
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiquid-crystalline TADF materials based on substituted carbazoles and terephthalonitrile†
Alfiya F.
Suleymanova
 a,
Marsel Z.
Shafikov
bc,
Adrian C.
Whitwood
a,
Rafał
Czerwieniec
*b and
Duncan W.
Bruce
*a
a,
Marsel Z.
Shafikov
bc,
Adrian C.
Whitwood
a,
Rafał
Czerwieniec
*b and
Duncan W.
Bruce
*a
aDepartment of Chemistry, University of York, Heslington, YORK YO10 5DD, UK. E-mail: duncan.bruce@york.ac.uk; Tel: +44 1904 324085
bInstitut für Physikalische Chemie Universität Regensburg, Universitätsstrasse 31, Regensburg 93053, Germany. E-mail: rafal.czerwieniec@chemie.uni-regensburg.de; Tel: +49 941 943 4463
cUral Federal University, Mira 19, Ekaterinburg, 620002, Russia
First published on 30th April 2021
Abstract
By functionalising 2,5-di(N,N′-carbazolyl)terephthalonitrile or 2,3,5,6-tetra(N,N′-carbazolyl)terephthalonitrile with alkoxy chains located on the carbazole moiety, a family of materials is realised, all of which show a TADF response and two of which are also liquid crystalline.
Introduction
There is great interest in luminescent liquid crystals as multifunctional materials, as they combine emissive properties with the ability to self-organise over extended length scales. This combination of properties suggests potential applications in the general field of electronic devices, such as polarised emitters,1,2 organic field-effect transistors (OFETs),3,4 organic photovoltaic devices5,6 and organic light emitting diodes, OLEDs.7Because of their ability to harvest 100% of the excitons produced in the OLED device, phosphorescent materials are much more effective emitters comparing to fluorescence equivalents and those based on iridium(III) in particular are used successfully in the current generation of commercial OLED displays.8,9 However, more recently there has been increasing interest in a new family of emissive materials based on fluorescence which, in common with phosphorescent materials, can utilise 100% of device excitons.10 This is possible when the lowest excited singlet and triplet states, S1 and T1, are close in energy. When the S1–T1 separation is sufficiently small, the singlet state can be populated thermally from the lower-lying triplet state generated in course of electroluminescent excitation in the OLED. Thus, thermally activated delayed fluorescence, TADF, is observed. The small separation of the S1 and T1 states can be achieved when the HOMO and LUMO orbitals are separated significantly in space. In such a case, the exchange energy is small and the states resulting from the HOMO → LUMO excitation are close in energy. The ability to realise high electroluminescence efficiency then no longer relies on the incorporation of a heavy metal to facilitate spin–orbit coupling.
However, while there has been interest in the design and synthesis of liquid-crystalline materials that are either phosphorescent or emit through conventional fluorescence mechanisms, to the best of our knowledge there are currently no examples of liquid-crystalline TADF materials.
Pioneered by Adachi and co-workers,11 the use of carbazole donors with terephthalonitrile (1,4-dicyanobenzene) acceptors represents an archetypal class of purely organic TADF materials. In these compounds, the HOMO is essentially centred at the carbazole moieties whereas the LUMO is localised on the dicyanobenzene moiety. The separation of the frontier orbitals arises as the donor and acceptor rings are essentially orthogonal to each other.
