
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn up-conversion luminophore with high quantum yield and brightness based on BaF2:Yb3+,Er3+ single crystals†
Eduard I.
Madirov
ab,
Vasilii A.
Konyushkin
c,
Andrey N.
Nakladov
c,
Pavel P.
Fedorov
c,
Thomas
Bergfeldt
 d,
Dmitry
Busko
b,
Ian A.
Howard
be,
Bryce S.
Richards
be,
Sergey V.
Kuznetsov
*c and
Andrey
Turshatov
*b
d,
Dmitry
Busko
b,
Ian A.
Howard
be,
Bryce S.
Richards
be,
Sergey V.
Kuznetsov
*c and
Andrey
Turshatov
*b
aKazan Federal University, Kremlyovskaya str, 18, Kazan, 420008, Russia
bInstitute of Microstructure Technology, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, Eggenstein-Leopoldshafen, 76344, Germany. E-mail: andrey.turshatov@kit.edu
cProkhorov General Physics Institute of the Russian Academy of Sciences (GPI RAS), Vavilov str, 38, Moscow, 119991, Russia. E-mail: kouznetzovsv@gmail.com
dInstitute for Applied Materials, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, Eggenstein-Leopoldshafen, 76344, Germany
eLight Technology Institute, Karlsruhe Institute of Technology (KIT), Engesserstrasse 13, Karlsruhe, 76131, Germany
First published on 27th January 2021
Abstract
Up-conversion (UC) of near-infrared radiation to visible light has received much attention because of its use in the conversion of solar radiation, luminescence thermometry, biosensing, and anti-counterfeiting applications. However, the main issue hindering the successful utilization of UC is the relatively low quantum efficiency of the process. In order to design new UC systems with high quantum yield (ϕUC) values, we synthesized two series of co-doped BaF2 single crystals with nominal concentrations of Yb3+ (2–15 mol%)/Er3+ (2 mol%) as well as Yb3+ (3 mol%)/Er3+ (2–15 mol%). The highest ϕUC value of 10.0% was demonstrated for the BaF2:Er3+ (2 mol%) and Yb3+ (3 mol%) sample under 490 W cm−2 of 976 nm excitation. To study the natural limit of UC efficiency, quantum yield values upon direct excitation (ϕDS) of the 4S3/2 (ϕDS ≤ 4%) and 4F9/2 (ϕDS ≤ 26%) levels were measured. Comparison of experimental values of quantum yields to the ones obtained using Judd–Ofelt theory reveals strong quenching of the 4S3/2 state for all investigated compositions. In addition, we observed an unusually strong contribution of the Er3+:4I9/2 excited state to both UC and down-shifting luminescent processes. This contribution becomes possible due to the very low maximum phonon energy of BaF2 crystals (240 cm−1).
Introduction
Luminescent materials based on lanthanide ions – ranging from molecular complexes to inorganic phosphors – remain not only interesting from a scientific point of view but also relevant for many new applications.1,2 These applications include optical nanoprobes for medical usage,3–7 colour conversion materials for light emitting diodes,8,9 organic light emitting diodes,10 solar radiation converters,11–13 luminescent thermometers14–17 and inks used for anti-counterfeiting purposes.18,19 In general, the luminescence of lanthanide-based materials can be divided into two main types: Stokes and anti-Stokes. The majority of known luminescence materials exhibit Stokes emission, also known as down-shifted (DS) emission, meaning that emitted photons have lower energy than absorbed ones. Fewer materials exhibit anti-Stokes emission, where emitted photons have higher energy than the absorbed ones.Anti-Stokes emission, the so called the up-conversion (UC) process, based on trivalent lanthanide ions (Ln3+) can reach high photoluminescence quantum yields of 5–11% under relatively low excitation intensity (<40 W cm−2),20–23 in contrast to high light intensity (>106 W cm−2) required for other prominent anti-Stokes processes such as multi-photon absorption and multi-harmonic generation.24 Thus, lower-power light emitting diodes25 or even xenon lamp (for a special case of dye-synthesized UC)26 can be used as excitation sources in lanthanide based UC. Four main UC mechanisms generally considered are ground state absorption with a subsequent excited state absorption (GSA/ESA), energy transfer UC (ETU), cooperative UC and photon avalanche UC.27,28 The ETU is the most efficient mechanism among these four and occurs at high Ln3+ concentrations (>2 mol%) and moderate excitation intensity.29,30 At lower doping concentrations or higher excitation intensities, GSA/ESA can occur simultaneously with the ETU process or even start playing a dominant role.31
In order to increase the efficiency of the UC, co-doping with a material (called sensitizer) that has a high absorption cross-section can be utilized. If the goal is to achieve UC from the near-infrared (NIR) to visible (Vis) region, then co-doping with Yb3+ ions is often applied. The 2F7/2 → 2F5/2 transition of the Yb3+ is resonant with the f–f transitions of Er3+, Tm3+, and Ho3+ ions, thus providing efficient energy transfer. Thereby, Er3+/Yb3+, Ho3+/Yb3+, and Tm3+/Yb3+ pairs are often used in the NIR-to-Vis UC systems.32–37
Another important factor for the efficient UC process is the host matrix, as it affects the environment around the optical centres. The host matrix has to have low phonon energy in order to minimize the non-radiative losses and favour radiative transitions. In a wide range of UC materials, fluorides are optimal candidates for use as the host due to their relatively low phonon energies and good chemical stability.38,39 Recently, we investigated SrF2:Yb3+,Er3+ single crystals and reported a very high UC quantum yield of 6.5%.23 Inspired by this work, we assumed that a BaF2 single crystal with maximum phonon energy of 240 cm−1, which is significantly lower than the phonon energy of other prominent fluoride hosts (β-NaYF4 – 360 cm−1,40 LaF3 – 350 cm−1,41 CaF2 – 320 cm−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 42 and SrF2 – 284 cm−142), is a good candidate for further improving the UC efficiency.
42 and SrF2 – 284 cm−142), is a good candidate for further improving the UC efficiency.
It is known that the normally forbidden f–f transitions in rare-earth element (Ln3+) ions become partially allowed in materials with a low crystal symmetry and strong local distortion of the crystal field. Though, the BaF2 crystal (as well as SrF2 and CaF2 hosts) exhibits a high cubic crystal symmetry with a fluorite structure which is usually not favourable for an efficient UC process.43 However, co-doping with Yb3+ and Er3+ occurs via the substitution of the divalent cation and requires charge compensation via negative fluorine ions (F−) in interstitial positions. These interstitial anions reduce the symmetry of Ln3+ single ion centres giving rise to trigonal and tetragonal symmetry and, thus, increase the probability of radiative transitions.44 Moreover, at higher dopant concentrations preferential clustering of lanthanide ions occurs, which can reduce inter-ionic distances and enhance both ETU and cross-relaxation processes.45,46
There have been a number of studies dedicated to the optical properties of BaF2 doped with Er3+ and Yb3+ ions. The majority of works have used glasses or glass ceramics.47–50 Although these studies provide some insight into their UC behaviour, a more extensive study of the optical properties is required in order to get a more detailed picture of UC properties of BaF2-based materials. Thus, the focus of this work is (i) to assess how efficient UC in the BaF2 host can be via measurements of the absolute quantum yield in an integrating sphere (ϕUC) for different concentrations of doping ions and (ii) to provide a more detailed understanding of UC mechanism in the BaF2 host (via the study of both UC and DS luminescent properties). In this context, single-crystals of BaF2 are great study objects because of two reasons (i) lack of grain boundaries reduces light scattering and ensures efficient dissipation of heat produced within non-radiative relaxation of excited ions51–55 and (ii) large volume to surface ratio allows neglecting the surface quenching effect and improves chemical stability of the samples.
