
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural self-sorting of pseudopeptide homo and heterodimeric disulfide cages in water: mechanistic insights and cation sensing†
Marcin
Konopka
ab,
Piotr
Cecot
ab,
Jack M.
Harrowfield
 c and
Artur R.
Stefankiewicz
*ab
c and
Artur R.
Stefankiewicz
*ab
aFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland. E-mail: ars@amu.edu.pl
bCenter for Advanced Technologies, Adam Mickiewicz University, Uniwersytetu Poznańskiego 10, 61-614 Poznań, Poland
cInstitut de Science et d’Ingénierie Supramoléculaires, Université de Strasbourg, 8 allée Gaspard Monge, 67083 Strasbourg, France
First published on 10th June 2021
Abstract
The molecular components of biological systems self-sort via different mechanisms to act in a cooperative manner and to avoid interfering with each other. Herein we describe mechanistic insights and a versatile strategy for the synthesis of water-soluble, pseudopeptide molecular cages based on disulfide bonds. The use of trifunctional thiols led to a dynamic combinatorial library (DCL) of readily isolable, multi-component homo and hetero cage-like architectures showing a degree of self-sorting related to the symmetry and size of the trithiol. The work provided detailed kinetic studies and DFT molecular modeling giving original insights into the disulfide cages' properties. We also applied the selected cage system in the fluorometric detection of La3+ cations, which led to the generation of a strongly luminescent metal–organic assembly.
Introduction
The synthesis of functional molecular cages continues to pose an intriguing challenge due to the numerous applications of such structures1 as chemical receptors,2–4 nanocapsules for molecular transport,5–7 or in molecular recognition.8 Despite enormous progress in the development of this type of architecture,9–12 examples of stable, yet dynamic and water-soluble structures are still very scarce.13–16 Dynamic combinatorial chemistry (DCC) of disulfides is an effective approach to generate artificial biocompatible systems in water.17,18 Disulfides have been extensively studied with linear,19,20 macrocyclic21–25 or interlocked topologies26–31 but far less is known about disulfide cages. To date, several examples have been reported of disulfide cages based on trifunctional thiols,32–35 although only a few are water-soluble.36–39 There is a real lack of information regarding the impact of the thiol component structure on the tendency of dynamic system to favor exclusive formation of cage-like structures through self-assembly and self-sorting in water-soluble systems.40–42 Hetero-component cages seem to be particularly understudied, a situation, which is probably due to the difficulties in isolating pure cages from post-reaction mixtures. Our objective thus was to fill these gaps by developing an efficient synthesis of pure homo- and hetero-dimeric pseudopeptide, water-stable molecular cages, by using thio-amino-acid functionalized organic platforms. This was expected to provide a robust workshop to explore the phenomenon of self-assembly and self-sorting in a dynamic multi-disulfide system. We also attempted to draw some new insights into the structural and geometric factors crucial for disulfide cage formation based on experimental data and DFT calculations. Based on this, we propose a theoretical model that could be applied for the effective design of next mono and bicomponent disulfide cages. In addition, we describe a preliminary investigation of a strongly fluorescent metal–organic material obtained using a disulfide cage as poly-anionic ligand on La(III).Results and discussion
Design of building blocks
The three core platforms used were based on triamides formed from aromatic tricarboxylic acids and L-cysteine. The use of natural L-cysteine ensured the absence of diastereomeric products and the formation of chiral cages with stable absolute configurations. The set of aromatic platforms was selected to gradually increase the size (diameter) of the rigid core. Each was functionalised by amide formation with L-cysteine in order to introduce –SH (for disulfide formation) and –COOH (for water solubility) groups. All building blocks were obtained by a slightly modified three-step synthetic route36 (see ESI,† Fig. S1). In the first stage, the reaction of each tricarboxylic acid with N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl) provided the corresponding NHS activated esters. Next, formation of the amides by reaction with L-S-trityl-cysteine followed by hydrolysis of the trityl-moieties in acidic conditions led to the desired trifunctional components 1, 2 and 3, which have planar sizes in the order 3 > 2 > 1 (Fig. 1). | ||
| Fig. 1 General scheme of the dynamic covalent DCL. Three building blocks react together and give five different disulfide cages, three homodimeric (1-1, 2-2 and 3-3) and two heterodimeric cage-like architectures (1-2 and 2-3). | ||
Synthesis of homodimeric cages
