
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence((R)-(−)-3-Hydroxyquinuclidium)[FeCl4]; a plastic hybrid compound with chirality, ferroelectricity and long range magnetic ordering†
Palmerina
González-Izquierdo
*ab,
Oscar
Fabelo
 *b,
Laura
Cañadillas-Delgado
b,
Garikoitz
Beobide
cd,
Oriol
Vallcorba
e,
Jorge
Salgado-Beceiro
f,
Manuel
Sánchez-Andújar
f,
Carmen
Martin
g,
Javier
Ruiz-Fuentes
a,
José Eduardo
García
h,
María Teresa
Fernández-Díaz
b and
Imanol
de Pedro
*a
*b,
Laura
Cañadillas-Delgado
b,
Garikoitz
Beobide
cd,
Oriol
Vallcorba
e,
Jorge
Salgado-Beceiro
f,
Manuel
Sánchez-Andújar
f,
Carmen
Martin
g,
Javier
Ruiz-Fuentes
a,
José Eduardo
García
h,
María Teresa
Fernández-Díaz
b and
Imanol
de Pedro
*a
aCITIMAC, Facultad de Ciencias, Universidad de Cantabria, 39005 Santander, Spain. E-mail: depedrovm@unican.es
bInstitut Laue-Langevin, BP 156X, F-38042 Grenoble Cedex, France. E-mail: fabelo@ill.fr; gonzalez-izquierdo@ill.fr
cDepartamento de Química Inorgánica, Facultad de Ciencia y Tecnología, Universidad del País Vasco, Apartado 644, E-48080, Bilbao, Spain
dBasque Ctr Mat Applicat & Nanostruct, BCMat, UPV EHU Sci Pk, Leioa 48940, Spain
eALBA Synchrotron Light Source, Cerdanyola del Vallés, Barcelona, Spain
fQuiMolMat Group, Department of Chemistry, Faculty of Science and Advanced Scientific Research Center (CICA), Zapateira, University of A Coruna, 15071 A Coruna, Spain
gDepartamento de Química Física, Facultad de Química, Universidad de Sevilla, c/Profesor García González s/n, 41012 Sevilla, Spain
hDepartment of Physics, Universitat Politècnica de Catalunya—BarcelonaTech, 08034 Barcelona, Spain
First published on 16th February 2021
Abstract
Quinuclidinium salts and their derivatives are now in the focus of materials science as building units of multifunctional materials. Their properties can be easily switchable, allowing their use in a wide range of physical applications. One type of these kinds of materials, the homochiral hybrid halometallate ferroelectric compounds, is not well understood. In this work, (R)-(−)-3-quinuclidinol hydrochloride was used in the synthesis of ((R)-(−)-3-hydroxyquinuclidium)[FeCl4]. The use of this enantiomeric cation forces crystallographic non-centrosymmetry, which was confirmed by polarimetry and circular dichroism spectroscopy. We studied the physical properties of this compound at different temperatures by single crystal, synchrotron and neutron powder X-ray diffraction, which showed a rich series of structural and magnetic phase transitions. From synchrotron powder X-ray diffraction data, a plastic phase was observed above 370 K (phase I). Between 370 K and ca. 310 K, an intermediate polar phase was detected, solved in a non-centrosymmetric polar space group (C2) (phase II). Below ca. 310 K, the compound crystallizes in the triclinic P1 non-centrosymmetric space group (phase III) which is maintained down to 4 K, followed by phase IV, which shows tridimensional magnetic ordering. The temperature evolution of the neutron diffraction data shows the appearance of new reflections below 4 K. These reflections can be indexed to a commensurate propagation vector k = (0, 0, ½). The magnetic structure below TN was solved in the Ps1 Shubnikov space group, which gives rise to an antiferromagnetic structure, compatible with the magnetometry measurements. Near room temperature, the crystal phase transition is associated with a dielectric change. In particular, the phase transition between phase III (S.G.:P1) and phase II (S.G.:C2) involves an increase of symmetry between two non-centrosymmetric space groups. Therefore, it allows, by symmetry, the emergence of ferroelectric and ferroelastic ordering. Piezoresponse force microscopy (PFM) imaging measurements provided evidence for polarization switching and a local ferroelectric behavior of phase III at room temperature. Additionally, the obtained butterfly curve and hysteresis loop by PFM exhibits a low coercive voltage of ∼10 V. This value is remarkable, since it approaches those obtained for materials with application in ferroelectric random access memories (FeRAMs).
