
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the architectures and luminescence properties of Cu(I) compounds of phenyl and carboranyl pyrazoles: the impact of 2D versus 3D aromatic moieties in the ligand backbone†
Joan
Soldevila-Sanmartín
 ab,
Eliseo
Ruiz
c,
Duane
Choquesillo-Lazarte
d,
Mark E.
Light
e,
Clara
Viñas
a,
Francesc
Teixidor
a,
Rosario
Núñez
*a,
Josefina
Pons
b and
José
G. Planas
*a
ab,
Eliseo
Ruiz
c,
Duane
Choquesillo-Lazarte
d,
Mark E.
Light
e,
Clara
Viñas
a,
Francesc
Teixidor
a,
Rosario
Núñez
*a,
Josefina
Pons
b and
José
G. Planas
*a
aInstitut de Ciència de Materials de Barcelona, ICMAB-CSIC, Campus UAB, 08193 Bellaterra, Spain. E-mail: rosario@icmab.es; jginerplanas@icmab.es
bDepartament de Química, Universitat Autònoma de Barcelona, Bellaterra, 08193 Barcelona, Spain
cDepartament de Química Inorgànica i Orgànica and Institut de Recerca de Química Teòrica i Computacional, Universitat de Barcelona, Diagonal 645, 08028 Barcelona, Spain
dLaboratorio de Estudios Cristalógraficos, IACT (CSIC-Universidad de Granada), Avda. de las Palmeras 4, Armilla, Granada, 18100, Spain
eSchool of Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK
First published on 28th May 2021
Abstract
Incorporation of one or two o-carborane moieties at the backbone of the pyrazole ring was achieved by lithiation and nucleophilic addition onto the corresponding 3,5-dimethyl-1-(2-toluene-p-sulfonyloxyethyl)pyrazole. Two monosubstituted carboranyl pyrazoles (L2 and L3) and one disubstituted carboranyl pyrazole (L4) were synthesized and fully characterized. All new compounds, and the corresponding monosubstituted phenylderivative (L1) behave as N-type ligands upon coordination with CuI to afford different polynuclear Cu(I) compounds 1–4. Compounds 1–4 were fully characterized and their molecular structures were determined by X-ray diffraction. It is noteworthy that whereas the pyrazolylphenyl ligand L1, without o-carborane, provides a 1D coordination polymer (1), ligands containing carborane, L2–L3, affords 0D coordination compounds 2 and 3, and disubstituted carboranyl pyrazole ligand L4 gives rise to a 3D coordination polymer. The photoluminescence behaviour of compounds 1–4 has been investigated in the solid state and by TDDFT calculations for molecular compounds 2 and 3. Complex 2 exhibits blue emission with a maximum at 483 nm and a high fluorescence quantum yield of 66.5%. According to TDDFT calculations the emission occurs from LUMO to HOMO−1 and HOMO−2 and deexcitation could be described as cluster-centred excited state of d–s transition in origin. This result contradicts previous studies of scarce tri-coordinated rhombohedral Cu(I) clusters, where it was assumed the origin of their emissions is (X + M)LCT in nature by analogy with tetra-coordinated rhombohedral Cu(I) clusters. Complex 3 exhibits very weak emission (ΦF of 5%) in the green region with a maximum at 517 nm, which according to TDDFT is through a 3CC state. Calculations also show that, upon excitation, 3 suffers a notable distortion resulting in the total cleavage of the Cu4I4 framework. This cleavage could be the cause of the relatively large Stokes shift observed for 3. To the best of our knowledge, this is the first time that such behaviour is observed for this type of octahedral compounds. Additionally, the 1D polymer 1 exhibits weak fluorescence emission in the orange range with a maximum at 609 nm and a remarkable Stokes shift, whereas the 3D polymer 4 exhibits a similar emission to compound 2, with a moderate quantum yield (ΦF of 13.7%).
Introduction
Polynuclear copper(I) halide clusters have been widely investigated over decades due to their intriguing photophysical properties.1–10 Potential applications for these materials include light-emitting devices (OLEDs),11–13 light-emitting electrochemical cells (LECs),14 solar cells15 and luminescent sensors.16,17 The interest in this family of compounds does not only stems from their photoluminescent properties, but also from the great diversity of structural motifs of the CuxXyLz (X = Cl, Br, I) skeleton.4,18 Thus, several structural motifs have been reported, ranging from 0D (coordination compounds) to 1-3D aggregates (coordination polymers).4,12,18 For instance, Cu2X2 rhomboid dimer or the Cu4X4 cubane and stepped-cubane are among the most common 0D structural motifs,4,18 while octahedral Cu4X4 motifs are less common19 (Chart 1). Regarding higher dimension aggregates, typical structural motifs include single-, double-, looped-, helical-chains, ribbons, columns, etc.16,18,20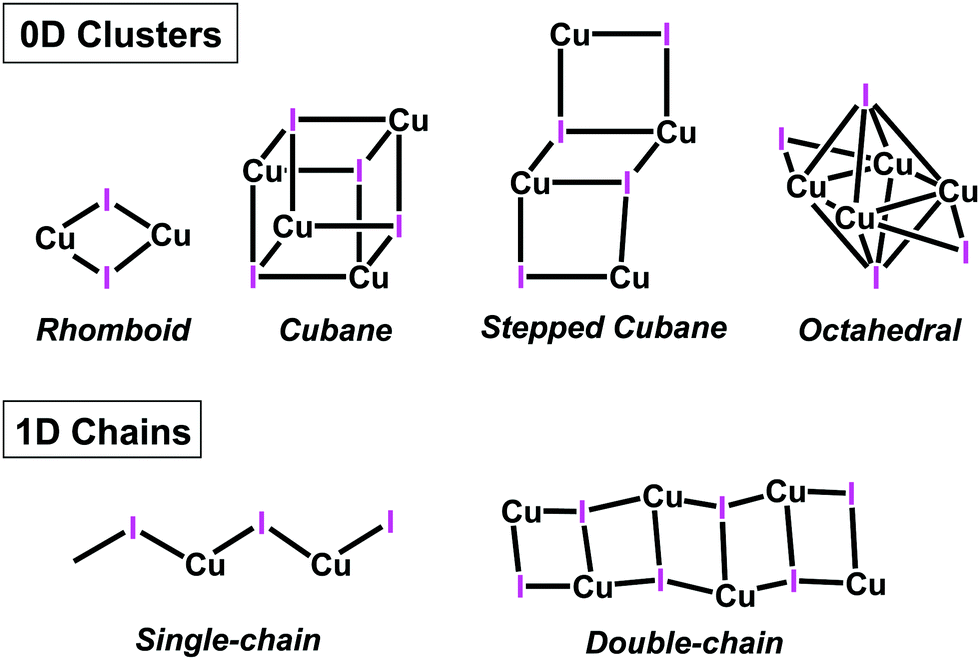 | ||
| Chart 1 Schematic representation of selected CuI aggregates in copper(I) coordination compounds and polymers. | ||
These different nuclear multiplicities and aggregate dimensionalities are not only intriguing per se, but are also interesting because their structural parameters greatly affect the photophysical properties of the resulting compounds. For instance, in cubane-like clusters a dual emission is observed arising from two different excited states. An halide-to-ligand charge transfer (3XLCT, sometimes mixed with metal-to-ligand charge transfer, 3MLCT) gives rise to a high energy (HE) emission (ca. 450 nm) while Cu4I4 cluster-centred (3CC) excited state yields a low energy (LE) emission (ca. 600 nm), which is related to the Cu–Cu distances.4,21 In fact, this relationship between Cu–Cu distance and the LE emission gives raise to one of the most interesting properties of cubane compounds: thermochromism.22 However, this LE emission can be quenched using rigid ligands that prevent cluster relaxation, thus “blocking” this emission,23 or fine-tuned using different heterocyclic donors.24 In contrast, stair-step clusters only show HE emission, due to having longer Cu–Cu distances.25 Regarding octahedral compounds, reports including photophysical studies are much scarcer, with fewer than a dozen papers published on the subject.19,23,25–31 This can be attributed to the fact that octahedral cores are energetically unfavourable compared to the cubane cores, as demonstrated by theoretical calculations.28 Optical properties of these octahedral compounds have also been studied and various reports suggest that the rigidity and steric demands of the ligands seems to play a more significant role in the photophysical properties than in the cubane type clusters.19
With regards to the ligands employed in the above mentioned polynuclear Cu(I) halides, nitrogen- (N-), sulfur- (S-) and phosphorus- (P-ligands) containing ligands are commonly found.4,5,18,32 Structures with N-ligands (either monodentate or multidentate) are by far more common and show intriguing luminescent properties.25,33 Reported examples on octahedral motifs include bidentate ligands bearing small angles (e.g., P^N,23,25,27,28,30 P^P,26,29,31,34–37 C^P38 and N^N19), with only one example based on a monodentate N-heterocyclic carbene ligand.39 Within the family of polynuclear Cu(I) compounds containing N-ligands, pyrazoles are significantly less studied than the overwhelming family of pyridines.32 Furthermore, pyrazole ligands employed in such polynuclear Cu(I) compounds are deprotonated (pyrazolate ligands) or neutral pyrazolyl-pyridine/pyrimidine combinations.32,40–44 To our knowledge, there is only one report on a neutral monodentate only-pyrazole (not having other nitrogenated substituents) based Cu(I) complex.45 There are however some more reports on neutral polypyrazolyl ligands, leading to a wide variety of coordination polymers.46–49
As part of our on-going research on pyrazole50–55 and carborane56–63 based ligands for functional metal compounds, we were interested in exploring the impact of adding the icosahedral carborane clusters to the pyrazole fragment on the coordination to Cu(I) ions and the corresponding luminescence properties. Icosahedral carboranes 1,n-C2B10H12 (n = 2, 7 or 12) are a class of commercially available and exceptionally stable 3D-aromatic boron-rich clusters that possess material-favourable properties such as thermal and chemical stability and high hydrophobicity.64–66 Neutral carboranes are remarkably robust boron clusters with two carbon atoms and possess 26 electrons for 12 vertices. The delocalized electron density is not uniform through the cage, giving rise to extraordinary differences in the electronic effects of the cluster.67 This unusual electronic structure is often highlighted by regarding carboranes as inorganic three-dimensional “aromatic” analogues of arenes.68,69 The average size of the o-carborane (148 Å3) is comparable to that of adamantane (136 Å3) and is significantly larger (40%) than the phenyl ring rotation envelope (102 Å3).70 Regarding the electronic effect, o-carborane behaves as a strong electron-withdrawing group (similar to fluorinated aryl) on a substituent at one of the cluster carbons.70–74 Thus, introduction of the carborane moiety into a ligand backbone is expected to exert a weaker metal–heteroatom interaction with respect to the related phenyl derivative.58,74,75 In addition, and different from classical rigid flat aromatic ligands, spherical-based linkers have access to extensive conformational space by a combination of low-energy torsion of the substituents (e.g., aliphatic) and by the spherical core of the linker.76 Thus, for example, a spherical ligand shape such as carborane could prevent the formation of intermolecular π–π staking interactions while favouring the weak dihydrogen bond interactions.77,78 Furthermore, a remarkable influence of the carboranes on the photophysical properties of luminescence materials has also been demonstrated.79,80 When o-carborane is linked to an aryl donor group, an intramolecular charge transfer (ICT) process occurs from the donor moiety to the acceptor cluster, which is influenced by the Ccluster–Ccluster (Cc–Cc) bond vibration,81 usually producing a quenching of the fluorescence in solution. Moreover, these systems exhibit aggregation-induced emission (AIE) in aggregate and solid states, due to a restriction of the molecular motion, which produces an important increase of the emission quantum yield.82–85 It is also noteworthy that the design of carborane–Cc-containing fluorophores whose fluorescence quantum yield in solution can be tailored by the cluster isomer (ortho- or meta-carborane) and the substituent at the Cc atom. Some of these systems show moderate to good fluorescence emission efficiency in solution and solid state, giving rise to fluorescent materials in both states.86–91
Herein we report the synthesis of three new pyrazolyl-carborane derivatives and their Cu(I) compounds. We also report a new Cu(I) derivative of a commercially available N-substituted phenyl pyrazole ligand (L1) that was taken for comparison between 2D and 3D aromatic groups (Scheme 1). We present the synthesis, structural characterization, and photophysical properties in solid state for all compounds. Additionally, photophysical properties for the molecular coordination compounds have been analysed by TDDFT calculations.