Solutions of each component (5 mM) were prepared in an aqueous 0.01 M NaOH solution and its pH was adjusted to 8.0 by addition of dilute HCl. The presence of thiolates in solution is obligatory for their oxidation to disulfides as well as for disulfide dynamic exchange. These solutions of the basic components were then placed in loosely capped 2 mL vials and allowed to oxidize and equilibrate for 5 days. To ensure that all DCLs reached thermodynamic equilibrium, each sample was further analysed by HPLC one week and one month after the end of the 5 day reaction time. Control analyses showed no quantitative or qualitative changes in any DCL. After this time, LC-MS analysis was performed. This showed that each post-reaction mixture contained only one product (more polar than the substrate), which was identified by mass spectroscopy as the monoprotonated species with a mass of twice that of the reactant less the 6 hydrogen atoms corresponding to those lost in formation of 3 disulfide links, i.e. the tris-disulfide dimeric cage. The compound 1-1 is a known species, identified previously by LC-MS and 1H NMR.37 However, we decided to include cage 1-1 in our studies for comparison and to establish its full characteristics. Further details of the structure 1-1 were established from its 1H NMR spectrum, which showed the D3 symmetry expected for the homodimeric product (see ESI,† Fig. S19 and S20). The 1H NMR spectra of the dimeric cages, 2-2 and 3-3 showed the same symmetry. Comparison of the 1H NMR spectra of the respective components with those of the cages and analysis of optimised structures revealed that the aromatic proton signals of the cages are shifted upfield, presumably as a result of close proximity of the aromatic platforms in the cage structure. In contrast, the signals originating from cysteine CH and CH2 groups were found to be deshielded due to their position outside the cages (see ESI,† Fig. S19, S24, S29 and S60–S65). Acidification to pH 2 of the post-reaction mixtures yielded the three cages as the water-insoluble hexa-carboxylic acids, which could be easily isolated by filtration. However, down to the slightly acidic region of pH 5–6 the cages remained soluble, which extends the range of their applications.Synthesis of heterodimeric cages
The next step was to investigate if the different building blocks would react with each other and form the hetero-component cage structures. Equimolar solutions containing pairs of components were prepared at pH 8.0 and then stirred for five days in air. Three different products in equal proportions were detected in the post-reaction mixture derived from components 1 and 2 (Fig. 2). Based on the LC-MS analysis two homodimeric cages, 1-1 and 2-2, were identified while the third product had the composition expected for the heterodimeric cage 1-2. Similar results were obtained for the mixture of 2 and 3, which after the reaction contained cages 2-2, 3-3 and heterodimeric cage 2-3. For the mixture of 1 and 3, only homodimeric structures were found in the post-reaction mixture, with none of the possible cage 1-3. We observe here a kind of structural self-sorting based on the components' C3 symmetry and size factors favoring the formation of the more symmetrical products in the final DCL. No linear or macrocyclic products were observed in any of the post-reaction mixtures. Each of the DCL samples was completely transparent before and after the reaction and was not additionally filtered before LC-MS analysis. For this reason, we exclude the possibility that polymeric or insoluble products have formed in the DCL that might not be detected by the HPLC DAD detector. To isolate the heterodimeric cages, the experiment with pairs of components 1 + 2 and 2 + 3 was repeated on a larger scale and at a higher concentration (10 mM). Heterodimeric cages were isolated by semi-preparative HPLC on a few milligrams scale and characterized by NMR spectroscopy. 1H and COSY NMR spectra confirmed their expected structures and C3 symmetry. Comparison of the 1H NMR spectra of the hetero cage 1-2 and the components 1 and 2 is also in line with the cage structure (Fig. 3). The upfield shifts of the doublets (cyan-green) from the triphenylamine moieties and the singlet (blue) from the central phenyl ring clearly indicate a strong interaction between aromatic hydrogens and the proximity of organic platforms of 1 and 2. This explanation is supported by analysis of the optimized structure of the 1-2 cage. In this structure, all of the aromatic hydrogens are additionally shielded by the accumulation of electron-rich sulfur (S–S bonds) and oxygen (amides) atoms. | ||
| Fig. 2 (a) HPLC chromatograms showing the formation of heterodimeric cages (1-2 and 2-3) in the component pairs mixtures. For the pair of 1 + 3, cage 1-3 was not observed. Due to significant differences in molar extinction coefficients of the components, the chromatograms do not show the real amounts of the products. The dashed line shows the actual content of each (see ESI,† Fig. S57 and S58), (b) comparison of experimental and simulated mass spectra (ESI-MS) of all five cages. | ||
 | ||
| Fig. 3 Comparison of 1H NMR spectra (D2O, 298 K, 600 MHz) of heterodimeric cage 1-2 and components 1 and 2. Dashed lines show chemical shifts of analogous signals from aromatic hydrogens from both organic platforms. | ||
The hydrodynamic radii deduced from DOSY experiments (see ESI† for details) coincide closely with the calculated results (Table 1). Finally, we performed an experiment with an equimolar mixture of all three components at once to check if in this three component DCL any new architectures would emerge. The resulting DCL contained an almost equimolar mixture of all five cages, without any traces of new species (see ESI,† Fig. S44). As in previous experiments, no products with a different architecture were found. This clearly established that our system based on well-defined C3 symmetric components spontaneously selects the stable cage-like structures from the numerous possibilities of combinatorial connections between the three trifunctional components (see ESI,† Fig. S70).
