
Reduced graphene oxide in perovskite solar cells: the influence on film formation, photophysics, performance, and stability†
Paulo Ernesto
Marchezi
 a,
Francineide Lopes
de Araújo
a,
Rodrigo
Szostack
a,
José Carlos
Germino
a,
Eralci M.
Therézio
b,
Alexandre
Marletta
c and
Ana Flavia
Nogueira
*a
a,
Francineide Lopes
de Araújo
a,
Rodrigo
Szostack
a,
José Carlos
Germino
a,
Eralci M.
Therézio
b,
Alexandre
Marletta
c and
Ana Flavia
Nogueira
*a
aUniversity of Campinas (UNICAMP), Laboratório de Nanotecnologia e Energia Solar, Chemistry Institute, Campinas, PO Box 6154, 13083-970, Brazil. E-mail: anafla@unicamp.br
bPhysics Institute, Federal University of Mato Grosso, Cuiabá, Brazil
cPhysics Institute, Federal University of Uberlândia, Uberlândia, Brazil
First published on 30th June 2021
Abstract
Mixed-halide perovskites of the type CsxFA1−xPb(BryI1−y)3 are promising materials for highly efficient single junction and tandem solar cells. This work details how RGO interacts with the perovskite, influencing film formation and its physico-chemical properties. RGO was introduced into the precursor solution to study how reduced graphene oxide (RGO) interferes with the crystallization, morphology, and optoelectronic properties of the perovskite film. By in situ GIWAXS experiments, a decrease in the rate of formation of the bulk perovskite phase was observed during the spin-coating and annealing processes. SEM-FEG images confirmed an increase in the grain size of the perovskite film that contained RGO. The most outstanding results were related to the stability of the devices. Devices with RGO-20 h (reduced for 20 hours) exhibited a higher stability, confirming that the films formed by larger grains have a slower rate of degradation.
Introduction
Over the past few years, thin-film photovoltaic (PV) technology has been quickly developed, and most research is aimed at reducing the cost per unit of electrical energy generated from sunlight compared with conventional silicon solar cells. In this way, many approaches have been developed, such as amorphous and nanocrystalline Si,1 inorganic compound semiconductors,2,3 quantum dot solar cells (QDSCs),4,5 dye-sensitized solar cells (DSSCs),6 organic solar cells (OSCs),7 and, more recently, perovskite solar cells (PSCs).8 Since the pioneering work by Miyasaka and co-workers in 2009,9 PSCs have emerged at the forefront of thin-film PVs due to their high power conversion efficiency and low-cost fabrication.8,10 This technology has witnessed impressive advances, with the solar-to-electrical power conversion efficiency (PCE) being increased from 3.8% to a certified value of 25.5% on a laboratory scale.11 Metal halide perovskites (MHPs) are an extensive class of materials that have the same crystal structure as calcium titanate, with the general chemical formula ABX3, where A is an monovalent organic or/and inorganic cation (methylammonium (MA+), formamidinium (FA+), Cs+ or Rb+), B is a divalent cation (Pb2+ or Sn2+), and X is an anion (Cl−, Br−, or I−).12 The chemical composition of these structures is quite vast, as it is possible to alloy multiple cations and anions in the same crystalline structure.13–16Despite PSCs with high efficiencies being constantly reported, there is still room for improvements before the commercialization and widespread deployment of this technology. Understanding film formation using different deposition techniques and parameters is essential to improve the device performance and stability.17–19 A poor perovskite film quality with small grain sizes is related to the quick reaction between lead halide and the cations during crystallization of the perovskite from the blended precursor solution.20 Therefore, control of the nucleation and crystallization processes is an important tool towards improving the perovskite file morphology and, hence, its efficiency. Because of the polycrystalline nature of the perovskite, grain interfaces act as recombination centers, influencing the device performance. Strategies to minimize the surface and grain-boundary defects include passivation using thiophene and Lewis bases,21 iodopentafluorobenzene,21 fullerene and other carbonaceous materials,22,23 and even PbI2.24
Graphene and its analogs have been used widely for optoelectronic applications because of their unique properties.25,26 Graphene oxide (GO), an oxidized derivative of graphene, shows hydrophilicity, while graphene exhibits a hydrophobic nature.27 Although the hydrophilic nature of GO enable solution processing with a polar solvent, the application of GO has been limited due to its inferior electronic properties compared with graphene.27,28 Consequently, reduced graphene oxide (RGO) has been proposed as a good substitute as it presents enhanced electronic properties compared with GO.29 Generally, RGO is applied in composites, improving the charge transport and efficiency of oxide and chalcogenide semiconductors.30,31 With an adjustable work function that ranges from 4.4 to 5.0 eV, RGO has a variety of applications, such as hole-transport material (HTM) and an electron-transport material (ETM).32–35 Furthermore, the electronic and optical properties of RGO can be varied by facile chemical functionalization, enabling RGO to meet the necessary demands.36,37
The introduction of graphene derivatives in perovskite films has already been reported. He et al.38 developed an in situ method to cover perovskite grains with RGO, leading to a significant improvement in the performance of a photodetector. Hadadian et al.23 studied the introduction of nitrogen-doped GO into the perovskite to passivate and improve the morphology of the film. More recently, Kim et al.39 reported an increase in the stability of PSCs by introducing RGO into the composition of the perovskite. Although they are remarkable studies, there is a lack of fundamental understanding about how RGO acts by interfering with the properties of the perovskite films. Vague explanations are used to explain the role of graphene when forming a perovskite composite. Herein, we report a detailed investigation on the formation mechanisms of Cs0.17FA0.83Pb(Br0.17I0.83)3 perovskite films that contain RGO. The perovskite composition was chosen based on previous reports in which higher efficiencies and photostabilities were found for devices where one-sixth of the iodine was replaced by bromine and one-sixth of FA was replaced by Cs.40–43 The volatile nature of the MA cation molecule itself is the reason that we chose to work with a composition without MA, to obtain a higher stability in the perovskite films.44In situ grazing incidence X-ray scattering (GIWAXS), scanning electron microscopy (SEM) and optical spectroscopy are used to understand the effect of RGO in the crystallization of perovskite films and to explain how the modifications are related to the optoelectronic properties, performance, and stability of the PSCs.
Experimental
Three different RGOs were synthesized by reacting 30 mg of GO dispersed in 40 mL of ethanol in the presence of 300 μL of 80% hydrazine (0.37 mol L−1, Merck, N2H4·H2O 50% v/v in H2O) and tetrabutylammonium hydroxide (TBAH, 0.087 mol L−1, Sigma-Aldrich). The dispersion was kept at room temperature under magnetic stirring. Different reduction times were used (5, 10 and 20 hours, for which the samples were named as RGO-5 h, RGO-10 h and RGO-20 h, respectively). GO was synthesized from natural graphite powder using the modified method of Hummers and Offeman method reported previously.45,46The RGO composition was also studied using X-ray photoelectron spectroscopy (XPS) at a reduced pressure of 10−9 Pa using a Scient Omicron ESCA+ spectrometer containing a high-performance hemispheric analyzer (EA-125). Monochromatic Al Kα (1486.6 eV) was used as the excitation source. Data analysis was performed using CasaXPS, setting the C 1s peak at 284.8 eV as the reference for calibration. The conductivity of the RGO films deposited in a glass substrate was measured using a four-probe method. The current was measured over the range of 0.1–1 V, and then the slope of the I–V plot was assigned as the resistance of the film.