Experimental
Synthesis and characterization
Barium fluoride, ytterbium fluoride and erbium fluoride were highly pure (99.99% LANHIT, Russia). The powders of the fluoride precursors were preliminarily melted under a CF4 fluorinating atmosphere. Afterwards, the fluoride single crystals were grown by the Bridgman technique in a vacuum furnace under a CF4 fluorinating atmosphere. Both the heater with a temperature gradient (60–80 K cm−1) and the crucible were made up of graphite. The temperature (1360 °C) and crystallization velocity (6.5 mm per hour) were chosen based on the phase diagrams of BaF2–Ln3+.56,57 The grown crystals are 5 cm long rods with 10 mm diameter. The crystals were cut in the direction perpendicular to the long axis and resulting discs (thickness of 2 mm and diameter of 10 mm) were polished for optical measurements.Two series of the single-crystal BaF2 crystals doped with Er3+ and Yb3+ ions were grown by the Bridgman technique. The first series consisted of BaF2 doped with nominal concentrations of 2 mol% of Er3+ ions and 2, 3, 5, 7, 10, 15 mol% of Yb3+ (hereafter the mol% represents the nominal concentration of the Ln3+ ions used in the synthesis of the crystals, whereas the exact compositions estimated via wavelength-dispersive X-ray fluorescence (WDXRF) spectroscopy are reported in Table S1, ESI†). The second series is doped with 3 mol% of Yb3+ ions as well as 3, 5, 10, 15 mol% of Er3+ ions. These concentration ranges were chosen because the previous research has revealed a strong concentration quenching and deteriorated UC luminescence at higher doping concentrations of both ions.58
The crystalline structure of the samples was determined using the powder XRD patterns recorded with a Bruker D2 PHASER diffractometer (CuKα radiation). For this purpose, a small part of the single crystal was ground into powder. The patterns were recorded in the 2 theta range from 10 to 70 degrees.
The concentration of elements Ba2+, Er3+ and Yb3+ were determined by WDXRF spectroscopy (Pioneer S4, from Bruker AXS). For the measurement, three replicates of each sample were analyzed. 25 mg of the sample material (accuracy ± 0.05 mg) were dissolved with 6 g EQF-TML-5050-5 (49.75% Li2B4O7 + 49.75% LiBO2 + 0.5% LiBr) in a platinum crucible at 1373 K. After cooling in a platinum stencil the fusion tablet was analyzed. For the calibration, four fusion tablets with matrix-adapted standards (BaF2, Er2O3, and Yb2O3) were melted. Two to three energy lines of the elements were used for the calculation. The standard deviation in the determination of the chemical composition did not exceed 0.6 wt% for barium, 0.07 wt% for erbium, and 0.05 wt% for ytterbium.
Optical methods
The Raman spectrum for the undoped BaF2 sample was recorded with an i-Raman device by Polytec (785 nm excitation, 3.5 cm−1 resolution).The refractive indices of the samples were measured with a Metricon 2010/M prism coupler using 1550 nm laser radiation (Thorlabs, TLK-L1550R). The detailed description of the setup was reported earlier.59
Absorption spectra were recorded at room temperature using a UV-Vis spectrometer PerkinElmer Lambda 950 in the absorbance mode. The absorption coefficient was calculated using the following expression in eqn (1):
 | (1) |
The setup and the methodology for the estimation of ϕUC under 976 nm excitation have been described previously.23,60 To record ϕDS of the 4S3/2 → 4I15/2 and 4F9/2 → 4I15/2 transitions of the Er3+ ions a tunable CW laser (SolsTis with EMM-Vis, M-Squared Lasers Ltd) pumped by a 532 nm laser (Verdi-V18, Coherent) was utilized. The system was tuned to 522 nm for the direct excitation of the 4S3/2 level and to 652 nm for the direct excitation of the 4F9/2 level. For the measurement of ϕDS of the 4I13/2 → 4I15/2 transition upon direct excitation, a tunable laser kit (Thorlabs, TLK-L1550M) operating at 1520 nm was used as the excitation source. The remaining setup was the same as described in our earlier publication.60
Luminescence lifetimes of the emissive levels were measured with a home-built optical system described previously.61 Briefly, 525 nm, 976 nm, and 633 nm (Roithner) and 1550 nm (Thorlabs) laser diodes mounted in temperature stabilized mounts (TCLDM9, Thorlabs) and driven by a laser diode controller (ITC4001, Thorlabs) as well as 375 nm LED also driven by the controller (ITC4001, Thorlabs) were used as the excitation sources. The power of the laser beam was adjusted with a controlled rotatable neutral density filter (Thorlabs). The remaining setup was the same as described in our previous publication.59
Results and discussion
Crystal structure characterization
The measured powder XRD patterns are presented in Fig. 1 together with JCPDS card 04-0452 (BaF2). The unit cell parameters (a) calculated from the XRD data are given in Table S1 (ESI†). They are in a good agreement with the values of the BaF2 unit cell parameter (a = 6.200 Å) available in the literature.62 It is observed that the unit cell parameter decreases with the increase of doping concentration of both Yb3+ and Er3+. This may be attributed to the fact that ionic radii of Er3+ and Yb3+ ions are smaller than that of Ba2+.63 This discrepancy results in a lower volume of the unit cell and reduced distance between doping ions, which, in turn, allows for a higher local concentration of the doping ions. | ||
| Fig. 1 Powder XRD patterns of the BaF2:Yb3+, Er3+ samples. | ||
Raman spectroscopy was performed for the undoped BaF2 crystalline sample. The spectrum has one distinct peak at 240 cm−1 (Fig. S1, ESI†), which perfectly correlates to the value of ∼240 cm−1 observed earlier in several other publications,42,64 and reveals low phonon energy of the BaF2 host.
In addition, the exact chemical composition of the samples was studied by WDXRF spectroscopy. The obtained weight% of the doping ions, via the WDXRF method, allowed the calculation of the mol% values that represent the fraction of the Ba cations substituted with rare-earth ions. The resulting values are given in Table S1 (ESI†). For the sake of clarity, we will use the nominal concentrations of Er3+ and Yb3+ (related to the sample names) in further discussion. The uncertainties for the concentrations were 0.30 wt%, 0.02 wt%, and 0.02 wt% for barium, erbium, and ytterbium, respectively.
Optical characterization
Absorption and luminescence spectra
The absorption spectra shown in Fig. 2 demonstrate absorption bands in ultraviolet (UV), Vis and near infrared (NIR) ranges, characteristic of Er3+ and Yb3+ ions. The narrow absorption bands arise from the f–f transitions of the Er3+ and Yb3+ ions. The positions of the lines are in accordance with the literature data.23,32,33 The shape of the peaks remains the same in all samples. It demonstrates that the local environment of the doping ions is consistent and there are no strong local deformations of the crystal structure in the investigated range of doping concentrations. | ||
| Fig. 2 Absorption spectra of two series of co-doped BaF2 single crystals (a) samples with fixed Er3+ concentration whereas Yb3+ concentration varied from 2 to 15 mol% and (b) samples with fixed Yb3+ concentration where Er3+ concentration varies from 2 to 15 mol%. | ||
Table 1 displays the values of the peak absorption cross-sections of the most prominent absorption bands of Er3+ and Yb3+. For instance, the peak cross-section of the Yb3+:2F7/2 → 2F5/2 absorption band is 0.62–0.7 pm2 in the concentration range of Yb3+ of 3–15 mol%. The absorption cross-section of the Yb3+:2F7/2 → 2F5/2 transition was calculated only for the samples with 2% of Er3+ because at higher doping concentrations the contribution of the Er3+ absorption becomes significant at this wavelength.