| Cage | 1-1 | 2-2 | 3-3 | 1-2 | 2-3 | 1-3 |
|---|---|---|---|---|---|---|
| a For cage 1-3 only calculated properties are available. b Calculated from DOSY diffusion coefficients using the standard Stokes–Einstein equation. c Measured from optimised models using Gaussian software. d d – distance between platforms. e φ – average disulfide bonds dihedral angle. f ϑ – rotation angle between organic platforms. g ζ – trapezoidal angle. | ||||||
| Calculated structure |

|
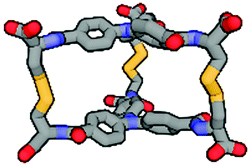
|

|
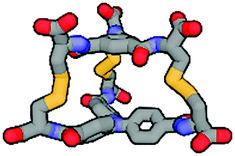
|

|

|
| Symmetry | D 3 | D 3 | D 3 | C 3 | C 3 | C 3 |
| D [m2 s−1 × 10−10] | 2.05 | 1.80 | 1.47 | 2.25 | 1.93 | n/a |
| r hyd [Å] | 9.68 | 11.02 | 13.50 | 8.02 | 10.28 | n/a |
| r calc. [Å] | 8.36 | 10.35 | 12.05 | 7.86 | 9.13 | 10.22 |
| V sph [Å3] | 3971 | 5612 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 304 304 |
2873 | 4553 | 4341 |
| d [Å] | 7.9 | 8.2 | 8.1 | 7.5 | 4.0 | 3.5 |
| ϕ | 107.0 | 136.6 | 105.5 | 105.4 | 85.5 | 101.7 |
| ζ | 95.1 | 93.7 | 96.3 | 106.9 | 99.8 | 120.8 |
| ϑ | 4.9 | 9.7 | 5.2 | 8.1 | 1.5 | 14.5 |
Although we have obtained pure products, we have been unable to obtain crystals suitable for X-ray structure determinations. For this reason, we decided to use computational tools to optimize the cages' structures (Table 1 and ESI,† Fig. S60–S65).43–45 Based on this it can be concluded that the cage molecules prefer an “open” conformation, where a small internal space of the molecule is present and the disulfide groups are directed towards the inside of the molecule. The S–S torsion angles are typical for this binding in all optimized cages and averaged 107° (in the range 85° to 136°). One structure with a “closed” conformation and disulfide groups directed outwards turned out to be that of the cage 2-3, which can be explained by the significant differences in the size of aromatic-organic platforms that enforce such arrangement of cysteine arms and disulfide bonds. Also, we found that this structure is additionally stabilized by stacking interactions, due to the very close proximity of the aromatic cores (3.0–4.0 Å). The cage 3-3 composed of two large aromatic platforms should also be stabilized in this way, since the angles between the arms and platforms are close to 90°, favoring extensive close overlap. For this reason, we have defined two geometric factors that can influence the structure.
One, termed the trapezoidal angle ζ (zeta), is the angle between the platform plane and the cysteine arm, which defines the relationship between the size of the platform and the angle imposed on the binding arm. The second, ϑ (theta), is the degree of rotation between the platforms about the principal z-axis. This angle is important for the measure of structural stress caused by the differences in the size of the platforms and for geometric considerations, e.g., steric hindrance. The angles were determined for four points: centroid of the smaller platform – carbon alpha of the arm of the smaller platform – alpha carbon of the larger platform – centroid of the larger platform. Centroids have been designated as the central nitrogen atom in platform 2 or the geometric center of the benzene ring in platforms 1 and 3. So, high values for both of these angles seem to be universal factors for determining the geometric barriers to formation of a given cage structure. For each homodimeric cage, the values of those angles are very similar (ϑ near 5° and ζ near 95°) and those compounds are formed smoothly. This two-angles factor is slightly more complicated for two observed heterodimeric cages 1-2 and 2-3, but the values do not exceed 8° theta and 115° zeta. Cage 1-3 is the only one not formed during the experiments, and the reason for this may be the difficulty involved in attaining the cage geometry. The zeta angle takes on the highest value of 121° due to the significant difference between the sizes of the aromatic platforms of 1 and 3. The platforms can rotate about the z-axis to compensate for this stretching effect but the optimal theta angle then turns out to be large enough to stop the cage from forming (Fig. 4).