RGO was incorporated into the perovskite film by dispersing the powder in the perovskite precursor solution containing FAI, CsI, PbBr2 and PbI2 (Sigma-Aldrich) dissolved in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 anhydrous mixture of N,N-dimethylformamide (DMF):dimethyl sulfoxide (DMSO) to yield the Cs0.17FA0.83Pb(Br0.17I0.83)3 composition. The concentration of RGO was fixed at 0.01 mg mL−1. This concentration used was the largest amount of RGO that we could disperse into the precursor solution without graphene precipitation. The perovskite films were then deposited using an anti-solvent engineering method.47 Following a two-step procedure, 10 s at 1000 rpm and 30 s at 6000 rpm, chlorobenzene was added dropwise, as the anti-solvent, into the spinning substrate after 25 s. The films were then annealed at 100 °C for 30 min, inside a N2 glove box. All the perovskite films were deposited in the same way. The samples were named according to the RGO used in the modification, e.g., “control” for the perovskite without modification and “control+RGO-xh” (x = 5, 10 and 20) for the modified samples.
:1 anhydrous mixture of N,N-dimethylformamide (DMF):dimethyl sulfoxide (DMSO) to yield the Cs0.17FA0.83Pb(Br0.17I0.83)3 composition. The concentration of RGO was fixed at 0.01 mg mL−1. This concentration used was the largest amount of RGO that we could disperse into the precursor solution without graphene precipitation. The perovskite films were then deposited using an anti-solvent engineering method.47 Following a two-step procedure, 10 s at 1000 rpm and 30 s at 6000 rpm, chlorobenzene was added dropwise, as the anti-solvent, into the spinning substrate after 25 s. The films were then annealed at 100 °C for 30 min, inside a N2 glove box. All the perovskite films were deposited in the same way. The samples were named according to the RGO used in the modification, e.g., “control” for the perovskite without modification and “control+RGO-xh” (x = 5, 10 and 20) for the modified samples.
Perovskite solar cells were assembled in a standard configuration, consisting of FTO/bl-TiO2/mp-TiO2/Cs0.17FA0.83Pb(Br0.17I0.83)3/spiro-OMeTAD/Au; all devices were measured with backward and forward scanning. Current versus voltage measurements were carried out using a current–voltage source (KEITHLEY 2410-c), controlled using an Arduino system developed in our laboratory, using a Python code to communicate with the computer and calculate the solar cell parameters. For stability tests, the devices were stored under ambient conditions (T ≈ 25 °C and RH ≈ 55%), in the dark, following the ISOS protocol ISOS-D-1.48
In situ GIWAXS measurements were performed on the XRD2 beam line at the National Synchrotron Light Laboratory (LNLS), Campinas, Brazil. The experiments performed during spin coating and annealing steps were conducted inside a homemade N2-filled acrylic chamber, to resemble the deposition of films made inside a glovebox, as described in our previous studies.49,50 The X-ray energy was 7 keV and the scatter signals were collected using a Pilatus 300 k detector with an integration time of 1 s, placed at 190 mm from the sample. The angle of incidence of the X-ray beam was 3°. Each GIWAXS image was calibrated using a AgBH standard, azimuthally integrated to obtain 1D X-ray diffraction patterns, and the reciprocal lattice maps were calculated using pigyx and py-FAI Python libraries. Ex situ X-ray diffractograms of the perovskite films with and without modification and of the RGO powder were obtained using a Shimadzu diffractometer, model XRD-6000, with Cu Kα radiation and with a wavelength of 0.154 nm.
The morphology of the RGO samples used in this work was investigated through scanning electron microscopy using a Quanta 250 microscope coupled with field emission-scanning (FEG-SEM). We studied the perovskite film morphology using the same FEG-SEM microscope.
Absorption spectra of the perovskite films with and without modification were acquired using an Agilent spectrophotometer, model Cary 60, in the 400–850 nm range. Steady-state photoluminescence (PL) spectra of the perovskite were acquired using a Kimmon He-Cd laser (λexc = 442 nm and power = 300 W) as the excitation source and an Ocean Optics USB2000+detector with LabView laboratory software of the interface.
The charge-carrier emission dynamics of the perovskite films were acquired via time-correlated single-photon counting (TCSPC) using an Edinburgh Analytical Instruments FL 900 spectrofluorometer with an MCP-PMT (Hamamatsu R3809U-50) and a PicoQuant pulsed laser operating at λexc = 440 (F = 24.3 nJ cm−2). The decay signals were collected at the maximum emission wavelength of the perovskite film. The instrument response was recorded using Ludox samples. At least 10000 counts in the peak channel were accumulated for the lifetime determination. High-order charge-carrier dynamics cannot be accessed due to the low fluence (F = 24.3 nJ cm−2) used in the TCSPC, which prevent access to high-density photogenerated charge-carriers (n0 < 1015 cm−3), where there is a strong dependence of the charge-carrier dynamics with the source fluence.51 A single exponential decay function should be adequate to fit the time-resolved photoluminescence (TrPL) decay under these conditions. However, a simple exponential decay function did not fit the TrPL data because of the disorder of the samples (i.e., polycrystallinity, alloy composition, grain size distribution, Br- and I-rich phase segregation, high-density of trap-states, etc.).52