| 378 nm | 406 nm | 450 nm | 522 nm | 650 nm | 1506 nm | 976a nm | |
|---|---|---|---|---|---|---|---|
| Er3+:4I15/2 → 4G11/2 | Er3+:4I15/2 → 2H9/2 | Er3+:4I15/2 → 4F5/2 | Er3+:4I15/2 → 2H11/2 | Er3+:4I15/2 → 4F9/2 | Er3+:4I15/2 → 4I13/2 | Yb3+:2F7/2 → 2F5/2 | |
| a The absorption cross-section of the Yb3+:2F7/2 → 2F5/2 transition was calculated only for the samples with 2% of Er3+, because at higher doping concentrations the contribution of the Er3+ absorption results in the overestimation of the absorption cross-section. | |||||||
| Er2Yb2 | 0.91 | 0.09 | 0.12 | 0.64 | 0.40 | 0.37 | 0.52 |
| Er2Yb3 | 0.98 | 0.11 | 0.13 | 0.68 | 0.42 | 0.40 | 0.62 |
| Er2Yb5 | 1.25 | 0.13 | 0.16 | 0.87 | 0.54 | 0.52 | 0.69 |
| Er2Yb7 | 1.23 | 0.14 | 0.18 | 0.89 | 0.55 | 0.56 | 0.70 |
| Er2Yb10 | 1.31 | 0.16 | 0.20 | 0.97 | 0.61 | 0.62 | 0.69 |
| Er2Yb15 | 1.28 | 0.15 | 0.19 | 0.94 | 0.58 | 0.62 | 0.66 |
| Er3Yb3 | 1.01 | 0.11 | 0.14 | 0.73 | 0.46 | 0.44 | |
| Er5Yb3 | 0.99 | 0.13 | 0.16 | 0.76 | 0.51 | 0.49 | |
| Er10Yb3 | 0.99 | 0.16 | 0.21 | 0.86 | 0.64 | 0.68 | |
| Er15Yb3 | 0.81 | 0.15 | 0.19 | 0.73 | 0.57 | 0.67 | |
The values are in line with the absorption cross-section values of the Er3+ and Yb3+ ions in hosts with a comparable structure available in the literature.23,45,65–67 Overall, Yb3+ ions in the BaF2 host demonstrate absorption cross-section comparable to values reported for CaF2 (0.55 pm2)66 and SrF2 (0.89 pm2),67 whereas absorption cross-section in oxide crystals is usually higher. For example, Yb3+ absorption cross-sections of 0.8 pm2 in YAG,68 1.7 pm2 in Gd2O3,69 and 8 pm2 in GdVO470 were previously reported. Another noticeable trend in the data is the significant increase in the absorption cross-section of Er3+ bands with the increase of both Er3+ and Yb3+ doping concentrations.
This phenomenon has already been reported by Auzel et al.71 and may be attributed to the fact that trivalent Er3+ and Yb3+ ions substitute divalent Ba2+ ions in the crystal. Higher amounts of the doping ions create stronger local distortion of the crystal field that favours the radiative transitions in the Ln3+ions.45
The emission spectra of co-doped BaF2 crystals are presented in Fig. 3. The DS emission spectra obtained under 375 nm excitation (4G11/2 level of the Er3+ ions) are given in Fig. 3a and b while the UC emission spectra obtained under 976 nm excitation (2F5/2 level of the Yb3+ ion) are shown in Fig. 3c and d. All spectra have typical emission bands of the Er3+ and Yb3+ ions with the emission of Er3+ ions located around 405 nm (2H9/2 → 4I15/2), 521 nm (2H11/2 → 4I15/2), 540 nm (4S3/2 → 4I15/2), 650 nm (4F9/2 → 4I15/2), 810 nm (4I9/2 → 4I15/2) and 850 nm (4S3/2 → 4I13/2) and the emission of {Er3+:4I11/2 & Yb3+:2F5/2} manifold at 1020 nm. The position of these transitions on the energy level diagram is additionally given in Fig. S2 (ESI†). Under 375 nm excitation, the relative intensities of the Er3+ emission bands do not exhibit a strong dependence on the doping concentrations of Yb3+ until it reaches 10 mol% (see Fig. 3a). At this point, the relative intensity at 668 nm strongly increases, indicating two possible effects: (i) a strong depopulation of the 4S3/2 level and/or (ii) an extra population of the 4F9/2 level. The 2H9/2 → 4F9/2 transition in Er3+ is resonant with the 2F7/2 → 2F5/2 transition in Yb3+72 (as shown in Fig. S2, ESI,† transition ①). If existing, these transitions lead to both a lower population of the 4S3/2 level and an increase of the 4F9/2 level population in line with the results presented in the Fig. 3a. The increase in the {Er3+:4I11/2 & Yb3+:2F5/2} manifold relative intensity at high doping concentrations can also be explained in a similar manner.
 | ||
| Fig. 3 Emission spectra of the samples under (a and b) – 375 nm (intensities of all spectra in the range from 900 to 1100 nm decreased to one-third of their original intensity) and (c and d) – 976 nm excitation (I = 490 W cm−2). All spectra were normalized using the intensity of the 4S3/2-4I15/2 (549 nm) peak. | ||
In addition, Fig. 3a displays a change of the shape of the blue edge of the 1020 nm emission bands. This observation suggests strong self-absorption of the {Er3+:4I11/2 & Yb3+:2F5/2} manifold starting from low Yb3+ doping concentration of 3 mol%. This behaviour is expected of Yb3+-doped materials as it was observed in materials with Yb3+ doping concentration as low as 1 mol%.73
The increase of the Er3+ concentration (Fig. 3b) results in a continuous increase of the relative intensity of the {Er3+:4I11/2 & Yb3+:2F5/2} manifold. However, the increase of the relative intensity at 668 nm is observed only for the highest concentration (15 mol%) of Er3+. This behaviour can be explained by two energy transfer processes (Fig. S2, ESI†): (i) 4F7/2 → 4I11/2 which is resonant with the 4I15/2 → 4I11/2 transition (Fig. S2, ESI,† transition ②) and (ii) 4F5/2 → 4F9/2 which is resonant with the 4I15/2 → 4I13/2 transition (Fig. S2, ESI,† transition ③).72 The contribution of the first energy transfer process can give rise to the {Er3+:4I11/2 & Yb3+:2F5/2} manifold emission. In turn, the contribution of the second process can be responsible for increase of the relative intensity of the 4F9/2 level. It is reasonable that the ground state of Er3+ is a weaker energy acceptor than the ground state of Yb3+ as quenching is observed at a significantly higher concentration of Er3+ (15 mol%) as compared to the Yb3+ concentration (10 mol%).
The relative ratio of UC emission peaks has a much weaker dependence on the concentrations of the doping ions (Fig. 3c and d). In contrast to UV excitation, the increase in the Yb3+ concentration results in a moderate decrease in the 668 nm relative emission. This is due to the fact that the upper level of Er3+ is less involved in the UC process. Thus, we assumed that the energy transfer processes from the upper levels of Er3+ are unlikely to occur for all investigated samples at excitation intensities up to 490 W cm−2. The significant increase in the emission at around 800 nm at high Er3+ concentrations (Fig. 3d) can be explained by the increasing population of the 4I9/2 level from the 4I13/2 state due to the resonance of this transition to the 4S3/2 → 4I9/2 transition, as shown in Fig. S3 (ESI†) (transition ①). However, these cross-relaxation processes become significant only if the concentration of Er3+ is high (>5 mol%). The proposed pathways for the deactivation of upper Er3+ levels were also confirmed via measurements of excitation spectra for the Er3+: 4F9/2 energy level monitored at 660 nm (Fig. S4, ESI†).