 | ||
| Fig. 4 Diagram of ϑ (theta) and ζ (zeta) angles for the six optimized dimeric cages (blue squares – homodimeric cages, triangles – heterodimeric cages, green – observed cages, red indicates no cage formation for 1-3). | ||
Kinetic studies
Because the calculations indicated that the formation of cage 1-3 is the least preferred for geometric reasons, but structurally possible, we decided to supplement our work with kinetic studies to find any additional reasons for the absence of 1-3. We employed HPLC to monitor cage formation over time. Reactions were performed in 2 mL vials at pH 8 in 5 mM concentration in room temperature. Each reaction mixture was observed for 30 hours and monitored every 30 minutes. The first HPLC injection (t0) for every sample was done immediately after dissolving the component in basic water and setting the pH to 8.0. Thus, we obtained a set of chromatograms illustrating component decay and cage growth over time based on the relative peak area (RPA). To ensure that the method was quantitative, absorption spectra were recorded from the solutions of each component and the corresponding cage at the same concentration. The difference in registered absorbance between the component/cage pairs did not exceed 5%. Each reaction was repeated and monitored three times to ensure that results were the same. For the homodimeric cages, the material distribution curves are shown in Fig. 5a–c. These show that in mono-mixtures the time for conversion to 100% product within experimental error was shortest for cage 3-3 (10 h), followed by 1-1 (20 h) and longest for 2-2 (26 h). We attribute this to the influence of subtle effects such as the differences in solvation of polar cysteine moieties, in surface polarity and in the size of each component. Cooperativity between these factors appears to result in the largest aromatic species 3 having the fastest cage formation.46–48 | ||
| Fig. 5 Normalized rate plots of mono-component DCL (a–c) and multi-component DCL equilibration (d) 1 + 2 mixture, (e) 2 + 3 and (f) 1 + 2 + 3. | ||
LC-MS measurements taken 60 minutes after the start of reactions provided a deeper understanding of the cage formation process. These measurements (ESI,† Fig. S45–S50) showed that aside from unreacted trithiol, the dominant species present corresponded to the linkage of two trithiol molecules through one, two or three disulfide bonds. No species derived from more than two trithiol units were detectable. This is consistent with an initially intermolecular reaction to give the mono-disulfide being followed by rapid intramolecular reactions giving the macrocyclic bis(disulfide) and then the cage tris(disulfide). Competition by intermolecular processes after the first step appears to be ineffective. The fact that the cage dominates over the macrocycle at a time when the mono-disulfide is still present indicates that restrictions of motion in the macrocycle must lead to faster disulfide formation relative to that giving the macrocycle. Thus, in the first step, two molecules of trithiol form open-chain mono-disulfide adducts (1A, 2A, 3A). These are then further oxidized to the macrocyclic bis-disulfide products (1B, 2B, 3B), which in the third step are finally oxidized to the cages. (Intermediates are shown as gray and black lines in Fig. 5a–c). In the case of cage 2-2, the bis(disulfide) intermediate was more abundant than the mono-(disulfide) intermediate at all points where the reaction mixture was sampled (Fig. 5b). This is consistent with acceleration in rate due to the intramolecular nature of the second reaction step, which is largely dependent on the chemical structure and shape of the component employed. This is supported by the molecular modeling results that showed smaller conformational restrictions in the macromonocyclic bis(disulfide) intermediate 2B than those found for 1B and 3B, see ESI,† Fig. S66–S69). For cages 1-1 and 3-3, the observations are less complicated and the disappearance of the reactant in both cases can be well fitted to a single exponential decay. This implies that the reactant loss is a first-order process (seemingly irreversible), a situation which might arise if the rate-determining step of the reaction were to be the very first step involving the trithiol and dissolved oxygen. Although formally a second order process, if the agitation of the solution and its contact with the normal atmosphere were sufficient to maintain a constant concentration of dissolved oxygen, it would obey pseudo-first-order kinetics. Once the first species is formed and then dimerizes, all subsequent reaction steps, as noted above, would be intramolecular and probably greatly accelerated as a result. Examination of the kinetics of cage formation in the mixed 1 + 3 DCL (Fig. 6a) provided evidence for one of the complications in these reactions. In this system cage 3-3 is exclusively formed first, then 1-1. Formation of 1-1 shows an induction period lasting until the essentially complete conversion of 3 to 3-3, and an intermediate species decaying to 3-3 is very rapidly formed in a substantial amount.