Results and discussion
Starting from GO as the precursor material and following a time-dependent hydrazine-assisted reduction method, three RGO samples with different degrees of reduction were prepared (RGO-5 h, RGO-10 h and RGO-20 h). The elemental composition of the RGO sheets was confirmed by XPS (Fig. S1A, ESI†). According to the elemental analysis of the samples, the oxygen percentage decreased from 29.6% to 12.3% and the carbon percentage increased from 70.4% to 84.1% (from GO to RGO-20 h). In addition, the nitrogen content increased from 0% to 3.6%. The increase in the amount of nitrogen was related to the method applied to reduce the GO, which uses hydrazine. An increase in the peak at a binding energy of 285.5 eV (Fig. 1A and Fig. S1B, ESI†) was observed, attributed to carbon with sp2 hybridization. The contribution of this peak to the total C 1s signal was 39% for RGO-5h, 45% for RGO-10h, and 47% for RGO-20 h. Along with this change, a significant decrease in the peaks associated with oxygenated carbons was observed for all RGO samples. A sharp decrease in the intensity of the peak at 533.7 eV (Fig. 1B and Fig. S1C, S2, ESI†) revealed that oxygen single-bonded to carbon is preferentially removed upon reaction with hydrazine. These values suggested that the reaction with hydrazine not only promotes additional removal of oxygen but also improves the restoration of sp2 conjugation. A more detailed discussion of the XPS results is shown in ESI† Text 1. | ||
Fig. 1 GO and RGO characterization. (A) C 1s high resolution XPS core-level spectra of GO and RGO-20 h; deconvolution peaks are attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (blue), C–C (orange), C–O (green), C–N (purple) and C–OOH (yellow). (B) O 1s high resolution XPS core-level spectra of GO and RGO-20 h; deconvolution peaks are attributed to quinone (orange), CO (blue) and C–O (green). (C) D- and G-band regions of Raman spectra for GO and all RGO is synthesized in this work. (D and E) Elemental concentration of the samples GO, RGO-5 h, RGO-10 h and RGO-20 h, obtained from XPS analysis. (F) Ratio between the intensities of the D and G bands (ID/IG) and the conductivity of the RGO films. C (blue), C–C (orange), C–O (green), C–N (purple) and C–OOH (yellow). (B) O 1s high resolution XPS core-level spectra of GO and RGO-20 h; deconvolution peaks are attributed to quinone (orange), CO (blue) and C–O (green). (C) D- and G-band regions of Raman spectra for GO and all RGO is synthesized in this work. (D and E) Elemental concentration of the samples GO, RGO-5 h, RGO-10 h and RGO-20 h, obtained from XPS analysis. (F) Ratio between the intensities of the D and G bands (ID/IG) and the conductivity of the RGO films. | ||
The Raman spectra (Fig. 1C and Fig. S3A, ESI†) present characteristic peaks at 1593 and 1350 cm−1, corresponding to the G and D bands, respectively.53 The G band, also known as the graphitic band, is related to the vibrational mode from C sp2 atoms in the hexagonal structure, and the D band is related to the defects associated with C sp3 atoms. The relationship between the intensities of the D and G bands (ID/IG) is used to analyze the quality of the graphene sheets in relation to the defects. This ratio is reduced from 1.5 to 1.0, from RGO-5 h to RGO-20 h, respectively, (Fig. 1F). This confirms that a restoration of the sp2 lattice is more effective when increasing the reduction time, and this is reflected in an increase in the conductivity of the samples. The electrical properties were evaluated by determining the conductivity of each RGO using the four-probe method. Fig. 1F shows a continuous increase in the conductivity for RGO-5 h, RGO-10 h and RGO-20 h. According to the XRD diffractograms, there is a reduction in the crystallinity of the material, which is related to the exfoliation of the reduced graphene oxide (Fig. S4, ESI†). The effect of the reduction degree on the RGO morphology was examined by scanning electron microscopy (SEM), as depicted in Fig. S4 (ESI†). The samples were prepared by drop casting the RGO suspension onto glass substrates. The morphology is similar for all RGO samples, presenting a mix of flake-like structures and sheets, with sizes varying from a couple of nanometers to tens of micrometers (Fig. S5, ESI†). The small size can be associated with the ultrasonication process used to disperse the RGO in the precursor solution.
To understand the influence of RGO on the perovskite formation rate, in situ GIWAXS measurements were performed during the spin coating of the solution using chlorobenzene as the anti-solvent (Fig. S6, ESI†). The synchrotron X-ray beam was set to hit the sample at an angle of incidence of 3° during the spin coating process, and the diffraction patterns were collected using an area detector. The antisolvent was dropped onto the sample in the last 15 s of the spin-coating process and then the diffraction spectrum was collected for an additional 3.5 min while the film was standing still. For the sample without RGO (Fig. 2B), the appearance of a diffraction peak at q = 10 nm−1 was observed, assigned to the (100) diffraction plane of the cubic perovskite, right after the dropping of the antisolvent. A peak related to the 2H phase at ∼8.1 nm−1 was also observed, in agreement with our previous work.54 Looking at Fig. 2C, it was possible to observe that the intensity of this peak changes at different rates for the samples with RGO. The intensity of the (100) peak of the samples containing RGO-10 h and RGO-20 h took more time to reach than the equivalent intensity for the standard sample. To investigate this behavior, the variation in the peak area at q = 10 nm−1 was plotted as a function of time (Fig. 2D). This plot reflects the kinetics of perovskite formation and it is possible to observe that the formation rate decreases with the increasing reduction time of RGO.