As demonstrated by Giroud–Godquin12 and by Swager,13,14 and then later used by ourselves with octahedral iridium(III) complexes,15,16 judicious functionalisation of the periphery of a more three-dimensional molecule by flexible alkyl chains can generate an anisotropic motif capable of being stabilised in a liquid-crystal phase. Indeed, fullerenes can also be made liquid crystalline.17 Therefore, using this general approach, we generated a family of four related compounds (3a, 3b, 4a, 4b) whose preparation and formulae are shown in Scheme 1. The mono-(1a) or di-(1b) alkoxyphenyl dioxaborolanes were obtained directly from the corresponding alkoxybromobenzene and were then reacted with 3,6-dibromocarbazole using a Suzuki–Miyaura protocol to give the substituted carbazoles 2. Target compounds 3 and 4 were then obtained in a nucleophilic substitution reaction with 2,5-difluoroterephthalonitrile or 2,3,5,6-tetrafluoroterephthalonitrile, respectively. Full details are found in the ESI.†
 | ||
| Scheme 1 Synthetic route to the target products 3 and 4. Conditions: (i) and (ii) K2CO3 (10 mol equiv.), [Pd3(OAc)6] (0.2 mol equiv.), S-Phos (0.4 mol equiv.), toluene/ethanol/water, 80 °C, 24–48 h; (III) and (iv) NaH (6 mol equiv.), DMF or THF, 75 °C, 4 h. | ||
It was possible to obtain single crystals of the methoxy analogues of 3b and 4b (labelled 3b-Me and 4b-Me, respectively) and their structures were determined using X-ray methods; the data are recorded in Table S1 (ESI†), while the structures are shown as Chart 1. Analysis of the structure of 3b-Me shows that the plane described by the central phenyl ring of the terephthalonitrile subtends an angle of 54.84° to a plane described by the five atoms in the pyrrolic ring of the carbazole. This is somewhat smaller than that found in related materials functionalised with more carbazole groups, reflecting the lower steric crowding. In terms of intermolecular interactions, there is a distance of 3.985 Å between the planes of phenyl groups bearing the methoxy groups (Fig. S1, ESI†), but no short contacts between carbazole units.18
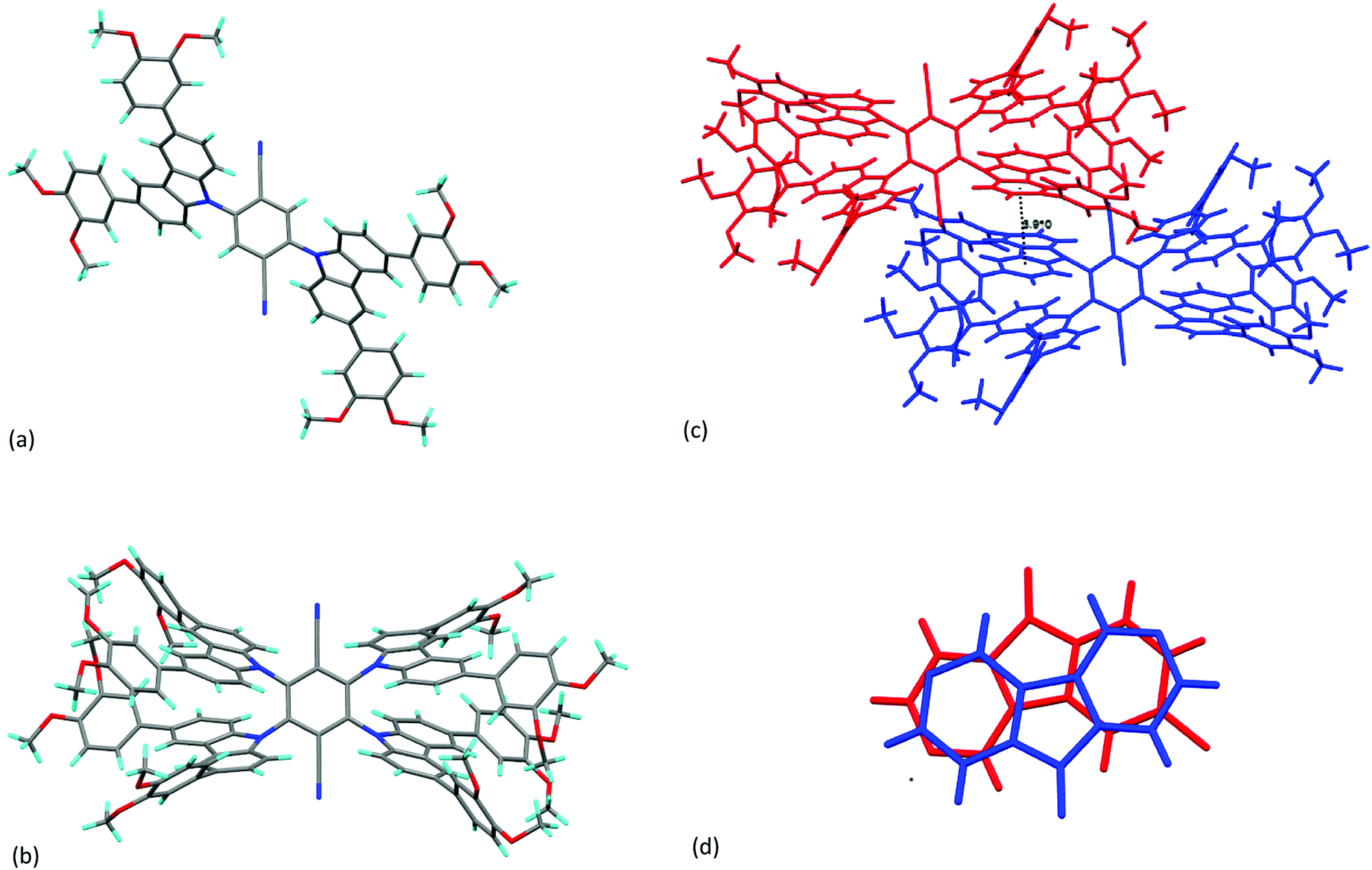 | ||