 | ||
| Fig. 6 (a) normalized kinetics plot of 1 + 3 DCL equilibration (substrates and products), (b) schematic representation of possible disulfide exchanges in the 1 + 3 mixture. | ||
More significantly, the homodimeric cages 1-1 and 3-3 form almost twice as fast as in the single component systems. Complete conversion of 3-3 takes about 4 h, while, once initiated, complete formation of 1-1 takes about 10 h. Since the thiol oxidation is slower process than the disulfide exchange,49,50 formation of the first S–S linkage between two tri-thiols must be the rate-determining step in the entire cage formation process.51 What distinguishes both systems (single vs. mixed component) is the formation of intermediate species e.g.1-3A (reaction III, Fig. 6b), and we assume that the presence of the latter is related to observed acceleration of cage formation. Thus, the nucleophilic attack of thiolate on the 1-S–S-3 disulfide (1-3A), results in the formation of linear S–S–S transition state, which decomposes to a more stable disulfide product.50 If 1 is a weaker acid than 3 then under the same pH conditions, there must be a lower 1-S− thiolate concentration, which results in both a slower radical formation during oxidation and a preferred attack of 3-thiolate on the intermediate 1-3A. In this case, the synthesis of 1-1 through either the 1-3A path (reactions III and V) or path IV can be effectively stopped until the system is essentially drained of 3. However, considering the complexity of this system, identifying the exact cause of this phenomenon is very challenging.52
Where hetero-cage formation occurs as in reactions involving 2, the kinetics become more complicated to analyze because both reactants are involved in competitive processes. By LC-MS analysis we also observed all the expected hetero-component intermediates, but at much lower concentrations (<5% RPA, LS-MS, ESI,† Fig. S51–S56). A schematic representation of possible reactions between a pair of different components leading to a mixture of homo and heterodimeric cages is shown in Fig. 6a. We assume that for pairs 1 + 2 and 2 + 3, reactions IV, VII and XI occur with a similar probability, resulting in the observed equimolar mixture of homo and hetero products. In the case of the pair 1 + 3, reaction intermediates III (1-3A) and VIII (1-3B) were found at a low concentration (<2% RPA, LC-MS). Normalized concentration-time plots for the reactions 1 + 2 and 2 + 3 are shown in Fig. 5d and e. In the 1 + 2 system, cage 2-2 forms most rapidly, then comes 1-2 and the slowest is 1-1. The total reaction time is much longer than in the 1 + 3 DCL and takes about 30 hours. 2-2 reaches 50% of the target conversion after 4 hours compared to 8 hours in the DCL with only 2. As in the 1 + 3 system, the products involving a geometrically larger component (1-2 and 2-2) are created at higher rates. This effect is even more visible in the 2 + 3 system (Fig. 5e). Here, cages 2-3 and 3-3 are formed at almost the same rate and the initial rate is much higher than in the 1 + 2 DCL. Only when the DCL is dominated 80% by 2-3 and 3-3 (after about 4 hours), does the rate of formation of 2-2 increase.
Observations on the system with all three components present (1 + 2 + 3) were consistent with the results from the two-component systems, once again showing an apparent size effect on the cage formation (Fig. 5d–f). Products containing the largest component 3 formed faster than those containing 2 and the slowest formed were those with 1. Cages built of 1 and 2 form preferentially only when the DCL has already consumed most of the available 3.