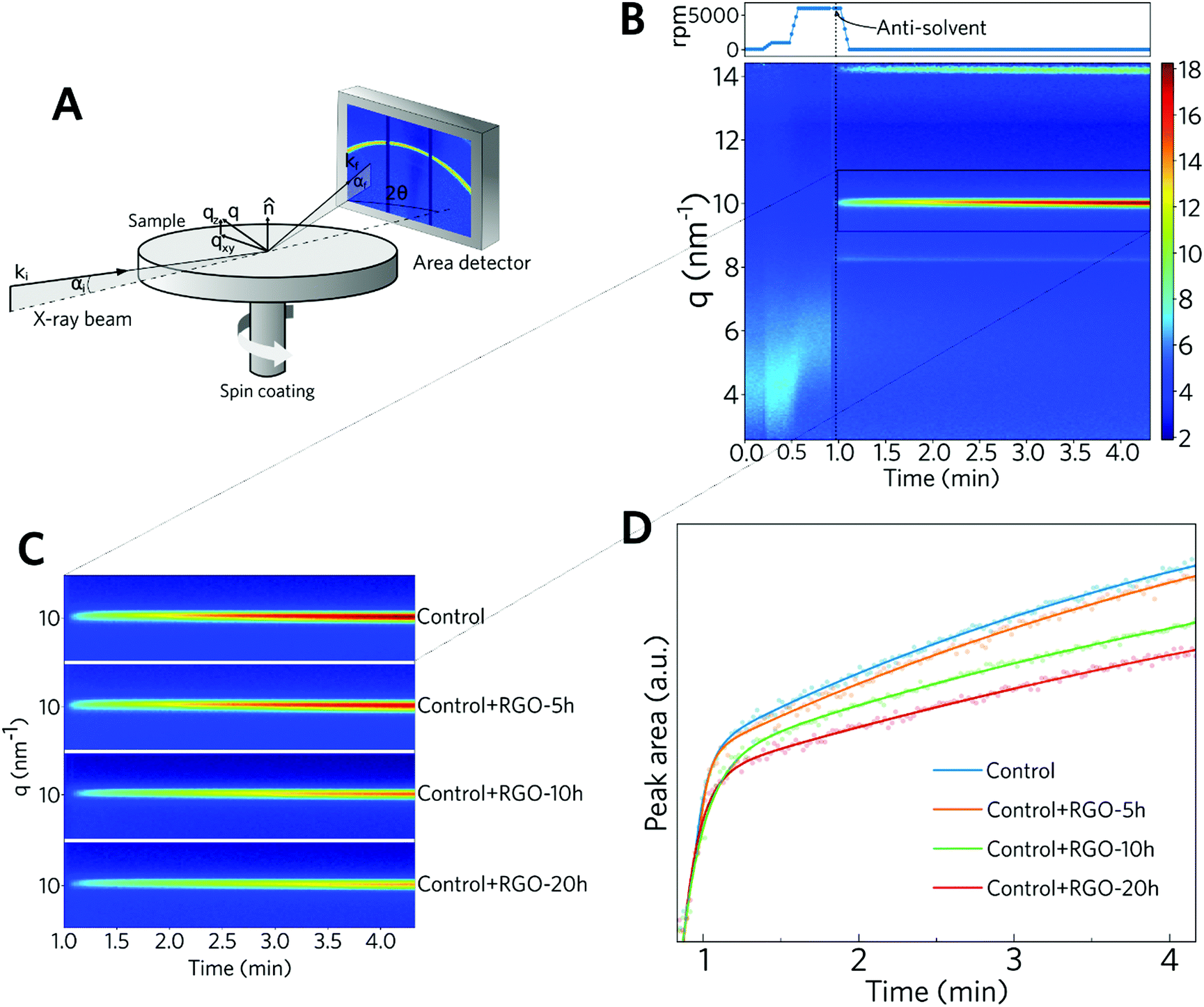 | ||
| Fig. 2 In situ GIWAXS data for the perovskite films containing RGO with different reduction times. (A) Schematic representation of the in situ GIWAXS measurements carried out during the spin-coating step of the perovskite films prepared using an antisolvent engineering method. (B) GIWAXS diffractogram map over time along the spin-coating conditions during the measurements of the control sample. (C) Expanded view at q = 10 nm−1 for each perovskite film with different RGOs in the precursor solution. (D) Area of the q = 10 nm−1 peak over time. All GIWAXS reciprocal lattice maps are presented in Fig. S8–S11 (ESI†). | ||
To complete the perovskite crystallization, the films were thermally annealed. This step was also probed via in situ GIWAXS. The samples were measured over 35 min of annealing to study both the crystallization and possible degradation caused by heating. Fig. 3A–D show the evolution of q = 10 nm−1 during the first 8 min of the annealing step, and after this time the intensity of the peak reaches a plateau. Looking at the evolution of the peak for the sample without RGO (Fig. 3A and F), the peak intensity did not increase significantly during the annealing step. In other words, the perovskite was almost completely formed without thermal annealing. Interestingly, for samples with RGO (Fig. 3B–D and F), the intensity of this peak continues to increase over time, reaching the same plateau over a much longer time. This is because the perovskite crystallization was not completed during deposition; instead, its crystallization continues during the annealing step. Besides the fact that incorporation of reduced RGO into the perovskite precursor solution decreases the perovskite formation rate, it appears that the more reduced the RGO, the more pronounced is the effect in the film formation. This becomes clearer in Fig. 3F, where RGO-10 h and RGO-20 h caused a significant delay in the increase of the (100) peak during the annealing process. Here, we conclude that the introduction of RGO with a higher reduction degree can retard both the formation of the perovskite during spin coating and its crystallization. The influence of the reduction degree on the formation rate and how it affects the grain size is discussed below. From the XRD patterns of the perovskite films (Fig. 3E), it is possible to infer that the perovskite films in the absence and presence of RGO have cubic perovskite phases with an almost unchanged crystal structure after annealing.55 Another feature observed is a change in the preferential crystalline orientation (Fig. 3E and G). It is observed that, in the XRD patterns for the sample without RGO (Fig. 3E), the peak at q = 14 nm−1 (110) appears more intense than the peak related to the (100) diffraction plane. With the addition of more reduced RGO, the ratio between peaks (100) and (110) changes with preferred orientation in the (100) direction for samples containing RGO, and which is most pronounced for sample control+RGO-20 h sample (Fig. 3G). However, we did not see this trend in the GIWAXS experiments performed during the spin coating and annealing steps (Fig. S7 and S16, ESI†). As reported by Muscarella et al.,56 neither the grain orientation nor the grain size is the dominant factor determining the optoelectronic properties of perovskite thin films. The main factors determining the efficiency of a device are the defects within the bulk and at the interface as well as the impurities within the materials.
 | ||
| Fig. 3 XRD analysis of the perovskite films. (A–D) XRD (100) peak (q = 10 nm−1) measured by in situ GIWAXS for control, control+RGO-5 h, control+RGO-10 h, and control+RGO-20 h perovskite films, respectively (all GIWAXS reciprocal lattice maps are presented in Fig. S12–S15, ESI†). (E) Full XRD patterns of the perovskite films after annealing. (F) Evolution of the (100) peak over time, from the GIWAXS experiment. (G) Ratio for the (100)/(110) XRD peaks. | ||
FEG-SEM images were obtained to investigate the morphology of the perovskite films. Fig. 4 and Fig. S17 (ESI†) show the results of FEG-SEM analysis for the same perovskite films measured using the GIWAXS setup. The control+RGO-xh films show full surface coverage, with larger grains in comparison with the reference (control) perovskite. Fig. 4E shows the grain size distribution in the film. The distribution of the grain size shifts to higher values for the modified perovskites and becomes wider when RGO-10 h and RGO-20 h are added. The average grain size (Fig. 4H) was estimated to be ∼97 nm for the pristine perovskite, ∼128 nm for control+RGO-5 h, ∼278 nm for control+RGO-10 h and ∼330 nm for control+RGO-20 h. Furthermore, few grains were measured to be larger than 900 nm for the perovskite/RGO-20 h sample.