| Chart 1 (a) Molecular structure of compound 3b-Me; (b); molecular structure of compound 4b-Me (c) figure showing the close approach of two molecules of 4b-Me in the solid state giving rise to the intermolecular ring–ring separation of 3.910 Å (different molecules color-coded for ease of observation); (d) figure to show the detail of the overlap of the carbazole ring systems. | ||
In the structure of 4b-Me, the plane described by the central phenyl ring of the terephthalonitrile subtends a somewhat larger angle of 63.48 ± 0.05° to the planes described by the five atoms in the pyrrolic ring of the carbazole. Steric effects open this angle up compared to that found in 4a-Me and indeed, the larger angle is similar values to those found in related structures described by Etherington et al.18 Also in common with their observations, a close intermolecular ring–ring contact of about 3.9 Å is found between carbazole phenyl rings (Chart 1b) and an illustration of the overlap is shown in Chart 1c. Such interactions were proposed as the origin of changes in emission characteristics, sometimes also as a function of concentration in the emissive layer, in the solid state and devices on the basis of the existence of persistent dimer states.
Liquid-crystalline properties
The thermal properties of compounds 3 and 4 were first determined by polarised optical microscopy, which showed that neither 3a nor 4a was a liquid crystal, each simply melting to give an isotropic fluid at 242 and 249 °C, respectively. Being (short-chain) methoxy derivatives, neither 3b-Me nor 4b-Me shows liquid crystal properties. However, for 3b and 4b the situation was different, and both were found to show a columnar hexagonal (Colh) mesophase; the observations are the same for both compounds. Placing the as-obtained material on the microscope slide, it was found to be rather viscous with the viscosity decreasing as the temperature increased and while there was an overall birefringence to the sample, there was no discernible texture to help identify the phase. However, at 181 °C (3b) or 191 °C (4b) the birefringent fluid gave way to an isotropic fluid which, on cooling developed well-defined optical textures as shown in Fig. 1a and b, both being characteristic of a Colh mesophase. The transitions were detected also by DSC (Fig. 1c and d) | ||
