revealed a structural phase transition in the region 5.20 → 7.15 GPa from monoclinic I2/a to a triclinic P supercell (Table 1), accompanied by a significant volume contraction as shown in Fig. 2a. We label this high pressure phase the γ-phase, which we note is metrically different from the P phase of [(CH3)2NH2]Cu(HCOO)3 previously discovered at 5.96 GPa by Collings and co-workers (the β-phase).26
 | ||
| Fig. 2 (a) Volume and (b) dimensions of coordination octahedra (O–O length) as a function of pressure. JT axes are shown as dotted sticks. | ||
Structural refinements against the 7.15 GPa data showed that the axial (JT elongated) Cu–O bonds initially along the a + b direction in the α-phase switched to lie along the c-axis in the γ-phase, while axial bonds initially along the a − b direction maintained their orientation (Fig. 1). This dramatic structural switch preserved anti–anti bonding of formate along the a + b direction (now involving only equatorial Cu–O bonds), but is accompanied by syn–anti bonding of formate in all other directions (Fig. 1 and 2b). Importantly, this directional switch of the JT axes can be understood in terms of a reordering of the CuII d-orbitals depicted by the equatorial planes drawn in Fig. 1 (see also Fig. S3 and S4 in ESI†). The GKA rules have been shown to correctly predict the ambient-pressure A-type magnetic ground state of [(CH3)2NH2]Cu(HCOO)3,12 in which CuII ions form FM layers extending in the ab plane, which stack antiferromagnetically along c. If we apply the same GKA rules for formate-mediated exchange (as outlined in ref. 12) to the high-pressure γ-phase described above, one predicts strong AFM superexchange pathways along a + b, and weak FM superexchange pathways along c and a − b; i.e. a different A-type magnetic structure in which the AFM stacking direction has switched from being along c in the α-phase to a + b in the γ-phase.
Pressure induced orbital reordering due to a JT switch has been observed in CrII and CuII Tutton salts27,28 and in a Mn12 single-molecule magnet cluster.29 Such transitions have also been found in a relatively small number of coordination polymers composed of at least two different ligand species that disobey the spectrochemical series in the low-pressure phase, hence enabling the different ligand strengths to act as the driving force behind the phase transition.30–33 Interestingly, in these coordination polymers a dramatic change in the magnetic structure was caused by the orbital reordering,30,32,34–36 in analogy with our predictions for [(CH3)2NH2]Cu(HCOO)3. To the best of our knowledge, pressure induced orbital reordering due to a JT switch has never been observed in a homoleptic coordination polymer. Hence, this observation points towards an altogether novel mechanism for orbital reordering that may apply to a much wider family of materials.
To untangle the underlying structural mechanism responsible for orbital reordering in [(CH3)2NH2]Cu(HCOO)3 we refined the crystal structure in terms of symmetry adapted displacive modes defined with respect to a hypothetical high-symmetry parent common to both α- and γ-phases. The adopted P4/mmm parent allows decomposing structural distortions relevant to this class of materials (e.g. JT distortion and octahedra tilts) into independent symmetry adapted modes. Moreover, compared to the standard choice Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, P4/mmm symmetry has the advantage of describing all possible structural distortion, including those pertinent to [(CH3)2NH2]+ cations, exclusively in terms of displacive modes without having to include occupational modes.24 The active distortion modes of all relevant Wyckoff sites were identified by decomposing the experimental α- and γ-phases using ISODISTORT,24 and classified according to the irreducible representation by which they transformed. The α-phase decomposes into modes of three Γ-point irreducible representations (Γ1+, Γ4+, and Γ5+) and modes of three A-point irreducible representations (A2+, A3+, and A5+). The largest distortions of the {Cu(HCOO)3−}n perovskite framework were associated with the (a−a−c−) octahedral tilts, which transform by A3+ and A5+ (Fig. 3a). These modes alone establish the I2/a α-phase symmetry and as such they can be considered primary modes with respect to our hypothetical parent. The Jahn–Teller distortions transform by A2+, and can be considered secondary modes–that is to say they are compatible with the crystal symmetry established by the octahedral tilts.