 | ||
| Fig. 4 FEG-SEM images of the perovskite films prepared with precursors containing RGO. (A–D) Top view of control, control+RGO-5 h, control+RGO-10 h, and control+RGO-20 h samples, respectively. (E) Grain size distributions. (F) Mean grain size of the perovskite films. (G–J) Cross-section of control, control+RGO-5 h, control+RGO-10 h, and control+RGO-20 h samples, respectively. (K) Film thickness of the different samples. | ||
The increase in the average grain size reflects the decrease in the formation rate of the perovskite observed in the in situ GIWAXS experiments. However, the wider distribution of the grain sizes observed for the modified perovskite films can be a consequence of the introduction of a heterogeneous component in the precursor solution. According to Fig. S5 (ESI†), the RGO used in this modification has sheets of several different sizes, related to the method of synthesis. We theorize that the interaction of the perovskite precursors with the RGO sheets decreases the mobility of the precursors inside the solution, impacting the crystallization. A retardation of the nucleation steps is expected, enabling the growth of fewer crystals with larger sizes. The effect is more pronounced with RGO-20 h due to a much stronger interaction of the sp2 lattice of the RGO with the 2D layered PbI2. Sinha et al.55 synthesized a monolayer of PbI2 with epitaxial alignment on graphene and demonstrated that this coupling is indeed favored due to van der Waals interactions. Zhou et al.57 studied theoretically the interaction of graphene with PbI2 and confirmed that this interaction is favorable. It is possible that the interaction of PbI2 with the RGO is increased when it becomes more reduced due to a much larger conjugated system. The restored sp2 conjugation provides more interaction with the hexagonal phases formed during the crystallization,54,58 leaving ions less available for the formation of the perovskite. These effects can be related to the reduction in the formation rate of the perovskite. An additional effect can also be assigned to the nitrogen groups on graphene that can be basic, depending on their positions in the graphene structure. Therefore, the nitrogen groups, as special pyrrolic N in the RGO (since the XPS spectrum of RGO samples shows that pyrrolic N is the main type among different nitrogen species in our RGO samples) make a different environment around the formamidinium hydrogen atoms, slowing down the crystallization, which also leads to an increase in the perovskite grain size.23 But, regarding the small amount of nitrogen present in the RGO synthesized in this work, we believe that the greatest influence in the reduction of the formation rate can be related to the restoration of the sp2 conjugation in the RGO, leading a higher interaction between the RGO sheets and the perovskite precursors. A gradual increase in the film thickness from the pristine sample (∼423 nm) to the control+RGO-20 h sample (∼641 nm) (Fig. 4K) was observed with a concomitant increase in the absorbance of the films, without any significant modification in the band gap of the perovskite (Fig. S18, ESI†). The increase in the thickness of the films implies a more efficient light-harvesting ability. One possible explanation for the thickness increase could be the lower relative amount of solvent in comparison with the control solution. Besides, it is well known that both DMSO and DMF interact with the RGO sheets.59–62 So, the increase in thickness can also be associated with the interaction of RGO with the precursors and solvents, acting as a barrier against their expulsion.
Fig. 5A shows the steady-state photoluminescence (PL) of the perovskite films deposited on glass substrates taken during 300 s of continuous laser illumination. Mixed-halide perovskites suffer from photoinduced segregation, where iodine and bromine regions are established upon irradiation.63 Here, the characteristic redshift in the PL spectra upon illumination in all samples is observed. The stationary PL emissions of RGO-modified samples are already shifted to higher wavelengths, compared with the initial emission of the pristine sample (Fig. 5A). This behavior is more evident for samples with RGO-10 h and RGO-20 h. RGO interacts more strongly with iodide than bromide in solution,45,64 and this can cause the formation of I-rich phases during the crystallization, which can be responsible for the red shift observed in the stationary PL results. It is worth noting that there is an increase in the PL intensity for all samples, associated with the filling of the halide vacancies by O2 and, mostly, H2O present in the atmosphere.65 For samples with RGO-10 h and RGO-20 h, the increase of the PL intensity over time is less pronounced, which could be a consequence of the hydrophobicity of these samples. This indicates that the oxygen groups present in the RGO cannot be ruled out as trap sites.
 | ||
| Fig. 5 Photophysical characterization of the PSCs. (A) Stationary PL evolution (λexc = 442 nm and power = 300 W) with time for control, control+RGO-5 h, control+RGO-10 h and control+RGO-20 h samples. The dashed line represents the band gap energy (EG). (B) TrPL decay (λPLE = 445 nm; F = 24.3 nJ cm−2; and λPL = 755 nm) of the control and RGO-modified samples deposited onto a glass substrate. Stretched exponential fit decays are shown in red curves. | ||
We employed time-resolved photoluminescence (TrPL) measurements in perovskite films deposited on glass substrates (Fig. 5B) to study the charge-carrier recombination dynamics in perovskite films modified with RGO. Due to the disorder in this kind of sample (i.e., polycrystallinity, alloy composition, grain size distribution, Br- and I-rich phase segregation, high-density of trap-states, etc.), a simple exponential decay function did not fit the TrPL data.51,52 Therefore, a stretched exponential decay function was chosen to fit the TrPL results66,67 and the data are summarized in Table 1. All samples containing RGO displayed shorter lifetimes compared with the pristine sample. This might indicate, among other processes, charge transfer from the perovskite to the RGO sheets in the films. The quenching in the PL signal is more pronounced in the sample containing RGO-5 h (Fig. 5B and Table 1). However, the disorder parameter of the decays (β factor) increases close to β = 1 with the addition of RGO in the perovskite and with increasing reduction time. This behavior is completely in accordance with the increase of the grain size and the reduction of the grain boundaries observed in the FEG-SEM top-view and cross-section images of the samples (Fig. 4).
| Sample | τ s | β | R 2 |
|---|---|---|---|
| Control | 836 | 0.64 | 0.98 |
| Control+RGO-5 h | 227 | 0.65 | 0.99 |
| Control+RGO-10 h | 499 | 0.72 | 0.99 |
| Control+RGO-20 h | 473 | 0.78 | 0.99 |
The same experiment was repeated with the films containing a Spiro-OMeTAD layer deposited on the top of the films (Fig. S19A, ESI†) and with the films deposited on a mesoporous TiO2 layer (Fig. S19B, ESI†). The quenching effect observed for all RGO-modified samples was even more evident when an ETL (TiO2) is present (Table S1, ESI†). In addition, a gradual decrease in the charge-carriers lifetime was observed with the increase in the reduction degree of the RGO used in the modification. This corroborated the restoration of the sp2 lattice of the RGO, which increased the electron-extraction properties of the RGO.68 In the presence of Spiro-OMeTAD (HTL), a decrease in the charge-carrier lifetime was observed (Table S1, ESI†),although less pronounced as with TiO2. The small difference between the pristine sample and the samples with RGO might account for the more electron-accepting than hole-accepting character of our RGO samples.68,69 Another result that corroborated this is the increase of the lifetime when increasing the reduction degree of the RGO (Table S1, ESI†), reflecting the increase in the electron-accepting character of the RGO.