| Fig. 1 Polarising optical microscopy images: (a) compound 3b at 177 °C on cooling; (b) compound 4b at 186 °C on cooling. DSC traces (second heat and second cool, scan rate of 5 °C min−1): (c) compound 3b; (d) compound 4b. | ||
To confirm the symmetry of the observed mesophases, small-angle X-ray scattering (SAXS) measurements were performed for both compounds and the diffraction patterns are shown in Fig. 2. Compound 3b showed a strong, low-angle (10) reflection at 2θ = 2.52° (Fig. 2a) corresponding to a d-spacing of 35 Å and a lattice (a) parameter of 40.4 Å. Much weaker reflections at 2θ = 4.33° and 4.93° were assigned as the corresponding (11) and (20) reflections, confirming hexagonal symmetry. For compound 4b, the strong reflection was found at a very similar angle (2θ = 2.79°, Fig. 2b), although higher-order reflections were not readily evident. The assignment of 4b as showing a Colh phase was confirmed as it was found to be continuously miscible with 3b (Fig. S2, ESI†). From the single crystal structure of 4b-Me and from literature data for all-trans C12 chains, it is possible to estimate the length of 3b and 4b to be about 53 Å, which is significantly greater than the lattice parameter evaluated for 3b (≈ 40 Å) or 4b (≈ 36 Å), suggesting some tilting of the molecular units within the columns.19
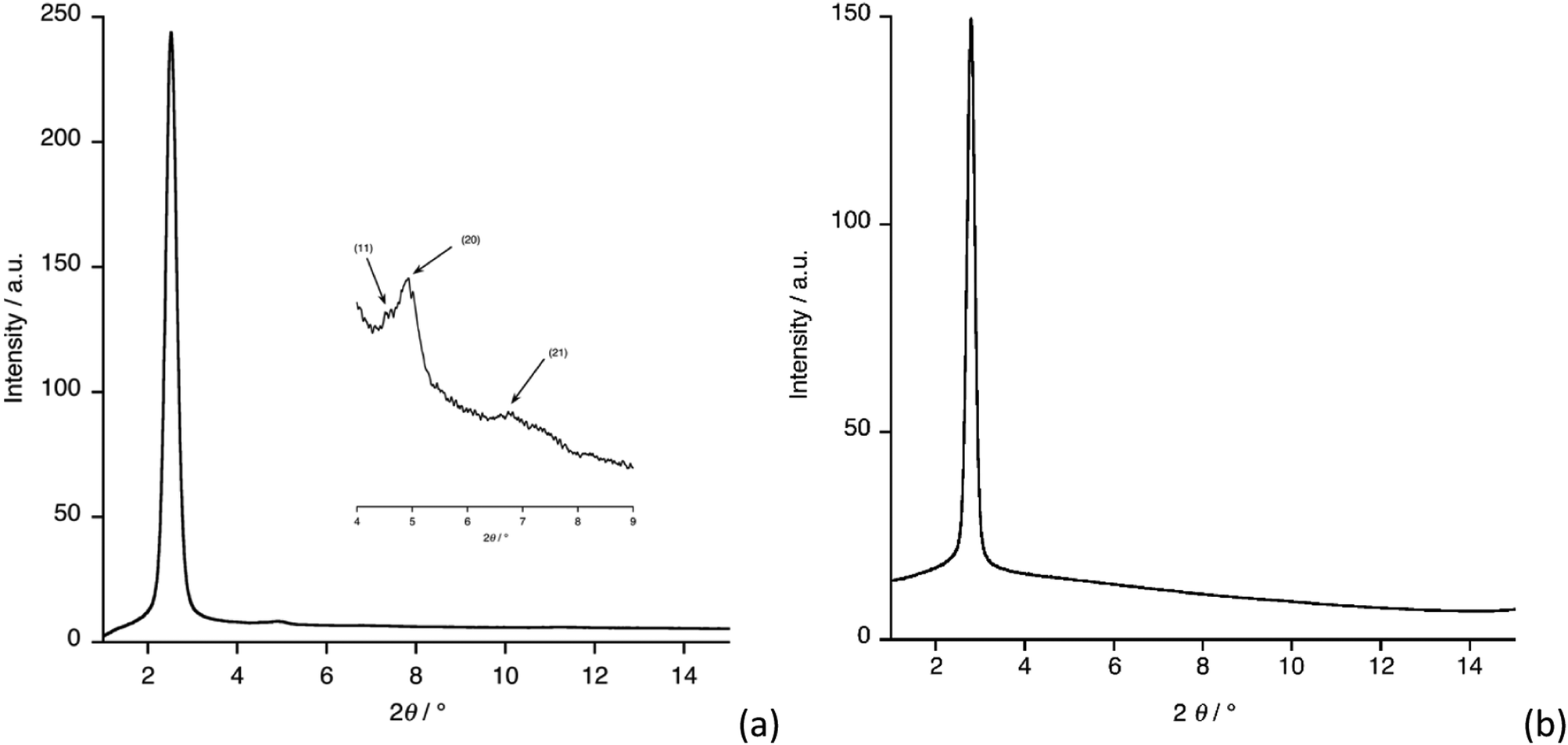 | ||
| Fig. 2 SAXS patterns for: (a) compound 3b at 39.4 °C; (b) compound 4b at 45.5 °C. | ||
Photophysical properties
UV-Vis absorption spectra of compounds 3a, 3b, 4a, and 4b reveal distinct, low-energy absorption bands due to charge-transfer transitions. Respective band maxima are, for example, 440 nm for 3a and 3b and 510 nm for 4a and 4b, respectively. (Fig. 3 and Fig. S3 in ESI†). The charge-transfer character of the excited states, resulting from an electron density shift from carbazole moieties to terephthalonitrile, is in accordance with literature data for analogous carbazole-cyanobenzenes11 and is also consistent with the results from TD-DFT calculations (below). | ||
| Fig. 3 UV-Vis absorption spectra of compounds 3a, 3b, 4a and 4b in toluene. | ||
All of the compounds are luminescent at ambient temperature both as dilute solutions in organic solvents and as solid samples and their emission spectra recorded in toluene are shown in Fig. 4. The band maxima, decay times, and quantum yields are listed in Table 1.