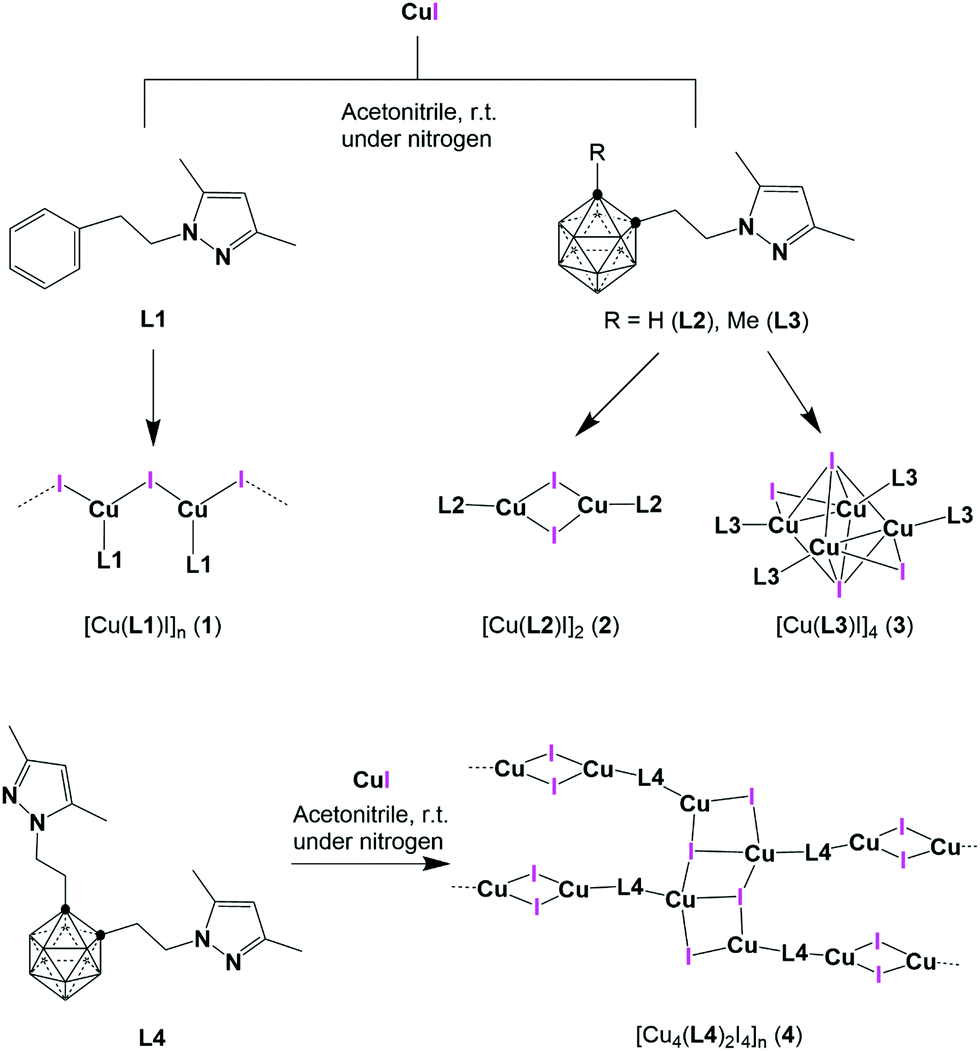 | ||
| Scheme 1 Representation of pyrazole ligands L1–L4 and their Cu(I) compounds 1–4. | ||
Results and discussion
Synthesis and characterization
Three new N-substituted carboranyl pyrazole ligands, derived from o-carborane, L2–L4 (Scheme 1), were prepared by the reaction of lithiated o-carborane with the corresponding 3,5-dimethyl-1-(2-toluene-p-sulfonyloxyethyl)pyrazole at low temperature (see ESI† for details). All carboranyl pyrazole ligands were fully characterized by standard spectroscopic and analytical techniques (ESI,† Fig. S1–S16) and the structure of L3 has been solved by single crystal X-ray diffraction (SCXRD; Fig. S17, ESI†). Reactions of the phenyl pyrazole ligand L1 or the carboranyl pyrazole ligands (L2–L4) with anhydrous CuI in dry acetonitrile under nitrogen provided crystalline compounds 1–4 in good yields (Scheme 1). FTIR-ATR spectra showed the characteristic signals in the region from 1556 to 650 cm−1 that belong to vibrations of the pyrazole ring (Pz) and of the N-substituted pyrazole derivatives.92,93 Carborane based compounds 2–4 show the characteristic broad B–H stretching bands from the carborane (in the range 2572–2563 cm−1; Fig. S18–S21, ESI†). Crystal structures of these Cu(I) compounds were determined by SCXRD (vide infra), and their simulated PXRD patterns were compared with their experimental ones, confirming their phase purity (Fig. S22–S24, ESI†). As shown in Scheme 1, compounds 1 and 4 are insoluble coordination polymers, while compounds 2 and 3 are molecular coordination compounds and therefore soluble in most organic solvents. All spectroscopic and analytical data is consistent with the solid-state structures determined by single crystal X-ray diffraction methods (vide infra and Fig. S25–S32, ESI†)Crystal structures
Suitable crystals for SCXRD were obtained for all compounds (Table 1). Coordination polymers 1 and 4 crystallized in the monoclinic P21/c space group, while molecular compounds 2 and 3 crystallized in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Fig. 1–3).
space group (Fig. 1–3).
| Compound | L3 | 1 | 2 | 3 | 4 |
|---|---|---|---|---|---|
| Empirical formula | C10H24B10N2 | C13H16CuIN2 | C18H44B20Cu2I2N4 | C20H48B20Cu2I2N4 | C34H67B20Cu4I4N9 |
| Formula weight | 280.41 | 390.72 | 913.65 | 941.70 | 1579.92 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Triclinic | Monoclinic |
| Space group | P21/c | P21/c |
P |
P |
P21/c |
| CCDC | 2048361 | 2050921 | 2050922 | 2050923 | 2050924 |
| Unit cell dimensions | |||||
| a (Å) | 10.8644(2) | 4.2749(3) | 10.361(2) | 12.0222(8) | 13.3779(13) |
| b (Å) | 20.6842(3) | 19.9244(15) | 12.195(2) | 12.9633(8) | 19.272(2) |
| c (Å) | 7.79360(10) | 16.0826(11) | 15.412(3) | 14.3391(9) | 23.825(2) |
| α (°) | 98.412(6) | 72.561(2) | 90.0 | ||
| β (°) | 106.795(2) | 97.106(2) | 91.117(6) | 78.697(2) | 9.679(2) |
| γ (°) | 96.772(6) | 64.922(2) | |||
| V (Å3) | 1676.68(5) | 1359.31(17) | 1911.6(6) | 1924.9(2) | 6055.0(10) |
| Z | 4 | 4 | 2 | 2 | 4 |
| F(000) | 592 | 760 | 888 | 920 | 3048 |
| θ (range) | 3.107–29.515° | 1.678–25.718° | 1.981–27.522° | 2.555–27.28° | 2.285–27.522° |
| Max./min. transmission | 1.000/0.6042 | 0.4920/0.2737 | 0.7456/0.4209 | 0.0285/0.0064 | 0.7456/0.3935 |
| Ind refln (Rint) | 4322(1) | 2381 (0.0937) | 8766 (0.0594) | 6686 (0.0917) | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 904 (0.1346) 904 (0.1346) |
| Final R indices [I > 2σ(I)] | R 1 = 0.0703, wR2 = 0.2152 | R 1 = 0.0451, wR2 = 0.1262 | R 1 = 0.0408, wR2 = 0.0774 | R 1 = 0.0541, wR2 = 0.1341 | R 1 = 0.0509, wR2 = 0.1006 |
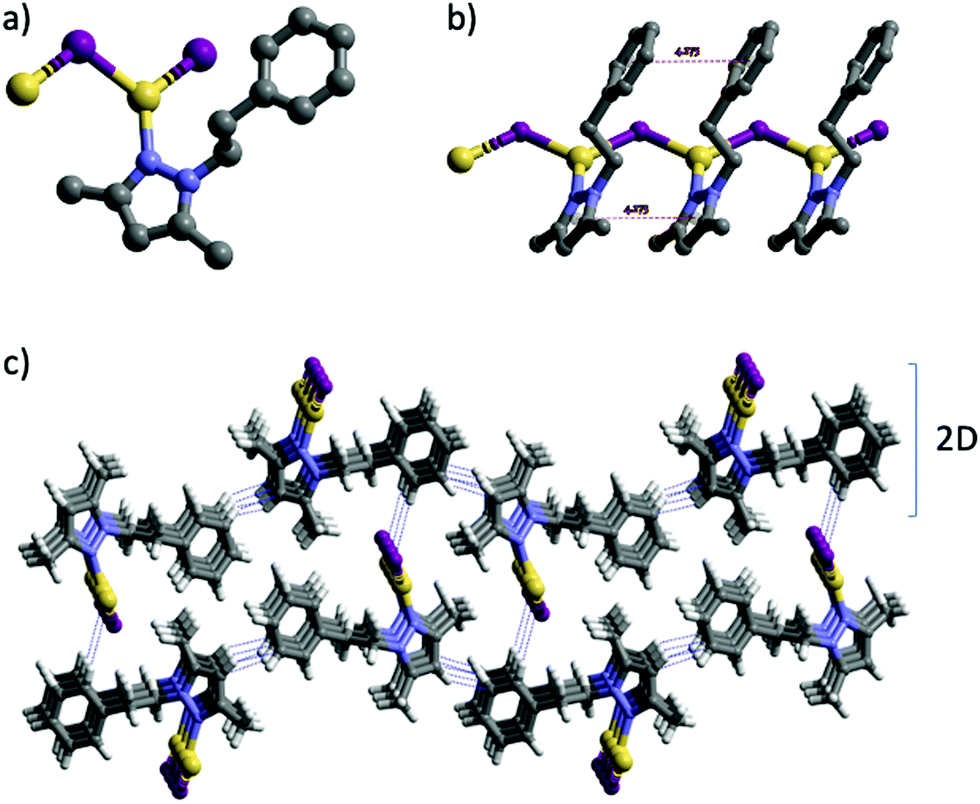 | ||
| Fig. 1 Crystal structure of 1: (a) view of the coordination environment of the Cu(I) centers. (b) Fragment of the 1D network of 1 showing the π–π interactions. (c) View of the 3D structure. H atoms have been omitted for clarity in (a) and (b). Intermolecular contacts are shown as dotted blue lines. Color codes: H pale grey; C grey; N blue; I violet; Cu yellow. | ||
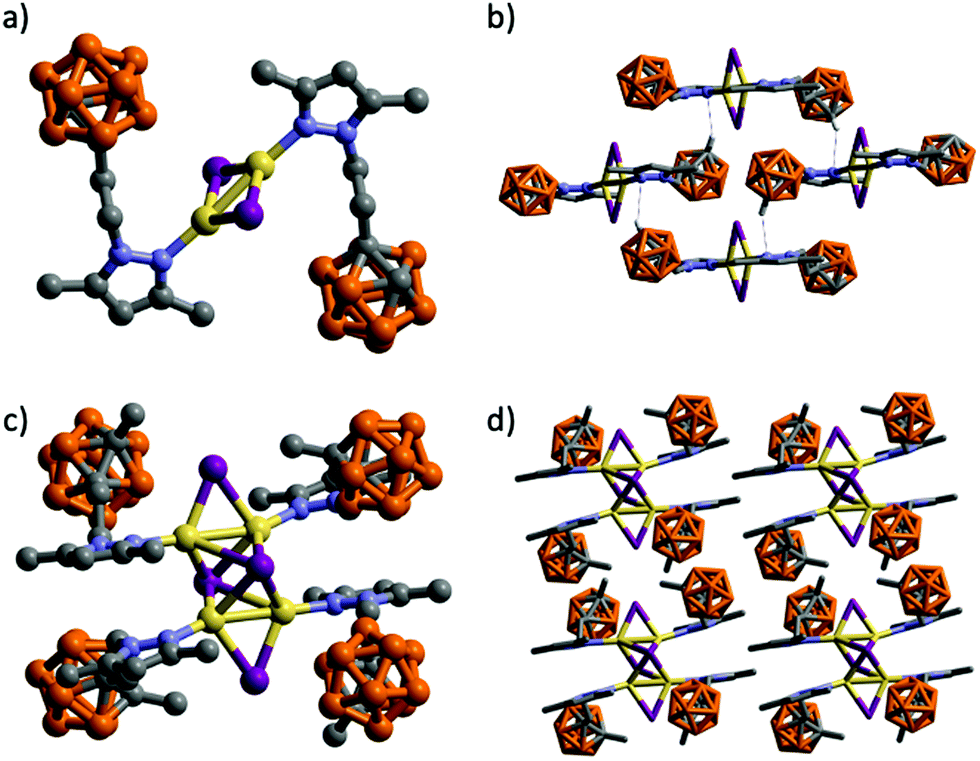 | ||
| Fig. 2 Molecular structures of 2 (a) and 3 (c) and two perspective views of the packing of four molecules (b) and (d). H atoms have been omitted for clarity. Intermolecular contacts are shown as dotted blue lines. Color codes: B orange; C grey; N blue; I violet; Cu yellow. | ||
The structure of 3 consists of a tetranuclear complex containing a Cu4I4 cluster and four molecules of ligand (Fig. 2c). Compound 3 represents a novel example of the rare octahedral clusters (Chart 1).19 The four Cu(I) atoms are arranged in a rectangular array with two significantly different Cu⋯Cu distances [2.5917(8) and 2.9289(7) Å]. The L3 and iodide ligands bridge each edge of the Cu4 plane to form a Cu4(L3)4I4 core. The Cu4 plane is further μ4-capped by two iodine atoms providing a distorted octahedron (Fig. 2c). The Cu–I bond lengths in μ4-bridging Cu–I lengths vary from 2.6985 to 3.1044 Å, which are significantly longer than the ones bridging the edges of the Cu4 plane (2.5662 and 2.6475 Å). The carboranyl pyrazole ligands coordinate to the four Cu(I) atoms through the ligand's pyrazole N atom (Fig. 2c). Cu–N distances vary from 1.985(4) to 1.990(4) Å. Selected bond lengths and angles are listed in Table S2 (ESI†). The geometric parameters have been compared with other reported examples. There are only fifteen entries in the CSD95 for eleven such type of octahedral cluster compounds and all of them bear two bidentate (neutral P^N25,31,96,97 or anionic N^N19) ligands. Thus, complex 3 represents the first example of such a family having monosubstituted neutral N-donor ligands. The 3D structure in this case is formed by self-assembly of molecules by weak intermolecular C–H⋯I (H⋯I 3.019 Å, C–H⋯I 161.7°) and C–H⋯H–B (H⋯H 2.448 Å, C–H⋯H 130.5°, B–H⋯H 110.4°) contacts (Fig. 2d and Fig. S33, ESI†).