To evaluate the influence of RGO on the PSC performance, we fabricated PSCs with the n–i–p configuration. Fig. 6, Table 2, and Fig. S20 (ESI†) display the results for the best cells and the statistical data of solar cell parameters, respectively. The best pristine PSC (control) reached 18.3% (16.0 ± 1.3%) and 17.0% (14.8 ± 1.5%) efficiency (PCE) for backward and forward scans, respectively. The devices with RGO presented lower PCE values. The device containing RGO that presented the highest efficiency was the one with RGO-5 h, which reached 16.5% (14.1 ± 1.5%) and 13.4% (12.8 ± 1.2%) PCE for the backward and forward scans, respectively. The devices assembled with RGO-20 h achieved a PCE of 14.9% (13.7 ± 1.1%) for the backward scan and 14.2% (12.7 ± 1.2%) for the forward scan. Comparing the average values, the VOC was slightly affected by the addition of RGO: a decrease from 1.09 ± 0.02 V to 1.04 ± 0.02 V (backward scan) when adding RGO to the precursor solution. The small reduction in the average VOC value is related to RGO sheets acting as localized electron acceptors in the perovskite films. Our findings are different from those observed recently by Kim et al.,39 where they observed a slight increase in the VOC values after the addition of minimal amounts of RGO. They also affirm that RGO plays a negligible role in the crystallization of the perovskite film. According to in situ GIWAXS experiments, the RGO retards the crystallization of the perovskite. This is not a surprise, and it is indeed expected, considering the size of the graphene flakes, which is by the way the same size as in the study of Kim et al.39
 | (1) |
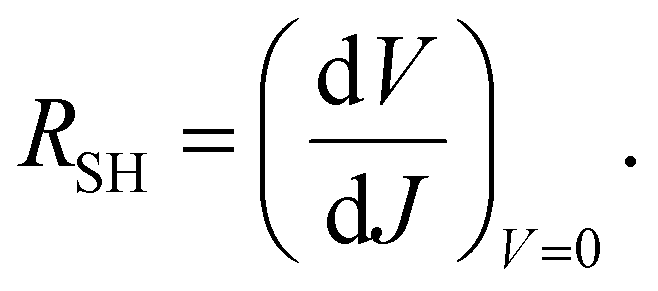 | (2) |
 | ||
| Fig. 6 Photovoltaic characterization of the PSCs. (A) J–V curves of all devices (forward measurement). (B) J–V curves of all devices (reverse measurement). | ||
| Sample | V OC (V) | J SC (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|
| Forward | ||||
| Control | 1.14 | 22.9 | 65.2 | 17.0 |
| Control+RGO-5 h | 1.00 | 24.1 | 56.1 | 13.4 |
| Control+RGO-10 h | 1.07 | 22.4 | 57.0 | 13.7 |
| Control+RGO-20 h | 1.04 | 22.8 | 59.8 | 14.2 |
| Sample | V OC (V) | J SC (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|
| Reverse | ||||
| Control | 1.15 | 23.1 | 68.8 | 18.3 |
| Control+RGO-5 h | 1.03 | 23.7 | 67.3 | 16.5 |
| Control+RGO-10 h | 1.09 | 22.3 | 60.1 | 14.6 |
| Control+RGO-20 h | 1.05 | 22.3 | 63.4 | 14.9 |
The values of the series resistance (RS), signifying losses related to the charge resistance of the device materials and resistive contacts, and the shunt resistance (RSH), representing the loss by recombination of the charge carriers, particularly on the interfaces, were calculated to obtain a deeper understanding of the device performance. These values can be estimated by the analysis of the variation of the current versus the potential through eqn (1) and (2). It is important to notice that, to optimize the performance of the solar cells, RS → 0 and RSH → ∞. The calculated resistance values, from the forward J–V measurements, are displayed in Table 3. The values of RS increase slightly with the modifications. On the other hand, the RSH value for only the control+RGO-5 h sample was higher than the control device. This reflects the decrease in the photovoltaic parameters observed for all the modified devices. It can be concluded that RGO is acting by disturbing the overall charge mobility within the device. The decrease in the PCE values is mainly related to the decrease in VOC and FF values. The decrease in FF is attributed mainly to the increase in the RS value, which reflects the increase in the resistance of charge transfer provoked by the electron-acceptor character of the RGO present in the film, which can act as a barrier for the overall mobility of the charges. Thus, there is a trade off when using these materials in PSCs, and the balance between JSC, VOC and FF must be maximized.
| Sample | R S (Ω cm2) | R SH (Ω cm2) |
|---|---|---|
| Control | 8.4 | 835.9 |
| Control +RGO-5 h | 11.3 | 1001.7 |
| Control +RGO-10 h | 11.4 | 595.0 |
| Control+RGO-20 h | 11.8 | 511.1 |
An important aspect for the commercialization of PSCs is their intrinsic stability. The stability of the devices under the dark storage conditions was evaluated to provide information on the tolerance of the solar cells to oxygen, moisture, and even pollution present in the air.48 The devices were stored under ambient conditions (T ≈ 25 °C) in the dark, following the ISOS protocol ISOS-D-1.48 It is important to mention that the devices were not sealed. Fig. 7 shows the normalized changes in PCE measured over 720 hours. The results show that the sample without RGO degraded faster than the other samples and that graphene is a good inhibitor of degradation possibly because of its hydrophobic character. The stability improvement promoted by the modification with RGO has also been reported recently by Kim et al.,39 where they observed an increase in the thermal stability of the devices, mainly where RGO is introduced in the spiro-OMeTAD layer. Here, we show that the modification of the perovskite precursor solution with RGO can lead to an improvement in the stability of the devices as well. The sample that had the lowest PCE decrease was the sample with RGO-20 h. This sample showed more than 80% stability for up to 100 hours of dark storage, and even after 720 hours the sample remained more stable than the others. This can be related to the increase in the perovskite grain size observed for the modified samples. With larger grains, there is a decrease in the superficial area, which inhibits the reaction with atmospheric oxygen and humidity. The stability improvement can also be associated with the increase in the hydrophobic nature of the RGO with the higher reduction degree.59,60 The sum of all these factors promoted an increase in the overall stability of the PSCs.