m, P4/mmm symmetry has the advantage of describing all possible structural distortion, including those pertinent to [(CH3)2NH2]+ cations, exclusively in terms of displacive modes without having to include occupational modes.24 The active distortion modes of all relevant Wyckoff sites were identified by decomposing the experimental α- and γ-phases using ISODISTORT,24 and classified according to the irreducible representation by which they transformed. The α-phase decomposes into modes of three Γ-point irreducible representations (Γ1+, Γ4+, and Γ5+) and modes of three A-point irreducible representations (A2+, A3+, and A5+). The largest distortions of the {Cu(HCOO)3−}n perovskite framework were associated with the (a−a−c−) octahedral tilts, which transform by A3+ and A5+ (Fig. 3a). These modes alone establish the I2/a α-phase symmetry and as such they can be considered primary modes with respect to our hypothetical parent. The Jahn–Teller distortions transform by A2+, and can be considered secondary modes–that is to say they are compatible with the crystal symmetry established by the octahedral tilts.
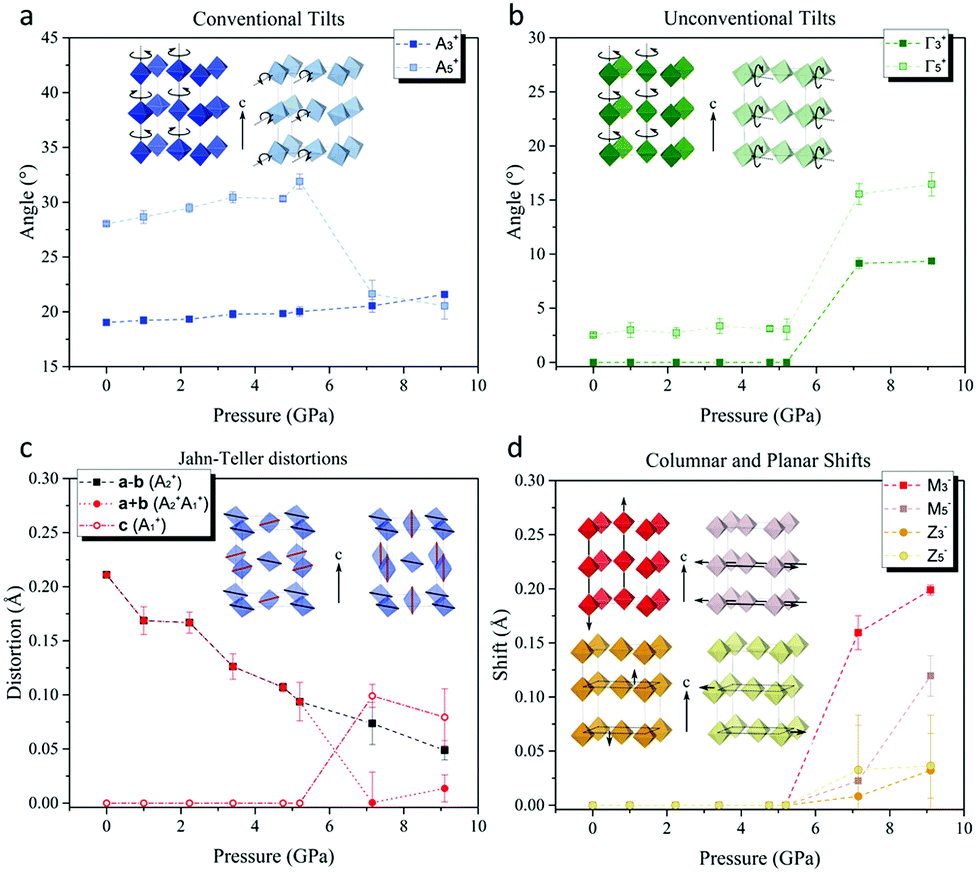 | ||
| Fig. 3 Pressure dependence of (a) conventional octahedra tilts rotation angles, (b) unconventional octahedra tilts rotation angles, (c) Jahn–Teller distortions and (d) columnar and planar shifts of the framework. | ||
Interestingly, the I2/a crystal symmetry also allows unconventional octahedral tilts (uniform rotations about the I2/ab-axis, Fig. 3b) that transform by Γ5+, however the respective mode amplitudes are negligible in the α-phase. These unconventional tilts would result in a departure from anti–anti type bonding of every formate anion, towards a syn–anti configuration. The fact that these distortions are allowed by symmetry yet found to be small implies the presence of microscopic interactions that energetically favour anti–anti bonding over syn–anti bonding in the perovskite framework, as reported in the study by H. L. Bostrom,37 thus prompting the structure to adopt conventional octahedra tilts only.