Luminescence decay
The luminescence decay kinetics of the 2H9/2, 4S3/2, 4F9/2, 4I13/2 levels of the Er3+ ions as well as the {Er3+:4I11/2 & Yb3+:2F5/2} manifold are recorded using two excitation sources: a 976 nm diode laser (excitation of the 2F5/2 level of the Yb3+ ions) and a 375 nm LED (excitation of the 4G11/2 level of the Er3+ ions). The obtained curves are given in Fig. S5–S8 (ESI†). All of the luminescence decay curves except those at 540 nm exhibit mono-exponential behaviour under 375 nm and 976 nm excitations. In the case of 4S3/2 → 4I15/2 (540 nm) transition, the decay demonstrates strong non-mono exponential behaviour (see Fig. S6a and b, ESI†) and therefore it was fitted with a double exponential function (eqn (2)) that gives a good level of conformity between the fit and the experimental data. | (2) |
The mean decay times presented in Fig. 4 and Table S2 (ESI†) are calculated using eqn (3)74
 | (3) |
 | ||
| Fig. 4 Luminescence decay time of 4S3/2 → 4I15/2 (green symbols) and 4F9/2 → 4I15/2 (red symbols) transitions of the Er3+ ions as well as decay time of {Er3+:4I11/2 & Yb3+:2F5/2} manifold (brown symbols) under 375 nm (empty symbols) and 976 nm (filled symbols) excitation as a function of Yb3+ (a) and Er3+(b) nominal concentrations. The dashed lines are a guide to the eye. The exact decay times are presented in Table S1 (ESI†). | ||
Fig. 4 indicates that under 375 nm excitation the increase in Yb3+ and Er3+ concentration leads to a decrease in the decay times of both 4S3/2 and 4F9/2 energy levels. This decrease can be explained by the excitation energy migration within an excited state manifold. If an excitation migrates until it meets a quenching centre, the migration process reduces the decay time of the excited state.75 The quenching process can be based on cross-relaxation or interaction with a ground state (for instance the cross-relaxation with the resonance between 4S3/2 → 4I9/2 and 4I13/2 → 4I9/2 transitions (Fig. S3, ESI,† transition ①)) or interaction of the 4H11/2 level with the Er3+ground state: 4H11/2 → 4I13/2 is resonant with 4F15/2 → 4I9/2 (Fig. S3, ESI,† transition ②). In addition, the observed decay time can also decrease, if the increased Ln3+ ion concentration affects the crystal lattice and thereby the lifetimes of radiative transitions. For instance, previously we observed an increase in the absorption cross-section (absorption enhancement) with the increase of the dopant concentration (Table 1). We can assume that the radiative rate also increases with the increase of Yb3+ and Er3+ concentration that results in an additional drop in the decay time.
Under 976 nm excitation the decay times of the 4S3/2 and 4F9/2 energy levels are extended compared to the decay times obtained under 375 nm excitation. This proves that in the case of UC excitation, the population of the higher states of the Er3+ ions is governed by energy transfer from long-lived intermediate states with lower energy. For instance, the decay time of the 4S3/2 level reflects the long decay time of the {Er3+:4I11/2 & Yb3+:2F5/2} manifold. At higher Yb3+ concentrations the decay time of the {Er3+:4I11/2 & Yb3+:2F5/2} manifold decreases due to ETU enhancement. This effect leads to the shortening of 4S3/2 decay time at a high Yb3+ content. In contrast, at higher Er3+ concentrations the decay time of the 4S3/2 level under 976 nm excitation becomes longer. This again reflects the increase of the {Er3+:4I11/2 & Yb3+:2F5/2} manifold decay time with the increase of the Er3+ concentration.
This trend in the decay time behaviour for the {Er3+:4I11/2 & Yb3+:2F5/2} manifold – it decreases with the increase in the Yb3+ concentration, but increases with the increase in the Er3+ content for both excitation types – is quite an interesting observation, as both ions are from the same {Er3+:4I11/2 & Yb3+:2F5/2} manifold.
Under certain conditions, the prolongation of the decay time can be attributed to the effect of reabsorption, when luminescence is reabsorbed and reemitted several times within the same crystal. However, the reabsorption of 1020 nm emission can have only a minor effect (Fig. 3b) and cannot explain the increase of {Er3+:4I11/2 & Yb3+:2F5/2} manifold decay time with the increase in the Er3+ concentration. Alternatively, the increase of {Er3+:4I11/2 & Yb3+:2F5/2} manifold decay time observed under 976 nm excitation can reflect the fact that the lifetime of Er3+:4I11/2 is much longer than the lifetime of the Yb3+:2F5/2 state. For instance, the lifetimes of 7.41 ms and 0.77 ms were measured for single Er3+doped (5 mol%) and single Yb3+ doped (5 mol%) BaF2 crystals, respectively (Fig. S9, ESI†). Thus, an increase in the Er3+ concentration should increase the contribution of the long-lived Er3+:4I11/2 state to the decay time of the {Er3+:4I11/2 & Yb3+:2F5/2} manifold, and increase it.
The additional prolongation of the {Er3+:4I11/2 & Yb3+:2F5/2}) manifold decay time observed under 375 nm excitation at a high Er3+ concentration can be explained by the increasing role of the Er3+:4I9/2 level in the {Er3+:4I11/2 & Yb3+:2F5/2}) manifold population. Fig. S3 (ESI†) demonstrates a number of possible ways (① and ②) by which the Er3+:4I9/2 level can be populated by a further transition to the {Er3+:4I11/2 & Yb3+:2F5/2}) manifold. In the crystals with low maximum phonon energy (240 cm-1 for BaF2), the rate of multiphonon relaxation for the 4I9/2 → 4I11/2 transition (with the energy gap of ΔE = 2000 cm- 1) is very slow and, thus, the decay time approaches the radiative lifetime of the 4I9/2 state after 10 ms.76 Thus, this weakly emissive, but long-lived state can be considered as an additional reservoir (in parallel with the {Er3+:4I11/2 & Yb3+:2F5/2} manifold and the Er3+:4I13/2 state) of metastable excited states influencing the DS and UC processes. Under this circumstance, the moderate and high concentrations of Er3+ contribute to the population of the 4I9/2 state increasing the lifetime of 4S3/2, 4F9/2, and {Er3+:4I11/2 & Yb3+:2F5/2} states.
Quantum yield
The UC quantum yield ϕUC is the main figure-of-merit parameter, which can help to understand the physical mechanism and practical value of the UC process. The ϕUC, as it was introduced previously, is the internal quantum yield. The internal quantum yield characterizes the conversion efficiency of absorbed photons into emitted photons. However, the parameter of brightness (B), which depends also on a number of absorbed photons, is more important for some applications.77 It can be calculated as: | (4) |
The highest ϕUC and brightness values for the UC emission integrated in the 400–900 nm range as well as the ϕUC values of certain emission bands are given in Table 2. Additionally, the ϕUC values of the 2H11/2–4I15/2, 4I9/2–4I15/2 and 4S3/2–4I13/2 are presented in Table S3 (ESI†) and ϕDS values under 375 nm excitation are noted in Table S4 (ESI†). The highest measured ϕUC values reach 9.9% and 10.0% under an excitation intensity of 490 W cm−2 in the samples doped with 2% of Er3+ as well as 2 and 3% of Yb3+ ions, respectively. These results significantly exceed the ϕUC values of 6.5% observed in the SrF2 single crystals23 as well as 2.8% observed in SrF2 nano- and micro- particles61 doped with Er3+ and Yb3+ ions.22,23 At the same time the sample doped with 2% of Er3+ and 10% of Yb3+ demonstrates the highest brightness value.
| 4S3/2 → 4I15/2 | 4F9/2 → 4I15/2 | Total | Brightness, cm−1 | |
|---|---|---|---|---|
| Er2Yb2 | 0.021 | 0.063 | 0.099 | 0.121 |
| Er2Yb3 | 0.023 | 0.061 | 0.100 | 0.192 |
| Er2Yb5 | 0.021 | 0.053 | 0.088 | 0.262 |
| Er2Yb7 | 0.023 | 0.043 | 0.081 | 0.374 |
| Er2Yb10 | 0.019 | 0.036 | 0.068 | 0.495 |
| Er2Yb15 | 0.013 | 0.027 | 0.048 | 0.419 |
| Er3Yb3 | 0.018 | 0.043 | 0.064 | 0.120 |
| Er5Yb3 | 0.017 | 0.03 | 0.062 | 0.148 |
| Er10Yb3 | 0.008 | 0.01 | 0.026 | 0.121 |
| Er15Yb3 | 0.002 | 0.004 | 0.011 | 0.065 |
This composition should provide the largest number of emitted photons per volume and can be optimal for UC applications of luminescent BaF2:Yb3+, Er3+ materials.