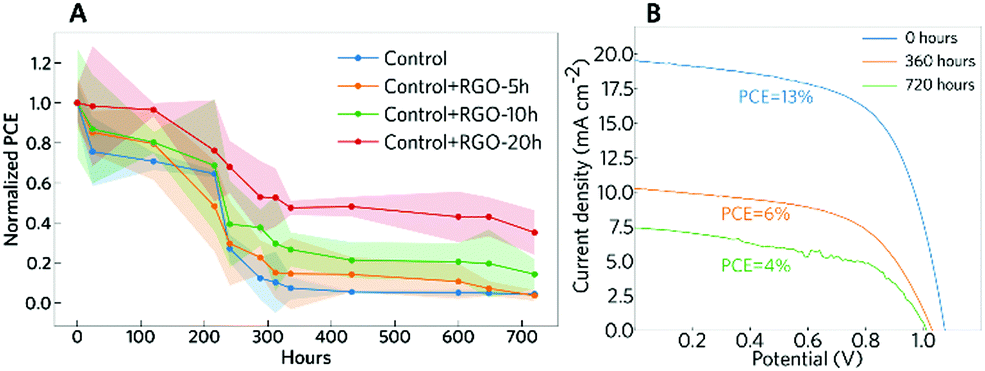 | ||
| Fig. 7 Stability of the PSCs. (A) Normalized average PCE changes of PSCs stored in the dark but exposed to ambient conditions (T ≈ 25 °C and RH ≈ 55%), following the ISOS protocol ISOS-D-1. (B) J–V curves measured at the first day, after 360 hours and after 720 hours (sample, control+RGO-20 h). | ||
Conclusions
In summary, we investigated the effect of the combination of two emerging materials, reduced graphene oxide and organic–inorganic lead halide perovskite, in the active layer of solar cells. The influence of the degree of reduction for RGO on the formation rate of the mixed perovskite was studied by systematically adding RGO with different sp2 characters (RGO-5 h, RGO-10 h and RGO-20 h) into the perovskite precursor solution. The formation of the perovskite was monitored by in situ GIWAXS experiments. It was possible to observe a relative delay in the appearance and an increase in the intensity of the (100) peak of the perovskite during the spin-coating process. This delay was more pronounced in the deposition of films modified with RGO with a higher degree of reduction (RGO-10 h and RGO-20 h). This experiment showed that the components of the precursor solution might interact with the restored sp2 conjugation of the RGO and then delay the formation of the perovskite nuclei. The annealing process was also studied by in situ GIWAXS, and it was observed that RGO also plays a role in the final crystallization step. This delay in the formation and crystallization of the perovskite promoted the formation of perovskite films with larger grains.The RGO sheets could also act as points of nucleation; this effect led to obtaining perovskite films with larger grains. The main improvement of the increase in the grain sizes of the perovskite was the improvement in the stability of the solar cells. We can observe that the device modified with RGO-20 h presented the highest stability. A sum of diverse effects is responsible for the improved stability: the increase in the grain size, which decreases the availability of the films to react with ambient components, and the increase in the hydrophobicity of the films as the reducing character of the RGO is increased.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the Laboratorio Nacional de Luz Sincrotron (LNLS), XRD2 facility. The authors are grateful to Dr Renato V. Gonçalves for providing access to the XPS instrument. A.F.N., P. E. M., F. L. A., J. C. G. and R. S gratefully acknowledge support from FAPESP (the Sao Paulo Research Foundation, Process 2017/11986-5) and Shell and the strategic importance of the support given by ANP (Brazil's National Oil, Natural Gas and Biofuels Agency) through the R&D levy regulation. F. L. A. acknowledges FAPESP (20/14451-8). E. M. T. acknowledges CNPq (150043/2018-0). P. E. M. and A. F. N. also acknowledge CNPq and CAPES.References
- M. A. Green, Y. Hishikawa, E. D. Dunlop, D. H. Levi, J. Hohl-Ebinger, M. Yoshita and A. W. Y. Ho-Baillie, Prog. Photovoltaics Res. Appl., 2019, 27, 3–12 CrossRef.
- First Solar Release, 2015, 0–1.
- J. Britt and C. Ferekides, Appl. Phys. Lett., 1993, 62, 2851–2852 CrossRef CAS.
- K. Tvrdy and P. V. Kamat, Comprehensive Nanoscience and Technology, North-Holland, 2010, vol. 1–5, pp. 257–275 Search PubMed.
- Z. Pan, H. Rao, I. Mora-Seró, J. Bisquert and X. Zhong, Chem. Soc. Rev., 2018, 47, 7659–7702 RSC.
- B. O’Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474–1476 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- H. S. Kim, C. R. Lee, J. H. Im, K. B. Lee, T. Moehl, A. Marchioro, S. J. Moon, R. Humphry-Baker, J. H. Yum, J. E. Moser, M. Grätzel and N. G. Park, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- NREL, NREL Best Res. Effic. Chart, https://www.nrel.gov/pv/cell-efficiency.html, accessed 6 March 2020.
- J. Breternitz and S. Schorr, Adv. Energy Mater., 2018, 1802366, 1802366 CrossRef.
- Z. Xiao, Z. Song and Y. Yan, Adv. Mater., 2019, 31, 1–22 Search PubMed.
- S. Svanström, T. J. Jacobsson, T. Sloboda, E. Giangrisostomi, R. Ovsyannikov, H. Rensmo and U. B. Cappel, J. Mater. Chem. A, 2018, 6, 22134–22144 RSC.
- K. Poorkazem and T. L. Kelly, ACS Appl. Energy Mater., 2018, 1, 181–190 CrossRef CAS.
- M. Zhou, J. S. Sarmiento, C. Fei, X. Zhang and H. Wang, J. Phys. Chem. Lett., 2020, 11, 1502–1507 CrossRef CAS PubMed.
- Z. Xiao, Q. Dong, C. Bi, Y. Shao, Y. Yuan and J. Huang, Adv. Mater., 2014, 26, 6503–6509 CrossRef CAS PubMed.
- N. Mohammadian, A. Moshaii, A. Alizadeh, S. Gharibzadeh and R. Mohammadpour, J. Phys. Chem. Lett., 2016, 7, 4614–4621 CrossRef CAS PubMed.
- M. Salado, L. Calió, L. Contreras-Bernal, J. Idígoras, J. A. Anta, S. Ahmad and S. Kazim, Materials, 2018, 11, 1073 CrossRef PubMed.
- C. Bi, Y. Shao, Y. Yuan, Z. Xiao, C. Wang, Y. Gao and J. Huang, J. Mater. Chem. A, 2014, 2, 18508–18514 RSC.
- N. K. Noel, A. Abate, S. D. Stranks, E. S. Parrott, V. M. Burlakov, A. Goriely and H. J. Snaith, ACS Nano, 2014, 8, 9815–9821 CrossRef CAS PubMed.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Nat. Commun., 2014, 5, 5784 CrossRef CAS PubMed.
- M. Hadadian, J.-P. Correa-Baena, E. K. Goharshadi, A. Ummadisingu, J.-Y. Seo, J. Luo, S. Gholipour, S. M. Zakeeruddin, M. Saliba, A. Abate, M. Grätzel and A. Hagfeldt, Adv. Mater., 2016, 28, 8681–8686 CrossRef CAS PubMed.
- V. Somsongkul, F. Lang, A. R. Jeong, M. Rusu, M. Arunchaiya and T. Dittrich, Phys. Status Solidi RRL, 2014, 08, 763–766 CrossRef CAS.
- K. S. Novoselov, V. I. Fal′ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- K. Kim, H.-B.-R. Lee, R. W. Johnson, J. T. Tanskanen, N. Liu, M.-G. Kim, C. Pang, C. Ahn, S. F. Bent and Z. Bao, Nat. Commun., 2014, 5, 4781 CrossRef CAS PubMed.
- D. Konios, M. M. Stylianakis, E. Stratakis and E. Kymakis, J. Colloid Interface Sci., 2014, 430, 108–112 CrossRef CAS PubMed.
- R. Aradhana, S. Mohanty and S. K. Nayak, Polymer, 2018, 141, 109–123 CrossRef CAS.
- T. S. Sreeprasad and V. Berry, Small, 2013, 9, 341–350 CrossRef CAS PubMed.
- J. D. Roy-Mayhew and I. A. Aksay, Chem. Rev., 2014, 114, 6323–6348 CrossRef CAS PubMed.
- X. Guo, G. Lu and J. Chen, Front. Energy Res., 2015, 3, 50 Search PubMed.
- Q.-D. Yang, J. Li, Y. Cheng, H.-W. Li, Z. Guan, B. Yu and S.-W. Tsang, J. Mater. Chem. A, 2017, 5, 9852–9858 RSC.