Fluorometric detection of La3+ cations and generation of a strongly luminescent metal–organic material
While working with the cages, we noticed that both 2 and cage 2-2, due to the triphenylamine core, show strong fluorescence. We decided to further explore this feature of 2-2 to preliminary investigate some useful properties for potential application of this particular cage. Comparison of the emission spectra of 2 and 2-2 shows almost four times stronger fluorescence of the cage (max. 456 nm) than that of the component (max. 466 nm) and the a blue-shift of the maximum emission of 2-2 by 10 nm (Fig. 7a). We assume that this is due to an AIE-like effect (aggregation-induced emission) because the closed cage structure prevents the chromophore NPh3 rings from rotating freely, which results in increased emission intensity. Moreover, each cage is a polyanion with six CO2− groups concentrated on a small molecule area and available for coordination through O-donors. | ||
| Fig. 7 Synthesis of a fluorescent 2-2-La material; (a) normalized emission spectra of the cage 2-2 and 2 (10−5–10−6 M, 1 × 1 cm cuvette, ex: 345 nm, water pH 8.0), (b) normalized solid-state emission spectra of 2-2 and 2-2-La (ex: 345 nm), images taken under 325 nm UV lamp, (c) TGA/DGA analysis of 2, 2-2 and 2-2-La, (d) SEM and SEM-EDS analysis of 2-2-La. See ESI† for details (Fig. S71 and S72). | ||
Encouraged by these cage features, we decided to synthesize a metal complex based on 2-2. Mixing an aqueous solution of 2-2 as sodium salt with aqueous La(NO3)3 results in the quantitative precipitation of highly insoluble, amorphous metal–organic 2-2-La material (see ESI† for details). Taking into account the number and location of the cage chelating groups as well as coordination preferences of La3+ cation, formation of cross-linked coordination assembly was expected. The composition and purity of the generated material in the solid state was confirmed by the elemental (NHCS) analysis, which indicated 1:1 metal:cage ratio (see ESI,† Fig. S72). This was further supported by the ICP-MS analysis, which showed 10.6 wt% of La in 2-2-La for expected 12.6 wt% (see ESI,† page 45). Unfortunately, despite many attempts, the solubility of the obtained material prevented the mass spectrum to be registered. To determine the thermal stability of 2-2-La, TGA in the range of 30–600 °C was conducted and showed high stability of this material, which lost only 40 wt% with fairly linear weight loss. In comparison, organic compounds 2 and 2-2 turned out to be much less stable and already at 250 °C showed a major weight loss of about 30 wt% (over the full range of heating lost of 65 wt% and 50 wt%, respectively was recorded; Fig. 7c). While the volatile products from thermal decomposition were not characterised, it would be expected that the organic parts of the materials would undergo progressive decomposition, probably in multiple steps, to give a residue of La2O3 plus, possibly, Na2SO4 or Na2CO3 from the 2-2-La and the sodium salts alone from 2-2, so that the overall % changes in mass should be different. To get visual insights into the morphology of the 2-2-La complex, SEM imaging was performed and revealed the amorphous state of the powdered material. The close-ups show a spongy porous structure with numerous pores, about 0.1 μm in diameter, presumably due to the three-dimensional architecture of cage 2-2 with internal hydrophobic voids (Fig. 7d). Additionally SEM-EDS analysis was used to map the surface distribution of elements in the 2-2-La. The obtained results confirmed the presence of all elements expected in the investigated material (Fig. 7d and ESI† Fig. S71, S72). Since in aqueous solution, 2-2 exhibits interesting fluorescence behavior, we decided to examine and compare this property with metal–organic 2-2-La measured in the solid state. These measurements show that described system is photo-responsive, which opens several avenues towards their application in various technological fields. As presented in Fig. 7b, after excitation at 345 nm, the emission spectrum of 2-2-La shows a broad band with its maximum at 445 nm, while in the emission spectrum of 2-2 apart from seemingly lower intensity, there is a notable blue-shift (55 nm) in the band maximum = 500 nm (for absorbance spectra see ESI,† Fig. S59). The difference in solid-state fluorescence of both materials under excitation at 325 nm is also clearly visible to the naked eye as a significant difference in color (Fig. 7b insets).