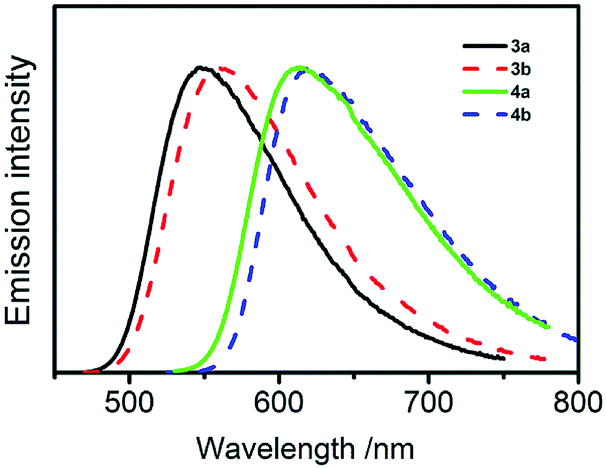 | ||
| Fig. 4 Luminescence spectra of compounds 3a, 3b, 4a, and 4b recorded in degassed diluted toluene solutions (ca 5 × 10−5 mol dm−3) at ambient temperature. | ||
| λ max/nm | τ/ns prompt flu. | τ/μs delayed em. | ϕ PL/% | k r/s−1 | k nr/s−1 | |
|---|---|---|---|---|---|---|
| a ‘int’ refers to the % emission intensity arising from the longer-lived emission. b Recorded in air. | ||||||
| 3a | 545 | 12.2 (9.9)b | 5.5 (10% int)a | 18 (12)air | 1.3 × 107 | 6.9 × 107 |
| (1.2 × 107)b | (8.9 × 107)b | |||||
| 3b | 560 | 6.9 (6.4)b | 3.5 (5% int)a | 5 | 0.6 × 107 | 14 × 107 |
| (0.8 × 107)b | (15 × 107)b | |||||
| 4a | 615 | 3.9 | ≤1 | 5 | 1.3 × 107 | 24 × 107 |
| 4b | 620 | 1.2 | ≤1 | 1.5 | 1.3 × 107 | 82 × 107 |
The emission bands are broad and structureless, reflecting the strongly charge-transfer character of the respective electronic transitions. Owing to the measured decay times on the order of several nanoseconds and partial spectral overlap with the lowest-energy absorption features, the emissions are identified as being dominated by prompt fluorescence (S1 → S0). However, in the decay profiles, longer-lived components with a decay time of several hundred ns (4a, 4b) up to 3.5 and 5.5 μs (3b and 3a, respectively) are present. The longer-lived emission component, which is on a μs timescale and originates from the T1 state, is the most evident in compound 3a (Fig. 5) and accounts for around 10% of the net intensity. Intrinsic phosphorescence (radiative transition T1 → S0) lifetimes for organic compounds normally amount to hundreds of milliseconds up to seconds. The T1 lifetime observed here, however, which is in the range <1 μs to 5.5 μs is much shorter and points to another relaxation mechanism, which, at ambient temperature, is most probably TADF. This assignment results straightforwardly from the analogy to related carbazole-terephthalonitrile derivatives, for which significant TADF was reported,11 and results of quantum chemical computations (below), predicting small S1–T1 energy separations of less than 0.1 eV – a prerequisite for efficient TADF.
 | ||
| Fig. 5 Emission decay curves for compound 3a recorded in toluene at ambient temperature. Degassed and air saturated samples are compared. | ||
The energy of the emitting singlet state depends on both the number of alkoxy groups attached to each carbazole moiety (two in compounds 3a and 4a and four in 3b and 4b, respectively) and the number of carbazole moieties in the molecule (two in 3a and 3b, and four in 4a and 4b, respectively). Thus, the emission maximum of compound 3a with just two carbazoles bearing two alkoxy groups is 545 nm while the emission maximum of compound 4b with four carbazoles bearing four alkoxy groups is 620 nm. The maxima of compounds 3b and 4a have intermediate values of 560 and 615 nm, respectively and each alkoxy group increases the donor strength of the carbazole moiety, thus shifting the S1 state to lower energy. Greater numbers of donor moieties in compounds 4a and 4b as compared to 3a and 3b increase the HOMO energy due to π–π interactions between adjacent carbazoles (in ortho position one to another) and decreases the LUMO energy, probably as an effect of the larger angles between the planes of terephthalonitrile acceptor and the carbazole donor moieties and the concomitantly reduced LUMO delocalisation in 4a and 4b. (cf. Fig. S8, ESI†) The other point of interest here is that while the effect of the addition of a methoxy group in the 4-position of the phenyl group is significant (e.g. λem = 615 nm for 4a compared to 577 nm for the equivalent compound with no methoxy groups–3,6-diphenylcarbazole donor),11 addition of the second methoxy group further red-shifts the emission by only 5 nm to 620 nm because the π-donor in the 3-position of the phenyl ring is not in conjugation with the carbazole.