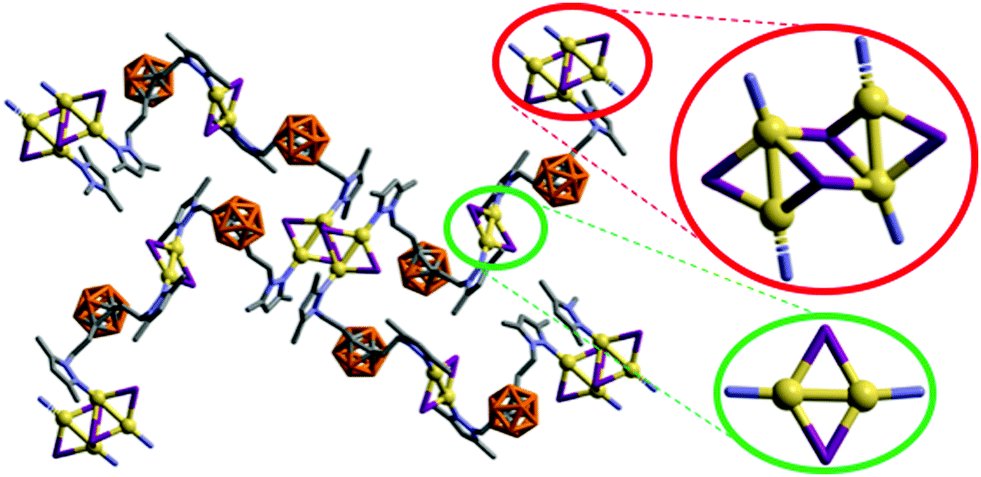 | ||
| Fig. 3 Fragment of the 3D structure of 4 showing rhombohedric [Cu2I2] (green) and stepped cubane [Cu4I4] (red) clusters. H atoms have been omitted for clarity. Color codes: B orange; C grey; N blue; I violet; Cu yellow. | ||
It is noteworthy that whilst the pyrazolylphenyl ligand (L1, Scheme 1) provides a Cu(I) coordination polymer, the corresponding ligands with carborane in place of phenyl (L2–L3, Scheme 1) provide 0D metal compounds (Scheme 1 and Fig. 1, 2). The π–π interactions observed between the pyrazole and phenyl rings of L1 in compound 1 indicate that most probably such interactions occur in solution and during the reaction with CuI to provide the observed single chain coordination polymer 1. Such π–π interactions are not possible when the phenyl ring is replaced by the carborane fragments and this obviously prevents the formation of coordination polymers. The latter can only be obtained by employing the disubstituted carboranyl pyrazole ligand L4 (Scheme 1 and Fig. 3).
Luminescence properties in the solid state
Compounds 1–4 are white crystalline solids under ambient light. The absorption spectra of 1–4 were measured in solid-state at room temperature (Fig. S35, ESI†). Polymer 1 shows three absorption bands at high energies (224, 269, 290 nm) and a shoulder at lower energy (369 nm). Complex 2 exhibits a maximum of absorption at 363 nm. The tetranuclear complex 3 shows three absorption regions, the first one at high energy in the range from 200 nm to 320 nm with a maximum at 306 nm, a second region in the range 320–410 nm and a the third one at lower energy from 410 tailing to 600 nm. Polymer 4 exhibits a very broad absorption band composed of two different regions, the high energy region from 210 nm to 320 nm with two maxima at 257 and 289 nm and a low energy region from 320 nm tailing to 600 nm. This broad absorption band of 4 seems to correspond to the combined absorption spectra for 2 and 3 (Fig. S35, ESI†). Absorption bands at higher energies have been associated with pyrazole ligand-centred π → π* transitions, and they correlate nicely with the UV-vis spectra of the free ligands (Fig. S16, ESI†). On the other hand, lower energy absorptions have been attributed to (X+M)LCT transfers.98 In order to further investigate the UV-vis spectra, theoretical calculations were performed for discrete coordination compounds 2 and 3 (vide infra).The normalised solid-state excitation and emission spectra for the four compounds are shown in Fig. 4. The emissive excited states observed in Cu(I)–halide compounds are known to originate from a variety of sources, such as halide-to-ligand charge transfer (XLCT),1,99 metal-to-ligand charge transfer (MLCT),1,99 intra-ligand charge transfer (ILCT),100 and, for Cu(I)-cluster compounds, a cluster-centred excited state of d–s character (CC).99 As such, their photophysical properties bear a strong intrinsic relationship with their structural features.
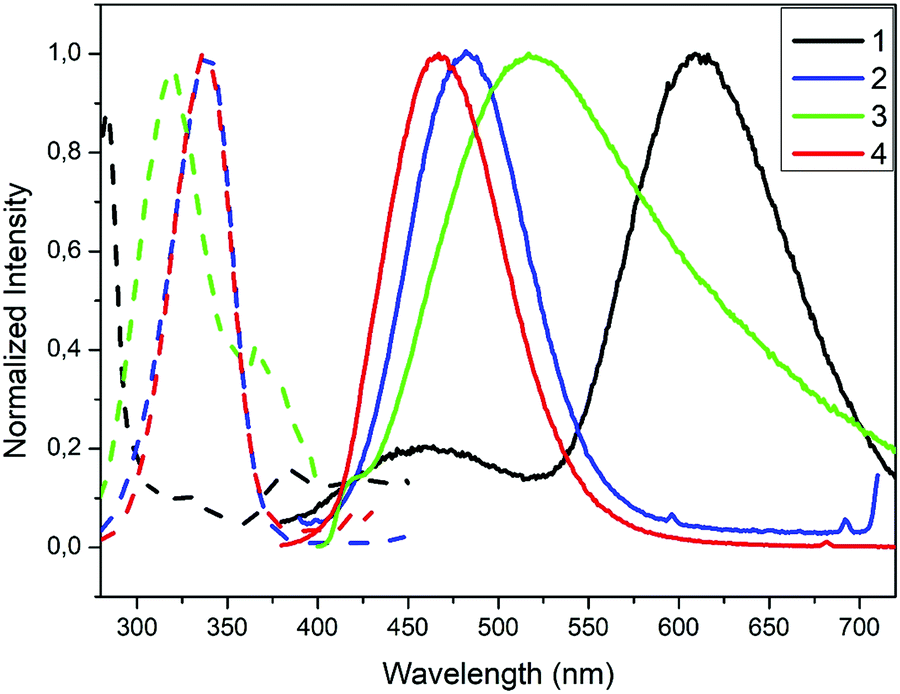 | ||
| Fig. 4 Excitation (dashed lines) and emission (solid lines) spectra for compounds 1–4 in the solid state at 298 K. | ||
The crystalline solid of polymer 1 exhibits dual emission in the blue (460 nm, HE) and orange (609 nm, LE) regions of the visible spectra upon excitation at 283 nm, with low absolute quantum yield (ΦF) of 4% (Table 2). The emission spectrum of 1 is broad and unstructured, which according to the literature on Cu(I) compounds is typical of charge-transfer (CT) emissions.101 Contrary to Cu(I)–iodide rhomboid dimers or tetramers, photophysical studies of single-stranded Cu(I)–iodide polymers are scarce. The first one was reported by Cariati et al. in 2002,3 and since, only few others have been reported.48,49,102,103 As the Cu–Cu bond length is the primary factor governing CC excited states in Cu(I) clusters,4 their long distances in 1 (4.275 Å, Table S4, ESI†), being larger than the sum of their van der Waals radii (2.8 Å), seem to preclude them as the source of its emission. Thus, according to the few reports on similar single-stranded Cu(I) polymers, the HE emission is attributed to a mixture of 3XLCT and 3MLCT,48,49,102 whereas LE emission to 3XMCT.104 Note that there are only two cases of double emission in single-stranded Cu(I) polymers, both reported by Cariati.3 Although similar results have been reported for double-stranded or grid-like Cu(I) polymers,105,106 direct extrapolation should be avoided, as the latter can possess 3CC based-emission104,107 and they show completely different structures. The remarkable Stokes shift for polymer 1 is striking in comparison with the other three compounds (Table 2). Such large shifts have been explained in 2D grid-like [CuBr(pyz)]n (pyz = pyrazine) and [CuBr(quinz)]n (quinz = quinazoline) compounds by a HOMO (based on a mix of halide 4p and metal 3d orbitals) to an antibonding LUMO (ligand based) charge transfer ((X + M)LCT).108 Charge transfer to an antibonding LUMO should cause a geometrical change in the bonds, resulting in a large Stokes shift. This fact is also consistent with the low measured quantum efficiency value for 1.
| Compound | UV-vis maximum absorbance (nm) | Excitation (λmax, nm) | Emission (λmax, nm) | Φ F (%) |
|---|---|---|---|---|
| 1 | 290 | 283 | 460, 609 | 3.8 |
| 2 | 363 | 340 | 483 | 66.5 |
| 3 | 306 | 318 | 517 | 4.9 |
| 4 | 349 | 336 | 467 | 13.7 |
Molecular compounds 2–3 emit at high energies, in the blue-to-yellow region (450–520 nm) of the visible spectrum (Fig. 4 and Table 2). A crystalline powder of complex 2 exhibits an outstanding blue emission compared to other analogues,103 with a maximum at 483 nm upon excitation at 340 nm, and an absolute quantum yield (ΦF) of 66.5%. The emission spectrum is also broad and unstructured, consistent with charge-transfer emissions.109 Among the reported Cu(I) rhomboid dimers, the majority corresponds to four coordinated Cu(I)-dimers, with only ca. 20% of the structures being three-coordinated Cu(I) compounds (such as in 2).110,111 Thus, photophysical properties of the tetra-coordinated Cu-dimers have been extensively studied; in particular the rhomboid dimers containing bidentate Npyridine,P-heteroleptic ligands have been extensively studied by Bräse's group.7,13,24,109,112 Their analyses have revealed that HOMO orbitals are mainly centred in the Cu2X2 framework and LUMO orbitals in the pyridine ligand. Thus, their emission stems mainly from (X + M)LCT, and can be tuned by slightly modifying the pyridine ligands.13,24,109,112 As mentioned above, dimeric Cu(I) compounds with tri-coordinated metal centres, such as 2, are much less studied than the corresponding tetra-coordinated compounds. Thus, the few reports in tri-coordinated compounds assume a similar origin for their emission than the tetra-coordinated one.113–115 We therefore decided to perform TDDFT calculations in order to verify or refute this hypothesis (vide infra). To the best of our knowledge, ours is the first report of photophysical studies of N-donor tri-coordinated Cu(I) rhomboid dimer compounds supported by TDDFT calculations.