Conclusions
We have described here a facile approach to the synthesis of pure disulfide, water-soluble molecular cages based on trifunctional organic platforms, and which can be formed easily in a one-pot reaction under very mild conditions. We have shown that high symmetry of its components pushes a DCL to a self-sorted selection of cage species. If the difference in component size is small, the DCL self-sorts into a mixture of homo and heterodimeric cages. The loss of geometrical compatibility with large size differences results in self-sorting of a mixture of two trithiols into a pair of two homodimeric cages, with no other products detectable. Based on these observations and DFT calculations, we created a two-factor theoretical model to predict the tendencies of trifunctional thiols to self-assembly into homo- or heterodimeric cages. We have also provided original kinetic studies giving new insight into the disulfide cages' formation mechanism through careful analysis of every intermediate product. As shown by the synthesis of 2-2-La in aqueous conditions, they have potential applications as novel lanthanides complexing agents and functional fluorescent materials. It has been shown that the cage-like structure of these compounds is very stable in aqueous conditions, a highly desirable property for applications in molecular transport and drug delivery. The anionic form of described cages at basic pH could be potentially used as an ionic receptor for molecular recognition. The applications of the cages could be easily extended to non-aqueous solvents by choosing the appropriate organic counter-ions while maintaining the ionic character of the entire system. Our methodological approach described here allows for easy and quick design of multi-functional disulfide cages with the promise of a wide range of applications in numerous branches of chemistry.Author contributions
MK performed all organic synthesis, all LC-MS, HPLC, NMR analysis and co-wrote the paper. PC performed molecular modeling, optimization of the dimeric cages and co-wrote the paper. JMH co-wrote the paper. ARS designed the project, interpreted the results and co-wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MK and PC thank the grant INNCHEM no. POWR.03.02.00-00-I023/17 co-financed by the European Union through the European Social Fund under the Operational Program Knowledge Education Development. PC thanks the Poznań Supercomputing and Networking Center (grant no. 401) for granting super-computers access. ARS thanks the National Science Centre (grant SONATA BIS 2018/30/E/ST5/00032) for financial support. We thank Prof. Tomasz Grzyb for his help with the solid-state fluorescence measurements.References
- S. Ulrich, Acc. Chem. Res., 2019, 52, 510–519 CrossRef CAS PubMed.
- J. W. Nial, R. B. Craig, J. G. Collins, K. Sharon and R. A.-W. Janice, Mini-Rev. Med. Chem., 2007, 7, 627–648 CrossRef PubMed.
- C. Bravin, A. Guidetti, G. Licini and C. Zonta, Chem. Sci., 2019, 10, 3523–3528 RSC.
- A. Brzechwa-Chodzyńska, W. Drożdż, J. Harrowfield and A. R. Stefankiewicz, Coord. Chem. Rev., 2021, 434, 213820 CrossRef.
- G. Markiewicz, A. Jenczak, M. Kolodziejski, J. J. Holstein, J. K. M. Sanders and A. R. Stefankiewicz, Nat. Commun., 2017, 8, 15109 CrossRef.
- S. Durot, J. Taesch and V. Heitz, Chem. Rev., 2014, 114, 8542–8578 CrossRef CAS PubMed.
- H. Jedrzejewska and A. Szumna, Chem. Sci., 2019, 10, 4412–4421 RSC.
- W. Drożdż, A. Walczak, Y. Bessin, V. Gervais, X.-Y. Cao, J.-M. Lehn, S. Ulrich and A. R. Stefankiewicz, Chem. – Eur. J., 2018, 24, 10802–10811 CrossRef PubMed.
- M. Mastalerz, Acc. Chem. Res., 2018, 51, 2411–2422 CrossRef CAS PubMed.
- G. Zhang and M. Mastalerz, Chem. Soc. Rev., 2014, 43, 1934–1947 RSC.
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS PubMed.
- F. Beuerle, S. Klotzbach and A. Dhara, Synlett, 2016, 1133–1138 CrossRef CAS.
- L. Catti, N. Kishida, T. Kai, M. Akita and M. Yoshizawa, Nat. Commun., 2019, 10, 1948 CrossRef PubMed.
- J. H. Jordan and B. C. Gibb, Chem. Soc. Rev., 2015, 44, 547–585 RSC.
- I. Pisagatti, L. Barbera, G. Gattuso, M. F. Parisi, S. Geremia, N. Hickey and A. Notti, Org. Chem. Front., 2019, 6, 3804–3809 RSC.
- B. Setner and A. Szumna, Beilstein J. Org. Chem., 2019, 15, 1913–1924 CrossRef CAS PubMed.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- A. R. Stefankiewicz and J. K. M. Sanders, Chem. Commun., 2013, 49, 5820–5822 RSC.
- W. Drożdż, A. Walczak and A. R. Stefankiewicz, Org. Lett., 2021, 23, 3641–3645 CrossRef.
- T. M. Postma, W. R. J. D. Galloway, F. B. L. Cougnon, G. D. Pantoş, J. E. Stokes and D. R. Spring, Synlett, 2013, 765–769 CAS.
- S. Otto, R. L. E. Furlan and J. K. M. Sanders, J. Am. Chem. Soc., 2000, 122, 12063–12064 CrossRef CAS.
- S. Otto, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590 CrossRef CAS PubMed.
- F. Ulatowski, A. Sadowska-Kuziola and J. Jurczak, J. Org. Chem., 2014, 79, 9762–9770 CrossRef CAS PubMed.
- W. Drożdż, M. Kołodziejski, G. Markiewicz, A. Jenczak and R. A. Stefankiewicz, Int. J. Mol. Sci., 2015, 16 Search PubMed.