The same trend found in solution is also observed for solid samples (Fig. S4 and Table S2 in ESI†) and frozen (77 K) solutions (Fig. S5 and Table S2 in ESI†). However, the magnitudes of each particular energy shift are not exactly the same, indicating that also other factors, such as local microenvironment, modulate excited state energies significantly.
The emissions recorded in liquid samples, with quantum yields from 1.5 to 18%, are moderate, whereas those determined for solid samples are significantly higher with the maximum value of 30% found for compound 3a. (Table S1, ESI†) This increase in quantum yield is a consequence of diminished non-radiative losses in a rigid microenvironments expressed by lower values of the non-radiative relaxation rate knr determined for solid samples (Table S1, ESI†) when compared to solution (Table 1). The rates found for different compounds increase with decreasing energy of the emitting excited state, resembling the trend expected according to the energy gap law.20 For example, for liquid samples of 3a and 4b, knr values of 6.9 × 107 s−1 and 82 × 107 s−1 are found, representing a difference by a factor of more than 10. On the other hand, the radiative rate of prompt fluorescence (kr in Table 1 and Tables S1 and S2, ESI†) is essentially invariant with compound composition and rigidity of the matrix.
Theoretical studies
To gain more insight into the luminescent properties of the materials, the electronic structure of the methoxy analogues 3b-Me, 4a-Me and 4b-Me was analysed theoretically. The computations utilised the Gaussian 09 code21 with the M11L22,23 density functional and def2-SVP basis set.24 Structures of 3b-Me, 4a-Me and 4b-Me were optimised in the ground state (S0) and the lowest excited triplet state electronic configurations. The corresponding molecular geometries can be found in the format of Cartesian coordinates in the ESI.† The relaxed ground-state geometries calculated for 3b-Me and 4b-Me are comparable to those determined by X-ray analysis and, for example, the calculated interplanar angles between the mean planes of carbazoles and mean plane of the phenyl ring of terephthalonitrile (ϕ) are rather similar: (Expt./Calc.): 3b-Me (54.84°/57.60°); 4b-Me (63.48°/65.97°). The notably larger values of parameter ϕ in 4a-Me and 4b-Me compared to 3b-Me, is due to the greater degree of substitution, where planarisation of the molecule by rotation of the carbazole moieties is hampered by increased intramolecular steric hinderance.According to the calculations, the lowest excited singlet (S1) and triplet (T1) states of 3b-Me, 4a-Me, 4b-Me, are of HOMO → LUMO orbital origin. The HOMOs of these compounds represent π-orbitals localised predominantly on the carbazole units (donors) whereas the LUMO is a π* orbital localised predominantly on the terephthalonitrile (Fig. 6 and Tables S3–S6 in the ESI†). Thus, transitions between the S1 and T1 states of 3b-Me, 4a-Me, 4b-Me can be described as π → π* in character (1,3ππ*) with strong charge transfer (1,3CT) from the donor carbazole to the acceptor terephthalonitrile. The unpaired electrons in these excited states are well separated spatially and have a weak exchange interaction resulting in a small triplet–singlet splitting ΔE(S1–T1). The ΔE(S1–T1) values, estimated from the energies of vertical (Frank–Condon) transitions S0 → T1 and S0 → S1 in the optimised T1 state geometries, are 0.05 eV (400 cm−1) for 3b-Me, 0.06 eV (480 cm−1) for 4a-Me and 0.04 eV for 4b-Me (320 cm−1). These values are comparable to those of other donor–acceptor (D–A) organic materials designed to exhibit TADF25 and at ambient temperature should allow efficient thermal activation of the T1 state to vibrational levels isoenergetic with the lower vibrational levels of the S1 state.