The crystalline solid of complex 3 shows a very broad emission band with a maximum at 517 nm upon excitation at 318 nm at room temperature with an absolute ΦF of around 5% (Fig. 4 and Table 2). The latter is significantly lower than those reported for a number of homologous compounds.26,28 For some reported octahedral Cu4I4 compounds, dual emission has been observed, that is a LE emission (550–600 nm) dominating at room temperature and a HE emission (400–500 nm) dominating at lower temperatures.23,28,31 By analogy with the better studied Cu4I4Lx (x = 2–4) cubane clusters, these have been attributed to 3CC transitions and 3XLCT transitions respectively.25,26,29 However, recent studies have shown that other octahedral clusters display single LE or HE emission bands which are largely temperature independent,116,117 a phenomenon known as rigidochromism, while others display only a single emission in the region of 500–560 nm, halfway between LE and HE emission.28,30 Complex 3 seems to fall under this behaviour, as no dual emission is observed. This disparity in the results of photophysical studies for octahedral clusters show that a universally accepted process for rationalizing its photoluminescent behaviour has yet to been achieved. Therefore, we have performed the TD-DFT calculations for 3 (vide infra).
The crystalline solid of multinuclear coordination polymer 4 exhibits blue emission with a maximum at 467 nm upon excitation at 336 nm and an absolute quantum yield of 13.7% (Fig. 4 and Table 2). It is noteworthy that both the emission and excitation spectra of 4 are similar to those of complex 2 (Cu2I2 units). In one of the few Cu(I) compounds containing different metal clusters, it was observed that its photoluminescent behaviour was a combination of that of its constituent parts.44 Thus, assuming a similar behaviour for 4, it seems that the emission originating in the Cu2I2 unit dominates over that originating in the open cubane Cu4I4 unit. This fact is also consistent with the high ΦF observed for 2, compared to the low ΦF observed for the tetranuclear cluster in 3. Thus, we assume the emission for both 4 and 2 originate in the dimeric units. On this basis, the TDDFT calculations performed for 2 also help shed some light on the photophysical results obtained for 4 (vide infra).
In order to analyze and fully understand the photophysical properties of the molecular systems, time-dependent density functional theory (TDDFT) calculations (see Computational details section) were performed using Gaussian16 program,118 with the B3LYP functional119,120 using the Def2TZV basis set.121 Complex 2, containing a rhombohedral shape Cu2I2 core, has absorption and excitation peaks at 363 and 340 nm, respectively (Table 2). The calculated TDDFT values at B3LYP level show the first non-zero extinction coefficient (second excitation) at 321 and 335 nm, respectively for the optimized and experimental structures. The orbitals involved in such processes are represented in the top of Fig. 5. The excitation is from the HOMO to the LUMO+1, and it can be described as a (X + M)LCT transfer from the Cu2I2 framework to the ligand, much in the same manner as the tetracoordinated rhomboid dimers studied by Bräse's group.13,24,109,112 Previously, it has been reported that the deexcitation in these kinds of systems are through triplet states with long excited state lifetimes.122 The theoretical analysis of the emission spectra was done by optimizing the structure of the wavefunction corresponding to the triplet state (see Experimental section for details). The structure of the triplet excited state from 2 is very distorted with non-bridging iodide ligands (see Fig. 5), as already reported for other Cu2I2 systems.122 The Cu⋯Cu distance was found to be 2.346 and 2.593 Å in the optimized triplet excited and singlet ground state, respectively, and 2.516 Å in the experimental structure. The results indicate that the emission is from the LUMO orbital to HOMO−1 and HOMO−2 (see Fig. 5 bottom). There are triplet excitations (542, 498 and 487 nm) that are close to the experimental emission peak (483 nm). The analysis of the orbitals involved in such deexcitation transitions reveals that they basically occur among the Cu2I2 orbitals. The LUMO is mainly a combination of 4s copper orbitals while HOMO−1 and HOMO−2 correspond to bonding d–d orbitals (for dx2−y2 and dz2 pairs) of copper mixed with large contributions of p orbitals of the iodine atoms. Thus, such deexcitation transitions could be described as cluster-centred excited state of d–s transition in origin, therefore contradicting the previous reports that hypothesized that they were (X + M)LCT in origin.113–115 A similar result was observed for an adduct containing Cu2I2 and Cu3-pyrazolate clusters.122 Those results suggest that tetra-coordinated and tri-coordinated Cu2I2 clusters follow different emission behaviour.
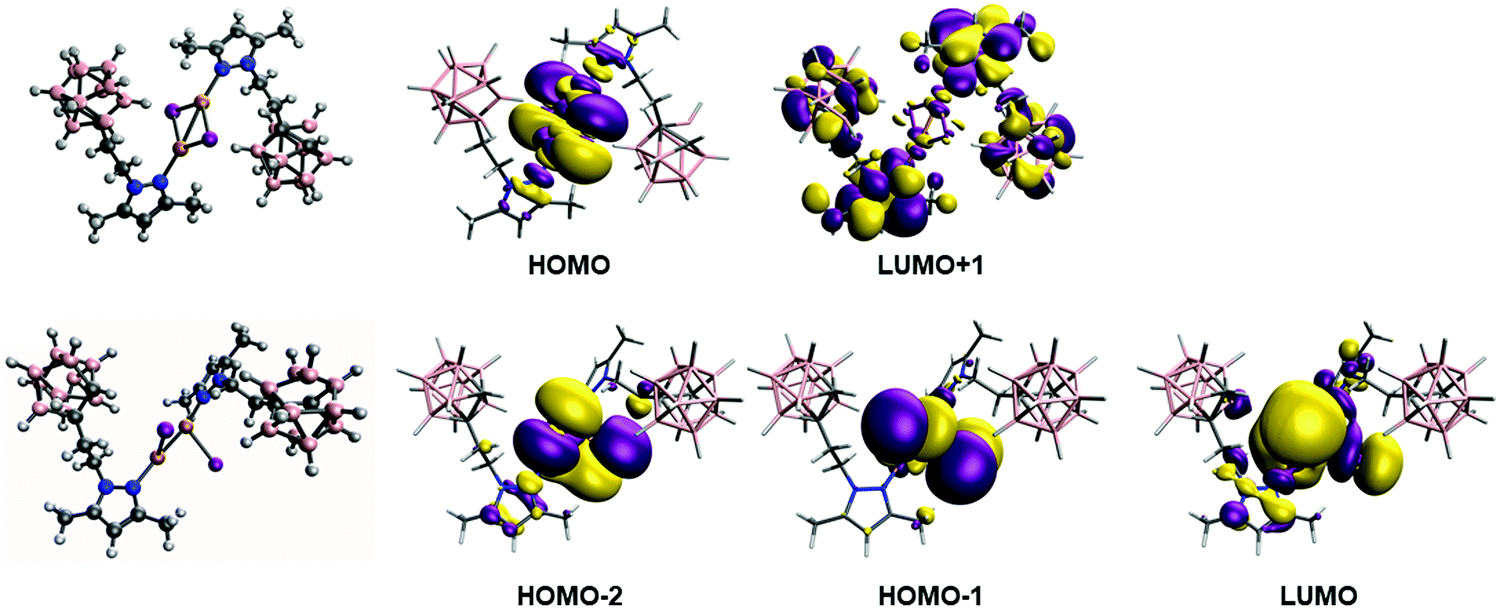 | ||
| Fig. 5 Top: Ground state structure and calculated orbitals at B3LYP level involved in the absorption process for 2. Bottom: Optimized structure of triplet state involved in the deexcitation process and the orbitals of the singlet state with such optimized structure. | ||
In the case of complex 3, there are four calculated excitations that have non-zero extinction coefficients in the same region as the experimental results. They correspond to transitions between a mixture of the three last occupied orbitals (HOMO, HOMO−1 and HOMO−2, Fig. 6 top) with large contribution of iodine and copper atoms to the first empty orbitals (LUMO, LUMO+1, LUMO+2 and LUMO+3, Fig. 6 bottom), which are mainly ligand-based. The calculated excitation values are 310, 305, 301, 295 nm for the experimental structure (second, third, sixth and seventh excitations) while for the optimized structure are 318, 312, 305 and 298 nm (first, third, fifth and seventh excitations). Such values are in good agreement with the experimental absorption and excitation peaks of 306 and 318 nm, respectively. The nature of such excitation is clearly a (X + M)LCT transfer from the Cu4I4 framework.
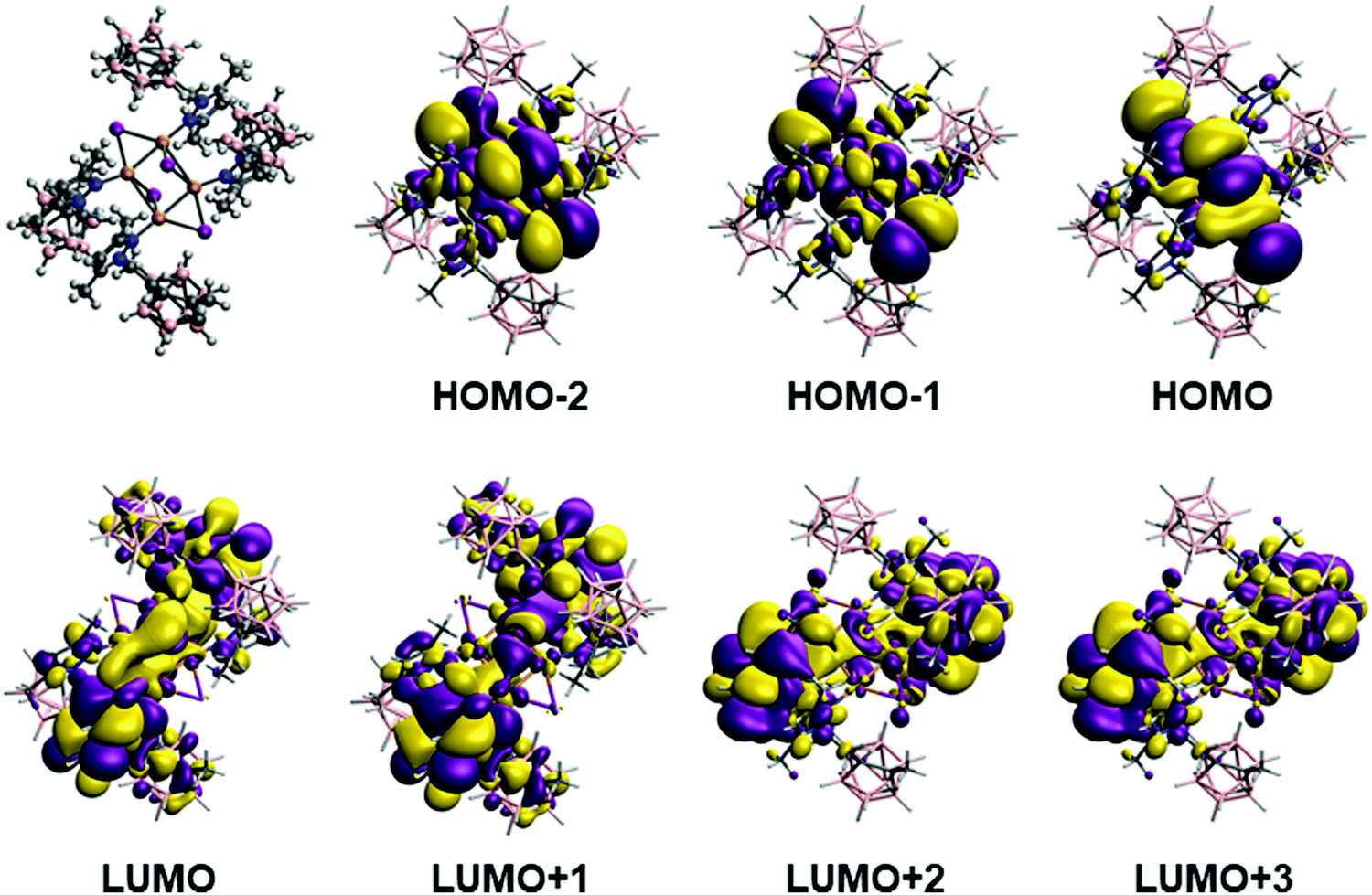 | ||
| Fig. 6 Optimized structure and calculated orbitals at B3LYP level involved in the absorption process that corresponds to transition between a mixture of the three last occupied orbitals to the first empty orbitals for complex 3. See the text for details. | ||
One of the most appealing features of the photophysical properties of 3 is the very large Stokes shift with an emission at 517 nm (Table 2). In order to analyse the origin of such a large shift in the emission, we have optimized the structure of the excited states of this molecule. The optimization of such state leads to a huge change in the structure of the Cu4I4 framework (see Fig. 7). As we have previously seen for 2, the structure is considerably distorted, especially the central Cu4I4 framework. One of the Cu2I2 units adopts a similar structure to that of the triplet state of complex 2 with a short Cu–Cu distance (2.363 Å) and with terminal iodide ligands (see Fig. 5 and 7). The second Cu2I2 moiety has the bridging iodide ligands and a relatively long Cu⋯Cu distance (2.655 Å). The calculation of the vertical excitations using TDDFT of the singlet ground state for such triplet optimized structure gives triplet deexcitation transitions at 538 and 531 nm in good agreement with the experimental emission band at 517 nm. The analysis of the orbitals involved indicates a deexcitation from the LUMO orbital (Fig. 7) mainly localized in the Cu2I2 moiety with short Cu⋯Cu distance to orbitals delocalized in the whole Cu4I4 framework (HOMO and HOMO−1, Fig. 7). Thus, as for 2, the deexcitation process can be described as a 3CC process.
