
Spin-ladder behaviour in molecular materials†
 a
and
Manuel
Almeida
*ab
a
and
Manuel
Almeida
*ab
Abstract
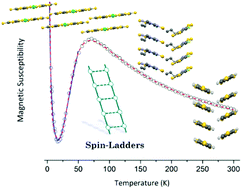
Spin-ladders are quantum magnetic systems at the crossroads between one and two dimensions that have attracted increasing interest during the last decades due to unique properties critically dependent on spin system topology. The first materials where spin-ladder behaviour was identified were transition metal oxides and copper(II) halides or coordination polymers. However, since the report of the first spin-ladder based in an organic molecule in 1997, an increasing number of molecular compounds showing spin-ladder behaviour have been reported. Their structural and magnetic properties are reviewed with emphasis on the possibilities offered by molecular units of different nature and crystal engineering tools to develop new spin-ladder systems.
- This article is part of the themed collection: Materials for molecular electronics and magnetism
 Please wait while we load your content...
Please wait while we load your content...