The amplitude of conventional octahedral rotations about the c-axis (A3+) varied little over the measured pressure range (Fig. 3a), while the conventional octahedral rotations about an axis lying perpendicular to the c-axis (A5+) are reduced in amplitude upon entering the γ-phase. Furthermore, their axis of rotation, constrained by symmetry to be parallel to the a-axis in the α-phase, now rotates by about 35° towards the b-axis. Throughout the α-phase one observes a significant, monotonic suppression of the JT distortion, as mentioned previously (Fig. 3c). In the γ-phase, new JT displacive modes that transform by the A1+ irreducible representation admix with the α-phase JT displacements (A2+), leading to the observed JT switch.
The number of modes that enter into the decomposition of the γ-phase structure is considerably greater than those appearing in the decomposition of the α-phase (see ESI†). In further contrast to the α-phase, our analysis showed that the low symmetry of the γ-phase can only be reached by no less than three primary, symmetry breaking modes. The possible combinations of primary modes is vast, yet all include either M-point or Z-point modes that do not appear in the decomposition of the α-phase. The relevant M-point primary modes transform by either the M2−, M3−, or M5− irreducible representations, and correspond to the ordering of A site dimethylammonium molecular orientations, unconventional c-axis columnar shifts parallel to c, and unconventional c-axis columnar shifts perpendicular to c, respectively (Fig. 3d). The relevant Z-point primary modes transform by either Z3− or Z5−, and correspond to ab-plane planar shifts parallel and perpendicular to c, respectively. We also note that the Γ5+ unconventional octahedral rotations, allowed by symmetry in the α-phase but found to have negligible amplitudes, now appear with large amplitude in the γ-phase (Fig. 3b).
It is well established that the unit cell volume of perovskite-type structures can be reduced through a linear-quadratic coupling of the lattice strain to conventional octahedra tilts. [(CH3)2NH2]Cu(HCOO)3 already hosts large conventional tilts at ambient pressure, and, somewhat surprisingly, the amplitude of these tilts varies little with increasing pressure (Fig. 3a). Instead, the volume contraction in the α-phase largely originates in a compression of the coordinative bonds (Fig. 3c, discussed above). Unconventional octahedra tilts and shifts can couple to lattice strain in the same way as the conventional octahedral tilts, hence they represent extra structural degrees of freedom by which the unit cell volume can be reduced. Since pressure stabilises structural deformations that lead to unit cell volume reduction, we suggest that unconventional octahedra tilts and shifts naturally play a leading role in the pressure-induced phase transition and associated Jahn–Teller switch. This scenario is further supported by the absence, within the same pressure regime, of a phase transition in KCuF3, in which the network of corner sharing octahedra forbid such unconventional structural degrees of freedom. All unconventional distortions introduce energetically unfavourable syn–anti bonding of the formate anions, hence the critical behaviour associated with the phase transition might be understood in terms of an energetic competition between formate bonding requirements and the steric contraction of the lattice. Finally, we note that the [(CH3)2NH2]+ cations at the A sites interact weakly with each other, and also weakly with the rest of the perovskite framework, hence the reconfiguration of their order is unlikely to drive the large structural distortions observed at the phase transition.