The power dependent ϕUC under 976 nm excitation is summarized in Fig. 5, where the following trends can be observed: (i) under lower excitation intensity (<100 W cm−2) the samples exhibit an increase of ϕUC with the increase of the Yb3+ concentration; (ii) in contrast, under high excitation intensity, a reduced concentration of Yb3+ is preferable for achieving higher ϕUC values; (iii) increase of Er3+ concentration results in increased ϕUC at intensity <10 W cm−2, and lowered ϕUC in the broad intensity range (10–490 W cm−2). A similar effect was also observed in β-NaYF4 doped with Er3+.78
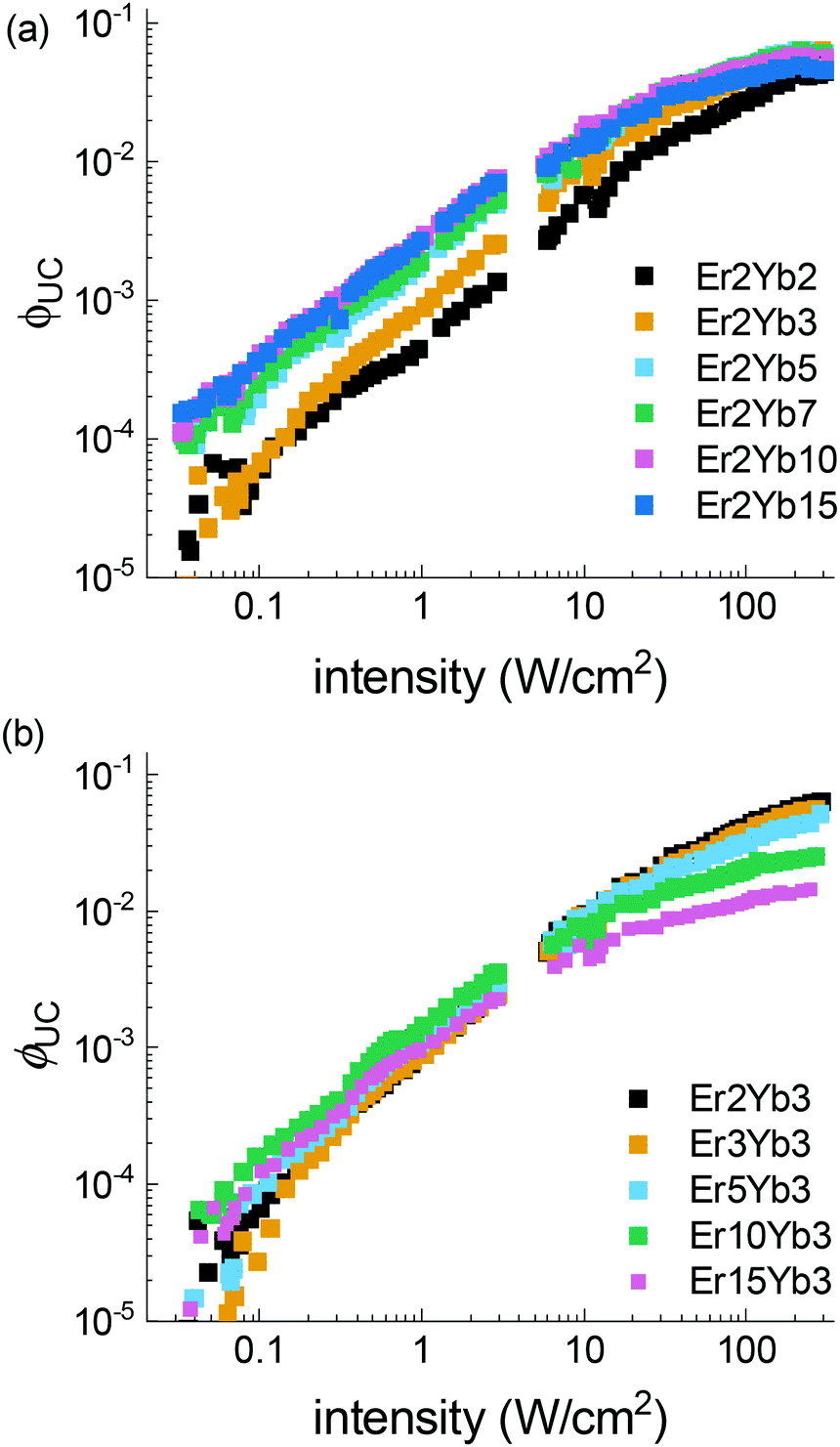 | ||
| Fig. 5 Intensity dependence of the UC quantum yield (ϕUC) under 976 nm excitation. | ||
Another UC figure-of-merit parameter, critical power density (CPD), is calculated for each sample using an earlier published method.60 This parameter describes the saturation ϕUC and facilitates the comparison of different UC materials. Combined with the maximum ϕUC value, it can provide a full set of characteristics required for the analysis of application perspectives of UC materials.
The beneficial low CPD values of the 4S3/2 → 4I15/2 transition of the Er3+ ions were earlier reported for the most efficient UC materials as 0.7 W cm−2 in β-NaYF4, 1.0 W cm−2 in YF3 and 1.0 W cm−2 in La2O3.60 The smallest CPD value of the 4S3/2 → 4I15/2 transition in the BaF2 crystal (with 2% of Er3+, 15% of Yb3+ ions) is 1.1 W cm−2, which is just fractionally higher than the values calculated for the best UC materials. Table S5 (ESI†) shows the results for the samples that provided the best fit.
Possible heating of samples was considered during the intensity-dependent measurements. To monitor a possible change of the sample temperature an approach from the literature79 was utilized. The ratio between 2H11/2–4I15/2 (521 nm) and 4S3/2–4I15/2 (545 nm) emission bands was calculated. The results are presented in Fig. S10 (ESI†). They show that noticeable heating is observable only at the highest power densities (>200 W cm−2) in the samples with high Yb3+ and Er3+ doping concentrations. We assume that the increase of sample temperature can be responsible for the drop of ϕUC observed at intensity >200 W cm−2 for samples with high doping concentrations.
A more detailed study of the down-shifting emission of the 4S3/2 and 4F9/2 levels at direct excitation (522 nm for 4S3/2 and 652 nm for 4F9/2) should help to have a deeper insight into the UC process efficiency. We observed that the ϕDS of the 540 nm emission under 522 nm excitation is in the range of 1–4% and ϕDS of the 660 nm emission under 652 nm excitation is in the range of 15–26% (Table S6, ESI†). Additionally, the ϕDS values of 4F9/2–4I15/2, 4I9/2–4I15/2 and 4S3/2–4I13/2 emission bands under 522 nm excitation can be found in Table S7 (ESI†).