 | ||
| Fig. 6 The DFT (M11L/def2SVP) calculated iso-surface contour plots (iso-value = 0.03) and energies of the frontier orbitals of 3a-Me (a), 4a-Me (b) and 4b-Me (c) in the T1 state optimised geometries. Hydrogens are omitted for clarity. | ||
Intersystem crossing S1 → T1 (ISC) and reverse intersystem crossing T1 → S1 (RISC) are non-radiative transitions occurring between isoenergetic vibrational levels of states S1 and T1. The rates of these transitions determine the efficiency of thermal equilibration of S1 and T1, which is essential for TADF to occur. The rates of transition where a change in multiplicity is involved (e.g. singlet ↔ triplet) is determined by the efficiency of the spin–flip of an unpaired electron during the transition and is efficient only with conservation of the total momentum of the electron (spin plus orbital–El-Sayed's rule26). In organic molecules, the main mechanism facilitating this conservation is spin–orbit coupling (SOC).27 For the S1 and T1 states of 3b-Me, 4a-Me and 4b-Me, 1ππ* ↔ 3 ππ* ISC and 3 ππ* → 1 ππ* RISC processes are spin-forbidden in the zeroth-order approximation. The forbidden nature in this approximation can, however, be relaxed with perturbations caused by molecular vibrations. These can induce vibronic and spin–orbit coupling of the S1 and T1 states with ππ* states resulting from local excitations at the carbazole unit and at the terephthalonotrile (3LE triplets) as well as with σπ* and πσ* states.28 For instance, the vibrations displacing the atoms out of the plane of π-conjugation electronically perturb the S1 state (charge transfer 1ππ*) admixing it with singlet states of 1σπ* and 1πσ* character (vibronic coupling), giving a non-vanishing SOC matrix element with a triplet state having (at least partial) 3ππ* character. Also, SOC of the S1 state with triplet states of 3σπ* and 3πσ* character or with 3ππ* character LE triplets, which may become close energetically to S1 during the molecular vibrations, relaxes the spin-forbidden nature of the S1 ↔ T1 transition. Similarly, the T1 state is perturbed by vibronic coupling to other triplet states and by SOC with electronically suitable singlet states. Therefore, the perturbed S1 and T1 states can have a non-vanishing SOC matrix element relaxing the otherwise forbidden zeroth-order ISC and RISC processes between them. It is noted that this relaxation is weak due to the comparatively large energy gap to the perturbing (admixing) states (σπ*, πσ* and LE ππ* states) and small spin–orbit coupling constants of the light elements (H, C, N)29 constituting 3b-Me, 4a-Me and 4b-Me.
Since the molecular vibrations facilitate the S1 ↔ T1 transition rates, it can be expected that ISC and RISC rates in 4a-Me and 4b-Me are larger compared to 3b-Me. This is due to the structural design of 4a-Me and 4b-Me with four carbazole derivatives affording much larger number of vibrational modes of the molecule (3N − 6) compared to 3b-Me with only two carbazole units. Therefore, in 4a-Me and 4b-Me, the thermal equilibration of the S1 and T1 states is more efficient than in 3a-Me. It is noted, however, that S1 → T1 ISC and T1 → S1 RISC processes compete with the direct relaxation processes (radiative and non-radiative) to the ground state, S1 → S0 and T1 → S0, respectively. This may be important in comparing the photophysical properties of 4a-Me and 4b-Me. Thus, with double the number of alkoxy π-donors on the carbazole unit of 4b-Me, its HOMO is higher in energy, which in turn results in lower energies of the S1 and T1 excited states (HOMO → LUMO), compared to 4a-Me. According to the energy gap law,20,30 a decrease in the energy of an excited state facilitates exponential increase of non-radiative relaxation rates to the ground state. Therefore, the T1 → S0 non-radiative decay in 4b-Me is probably faster than in 4a-Me, which may compromise the efficiency of competing T1 → S1 RISC and, consequently, the efficiency of TADF.