Conclusions
In summary, we have showed that quasi-hydrostatic compression in the region 5.20 → 7.15 GPa induced a structural phase transition in single-crystals of [(CH3)2NH2]Cu(HCOO)3–a model perovskite-like coordination polymer. At the transition the crystal symmetry was lowered from monoclinic to a previously unobserved triclinic phase in which half of the CuII Jahn–Teller axes had switched from the a + b to the c crystallographic direction. Such orbital reordering is unexpected in homoleptic coordination polymers and, to the best of our knowledge, has never been observed before. Symmetry analysis demonstrated that the phase transition is primarily driven by unconventional octahedral tilts and shifts in the framework, and/or a reconfiguration of A-site cation ordering. These structural instabilities are unique to the coordination polymer perovskites, which naturally explains the absence of a high-pressure phase transition in the canonical inorganic analogue KCuF3, and implies that such transitions may occur in a wide family of coordination polymers of current scientific interest. The unconventional distortions require a departure from energetically favourable anti–anti bonding of formate ligands towards the syn–anti limit. We suggest that this energetic cost competes with the steric contraction of the lattice, thereby resulting in a structural phase transition under pressure. Our results serve as a proof of principle, demonstrating that it is possible to control orbital order in coordination polymer perovskites by applied hydrostatic pressure. Importantly, the pressure induced switch of the Jahn–Teller axis is expected to reconfigure the main magnetic interactions in the material, hence magnetism and magnetic order under high pressure presents an interesting topic for future studies of [(CH3)2NH2]Cu(HCOO)3 and related materials.Author contributions
R. S. and P. M. contributed to conceptualisation and resources. Supervision was provided by P. M. and R. D. J., while R. S. administered the project. P. M. and R. S. acquired funding for the project. M. A. and R. S. carried out the investigation. M. A., R. S. and R. D. J. carried out the formal analysis of the data. R. S. and R. D. J. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. S. acknowledges support from the Swiss National Science Foundation (project Nr P2BEP2_188253). R. D. J. acknowledges support from a Royal Society University Research Fellowship. We acknowledge Paul Scherrer Institute, Villigen, Switzerland, for provision of synchrotron radiation beamtime at beamline X04SA of the SLS.Notes and references
- D. I. Khomskii and G. A. Sawatzky, Solid State Commun., 1997, 102, 87–99 CrossRef CAS.
- P. G. Radaelli, D. E. Cox, M. Marezio and S.-W. Cheong, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 3015–3023 CrossRef CAS.
- A. Maignan, C. Martin, S. Hébert and V. Hardy, J. Mater. Chem., 2007, 17, 5023–5031 RSC.
- M. W. Lufaso and P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 10–20 CrossRef PubMed.
- J. B. Goodenough, Phys. Rev., 1955, 100, 564–573 CrossRef CAS.
- J. Kanamori, J. Phys. Chem. Solids, 1959, 10, 87–98 CrossRef CAS.
- P. W. Anderson, Phys. Rev., 1950, 79, 350–356 CrossRef.
- C. Xu, Y. Li, B. Xu, J. Íñiguez, W. Duan and L. Bellaiche, Adv. Funct. Mater., 2017, 27, 1604513 CrossRef.
- L. Chen, C. Xu, H. Tian, H. Xiang, J. Íñiguez, Y. Yang and L. Bellaiche, Phys. Rev. Lett., 2019, 122, 247701 CrossRef CAS PubMed.
- J.-S. Zhou, J. A. Alonso, J. T. Han, M. T. Fernández-Díaz, J.-G. Cheng and J. B. Goodenough, J. Fluorine Chem., 2011, 132, 1117–1121 CrossRef CAS.
- A. M. Glazer, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 3384–3392 CrossRef CAS.
- R. Scatena, R. D. Johnson, P. Manuel and P. Macchi, J. Mater. Chem. C, 2020, 8, 12840–12847 RSC.
- H. L. B. Boström, J. A. Hill and A. L. Goodwin, Phys. Chem. Chem. Phys., 2016, 18, 31881–31894 RSC.
- L. Merrill and W. A. Bassett, Rev. Sci. Instrum., 1974, 45, 290–294 CrossRef.
- R. J. Angel, M. Bujak, J. Zhao, G. D. Gatta and S. D. Jacobsen, J. Appl. Crystallogr., 2007, 40, 26–32 CrossRef CAS.
- H. K. Mao, J. Xu and P. M. Bell, J. Geophys. Res., 1986, 91, 4673 CrossRef CAS.
- G. J. Piermarini, S. Block, J. D. Barnett and R. A. Forman, J. Appl. Phys., 1975, 46, 2774–2780 CrossRef CAS.
- P. Macchi, H.-B. Bürgi, A. S. Chimpri, J. Hauser and Z. Gál, J. Appl. Crystallogr., 2011, 44, 763–771 CrossRef CAS.