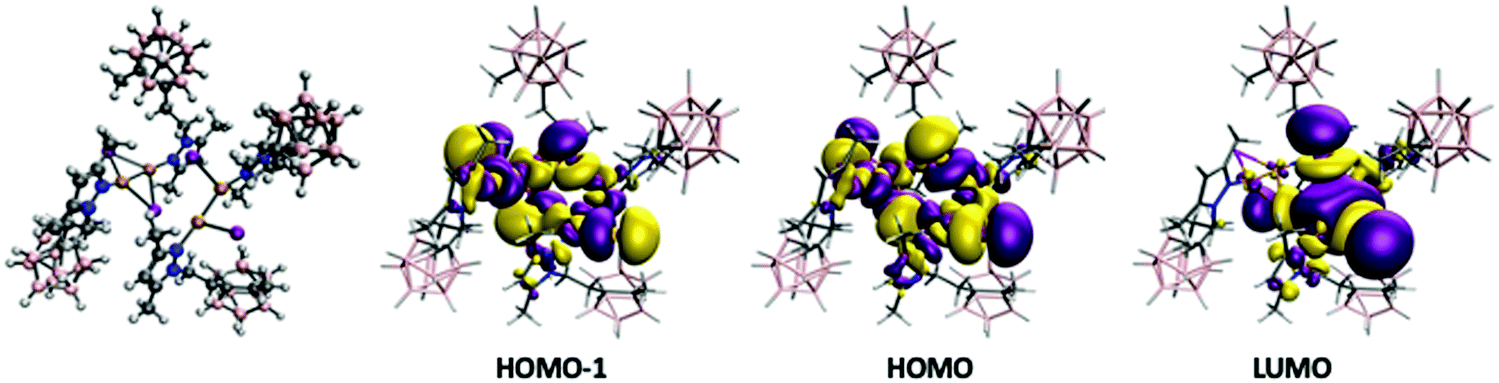 | ||
| Fig. 7 Optimized structure of triplet state of 3 involved in the deexcitation process and the orbitals of the singlet state with such optimized structure. See the text for details. | ||
Conclusions
In summary, we have synthesized and fully characterized two monosubstituted carboranyl pyrazole ligands (L2 and L3) and one disubstituted carboranyl pyrazole ligand (L4) by lithiation and nucleophilic addition onto the corresponding 3,5-dimethyl-1-(2-toluene-p-sulfonyloxyethyl)pyrazole. All new ligands, and the corresponding monosubstituted phenyl derivative (L1) behave as N-type ligands upon coordination with CuI to afford a series of polymeric ([Cu(L1)I]n (1) and [Cu4(L4)2I4]n (4)) or molecular ([Cu(L2)I]2 (2) and [Cu(L3)I]4 (3)) Cu(I) structures. Whereas 1, having a phenyl-based pyrazole, forms a coordination polymer, 2 and 3, both possessing a carboranyl-based pyrazole ligand, form molecular compounds. Lastly, the use of bidentate L4, results in the formation of another coordination polymer 4. 3 represents a novel example of the rare octahedral clusters, and the first one bearing a neutral monodentate ligand. 4 represents the first example where both dimeric and stepped cubane clusters are combined in a single coordination polymer.Absorption and emission properties of 1–4 have been studied in the solid state. Their emission covers a remarkable range of the visible spectrum, from the blue to the orange (460–609 nm). Noticeably, complex 2 exhibits an outstanding fluorescence efficiency of 66.5%, which is higher than that observed for reported analogues without the carborane units. TDDFT calculations performed for 2 and 3 allowed us to gain insight in the origin of their emission. For 2, they confirm that the emission origin is a cluster-centred excited state of d–s transition. This result is remarkable, as, to date, the origin of the emission in similar dimers has been assumed to be (X + M)LCT in nature. Regarding compound 3, TDDFT calculations indicate that the emission is through a 3CC state. Calculations also show that, upon excitation, 3 suffers a notable distortion resulting in the total cleavage of the Cu4I4 framework. To the best of our knowledge, this is the first time that such behaviour is observed for this type of octahedral compounds. These results depart from the classical assumption that octahedral Cu4I4 clusters behave as cubane clusters, highlighting that more effort is required to rationalise their emission.
We demonstrate that it is possible to tune the molecular structure and luminescence properties of these Cu(I) compounds by changing a phenyl ring (2D) by a carborane cluster (3D), which also allows introducing substituents at the cage. Herein, we report new understanding on some significant luminescence aspects for a rare type of Cu(I) complex for the first time and explore the relationship between the structural features and their luminescent behaviour, a knowledge that is key in the rational design of photoluminescent materials.
Experimental section
Materials and methods
All synthetic procedures were performed under dinitrogen atmosphere using standard Schlenk techniques unless otherwise noted. Tetrahydrofuran (THF) was purchased from Carlo Erba Reagents S.A. and distilled from Na/benzophenone prior to use. Commercial grade acetonitrile (CH3CN) was purchased from Merck and dried using 3 Å activated molecular sieves under dinitrogen atmosphere for 48 h. Spectroscopic grade methanol (MeOH) was purchased from ROMIL-SpS and used without further purification. 3,5-Dimethyl-1-(2-toluene-p-sulfonyloxyethyl)pyrazole was synthesized according to reported procedures.123 Ligand L1 and all other chemicals were commercially available and used as received.:4, Rf = 0.76; L4, eluent = ethyl acetate/hexane 9:1, Rf = 0.68). The products were extracted from the corresponding silica portions with CHCl3 and obtained as white powders on solvent evaporation. The products were washed with cold hexane (2 mL) and dried under vacuum.
L2 . Yield: 33% (0.622 g). Elem. anal. calc. for C9H22B10N2 (266.39): C 40.58, H 8.32, N 14.01. Found: C 40.49, H 8.23, N 13.85. M.p.: 172–176 °C. ATR-FTIR (wavenumber, cm−1): 2982–2917 (w) [ν(C–H)al], 2561 (s) [ν(B–H)], 2551 (s) [ν(B–H)], 1555 (s) [ν(C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/CN)ar], 1489 (w), 1466 (m), 1442 (m), 1425 (s) [δ(CC)/δ(CN)ar], 1377 (w), 1372 (m), 1321 (m), 1282 (w), 1218 (w), 1139 (w), 1071 (m), 1024 (s) [δ(C–H)ip], 988 (w), 937 (w), 903 (w), 835 (w), 789 (vs) [δ(C–H)oop], 719 (vs) [δ(C–H)oop], 673 (w), 656 (w), 640 (w), 556 (w), 540 (w). 1H NMR (CD3CN, 400.0 MHz): δ = 5.84 (s, 1H, CH(Pz)), 4.43 (br, 1H, Ccarb–H) 4.06 (m, 2H, NPzCH2CH2Ccarb), 2.75 (m, 2H, NPzCH2CH2Ccarb), 2.19 (s, 3H, Pz-CH3), 2.10 (s, 3H, Pz-CH3). 11B NMR (CD3CN, 128.6 MHz) δ = −2.75 (br d, 1JBH = 155.0, 1B), −5.67 (br d, 1JBH = 155.0, 1B), −9.66 (br d, 1JBH = 149.5, 2B), −10.2 to −13.6, overlapping signals (6B). 11B{1H} NMR (CD3CN, 128.6 MHz) δ = −2.88 (br s, 1B), −5.75 (br s, 1B), −9.65 (s, 2B), −11.15 to −13.0, overlapping signals (6B). 13C{1H} NMR (CD3CN, 100.6 MHz): δ = 147.6, 145.5 (Pz-C), 108.1 (Pz-CH), 73.0 (Ccarb–CH2R), 63.7 (Ccarb–H), 47.3 (Ccarb–CH2CH2NPz), 36.5 (Ccarb–CH2CH2NPz), 11.8, 11.2 (CH3(Pz)). UV-vis: (CH3CN, 6.5 × 10−5 M) λmax (ε (M−1 cm−1)) = 201 nm (6380).
C/CN)ar], 1489 (w), 1466 (m), 1442 (m), 1425 (s) [δ(CC)/δ(CN)ar], 1377 (w), 1372 (m), 1321 (m), 1282 (w), 1218 (w), 1139 (w), 1071 (m), 1024 (s) [δ(C–H)ip], 988 (w), 937 (w), 903 (w), 835 (w), 789 (vs) [δ(C–H)oop], 719 (vs) [δ(C–H)oop], 673 (w), 656 (w), 640 (w), 556 (w), 540 (w). 1H NMR (CD3CN, 400.0 MHz): δ = 5.84 (s, 1H, CH(Pz)), 4.43 (br, 1H, Ccarb–H) 4.06 (m, 2H, NPzCH2CH2Ccarb), 2.75 (m, 2H, NPzCH2CH2Ccarb), 2.19 (s, 3H, Pz-CH3), 2.10 (s, 3H, Pz-CH3). 11B NMR (CD3CN, 128.6 MHz) δ = −2.75 (br d, 1JBH = 155.0, 1B), −5.67 (br d, 1JBH = 155.0, 1B), −9.66 (br d, 1JBH = 149.5, 2B), −10.2 to −13.6, overlapping signals (6B). 11B{1H} NMR (CD3CN, 128.6 MHz) δ = −2.88 (br s, 1B), −5.75 (br s, 1B), −9.65 (s, 2B), −11.15 to −13.0, overlapping signals (6B). 13C{1H} NMR (CD3CN, 100.6 MHz): δ = 147.6, 145.5 (Pz-C), 108.1 (Pz-CH), 73.0 (Ccarb–CH2R), 63.7 (Ccarb–H), 47.3 (Ccarb–CH2CH2NPz), 36.5 (Ccarb–CH2CH2NPz), 11.8, 11.2 (CH3(Pz)). UV-vis: (CH3CN, 6.5 × 10−5 M) λmax (ε (M−1 cm−1)) = 201 nm (6380).
L4 . Yield: 13% (0.184 g). Elem. anal. calc. for C16H32B10N4 (388.56): C 49.46, H 8.30, N 14.01. Found: C 49.32, H 8.17, N 13.92. M.p.: 100–115 °C. ATR-FTIR (wavenumber, cm−1): 3128 (w) [ν(C–H)ar], 2996–2856 (w) [ν(C–H)al], 2563 (br, vs) [ν(B–H)], 1552 (s) [ν(C
C/CN)ar], 1450 (s), 1416 (s) [δ(CC)/δ(CN)ar], 1383 (s), 1365(s), 1315(s), 1258 (s), 1213 (m), 1173 (w), 1135 (w), 1080 (w), 1062 (w), 1020 (m), 981 (m), 980 (m), 788 (vs) [δ(C–H)oop], 717 (vs) [δ(C–H)oop], 670 (vs) [δ(C–H)oop], 632 (m), 534 (m).1H NMR (CD3CN, 400.0 MHz): δ = 5.78 (s, 2H, CH(Pz)), 4.09 (m, 4H, NPzCH2CH2Ccarb), 2.79 (m, 4H, NPzCH2CH2Ccarb), 2.21 (s, 12H, CH3(Pz)). 11B NMR (CD3CN, 128.6 MHz): δ = −4.71 (br d, 1JBH = 149.4, 1B), −9.64 to −11.64, overlapping signals (8B). 11B{1H} RMN (CD3CN, 128.6 MHz): δ = −4.71 (br s, 2B), −10.2 and −11.1 overlapping signals (8B). 13C{1H} NMR (CD3CN, 100.6 MHz): δ = 147.3 and 139.3 (Pz-C), 106.0 (CH(Pz)), 78.0 (Ccarb), 46.6 (NPzCH2CH2Ccarb), 33.9 (NPzCH2CH2Ccarb), 12.5 and 10.0 (CH3(Pz)). UV-vis: (CH3CN, 5.2 × 10−5 M) λmax (ε (M−1 cm−1)) = 214 nm (6231).