It is clear that the value of ϕDS gives an indication of the maximum achievable ϕUC for this particular level. It cannot exceed half of this value (ϕDS ≤ 4% for 4S3/2 → 4I15/2 transition and ϕDS ≤ 26% in case of 4F9/2 → 4I15/2 transition) due to the fact that the UC process involves at least two photons. The lack of any strong dependence on the Yb3+ concentration in both cases means that there are no transitions from 4S3/2 and 4F9/2 levels of Er3+ interacting with the ground state of Yb3+. However, in both cases, there is a strong drop in ϕDS values with the increase in the Er3+ concentration. This may prove that there is a strong energy migration and quenching within the Er3+:4S3/2 state even at relatively low Er3+ concentrations like 5% as it was assumed in the Luminescence Decay section.
Judd–Ofelt analysis
The experimental lifetimes are compared with radiative lifetimes of some levels of the Er3+ ions, which is calculated using the Judd–Ofelt theory by the standard procedure.80,81 The detailed description of transition probability calculation is presented in the corresponding section of the ESI.†Knowing the transition probabilities, it is possible to calculate the radiative decay time (τr) of a level and the corresponding branching ratio (β). These results together with experimentally measured decay times (τ) upon the direct excitation of the corresponding level in Er3+ ions (Fig. S11, ESI†) can help to predict the quantum yield value  for three transitions: 4S3/2 → 4I15/2, 4F9/2 → 4I15/2 and 4I13/2 → 4I15/2. The resulting
for three transitions: 4S3/2 → 4I15/2, 4F9/2 → 4I15/2 and 4I13/2 → 4I15/2. The resulting  values were calculated using eqn (8) and are summarized in Table S9 (ESI†) together with τ, τr, β.
values were calculated using eqn (8) and are summarized in Table S9 (ESI†) together with τ, τr, β.
 | (8) |
These results provide an insight into the possible application of the Judd–Ofelt theory to study UC and DS processes in Ln3+ co-doped systems. Altogether, the obtained values of the radiative decay times are close to the results of other studies devoted to optical properties of Er3+ ions in fluoride single crystals and micropowders.76,82–84
Although an acceptable level of conformity between theoretical prediction ( in Table S9, ESI†) and experimental results (
in Table S9, ESI†) and experimental results ( in Table S6, ESI†) for the 4F9/2 → 4I15/2 transition exists (the relative difference
in Table S6, ESI†) for the 4F9/2 → 4I15/2 transition exists (the relative difference  doesn’t exceed 20% in most cases), in the case of 4S3/2 → 4I15/2 and 4I13/2 → 4I15/2 transitions a discrepancy between quantum yields extracted from the Judd–Ofelt calculation and the experimental one is significant. The values of
doesn’t exceed 20% in most cases), in the case of 4S3/2 → 4I15/2 and 4I13/2 → 4I15/2 transitions a discrepancy between quantum yields extracted from the Judd–Ofelt calculation and the experimental one is significant. The values of  estimated via Judd–Ofelt analysis always exceed unity for the 4I13/2 → 4I15/2 transition, because the measured decay times (τ) are longer than predicted radiative lifetimes (τr). Unfortunately, it remains unclear whether Judd–Ofelt theory describes well the 4I13/2 → 4I15/2 transition with strong magnetic dipole contribution or there is another energy transfer process and/or strong emission reabsorption leading to the elongation of the decay time. For the 4S3/2 → 4I15/2 transition, the predicted values of
estimated via Judd–Ofelt analysis always exceed unity for the 4I13/2 → 4I15/2 transition, because the measured decay times (τ) are longer than predicted radiative lifetimes (τr). Unfortunately, it remains unclear whether Judd–Ofelt theory describes well the 4I13/2 → 4I15/2 transition with strong magnetic dipole contribution or there is another energy transfer process and/or strong emission reabsorption leading to the elongation of the decay time. For the 4S3/2 → 4I15/2 transition, the predicted values of  also overestimate the quantum yield in all investigated samples. We observed again the elongation of the decay times combined with rather small values of
also overestimate the quantum yield in all investigated samples. We observed again the elongation of the decay times combined with rather small values of  measured experimentally. This discrepancy is observed along with the strong deviation of 4S3/2 → 4I15/2 decays from the single-exponential behaviour (Fig. S11, ESI†) and can indicate the existence of an energy transfer pathway (energy migration85 thermal coupling between 4S3/2 and 2H11/2 states,86 for instance) and/or strong emission reabsorption which cannot be described using our simplified model. However, we believe that in our future work including Yb3+ and Er3+ single-doped crystals with a concentration range starting from very low dopant concentrations (0.1 mol%) we will be able to explain this interesting behaviour.
measured experimentally. This discrepancy is observed along with the strong deviation of 4S3/2 → 4I15/2 decays from the single-exponential behaviour (Fig. S11, ESI†) and can indicate the existence of an energy transfer pathway (energy migration85 thermal coupling between 4S3/2 and 2H11/2 states,86 for instance) and/or strong emission reabsorption which cannot be described using our simplified model. However, we believe that in our future work including Yb3+ and Er3+ single-doped crystals with a concentration range starting from very low dopant concentrations (0.1 mol%) we will be able to explain this interesting behaviour.
Conclusions
Optical properties of co-doped BaF2 single crystals were investigated for a broad range of Er3+ (2–15 mol%) and Yb3+ (2–15 mol%) doping concentrations. All samples demonstrate efficient UC emission under 976 nm excitation. A very high ϕUC value of 10.0% (at 490 W cm−2) was observed for the sample doped with 2% of Er3+ and 3% of Yb3+. This value exceeds previously reported ϕUC for SrF2 single crystals (6.5%) and approaches the efficiency of the best UC material known to date (NaYF4:Yb3+, Er3+ with a quantum yield of 11%21). The investigation of UC and DS luminescent spectra, lifetimes and quantum yields under multiple excitation wavelengths of 375, 522, 653, 976 and 1520 nm, as well as a comparison of the experimental results with predictions of Judd–Ofelt theory highlights the complexity of the UC process. More specifically, our results demonstrate a significant reduction of luminescence quantum yield of the Er3+:4S3/2 state in the DS regime, which in turn reduces the quantum yield of its emission in UC regimes. Due to the low maximum phonon energy of BaF2 crystals (240 cm−1), we observed an unusually strong contribution of the Er3+:4I9/2 state in the temporal behaviour of both UC and DS processes.
Author contributions
The manuscript was written through the contribution from all authors. E. M. and D. B. conducted spectroscopy experiments and E. M. wrote the paper. V. A. K. and A. N. N. grew the BaF2 single crystals. T. B. performed WDXRF analysis of the chemical composition. S. V. K., A. T. and C. W. developed the original concept of the paper. P. P. V., I. A. H. and B. S. R. contributed equally to scoping and structuring the paper and provided additional guidance on experimental methods. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The reported study was funded by the RFBR (project number 21-53-12017 of S. V. K., V. A. K., and A. N. N.) and DFG (project number TU 487/8-1 of A. T. and B. S. R.). B. S. R. acknowledges the financial support provided by the Helmholtz Association: (i) a Recruitment Initiative Fellowship to B. S. R.; (ii) the funding of chemical synthesis equipment from the Helmholtz Materials Energy Foundry (HEMF); and (iii) the Science and Technology of Nanosystems research programme. E. I. M. acknowledges the subsidy allocated to Kazan Federal University for the state assignment in the sphere of scientific activities No. 0671-2020-0051 and the scholarship of the President of the Russian Federation.Notes and references
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- J. Thirumalai, Luminescence - An Outlook on the Phenomena and their Applications, InTech, 2016 Search PubMed.
- X. Li, S. Lu, D. Tu, W. Zheng and X. Chen, Nanoscale, 2020, 12, 15021–15035 RSC.