Conclusions
Carbazole-substituted terephthalonitriles, functionalised on the periphery by alkoxy chains, have been prepared and studied in detail for their liquid-crystalline and photophysical properties, the latter underpinned by calculation. The challenge of generating an anisotropic molecule from a rather three-dimensional core necessitated the use of multiple peripheral chains; the synthetic chemistry dictated that these were attached as alkoxy chains. The consequence of using such good donors and in number is to red-shift emission and consequently to diminish the emission efficiency. Interestingly, the data show that the number of methoxy groups used affects the emission efficiency independent of their position whereas the position of the methoxy groups matters in relation to the degree of bathochromism in the emission. Clearly there are design criteria that emerge from this initial study that will inform future approaches, but this does not diminish the realisation of multifunctional materials with both liquid crystal and TADF properties.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the University of York (AFS), the Universität Regensburg (MZS) and the German Research Foundation (DFG) (Project No. 389797483) for financial support.References
- M. O’Neill and S. M. Kelly, Adv. Mater., 2011, 23, 566–584 CrossRef PubMed.
- A. Diez, S. J. Cowling and D. W. Bruce, Chem. Commun., 2012, 48, 10298–10300 RSC.
- H. Iino, T. Usui and J.-i. Hanna, Nat. Commun., 2015, 6, 6828 CrossRef CAS.
- S. Allard, M. Förster, B. Souharce, H. Thiem and U. Scherf, Angew. Chem., Int. Ed., 2008, 47, 4070–4098 CrossRef CAS PubMed.
- M. Kumar and S. Kumar, Polym. J., 2017, 49, 85–111 CrossRef CAS.
- E. Grelet, H. Bock, T. Brunet, J. Kelber, O. Thiebaut, P. Jolinat, S. Mirzaei and P. Destruel, Mol. Cryst. Liq. Cryst., 2011, 542, 182–189 CAS.
- Y. Wang, J. Shi, J. Chen, W. Zhu and E. Baranoff, J. Mater. Chem. C, 2015, 3, 7993–8005 RSC.
- S. Kappaun, C. Slugovc and E. J. W. List, Int. J. Mol. Sci., 2008, 9, 1527–1547 CrossRef CAS PubMed.
- H.-H. Kuo, Z.-l. Zhu, C.-S. Lee, Y.-K. Chen, S.-H. Liu, P.-T. Chou, A. K. Y. Jen and Y. Chi, Adv. Sci., 2018, 5, 1800846 CrossRef PubMed.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- A.-M. Giroud-Godquin and A. Rassat, C. R. Seances Acad. Sci., Ser. 2, 1982, 294, 241–243 CAS.
- A. Serrette, P. J. Carroll and T. M. Swager, J. Am. Chem. Soc., 1992, 114, 1887–1889 CrossRef CAS.
- A. G. Serrette and T. M. Swager, J. Am. Chem. Soc., 1993, 115, 8879–8880 CrossRef CAS.
- A. M. Prokhorov, A. Santoro, J. A. G. Williams and D. W. Bruce, Angew. Chem., Int. Ed., 2012, 51, 95–98 CrossRef CAS PubMed.
- A. Santoro, A. M. Prokhorov, V. N. Kozhevnikov, A. C. Whitwood, B. Donnio, J. A. G. Williams and D. W. Bruce, J. Am. Chem. Soc., 2011, 133, 5248–5251 CrossRef CAS PubMed.
- T. Chuard and R. Deschenaux, Helv. Chim. Acta, 1996, 79, 736–741 CrossRef CAS.
- M. K. Etherington, N. A. Kukhta, H. F. Higginbotham, A. Danos, A. N. Bismillah, D. R. Graves, P. R. McGonigal, N. Haase, A. Morherr, A. S. Batsanov, C. Pflumm, V. Bhalla, M. R. Bryce and A. P. Monkman, J. Phys. Chem. C, 2019, 123, 11109–11117 CrossRef CAS PubMed.
- B. Donnio, B. Heinrich, H. Allouchi, J. Kain, S. Diele, D. Guillon and D. W. Bruce, J. Am. Chem. Soc., 2004, 126, 15258–15268 CrossRef CAS PubMed.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- R. Peverati and D. G. Truhlar, J. Phys. Chem. Lett., 2012, 3, 117–124 CrossRef CAS.
- R. Peverati and D. G. Truhlar, Phys. Chem. Chem. Phys., 2012, 14, 11363–11370 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Saisalito, 2010, pp. 149–156 Search PubMed.
- B. R. Henry and W. Siebrand, J. Chem. Phys., 1971, 54, 1072–1085 CrossRef CAS.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, 3rd edn, 2006 Search PubMed.
- J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1983, 87, 952–957 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2056911 and 2056912. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc00443c |
| This journal is © The Royal Society of Chemistry 2021 |