:3, Rf = 0.47). The product was extracted from the corresponding silica portions with THF and obtained as white powders on solvent evaporation. Crystals suitable for SCXRD were obtained via slow evaporation of the latter THF solutions.
L3 . Yield: 36% (0.326 g). Elem. anal. calc. for C10H24B10N2 (280.42): C 42.83, H 8.63, N 9.99. Found: C 42.59, H 8.33, N 9.69. M.p.: 138–142 °C. ATR-FTIR (wavenumber, cm−1): 3134–3061 (w) [ν(C–H)ar], 2978–2858 (w) [ν(C–H)al], 2571 (s) [ν(B–H)], 1554 (s) [ν(C
C/CN)ar], 1482 (w), 1463 (m), 1452 (s) [δ(CC)/δ(CN)ar], 1421 (s), 1388 (s), 1372 (w), 1317 (w), 1299 (w), 1278 (m), 1218 (w), 1177 (w), 1161 (w), 1129 (w), 1088 (w), δ(C–H)ip: 1024 (s), 977 (w), 946 (w), 921 (w), 779 (s) [δ(C–H)oop], 726 (s) [δ(C–H)oop], 666 (m), 648 (m), 550 (m). 1H NMR (CD3CN, 400.0 MHz): δ = 5.78 (s, 1H, CH(Pz)), 4.07 (m, 2H, NPzCH2CH2Ccarb), 2.72 (m, 2H, NPzCH2CH2Ccarb), 2.21 (s, 3H, CH3(Carb)), 2.10 and 2.08 (s, 6H, CH3(Pz)). 11B NMR (CDCl3, 128.6 MHz): δ = −4.42 (br d, 1JBH = 157.3 Hz, 1B), −6.08 (br d, 1JBH = 151, 1B), −8.28 to −11.40, overlapping signals (8B). 11B{1H} NMR (CD3CN, 128.6 MHz): δ = −4.4 (br s, 1B), −6.2 (br s, 1B), −8.9 to −10.8, overlapping signals (8B). 13C{1H} NMR (CD3CN, 100.6 MHz): δ = 148.2 and 140.1 (Pz-C), 106.9 (CH(Pz)), 47.7 (NPzCH2CH2Ccarb), 36.1 (NPzCH2CH2Ccarb), 23.6 (CH3(Carb)), 13.6 and 11.1 (CH3(Pz)). UV-vis: (CH3CN, 2.1 × 10−5 M) λmax (ε (M−1 cm−1)) = 196 nm (1012).
1 . Yield: 60.1% (0.023 g) elem. anal. calc. for C13H16IN2Cu (390.72): C 39.96, H 4.13, N 7.17. Found: C 39.83, H 4.10, N 7.14. ATR-FTIR (wavenumber, cm−1): 3126–3030 (w) [ν(C–H)ar], 2978–2855 (w) [ν(C–H)al], 1550 (s) [ν(C
C/CN)ar], 1496 (m), 1488 (m), 1462 (m), 1453 (s) [δ(CC)/δ(CN)ar], 1421 (w), 1388 (m), 1371 (w), 1357 (m), 1310 (m), 1253 (m), 1152 (w), 1075 (w), 1048 (w), 1031 (w), 904 (w), 800 (s), 746 (s) [δ(C–H)oop], 696 (s) [δ(C–H)oop] and 687 (s) [δ(C–H)oop]. UV-vis: (solid state) λmax = 290 nm. Fluorescence (solid state): λex = 283 nm, λem = 609 nm.
2 . Yield: 88.6% (0.259 g) elem anal. calc. for C18H44B20I2N4Cu2 (913.69): C 23.66, H 4.85, N 6.13. Found: C 23.88, H 4.97, N 6.10. ATR-FTIR (wavenumber, cm−1): 3034 (w) [ν(C–H)ar], 2986–2913 (w) [ν(C–H)al], 2574 (vs) [ν(B–H)], 1555 (s) [ν(C
C/CN)ar], 1466 (m) [δ(CC)/δ(CN)ar], 1445 (m), 1421 (w), 1392 (m), 1377 (m), 1319 (m), 1283 (m), 1215 (w), 1136 (w), 1119 (m), 1075 (s), 1049 (m), 1032 (m), 1016 (m), 997 (w), 932 (w), 916 (w), 877 (w), 839 (w), 794 (vs) [δ(C–H)oop], 722 (vs) [δ(C–H)oop], 697 (w), 674 (w), 656 (m), 582 (w), 561 (w) and 541 (w). 1H NMR (CD3CN, 400.0 MHz): δ = 5.82 (s, 1H, CH(Pz)), 4.47 (br s, 1H, Ccarb–H) 4.13 (m, 2H, NPzCH2CH2Ccarb), 2.76 (m, 2H, NPzCH2CH2Ccarb), 2.19 (s, 3H, Pz-CH3), 2.16 (s, 3H, Pz-CH3). 11B NMR (CD3CN, 128.6 MHz) δ = −2.86 (br d, 1JBH = 146, 1B), −5.72 (br d, 1JBH = 146, 1B), −9.7 (br d, 1JBH = 158.9, 2B), −10.9 to −13.7, overlapping signals (6B). 11B{1H} NMR (CD3CN, 128.6 MHz) δ = −2.9 (br s, 1B), −5.80 (br s, 1B), −9.7 (br s, 2B), −11.5 (br s, 2B), −12.1 (br s, 2B), −13.0 (br s, 2B). 13C{1H} NMR (CD3CN, 100.6 MHz): δ = 148.6, 140.7 (Pz-C), 106.3 (Pz-CH), 74.2 (Ccarb–CH2R), 63.6 (Ccarb–H), 47.6 (Ccarb–CH2CH2NPz), 37.2 (Ccarb–CH2CH2NPz), 13.8, 11.0 (CH3(Pz)). UV-vis: (solid state) λmax = 363 nm. Fluorescence (solid state): λex = 340 nm, λem = 483 nm.
Suitable crystals for SCXRD were obtained via recrystallization of a solution of the white powder in dry CH3CN.
Yield: 58.5% (0.052 g) elem. anal. calc. for C40H96B40N8I4Cu4 (1883.48): C, 25.51; H, 5.14; N, 5.95. Found: C, 25.21; H, 4.98; N 5.85. ATR-FTIR (wavenumber, cm−1): 2984–2917 (w) [ν(C–H)al], 2563 (s) [ν(B–H)], 1556 (s) [ν(CC/CN)ar], 1467(w), 1445 (s), 1418 (s) [δ(CC/CN)ar], 1376 (m), 1324 (m), 1280 (m), 1041 (m), 1023 (m), 779 (s) [δ(C–H)oop], 729 (s) [δ(C–H)oop], 676 (w), 663 (w), 647 (w) and 633 (w). 1H NMR (CD3CN, 400.0 MHz): δ = 5.82 (s, 1H, CH(Pz)), 4.18 (m, 2H, NPzCH2CH2Ccarb), 2.71 (m, 2H, NPzCH2CH2Ccarb), 2.22 (s, 3H, CH3(Pz)), 2.15 (s, 3H, CH3(Pz)), 2.10 (s, 3H, CH3(Carb)). 11B NMR (CD3CN, 128.6 MHz): δ = −4.46 (br d, 1JBH = 185, 1B), −6.05 (br d, 1JBH = 143 Hz, 1B), −8.68 (br d, 1JBH = 143, 2B), −9.48 to −11.98 overlapping signals, (6B, B-H). 11B{1H} NMR (CD3CN, 128.6 MHz): δ = −4.40 (br s, 1B), −6.10 (br s, 1B), −8.90 to −10.8, overlapping signals (8B). 13C{1H} NMR (CD3CN, 100.6 MHz): δ = 148.6 and 140.6 (Pz-C), 106.2 (CH(Pz)), 77.4 and 76.8 (Ccarb-CR), 47.7 (NPzCH2CH2Ccarb), 35.0 (NPzCH2CH2Ccarb), 23.7 (CH3Ccarb), 13.7 and 11.0 (CH3(Pz)). UV-vis: (solid state) λmax = 306 nm. Fluorescence (solid state): λex = 318 nm, λem = 517 nm.
4 . Yield: 42.1% (0.068 g) elem. anal. calc. for C34H67B20N9I4Cu4 (1538.92): C, 25.85 H, 4.27; N, 7.98. Found: C, 25.67; H, 4.19; N 7.86. ATR-FTIR (wavenumber, cm−1): 3132 (w) [ν(C–H)ar], 2990–2918 (w) [ν(C–H)al], 2572 (vs) [ν(B–H)], 1555 (s) [ν(C
C/CN)ar], 1464 (m), 1436 (m), 1417 (s) [δ(CC/CN)ar], 1376 (s), 1317 (m), 1285 (w), 1248 (m), 1219 (w), 1154 (w), 1113 (w), 1071 (w), 1066 (w), 1036 (s), 998 (m), 982 (m), 798 (vs) [δ(C–H)oop], 795 (vs) [δ(C–H)oop], 730 (s) [δ(C–H)oop], 672 (s), 660 (s), 544 (m), 526 (m). UV-vis: (solid state) λmax = 349 nm. Fluorescence (solid state): λex = 336 nm, λem = 467 nm.
Powder X-ray diffraction patterns (PXRD) for 1–3 were measured using a Siemens D5000 apparatus (with 40 kW and 45 mA using Cu Kα radiation with λ = 1.5406 Å). Patterns were recorded from 2θ = 5° to 40° with a step scan of 0.02° counting for 1 s at each step. For 4, PXRD pattern was measured using a Malvern PANalytical X’pert PRO Material Powder Diffractometer (with 45 kW and 40 mA using Cu Kα radiation with λ = 1.5406 Å). The pattern was recorded in transmission mode from 2θ = 2.5° to 40° with a step scan of 0.03° counting for 200 s at each step. This diffractometer was equipped with a capillary spinner and the sample was placed inside a borosilicate glass capillary with an outer diameter of 0.7 mm. Elemental analyses (C, H, N) were carried out on a Thermo Scientific Flash 2000 CHNS Analyser. Melting points were measured on a Stuart Academic SMP10 melting point apparatus. FTIR-ATR spectra were recorded on a high-resolution spectrometer FT-IR PerkinElmer spectrum One equipped with a universal attenuated total reflectance (ATR) accessory with a diamond window in the range of 4000–500 cm−1. 1H NMR, 11B NMR, 11B{1H} NMR and 13C{1H} NMR spectra were recorded on a NMR-FT Bruker 400 MHz spectrometer using deuterated acetonitrile (CD3CN) at 25 °C. Chemical shifts are reported in ppm and referenced to the residual solvent peak for 1H and 13C{1H} NMR or to BF3·OEt2 as an external standard for 11B and 11B{1H} NMR. Chemical shifts are reported in ppm and coupling constants in Hertz. Multiplets nomenclature is as follows: s, singlet; d, doublet; t, triplet; br, broad; m, multiplet.
The electronic spectra in CH3CN (1.45 × 10−5–6.5 × 10−5 M) were run on a JASCO V-780 UV-visible/NIR spectrophotometer with a quartz cell having a path length of 1 cm in the range of 190–600 nm. Solid-state electronic spectra were run on a JASCO V-780 UV-visible/NIR spectrophotometer equipped with a 60 mm integrating sphere model ISN-901i/A001461872. Solid-state emission and excitation spectra were run on a Nanolog™ Horib Jobin Yvon IHR320 fluorimeter. Quantum yields for compounds 1–4 in the solid state were measured on a Hammatsu Absolute PL Quantum Yield Spectometer C9920-O2G.
Crystallographic data
Crystallographic data of L3 and compounds 1–4 is gathered in Table 1. Suitable crystals for X-ray elucidation were obtained via recrystallization of the products in THF (L3) or dry CH3CN (1–4).Crystals for X-ray Diffraction (XRD) were prepared under inert conditions immersed in perfluoropolyether or paratone as protecting oil for manipulation. Suitable crystals for L3, 1 and 3 were mounted on MiTeGen Micromounts™, and used for data collection at BL13 (XALOC)124 at the ALBA synchrotron with an undulator source and channel-cut Si(111) monochromator and Kirkpatrick–Baez focusing mirrors with a selected wavelength of 0.72932 Å. An MD2M-Maatel diffractometer fitted with a Dectris Pilatus 6M detector was employed. The sample was kept at 100 K with an Oxford Cryosystems 700 series Cryostream. Crystallographic data for 2 and 4 were collected with a Bruker D8 Venture diffractometer with graphite monochromated MoKα radiation (λ = 0.71073 Å).