- Y. Liu, D. Tu, H. Zhu and X. Chen, Chem. Soc. Rev., 2013, 42, 6924–6958 RSC.
- Y. I. Park, J. H. Kim, K. T. Lee, K. S. Jeon, H. B. Na, J. H. Yu, H. M. Kim, N. Lee, S. H. Choi, S. I. Baik, H. Kim, S. P. Park, B. J. Park, Y. W. Kim, S. H. Lee, S. Y. Yoon, I. C. Song, W. K. Moon, Y. D. Suh and T. Hyeon, Adv. Mater., 2009, 21, 4467–4471 CrossRef CAS.
- S. Xu, S. Huang, Q. He and L. Wang, Trends Anal. Chem., 2015, 66, 72–79 CrossRef CAS.
- D. V. Pominova, A. V. Ryabova, K. G. Linkov, I. D. Romanishkin, S. V. Kuznetsov, J. A. Rozhnova, V. I. Konov and V. B. Loschenov, Laser Phys., 2016, 26, 084001 CrossRef.
- Q. Dai, M. E. Foley, C. J. Breshike, A. Lita and G. F. Strouse, J. Am. Chem. Soc., 2011, 133, 15475–15486 CrossRef CAS.
- H.-A. Park, Y. K. Lee, W. B. Im, J. Heo and W. J. Chung, Opt. Mater., 2015, 41, 67–70 CrossRef CAS.
- R. C. Evans, P. Douglas and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093–2126 CrossRef CAS.
- J. C. Goldschmidt and S. Fischer, Adv. Opt. Mater., 2015, 3, 510–535 CrossRef CAS.
- B. M. van der Ende, L. Aarts and A. Meijerink, Phys. Chem. Chem. Phys., 2009, 11, 11081–11095 RSC.
- A. S. Nizamutdinov, S. V. Kuznetsov, E. I. Madirov, V. V. Voronov, A. R. Khadiev, A. D. Yapryntsev, V. K. Ivanov, V. V. Semashko and P. P. Fedorov, Opt. Mater., 2020, 108, 110185 CrossRef CAS.
- R. G. Geitenbeek, P. T. Prins, W. Albrecht, A. van Blaaderen, B. M. Weckhuysen and A. Meijerink, J. Phys. Chem. C, 2017, 121, 3503–3510 CrossRef CAS.
- J. Rocha, C. D. S. Brites and L. D. Carlos, Chem. – Eur. J., 2016, 22, 14782–14795 CrossRef CAS.
- M. S. Pudovkin, O. A. Morozov, V. V. Pavlov, S. L. Korableva, E. V. Lukinova, Y. N. Osin, V. G. Evtugyn, R. A. Safiullin and V. V. Semashko, J. Nanomater., 2017, 2017, 3108586 Search PubMed.
- D. Jaque and F. Vetrone, Nanoscale, 2012, 4, 4301–4326 RSC.
- J. Andres, R. D. Hersch, J.-E. Moser and A.-S. Chauvin, Adv. Funct. Mater., 2014, 24, 5029–5036 CrossRef CAS.
- P. Kumar, S. Singh and B. K. Gupta, Nanoscale, 2016, 8, 14297–14340 RSC.
- I. Etchart, A. Huignard, M. Bérard, M. N. Nordin, I. Hernández, R. J. Curry, W. P. Gillin and A. K. Cheetham, J. Mater. Chem., 2010, 20, 3989–3994 RSC.
- M. Kaiser, C. Würth, M. Kraft, I. Hyppänen, T. Soukka and U. Resch-Genger, Nanoscale, 2017, 9, 10051–10058 RSC.
- M. Pokhrel, G. A. Kumar and D. K. Sardar, J. Mater. Chem. A, 2013, 1, 11595–11606 RSC.
- D. Saleta Reig, B. Grauel, V. A. Konyushkin, A. N. Nakladov, P. P. Fedorov, D. Busko, I. A. Howard, B. S. Richards, U. Resch-Genger, S. V. Kuznetsov, A. Turshatov and C. Würth, J. Mater. Chem. C, 2020, 8, 4093–4101 RSC.
- M.-L. Zheng, K. Fujita, W.-Q. Chen, X.-M. Duan and S. Kawata, J. Phys. Chem. C, 2011, 115, 8988–8993 CrossRef CAS.
- Y. Zhong, I. Rostami, Z. Wang, H. Dai and Z. Hu, Adv. Mater., 2015, 27, 6418–6422 CrossRef CAS.
- A. Ishii, Y. Adachi, A. Hasegawa, M. Komaba, S. Ogata and M. Hasegawa, Sci. Technol. Adv. Mater., 2019, 20, 44–50 CrossRef CAS.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS.
- V. V. Ovsyankin and P. P. Feofilov, J. Exp. Theor. Phys., 1966, 3, 322 Search PubMed.
- D. C. Rodriguez Burbano, R. Naccache and J. A. Capobianco, Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 2015, vol. 47, pp. 273–347 Search PubMed.
- X. Zhang, X. Liu, J. P. Jouart and G. Mary, Chem. Phys. Lett., 1998, 287, 659–662 CrossRef CAS.
- F. Vetrone, J. C. Boyer, J. A. Capobianco, A. Speghini and M. Bettinelli, J. Phys. Chem. B, 2002, 106, 5622–5628 CrossRef CAS.
- J. F. Suyver, J. Grimm, K. W. Krämer and H. U. Güdel, J. Lumin., 2005, 114, 53–59 CrossRef CAS.
- H. Guo and Y. M. Qiao, Opt. Mater., 2009, 31, 583–589 CrossRef CAS.
- X. P. Chen, W. J. Zhang and Q. Y. Zhang, Phys. B, 2011, 406, 1248–1252 CrossRef CAS.
- A. Pandey, V. K. Rai, R. Dey and K. Kumar, Mater. Chem. Phys., 2013, 139, 483–488 CrossRef CAS.
- F. Vetrone, V. Mahalingam and J. A. Capobianco, Chem. Mater., 2009, 21, 1847–1851 CrossRef CAS.
- F. C. Guinhos, P. C. Nóbrega and P. A. Santa-Cruz, J. Alloys Compd., 2001, 323, 358–361 CrossRef.
- G. Buse, E. Preda, M. Stef, A. Pruna, F. Stef and I. Nicoara.
- Y. Tian, R. Xu, L. Hu and J. Zhang, Opt. Mater., 2011, 34, 308–312 CrossRef CAS.
- H. Wu, Z. Hao, L. Zhang, X. Zhang, Y. Xiao, G.-H. Pan, H. Wu, Y. Luo, H. Zhao and J. Zhang, J. Phys. Chem. C, 2018, 122, 9611–9618 CrossRef CAS.
- M. J. Weber, Phys. Rev., 1967, 157, 262–272 CrossRef CAS.
- D. G. Mead and G. R. Wilkinson, J. Phys. C: Solid State Phys., 1977, 10, 1063–1072 CrossRef CAS.
- S. Balabhadra, M. F. Reid, V. Golovko and J.-P. R. Wells, J. Alloys Compd., 2020, 834, 155165 CrossRef CAS.
- R. Reisfeld and C. K. Jørgensen, in Lasers and Excited States of Rare Earths, ed. R. Reisfeld and C. K. Jørgensen, Springer Berlin Heidelberg, Berlin, Heidelberg, 1977, DOI:10.1007/978-3-642-66696-4_1, pp. 1–63.
- C. Labbe, J. L. Doualan, P. Camy, R. Moncorgé and M. Thuau, Opt. Commun., 2002, 209, 193–199 CrossRef CAS.
- A. M. Golubev, L. P. Otroshchenko, V. N. Molchanov, L. E. Fykin and B. P. Sobolev, Crystallogr. Rep., 2009, 54, 423–430 CrossRef CAS.