- P. R. Willmott, D. Meister, S. J. Leake, M. Lange, A. Bergamaschi, M. Böge, M. Calvi, C. Cancellieri, N. Casati, A. Cervellino, Q. Chen, C. David, U. Flechsig, F. Gozzo, B. Henrich, S. Jäggi-Spielmann, B. Jakob, I. Kalichava, P. Karvinen, J. Krempasky, A. Lüdeke, R. Lüscher, S. Maag, C. Quitmann, M. L. Reinle-Schmitt, T. Schmidt, B. Schmitt, A. Streun, I. Vartiainen, M. Vitins, X. Wang and R. Wullschleger, J. Synchrotron Radiat., 2013, 20, 667–682 CrossRef CAS PubMed.
- CrysAlisPro version 171.38.46, 2017.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- B. J. Campbell, H. T. Stokes, D. E. Tanner and D. M. Hatch, J. Appl. Crystallogr., 2006, 39, 607–614 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- I. E. Collings, M. Bykov, E. Bykova, M. Hanfland, S. Van Smaalen, L. Dubrovinsky and N. Dubrovinskaia, CrystEngComm, 2018, 20, 3512–3521 RSC.
- C. J. Simmons, M. A. Hitchman, H. Stratemeier and A. J. Schultz, J. Am. Chem. Soc., 1993, 115, 11304–11311 CrossRef CAS.
- A. J. Schultz, M. A. Hitchman, J. D. Jorgensen, S. Lukin, P. G. Radaelli, C. J. Simmons and H. Stratemeier, Inorg. Chem., 1997, 36, 3382–3385 CrossRef CAS PubMed.
- P. Parois, S. A. Moggach, J. Sanchez-Benitez, K. V. Kamenev, A. R. Lennie, J. E. Warren, E. K. Brechin, S. Parsons and M. Murrie, Chem. Commun., 2010, 46, 1881–1883 RSC.
- G. J. Halder, K. W. Chapman, J. A. Schlueter and J. L. Manson, Angew. Chem., Int. Ed., 2011, 50, 419–421 CrossRef CAS PubMed.
- A. Lanza, C. Fiolka, M. Fisch, N. Casati, M. Skoulatos, C. Rüegg, K. W. Krämer and P. Macchi, Chem. Commun., 2014, 50, 14504–14507 RSC.
- B. Wehinger, C. Fiolka, A. Lanza, R. Scatena, M. Kubus, A. Grockowiak, W. A. Coniglio, D. Graf, M. Skoulatos, J.-H. Chen, J. Gukelberger, N. Casati, O. Zaharko, P. Macchi, K. W. Krämer, S. Tozer, C. Mudry, B. Normand and C. Rüegg, Phys. Rev. Lett., 2018, 121, 117201 CrossRef CAS PubMed.
- C. J. McMonagle, P. Comar, G. S. Nichol, D. R. Allan, J. González, J. A. Barreda-Argüeso, F. Rodríguez, R. Valiente, G. F. Turner, E. K. Brechin and S. A. Moggach, Chem. Sci., 2020, 11, 8793–8799 RSC.
- J. L. Musfeldt, L. I. Vergara, T. V. Brinzari, C. Lee, L. C. Tung, J. Kang, Y. J. Wang, J. A. Schlueter, J. L. Manson and M. H. Whangbo, Phys. Rev. Lett., 2009, 103, 1–4 CrossRef PubMed.
- A. Prescimone, C. Morien, D. Allan, J. A. Schlueter, S. W. Tozer, J. L. Manson, S. Parsons, E. K. Brechin and S. Hill, Angew. Chem., 2012, 124, 7608–7612 CrossRef.
- S. Ghannadzadeh, J. S. Möller, P. A. Goddard, T. Lancaster, F. Xiao, S. J. Blundell, A. Maisuradze, R. Khasanov, J. L. Manson, S. W. Tozer, D. Graf and J. A. Schlueter, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 241102 CrossRef.
- H. L. B. Boström, CrystEngComm, 2020, 22, 961–968 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: CIF file of the experimental structures as a function of pressure and PDF file with tables of the complete crystallographic information and refined mode amplitudes, and pictures of (i) the crystal in the diamond anvil cell, (ii) crystal structures, (iii) (0kl) diffraction plane and (iv) schematic representation of the irreducible representations used for the refinement of α and γ phases. CCDC 2067545–2067553. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc01966j |
| This journal is © The Royal Society of Chemistry 2021 |