The structure for L3 was solved with the ShelXT 2014/5 (Sheldrick, 2014) structure solution program using the direct phasing methods solution method and by using Olex2 as the graphical interface.125 The model was refined with version 2016/6 of ShelXL using Least Squares minimisation.126 The data for 1–4 were processed with APEX3 suite (Bruker APEX3. APEX3 V2019.1; Bruker-AXS: Madison, WI, USA, 2019). The structures were solved by Intrinsic Phasing using the ShelXT program,126 which revealed the position of all non-hydrogen atoms. These atoms were refined on F2 by a full-matrix least-squares procedure using anisotropic displacement parameter.126 All hydrogen atoms were located in difference Fourier maps and included as fixed contributions riding on attached atoms with isotropic thermal displacement parameters 1.2 or 1.5 times those of the respective atom. The OLEX2 software was used as a graphical interface.125 CCDC 204836 and 2050921–2050924.†
Computational details
To analyse the electronic structure and the nature of the excited states, time-dependent DFT calculations were performed by using the implementation of such approach in Gaussian16 program.106 After some test, B3LYP functional107,108 was selected because it provided a better agreement with the experimental data than other common exchange–correlation functional. An all electron Def2TZV basis set was employed in the calculations.109 The calculations were performed for the discrete molecules 2 and 3. To compare with the experimental absorption data, the calculations were carried out using both the experimental and also the optimized structures. The TDDFT calculations were employed only for the discrete molecules because for periodic structures would be mandatory to employ discrete models that usually lead to non-realistic results. The deexcitations energies are calculated by optimizing the structure of the excited triplet state and using such optimized geometry to perform a singlet calculation. Such procedure is employed because TDDFT calculations must be performed in the singlet ground state to extract the vertical excitation energies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by MICINN (PID2019-106832RB-I00, PGC2018-093863-B-C21), MICINN through the Severo Ochoa Program for Centers of Excellence for the FUNFUTURE (CEX2019-000917-S and MDM-2017-0767 projects) and the Generalitat de Catalunya (2017/SGR/1720 and SGR2017-1289). D. Ch.-L. acknowledges funding by project no. PGC2018-102047-B-I00 (MCIU/AEI/FEDER, UE). J. Soldevila-Sanmartín is enrolled in the UAB PhD program and acknowledges the PIF pre-doctoral Fellowship from the UAB for his pre-doctoral grant (no. 19032/2). E. R. acknowledges computer resources, technical expertise and assistance provided by the CSUC. We thank Prof. D. Ruiz and Dr C. Roscini for the quantum yield measurements. The present publication is dedicated to Prof. Elena Shubina on the occasion of her 70th birthday.References
- V. W.-W. Yam and K. K.-W. Lo, Chem. Soc. Rev., 1999, 28, 323–334 RSC.
- A. Barbieri, G. Accorsi and N. Armaroli, Chem. Commun., 2008, 2185 RSC.
- E. Cariati, D. Roberto, R. Ugo, P. C. Ford, S. Galli and A. Sironi, Chem. Mater., 2002, 14, 5116–5123 CrossRef CAS.
- P. C. Ford, E. Cariati and J. Bourassa, Chem. Rev., 1999, 99, 3625–3648 CrossRef CAS PubMed.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- J. Troyano, F. Zamora and S. Delgado, Chem. Soc. Rev., 2021, 50, 4606–4628 RSC.
- J. M. Busch, D. S. Koshelev, A. A. Vashchenko, O. Fuhr, M. Nieger, V. V. Utochnikova and S. Bräse, Inorg. Chem., 2021, 60, 2315–2332 CrossRef CAS PubMed.
- C. Gourlaouen, A. Hamano, K. Takano and C. Daniel, ChemPhysChem, 2021, 22, 509–515 CrossRef CAS PubMed.
- R. Giereth, A. K. Mengele, W. Frey, M. Kloß, A. Steffen, M. Karnahl and S. Tschierlei, Chem. – Eur. J., 2020, 26, 2675–2684 CrossRef CAS PubMed.
- J. Li, L. Wang, Z. Zhao, X. Li, X. Yu, P. Huo, Q. Jin, Z. Liu, Z. Bian and C. Huang, Angew. Chem., Int. Ed., 2020, 59, 8210–8217 CrossRef CAS PubMed.
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Coord. Chem. Rev., 2018, 373, 49–82 CrossRef CAS.
- W. Liu, Y. Fang and J. Li, Adv. Funct. Mater., 2018, 28, 1705593 CrossRef.
- D. Volz, Y. Chen, M. Wallesch, R. Liu, C. Fléchon, D. M. Zink, J. Friedrichs, H. Flügge, R. Steininger, J. Göttlicher, C. Heske, L. Weinhardt, S. Bräse, F. So and T. Baumann, Adv. Mater., 2015, 27, 2538–2543 CrossRef CAS PubMed.
- M. Alkan-Zambada, S. Keller, L. Martínez-Sarti, A. Prescimone, J. M. Junquera-Hernández, E. C. Constable, H. J. Bolink, M. Sessolo, E. Ortí and C. E. Housecroft, J. Mater. Chem. C, 2018, 6, 8460–8471 RSC.
- Y. Liu, S.-C. Yiu, C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2018, 375, 514–557 CrossRef CAS.
- J. Conesa-Egea, F. Zamora and P. Amo-Ochoa, Coord. Chem. Rev., 2019, 381, 65–78 CrossRef CAS.
- O. S. Wenger, Chem. Rev., 2013, 113, 3686–3733 CrossRef CAS PubMed.
- R. Peng, M. Li and D. Li, Coord. Chem. Rev., 2010, 254, 1–18 CrossRef CAS.
- Y.-D. Yu, L.-B. Meng, Q.-C. Chen, G.-H. Chen and X.-C. Huang, New J. Chem., 2018, 42, 8426–8437 RSC.
- C. Slabbert and M. Rademeyer, Coord. Chem. Rev., 2015, 288, 18–49 CrossRef CAS.
- Q. Benito, X. F. Le Goff, G. Nocton, A. Fargues, A. Garcia, A. Berhault, S. Kahlal, J.-Y. Saillard, C. Martineau, J. Trébosc, T. Gacoin, J.-P. Boilot and S. Perruchas, Inorg. Chem., 2015, 54, 4483–4494 CrossRef CAS PubMed.
- H. D. Hardt and A. Pierre, Z. Anorg. Allg. Chem., 1973, 402, 107–112 CrossRef CAS.
- E. I. Musina, A. V. Shamsieva, I. D. Strelnik, T. P. Gerasimova, D. B. Krivolapov, I. E. Kolesnikov, E. V. Grachova, S. P. Tunik, C. Bannwarth, S. Grimme, S. A. Katsyuba, A. A. Karasik and O. G. Sinyashin, Dalton Trans., 2016, 45, 2250–2260 RSC.
- D. M. Zink, T. Baumann, J. Friedrichs, M. Nieger and S. Bräse, Inorg. Chem., 2013, 52, 13509–13520 CrossRef CAS PubMed.
- L. Maini, P. P. Mazzeo, F. Farinella, V. Fattori and D. Braga, Faraday Discuss., 2014, 170, 93–107 RSC.
- W.-F. Fu, X. Gan, C.-M. Che, Q.-Y. Cao, Z.-Y. Zhou and N. N.-Y. Zhu, Chem. – Eur. J., 2004, 10, 2228–2236 CrossRef CAS PubMed.
- D. M. Zink, T. Grab, T. Baumann, M. Nieger, E. C. Barnes, W. Klopper and S. Bräse, Organometallics, 2011, 30, 3275–3283 CrossRef CAS.
- Z. Liu, P. I. Djurovich, M. T. Whited and M. E. Thompson, Inorg. Chem., 2012, 51, 230–236 CrossRef CAS PubMed.
- S. Naik, J. T. Mague and M. S. Balakrishna, Inorg. Chem., 2014, 53, 3864–3873 CrossRef CAS PubMed.
- K. Chen, J. Shearer and V. J. Catalano, Inorg. Chem., 2015, 54, 6245–6256 CrossRef CAS PubMed.
- E. W. Emerson, M. F. Cain, M. D. Sanderson, C. B. Knarr, D. S. Glueck, J. C. Ahern, H. E. Patterson and A. L. Rheingold, Inorg. Chim. Acta, 2015, 427, 168–172 CrossRef CAS.
- E. Cariati, E. Lucenti, C. Botta, U. Giovanella, D. Marinotto and S. Righetto, Coord. Chem. Rev., 2016, 306, 566–614 CrossRef CAS.
- X. Zhang, W. Liu, G. Z. Wei, D. Banerjee, Z. Hu and J. Li, J. Am. Chem. Soc., 2014, 136, 14230–14236 CrossRef CAS PubMed.
- N. Mézailles, P. Le Floch, K. Waschbüsch, L. Ricard, F. Mathey and C. P. Kubiak, J. Organomet. Chem., 1997, 541, 277–283 CrossRef.
- S. Daly, M. F. Haddow, A. G. Orpen, G. T. A. Rolls, D. F. Wass and R. L. Wingad, Organometallics, 2008, 27, 3196–3202 CrossRef CAS.
- R. Ahuja, M. Nethaji and A. G. Samuelson, Inorg. Chim. Acta, 2011, 372, 220–226 CrossRef CAS.
- M. Scherer, D. Stein, F. Breher, J. Geier, H. Schönberg and H. Grützmacher, Z. Anorg. Allg. Chem., 2005, 631, 2770–2774 CrossRef CAS.
- E. Kühnel, I. V. Shishkov, F. Rominger, T. Oeser and P. Hofmann, Organometallics, 2012, 31, 8000–8011 CrossRef.
- X. Han, Z. Weng, D. J. Young, G.-X. Jin and T. S. Andy Hor, Dalton Trans., 2014, 43, 1305–1312 RSC.
- K. A. Vinogradova, V. P. Krivopalov, E. B. Nikolaenkova, N. V. Pervukhina, D. Y. Naumov, M. I. Rakhmanova, E. G. Boguslavsky, L. A. Sheludyakova and M. B. Bushuev, Polyhedron, 2013, 57, 1–13 CrossRef CAS.
- J. C. Li, H. X. Li, H. Y. Li, W. J. Gong and J. P. Lang, Cryst. Growth Des., 2016, 16, 1617–1625 CrossRef CAS.
- M. Yang, X.-L. Chen and C.-Z. Lu, Dalton Trans., 2019, 48, 10790–10794 RSC.
- S.-Z. Zhan, T. Feng, W. Lu, M. R. Razali and D. Li, Cryst. Growth Des., 2018, 18, 7663–7673 CrossRef CAS.
- J.-H. Wang, M. Li, J. Zheng, X.-C. Huang and D. Li, Chem. Commun., 2014, 50, 9115–9118 RSC.
- F. Wu, H. Tong, K. Wang, X. Zhang, J. Zhang, W.-K. Wong and X. Zhu, J. Coord. Chem., 2016, 69, 926–933 CrossRef CAS.
- A. Tarassoli, V. Nobakht, E. Baladi, L. Carlucci and D. M. Proserpio, CrystEngComm, 2017, 19, 6116–6126 RSC.
- J. He, J. Duan, H. Shi, J. Huang, J. Huang, L. Yu, M. Zeller, A. D. Hunter and Z. Xu, Inorg. Chem., 2014, 53, 6837–6843 CrossRef CAS PubMed.
- W.-X. Wang, L. Wang, H.-X. Li, H.-Y. Li and J.-P. Lang, Z. Anorg. Allg. Chem., 2013, 639, 618–625 CrossRef CAS.
- H.-Y. Li, Y. Zhang, Y.-C. Ding, M. Wang, L.-X. Dai and J.-P. Lang, J. Mol. Struct., 2011, 996, 90–94 CrossRef CAS.
- J. Soldevila-Sanmartín, X. Montaner, T. Calvet, M. Font-Bardia and J. Pons, Polyhedron, 2020, 114686 CrossRef.
- M. Guerrero, S. Vázquez, J. A. Ayllón, T. Calvet, M. Font-Bardia and J. Pons, ChemistrySelect, 2017, 2, 632–639 CrossRef CAS.
- A. M. López Marzo, M. Guerrero, T. Calvet, M. Font-Bardia, E. Pellicer, M. D. Baró, J. Pons and J. Sort, RSC Adv., 2015, 5, 32369–32375 RSC.
- M. Guerrero, J. Pons, J. Ros, M. Font-Bardia, O. Vallcorba, J. Rius, V. Branchadell and A. Merkoçi, CrystEngComm, 2011, 13, 6457 RSC.
- M. Guerrero, J. Pons and J. Ros, J. Organomet. Chem., 2010, 695, 1957–1960 CrossRef CAS.
- M. Guerrero, J. Pons, M. Font-Bardia, T. Calvet and J. Ros, Aust. J. Chem., 2010, 63, 958 CrossRef CAS.