- A. Maaoui, M. Haouari, A. Bulou, B. Boulard and H. Ben Ouada, J. Lumin., 2018, 196, 1–10 CrossRef CAS.
- L. N. Ignatieva, N. V. Surovtsev, E. B. Merkulov, N. N. Savchenko, S. V. Adichtchev, Y. V. Marchenko and V. M. Bouznik, J. Non-Cryst. Solids, 2012, 358, 3248–3254 CrossRef CAS.
- K. Linganna, R. Narro-García, P. Manasa, H. Desirena, E. De la Rosa and C. K. Jayasankar, J. Rare Earths, 2018, 36, 58–63 CrossRef CAS.
- X. Qiao, X. Fan, M. Wang and X. Zhang, J. Non-Cryst. Solids, 2008, 354, 3273–3277 CrossRef CAS.
- S. Fischer, N. D. Bronstein, J. K. Swabeck, E. M. Chan and A. P. Alivisatos, Nano Lett., 2016, 16, 7241–7247 CrossRef CAS.
- C. Würth, M. Kaiser, S. Wilhelm, B. Grauel, T. Hirsch and U. Resch-Genger, Nanoscale, 2017, 9, 4283–4294 RSC.
- M. Kraft, C. Würth, V. Muhr, T. Hirsch and U. Resch-Genger, Nano Res., 2018, 11, 6360–6374 CrossRef CAS.
- R. Arppe, I. Hyppänen, N. Perälä, R. Peltomaa, M. Kaiser, C. Würth, S. Christ, U. Resch-Genger, M. Schäferling and T. Soukka, Nanoscale, 2015, 7, 11746–11757 RSC.
- P. A. Popov, P. P. Fedorov, S. V. Kuznetsov, V. A. Konyushkin, V. V. Osiko and T. T. Basiev, Dokl. Phys., 2008, 53, 353–355 CrossRef CAS.
- B. P. Sobolev and N. L. Tkachenko, J. Less-Common Met., 1982, 85, 155–170 CrossRef CAS.
- S. V. Kuznetsov and P. P. Fedorov, Inorg. Mater., 2008, 44, 1434–1458 CrossRef CAS.
- S. Wen, J. Zhou, K. Zheng, A. Bednarkiewicz, X. Liu and D. Jin, Nat. Commun., 2018, 9, 2415 CrossRef.
- S. Dottermusch, D. Busko, M. Langenhorst, U. W. Paetzold and B. S. Richards, Opt. Lett., 2019, 44, 29–32 CrossRef CAS.
- R. E. Joseph, C. Jiménez, D. Hudry, G. Gao, D. Busko, D. Biner, A. Turshatov, K. Krämer, B. S. Richards and I. A. Howard, J. Phys. Chem. A, 2019, 123, 6799–6811 CrossRef CAS.
- S. Kuznetsov, Y. Ermakova, V. Voronov, P. Fedorov, D. Busko, I. A. Howard, B. S. Richards and A. Turshatov, J. Mater. Chem. C, 2018, 6, 598–604 RSC.
- A. A. Kaminskii, H. Rhee, H. J. Eichler, L. Bohatý, P. Becker and K. Takaichi, Laser Phys. Lett., 2008, 5, 304–310 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- A. Anderson and F. F. Cleveland, Phys. Today, 1974, 27, 54–55 CrossRef.
- W. Ma, L. Su, X. Xu, J. Wang, D. Jiang, L. Zheng, X. Fan, C. Li, J. Liu and J. Xu, Opt. Mater. Express, 2016, 6, 409 CrossRef CAS.
- J. Xu, L. Su, H. Li, D. Zhang, L. Wen, H. Lin and G. Zhao, Opt. Mater., 2007, 29, 932–935 CrossRef CAS.
- J. L. Doualan, P. Camy, A. Benayad, M. Von Edlinger, V. Ménard and R. Moncorgé.
- S. Banerjee, J. Koerner, M. Siebold, Q. Yang, K. Ertel, P. D. Mason, P. J. Phillips, M. Loeser, H. Zhang, S. Lu, J. Hein, U. Schramm, M. C. Kaluza and J. L. Collier, Opt. Express, 2013, 21, A726–A734 CrossRef CAS.
- M. Velázquez, P. Veber, G. Buşe, Y. Petit, P. Goldner, V. Jubera, D. Rytz, A. Jaffres, M. Peltz, V. Wesemann, P. Aschehough and G. Aka, Opt. Mater., 2015, 39, 258–264 CrossRef.
- J. Liu, X. Mateos, H. Zhang, J. Wang, M. Jiang, U. Griebner and V. Petrov, Opt. Lett., 2006, 31, 2580–2582 CrossRef CAS.
- F. Auzel, K. E. Lipinska-Kalita and P. Santa-Cruz, Opt. Mater., 1996, 5, 75–78 CrossRef CAS.
- A. Baride, P. S. May and M. T. Berry, J. Phys. Chem. C, 2020, 124, 2193–2201 CrossRef CAS.
- A. Boccolini, J. Marques-Hueso, D. Chen, Y. Wang and B. S. Richards, Sol. Energy Mater. Sol. Cells, 2014, 122, 8–14 CrossRef CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, 2006 Search PubMed.
- J. Liu, T. Fu and C. Shi, J. Phys. Chem. C, 2019, 123, 9506–9515 CrossRef CAS.
- A. Bitam, S. Khiari, M. Diaf, H. Boubekri, E. Boulma, C. Bensalem, L. Guerbous and J. P. Jouart, Opt. Mater., 2018, 82, 104–109 CrossRef CAS.
- K.-L. Wong, J.-C. G. Bünzli and P. A. Tanner, J. Lumin., 2020, 224, 117256 CrossRef CAS.
- S. K. W. MacDougall, A. Ivaturi, J. Marques-Hueso, K. W. Krämer and B. S. Richards, Sol. Energy Mater. Sol. Cells, 2014, 128, 18–26 CrossRef CAS.
- R. E. Joseph, D. Busko, D. Hudry, G. Gao, D. Biner, K. Krämer, A. Turshatov, B. S. Richards and I. A. Howard, Opt. Mater., 2018, 82, 65–70 CrossRef CAS.
- M. P. Hehlen, M. G. Brik and K. W. Krämer, J. Lumin., 2013, 136, 221–239 CrossRef CAS.
- A. Lira C, I. Camarillo, E. Camarillo, F. Ramos, M. Flores and U. Caldiño, J. Phys.: Condens. Matter, 2004, 16, 5925–5936 CrossRef.
- E. Preda, M. Stef, G. Buse, A. Pruna and I. Nicoara, Phys. Scr., 2009, 79, 035304 CrossRef.
- Y. Zhang, B. Chen, S. Xu, X. Li, J. Zhang, J. Sun, X. Zhang, H. Xia and R. Hua, Phys. Chem. Chem. Phys., 2018, 20, 15876–15883 RSC.
- M. Luo, B. Chen, X. Li, J. Zhang, S. Xu, X. Zhang, Y. Cao, J. Sun, Y. Zhang, X. Wang, Y. Zhang, D. Gao and L. Wang, Phys. Chem. Chem. Phys., 2020, 22, 25177–25183 RSC.
- F. T. Rabouw, P. T. Prins, P. Villanueva-Delgado, M. Castelijns, R. G. Geitenbeek and A. Meijerink, ACS Nano, 2018, 12, 4812–4823 CrossRef CAS.
- Y. Zhang, B. Chen, S. Xu, X. Li, J. Zhang, J. Sun, X. Zhang, H. Xia and R. Hua, Phys. Chem. Chem. Phys., 2019, 21, 10840–10845 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc00104c |
| This journal is © The Royal Society of Chemistry 2021 |