- K. T. Cho, G. Grancini, Y. Lee, D. Konios, S. Paek, E. Kymakis and M. K. Nazeeruddin, ChemSusChem, 2016, 9, 3040–3046 CrossRef CAS PubMed.
- B. Lee, D. B. Buchholz and R. P. H. Chang, Energy Environ. Sci., 2012, 5, 6941 RSC.
- D. Konios, G. Kakavelakis, C. Petridis, K. Savva, E. Stratakis and E. Kymakis, J. Mater. Chem. A, 2016, 00, 1–12 Search PubMed.
- U. A. Méndez-Romero, S. A. Pérez-García, X. Xu, E. Wang and L. Licea-Jiménez, Carbon, 2019, 146, 491–502 CrossRef.
- S. Silvestrini, C. C. De Filippo, N. Vicentini, E. Menna, R. Mazzaro, V. Morandi, L. Ravotto, P. Ceroni and M. Maggini, Chem. Mater., 2018, 30, 2905–2914 CrossRef CAS.
- M. He, Y. Chen, H. Liu, J. Wang, X. Fang and Z. Liang, Chem. Commun., 2015, 51, 9659–9661 RSC.
- H.-S. Kim, B. Yang, M. M. Stylianakis, E. Kymakis, S. M. Zakeeruddin, M. Grätzel and A. Hagfeldt, Cell Rep. Phys. Sci., 2020, 1, 100053 CrossRef.
- T. Jesper Jacobsson, J.-P. Correa-Baena, M. Pazoki, M. Saliba, K. Schenk, M. Grätzel and A. Hagfeldt, Energy Environ. Sci., 2016, 9, 1706–1724 RSC.
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6, 613–617 RSC.
- Z. Wang, Q. Lin, F. P. Chmiel, N. Sakai, L. M. Herz and H. J. Snaith, Nat. Energy, 2017, 2, 17135–17145 CrossRef CAS.
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Hörantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz, H. J. Snaith, G. Sadoughi, B. Rech, M. B. Johnston, H. J. Snaith, M. T. Horantner, L. Korte, D. P. McMeekin, N. Sakai, A. Haghighirad, W. Rehman and G. E. Eperon, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- E. J. Juarez-Perez, Z. Hawash, S. R. Raga, L. K. Ono and Y. Qi, Energy Environ. Sci., 2016, 9, 3406–3410 RSC.
- P. E. Marchezi, G. G. Sonai, M. K. Hirata, M. A. Schiavon and A. F. Nogueira, J. Phys. Chem. C, 2016, 120, 23368–23376 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- Y. Rong, Z. Tang, Y. Zhao, X. Zhong, S. Venkatesan, H. Graham, M. Patton, Y. Jing, A. M. Guloy and Y. Yao, Nanoscale, 2015, 7, 10595–10599 RSC.
- M. V. Khenkin, E. A. Katz, A. Abate, G. Bardizza, J. J. Berry, C. Brabec, F. Brunetti, V. Bulović, Q. Burlingame and A. Di Carlo, et al. , Nat. Energy, 2020, 5, 35–49 CrossRef.
- R. Szostak, P. E. Marchezi, A. dos, S. Marques, J. C. da Silva, M. S. de Holanda, M. M. Soares, H. C. N. Tolentino and A. F. Nogueira, Sustainable Energy Fuels, 2019, 3, 2287–2297 RSC.
- A. A. Sutanto, R. Szostak, N. Drigo, V. I. E. Queloz, P. E. Marchezi, J. C. Germino, H. C. N. Tolentino, M. K. Nazeeruddin, A. F. Nogueira and G. Grancini, Nano Lett., 2020, 20, 3992–3998 CrossRef CAS PubMed.
- L. M. Herz, ACS Energy Lett., 2017, 2, 1539–1548 CrossRef CAS.
- S. D. Stranks, et al. , Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- D. López-Díaz, M. López Holgado, J. L. García-Fierro and M. M. Velázquez, J. Phys. Chem. C, 2017, 121, 20489–20497 CrossRef.
- R. Szostak, P. E. Marchezi, A. D. S. Marques, J. C. Da Silva, M. S. De Holanda, M. M. Soares, H. C. N. Tolentino and A. F. Nogueira, Sustainable Energy Fuels, 2019, 3, 2287–2297 RSC.
- S. Sinha, T. Zhu, A. France-Lanord, Y. Sheng, J. C. Grossman, K. Porfyrakis and J. H. Warner, Nat. Commun., 2020, 11, 1–13 CrossRef PubMed.
- L. A. Muscarella, E. M. Hutter, S. Sanchez, C. D. Dieleman, T. J. Savenije, A. Hagfeldt, M. Saliba and B. Ehrler, J. Phys. Chem. Lett., 2019, 10, 6010–6018 CrossRef CAS PubMed.
- M. Zhou, W. Duan, Y. Chen and A. Du, Nanoscale, 2015, 7, 15168–15174 RSC.
- M. Acik and S. B. Darling, J. Mater. Chem. A, 2016, 4, 6185–6235 RSC.
- J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- D. W. Johnson, B. P. Dobson and K. S. Coleman, Curr. Opin. Colloid Interface Sci., 2015, 20, 367–382 CrossRef CAS.
- H. R. Thomas, A. J. Marsden, M. Walker, N. R. Wilson and J. P. Rourke, Angew. Chem., Int. Ed., 2014, 53, 7613–7618 CrossRef CAS PubMed.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS.
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6, 613–617 RSC.
- G. Kalita, K. Wakita, M. Takahashi and M. Umeno, J. Mater. Chem., 2011, 21, 15209 RSC.
- D. J. Slotcavage, H. I. Karunadasa and M. D. McGehee, ACS Energy Lett., 2016, 1, 1199–1205 CrossRef CAS.
- M. B. Johnston and L. M. Herz, Acc. Chem. Res., 2016, 49, 146–154 CrossRef CAS PubMed.
- J. C. Germino, R. Szostak, S. G. Motti, R. F. Moral, P. E. Marchezi, H. S. Seleghini, L. G. Bonato, F. L. De Araújo, T. D. Z. Atvars, L. M. Herz, D. Fenning, A. Hagfeldt and A. F. Nogueira, ACS Photonics, 2020, 7, 2282–2291 CrossRef CAS.
- K. T. Cho, G. Grancini, Y. Lee, D. Konios, S. Paek, E. Kymakis and M. K. Nazeeruddin, ChemSusChem, 2016, 9, 3040–3044 CrossRef CAS PubMed.
- K. D. G. I. Jayawardena, R. Rhodes, K. K. Gandhi, M. R. R. Prabhath, G. D. M. R. Dabera, M. J. Beliatis, L. J. Rozanski, S. J. Henley and S. R. P. Silva, J. Mater. Chem. A, 2013, 1, 9922–9927 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc01360b |
| This journal is © The Royal Society of Chemistry 2021 |