- F. B. L. Cougnon, N. A. Jenkins, G. D. Pantoş and J. K. M. Sanders, Angew. Chem., Int. Ed., 2012, 51, 1443–1447 CrossRef CAS.
- N. Ponnuswamy, F. B. L. Cougnon, J. M. Clough, G. D. Pantoş and J. K. M. Sanders, Science, 2012, 338, 783–785 CrossRef CAS.
- L. Tapia, Y. Perez, M. Bolte, J. Casas, J. Sola, R. Quesada and I. Alfonso, Angew. Chem., Int. Ed., 2019, 58, 12465–12468 CrossRef CAS PubMed.
- M. Lafuente, I. Alfonso and J. Solà, ChemSystemsChem, 2019, 1, 25–31 CrossRef CAS.
- M. E. Dehkordi, V. Luxami and G. D. Pantoş, J. Org. Chem., 2018, 83, 11654–11660 CrossRef CAS PubMed.
- B. Bartolec, M. Altay and S. Otto, Chem. Commun., 2018, 54, 13096–13098 RSC.
- M. S. Collins, N.-M. Phan, L. N. Zakharov and D. W. Johnson, Inorg. Chem., 2018, 57, 3486–3496 CrossRef CAS PubMed.
- M. S. Collins, M. E. Carnes, B. P. Nell, L. N. Zakharov and D. W. Johnson, Nat. Commun., 2016, 7, 11052 CrossRef CAS PubMed.
- F. Brégier, J. Lavalle and J.-C. Chambron, Eur. J. Org. Chem., 2013, 2666–2671 CrossRef.
- M. A. Little, J. Donkin, J. Fisher, M. A. Halcrow, J. Loder and M. J. Hardie, Angew. Chem., Int. Ed., 2012, 51, 764–766 CrossRef CAS PubMed.
- A. R. Stefankiewicz, M. R. Sambrook and J. K. M. Sanders, Chem. Sci., 2012, 3, 2326 RSC.
- K. R. West, K. D. Bake and S. Otto, Org. Lett., 2005, 7, 2615–2618 CrossRef CAS PubMed.
- F. Brégier, O. Hudeček, F. Chaux, M.-J. Penouilh, J.-C. Chambron, P. Lhoták, E. Aubert and E. Espinosa, Eur. J. Org. Chem., 2017, 3795–3811 CrossRef.
- L. Krejčí, M. Buděšínský, I. Císařová and T. Kraus, Chem. Commun., 2009, 3557–3559 RSC.
- F. Wang, C. Han, C. He, Q. Zhou, J. Zhang, C. Wang, N. Li and F. Huang, J. Am. Chem. Soc., 2008, 130, 11254–11255 CrossRef CAS PubMed.
- S. Dong, X. Yan, B. Zheng, J. Chen, X. Ding, Y. Yu, D. Xu, M. Zhang and F. Huang, Chem. – Eur. J., 2012, 18, 4195–4199 CrossRef CAS PubMed.
- F. Wang, B. Zheng, K. Zhu, Q. Zhou, C. Zhai, S. Li, N. Li and F. Huang, Chem. Commun., 2009, 4375–4377 RSC.
- G. B. Rocha, R. O. Freire, A. M. Simas and J. J. Stewart, J. Comput. Chem., 2006, 27, 1101–1111 CrossRef CAS PubMed.
- J. J. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- D. Komáromy, M. C. A. Stuart, G. Monreal Santiago, M. Tezcan, V. V. Krasnikov and S. Otto, J. Am. Chem. Soc., 2017, 139, 6234–6241 CrossRef PubMed.
- S. Otto, Chem. Sci., 2013, 4, 2953–2959 RSC.
- W. Blokzijl and J. B. F. N. Engberts, Angew. Chem., Int. Ed. Engl., 1993, 32, 1545–1579 CrossRef.
- D. Luo, S. W. Smith and B. D. Anderson, J. Pharm. Sci., 2005, 94, 304–316 CrossRef CAS PubMed.
- P. Nagy, Antioxid. Redox Signaling, 2012, 18, 1623–1641 CrossRef PubMed.
- J. Atcher and I. Alfonso, RSC Adv., 2013, 3, 25605 RSC.
- M. Trujillo, B. Alvarez and R. Radi, Free Radical Res., 2016, 50, 150–171 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic protocols, NMR, LC-MS, HPLC data. See DOI: 10.1039/d1tc01445e |
| This journal is © The Royal Society of Chemistry 2021 |