- M. Y. Tsang, C. Viñas, F. Teixidor, J. G. Planas, N. Conde, R. SanMartin, M. T. Herrero, E. Dominguez, A. Lledos, P. Vidossich and D. Choquesillo-Lazarte, Inorg. Chem., 2014, 53, 9284–9295 CrossRef CAS PubMed.
- J. Planas, F. Teixidor and C. Viñas, Crystals, 2016, 6, 50 CrossRef.
- F. Di Salvo, M. Y. Tsang, F. Teixidor, C. Viñas, J. G. Planas, J. Crassous, N. Vanthuyne, N. Aliaga-Alcalde, E. Ruiz, G. Coquerel, S. Clevers, V. Dupray, D. Choquesillo-Lazarte, M. E. Light and M. B. Hursthouse, Chem. – Eur. J., 2014, 20, 1081–1090 CrossRef CAS PubMed.
- F. Di Salvo, F. Teixidor, C. Viñas and J. Giner Planas, Z. Anorg. Allg. Chem., 2013, 639, 1194–1198 CrossRef CAS.
- F. Di Salvo, F. Teixidor, C. Viñas, J. Giner Planas, M. E. Light, M. B. Hursthouse and N. Aliaga-Alcalde, Cryst. Growth Des., 2012, 12, 5720–5736 CrossRef CAS.
- G. Grach, G. Pieters, A. Dinut, V. Terrasson, R. Medimagh, A. Bridoux, V. Razafimahaleo, A. Gaucher, S. Marque, J. Marrot, D. Prim, R. Gil, J. Giner Planas, C. Vinas, I. Thomas, J.-P. Roblin and Y. Troin, Organometallics, 2011, 30, 4074–4086 CrossRef CAS.
- F. Di Salvo, J. G. Planas, C. Vinas, F. Teixidor, M. E. Light and M. B. Hursthouse, Acta Crystallogr., Sect. A: Found. Adv., 2011, 67, C368 Search PubMed.
- V. Terrasson, J. G. Planas, C. Viñas, F. Teixidor, D. Prim, M. E. Light and M. B. Hursthous, Organometallics, 2010, 29, 4130–4134 CrossRef CAS.
- J. Plesek, Chem. Rev., 1992, 92, 269–278 CrossRef CAS.
- F. Teixidor, C. Viñas, A. Demonceau and R. Nuñez, Pure Appl. Chem., 2003, 75, 1305–1313 CAS.
- R. N. Grimes, Carboranes, 2nd edn, 2011 Search PubMed.
- F. Teixidor, G. Barberà, A. Vaca, R. Kivekäs, R. Sillanpää, J. Oliva and C. Viñas, J. Am. Chem. Soc., 2005, 127, 10158–10159 CrossRef CAS PubMed.
- J. Poater, M. Solà, C. Viñas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed.
- J. Poater, C. Viñas, I. Bennour, S. Escayola, M. Solà and F. Teixidor, J. Am. Chem. Soc., 2020, 142, 9396–9407 CrossRef CAS PubMed.
- M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed.
- L. Schwartz, L. Eriksson, R. Lomoth, F. Teixidor, C. Viñas and S. Ott, Dalton Trans., 2008, 2379–2381 RSC.
- R. Núñez, P. Farràs, F. Teixidor, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2006, 45, 1270–1272 CrossRef PubMed.
- F. Teixidor, R. Núñez, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2000, 39, 4290–4292 CrossRef CAS.
- A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3, 590–596 CrossRef CAS PubMed.
- L. E. Riley, T. Krämer, C. L. McMullin, D. Ellis, G. M. Rosair, I. B. Sivaev and A. J. Welch, Dalton Trans., 2017, 46, 5218–5228 RSC.
- F. Tan, A. López-Periago, M. E. Light, J. Cirera, E. Ruiz, A. Borrás, F. Teixidor, C. Viñas, C. Domingo and J. G. Planas, Adv. Mater., 2018, 30, 1800726 CrossRef PubMed.
- M. A. Fox and A. K. Hughes, Coord. Chem. Rev., 2004, 248, 457–476 CrossRef CAS.
- J. G. Planas, C. Vinas, F. Teixidor, A. Comas-Vives, G. Ujaque, A. Lledos, M. E. Light and M. B. Hursthouse, J. Am. Chem. Soc., 2005, 127, 15976–15982 CrossRef CAS PubMed.
- R. Nunez, M. Tarres, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef CAS.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2016, 52, 1070–1093 RSC.
- F. Teixidor and C. Viñas, in Science of Synthesis, ed. D. E. Kaufmann and D. S. Matteson, Thieme, Stuttgart, 2005, vol. 6, pp. 1275–1325 Search PubMed.
- J. Ochi, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2020, 59, 9841–9855 CrossRef CAS PubMed.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2017, 56, 254–259 CrossRef CAS PubMed.
- D. Tu, S. Cai, C. Fernandez, H. Ma, X. Wang, H. Wang, C. Ma, H. Yan, C. Lu and Z. An, Angew. Chem., Int. Ed., 2019, 58, 9129–9133 CrossRef CAS PubMed.
- X. Wei, M.-J. Zhu, Z. Cheng, M. Lee, H. Yan, C. Lu and J.-J. Xu, Angew. Chem., Int. Ed., 2019, 58, 3162–3166 CrossRef CAS PubMed.
- J. Cabrera-Gonzalez, C. Vinas, M. Haukka, S. Bhattacharyya, J. Gierschner and R. Nunez, Chem. – Eur. J., 2016, 22, 13588–13598 CrossRef CAS PubMed.
- J. Cabrera-Gonzalez, S. Bhattacharyya, B. Milian-Medina, F. Teixidor, N. Farfan, R. Arcos-Ramos, V. Vargas-Reyes, J. Gierschner and R. Nunez, Eur. J. Inorg. Chem., 2017, 4575–4580 CrossRef CAS.
- J. Cabrera-González, M. Chaari, F. Teixidor, C. Viñas and R. Núñez, Molecules, 2020, 25, 1210 CrossRef PubMed.
- M. Chaari, Z. Kelemen, D. Choquesillo-Lazarte, F. Teixidor, C. Viñas and R. Núñez, Inorg. Chem. Front., 2020, 7, 2370–2380 RSC.
- M. Chaari, Z. Kelemen, D. Choquesillo-Lazarte, N. Gaztelumendi, F. Teixidor, C. Viñas, C. Nogués and R. Núñez, Biomater. Sci., 2019, 7, 5324–5337 RSC.
- M. Chaari, Z. Kelemen, J. G. Planas, F. Teixidor, D. Choquesillo-Lazarte, A. Ben Salah, C. Viñas and R. Núñez, J. Mater. Chem. C, 2018, 6, 11336–11347 RSC.
- M. T. Güllüoglu, M. Ozduran, M. Kurt, S. Kalaichelvan and N. Sundaraganesan, Spectrochim. Acta, Part A, 2010, 76, 107–114 CrossRef PubMed.
- G. Zerbi and C. Alberti, Spectrochim. Acta, 1962, 18, 407–423 CrossRef CAS.
- S. Demir, B. Eren and M. Hołyńska, J. Mol. Struct., 2015, 1081, 304–310 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- A. Neshat, R. B. Aghakhanpour, P. Mastrorilli, S. Todisco, F. Molani and A. Wojtczak, Polyhedron, 2018, 154, 217–228 CrossRef CAS.
- E. I. Musina, A. V. Shamsieva, I. D. Strelnik, T. P. Gerasimova, D. B. Krivolapov, I. E. Kolesnikov, E. V. Grachova, S. P. Tunik, C. Bannwarth, S. Grimme, S. A. Katsyuba, A. A. Karasik and O. G. Sinyashin, Dalton Trans., 2016, 45, 2250–2260 RSC.
- T. N. Sorrell and A. S. Borovik, Inorg. Chem., 1987, 26, 1957–1964 CrossRef CAS.
- M. Vitale and P. C. Ford, Coord. Chem. Rev., 2001, 219, 3–16 CrossRef.
- A. Acosta, J. I. Zink and J. Cheon, Inorg. Chem., 2000, 39, 427–432 CrossRef CAS PubMed.
- V. Balzani, G. Bergamini, S. Campagna and F. Puntoriero, in Photochemistry and Photophysics of Coordination Compounds I, ed. V. Balzani and S. Campagna, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, pp. 1–36 Search PubMed.
- J. Zhao, D. Shi, H. Cheng, L. Chen, P. Ma and J. Niu, Inorg. Chem. Commun., 2010, 13, 822–827 CrossRef CAS.
- A. V. Artem’ev, D. G. Samsonenko and O. V. Antonova, Polyhedron, 2018, 151, 171–176 CrossRef.
- A. Aguirrechu-Comerón, R. Hernández-Molina, P. Rodríguez-Hernández, A. Muñoz, U. R. Rodríguez-Mendoza, V. Lavín, R. J. Angel and J. Gonzalez-Platas, Inorg. Chem., 2016, 55, 7476–7484 CrossRef PubMed.
- I. L. Malaestean, V. C. Kravtsov, J. Lipkowski, E. Cariati, S. Righetto, D. Marinotto, A. Forni and M. S. Fonari, Inorg. Chem., 2017, 56, 5141–5151 CrossRef CAS PubMed.
- J. López, J. G. Platas, U. R. Rodríguez-Mendoza, J. I. Martínez, S. Delgado, G. Lifante-Pedrola, E. Cantelar, R. Guerrero-Lemus, C. Hernández-Rodríguez and P. Amo-Ochoa, Inorg. Chem., 2021, 60, 1208–1219 CrossRef PubMed.
- K. Hassanein, P. Amo-Ochoa, C. J. Gómez-García, S. Delgado, O. Castillo, P. Ocón, J. I. Martínez, J. Perles and F. Zamora, Inorg. Chem., 2015, 54, 10738–10747 CrossRef CAS PubMed.
- N. M. Khatri, M. H. Pablico-Lansigan, W. L. Boncher, J. E. Mertzman, A. C. Labatete, L. M. Grande, D. Wunder, M. J. Prushan, W. Zhang, P. S. Halasyamani, J. H. S. K. Monteiro, A. De Bettencourt-Dias and S. L. Stoll, Inorg. Chem., 2016, 55, 11408–11417 CrossRef CAS PubMed.
- D. Volz, D. M. Zink, T. Bocksrocker, J. Friedrichs, M. Nieger, T. Baumann, U. Lemmer and S. Bräse, Chem. Mater., 2013, 25, 3414–3426 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- K. Tsuge, Y. Chishina, H. Hashiguchi, Y. Sasaki, M. Kato, S. Ishizaka and N. Kitamura, Coord. Chem. Rev., 2016, 306, 636–651 CrossRef CAS.
- D. Volz, M. Wallesch, S. L. Grage, J. Göttlicher, R. Steininger, D. Batchelor, T. Vitova, A. S. Ulrich, C. Heske, L. Weinhardt, T. Baumann and S. Bräse, Inorg. Chem., 2014, 53, 7837–7847 CrossRef CAS PubMed.
- A. V. Artem’ev, D. G. Samsonenko and O. V. Antonova, Polyhedron, 2018, 151, 171–176 CrossRef.
- S. Demir, B. Eren and M. Hołyńska, J. Mol. Struct., 2015, 1081, 304–310 CrossRef CAS.
- S. Jana and S. Chattopadhyay, Polyhedron, 2014, 81, 298–307 CrossRef CAS.
- Y.-D. Yu, L.-B. Meng, Q.-C. Chen, G.-H. Chen and X.-C. Huang, New J. Chem., 2018, 42, 8426–8437 RSC.
- P. Boden, P. Di Martino-Fumo, J. Busch, F. Reahak, S. Steiger, O. Fuhr, M. Nieger, D. Volz, W. Klopper, S. Bräse and M. Gerhards, Chem. – Eur. J., 2021, 27, 5439–5452 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S.-Z. Zhan, X. Jiang, J. Zheng, X.-D. Huang, G.-H. Chen and D. Li, Dalton Trans., 2018, 47, 3679–3683 RSC.
- W. G. Haanstra, W. L. Driessen, J. Reedijk, U. Turpeinen and R. Hämäläinenn, J. Chem. Soc., Dalton Trans., 1989, 2309–2314 RSC.
- J. Juanhuix, F. Gil-Ortiz, G. Cuni, C. Colldelram, J. Nicolas, J. Lidon, E. Boter, C. Ruget, S. Ferrer and J. Benach, J. Synchrotron Radiat., 2014, 21, 679–689 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2048361 and 2050921–2050924. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc01395e |
| This journal is © The Royal Society of Chemistry 2021 |