1. Introduction
The existence of different functional properties, such as dielectric, magnetic, optical or caloric properties in hybrid halometallate compounds, composed of a metal halide anion and an organic cation, has attracted the interest of the scientific community in the last few years. The reasons are their potential applications in different fields like energy storage,1–5 separation of greenhouse gases,6–8 catalysis9–12 and semiconductor nanocrystals,13 among others. Their functionality is mostly related to structural solid-to-solid phase transitions, associated with the inorganic anions and organic cations. These structural units possess large orientational or conformational degree of freedom, which finally permits the possibility of appearance of soft “plastic” crystals above room temperature (RT). Although plastic crystals were described in the 1960's,14 there is no evidence about which cation and anion combinations will yield plastic crystalline materials or which will form salts that melt before any rotator phase is achieved. However, it is often found in molecules with globular structures.Hybrid halometallate compounds with spherical organic molecules, such as (Me4N)+, (Me4P)+, quinuclidine or dabco (1,4-diazabicyclo-[2.2.2]octane), with low rotational energy barriers, are promising candidates for inducing structural phase transitions and plastic phases. Another interesting feature is the possibility of presenting ferroelectric-type phase transitions. However, they do not always occur, since the high symmetry of these spherical molecules usually facilitates the crystallization of centrosymmetric structures. In this regard, the novel “quasi-spherical theory”15 proposes the decrease of the symmetry of the molecular constituents through modifications on the globular molecules in order to promote the crystallization in non-highly symmetric space groups, which generally preclude the emergence of ferroelectric phases. Thus, the combination of plastic crystals with the quasi-spherical approach can lead to systems in which the resulting cation freezes in a ferroelectric phase with specific polarization orientations. Remarkably, specific intermolecular interactions, such as halogen-bonding and hydrogen-bonding interactions, like the ones present in halometallate compounds, are of crucial importance to the arrangement of polar structures endowed with ferroelectricity.
For instance, molecular modifications of the (Me4N)+ cation, replacing one methyl with fluoro-methyl, chloromethyl, bromomethyl or iodomethyl, reduce the molecular symmetry from point group Td to C3v. When these modified organic cations are assembled with a metal halide anion, high-Tc perovskite molecular ferroelectrics are successfully obtained, such as (Me3NCH2Cl)[MCl3] (M = Mn and Cd),16 (Me3NCH2Cl)[CdBr3],17 (Me3NCH2Br)[MnBr3],18 (Me3NCH2X)[FeBr4] (X = F, Cl, Br, I)19 or hybrid halometallate antiperovskite ferroelectrics such as ((CH3)3NH)3− [MnX3][MnX4] (X = Cl and Br).20,21 Recently, this approach has been used by modifying the dabco cation to its isomer, 1,5-diazabicyclo[3.2.1]octonium ([3,2,1-dabco]), resulting in a polycrystalline molecular ferroelectric compound, [3,2,1-Hdabco](BF4), in which the ferroelectric signal has been increased significantly related to molecular ferroelectric [2.2.2-dabco]BF4.22 Moreover, the attachment of a hydroxyl or methyl group to dabco also modulates the ferroelectricity and brings about outstanding physical properties in the material, like in the case of (DMe-dabco)[CuCl4], which shows both ferroelectricity and thermochromism.23 An analogous strategy was also applied on the quinuclidinium cation, with the attachment of –CH3, –CH2, –OH or ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups.24 The incorporation of a O group into the (quinuclidinium)[ClO4] compound transformed the centrosymmetric space group (Pm
O groups.24 The incorporation of a O group into the (quinuclidinium)[ClO4] compound transformed the centrosymmetric space group (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), observed in the pristine sample at RT, into the orthorhombic polar space group Pna21, presenting eminent ferroelectric properties.25 In addition, the globular perchlorate anion combined with planar organic cations like acetamidinium26 or guanadinium27 cations display plastic phases with optic-electric duple bistabilities and multiaxial ferroelectric, respectively.
m), observed in the pristine sample at RT, into the orthorhombic polar space group Pna21, presenting eminent ferroelectric properties.25 In addition, the globular perchlorate anion combined with planar organic cations like acetamidinium26 or guanadinium27 cations display plastic phases with optic-electric duple bistabilities and multiaxial ferroelectric, respectively.
Homochirality also provides a reasonable strategy for designing new molecular ferroelectrics.28 For instance, the homochiral (R- and S-1-(4-chlorophenyl) ethylammonium)2[PbI4] halometallates display multiaxial ferroelectricity and semiconductor characteristics with a direct band gap of 2.34 eV.29 Moreover, chirality can generate interesting, valuable and unique physical effects, such as magneto-chiral dichroism (MChD), second harmonic generation (SHG) and chiral photonics. Remarkably, the halometallate compounds (S-CTA)2[CuCl4] and (R-CTA)2[CuCl4](CTA = 3-chloro-2-hydroxypropyltrimethylammonium) show switching properties in seven physical channels: dielectricity, conductivity, second harmonic generation (SHG), piezoelectricity, ferroelasticity, chirality, and thermochromism.30
Inspired by these works, we have selected the modified globular quinuclidine molecule, (R)-(−)-3-hydroxyquinuclidine, which presents an additional OH group, in order to reduce the molecular symmetry and introduce chirality in the quinuclidinium tetrachlorideferrate compound.31–33 Thus, a new halometallate compound, ((R)-(−)-3-hydroxyquinuclidium)[FeCl4] was synthetized. The use of this homochiral cation can help in achieving a ferroelectric compound. (see Scheme 1). Moreover, the high flexibility of this compound induces an intricate series of structural phase transitions, from a triclinic crystal system, at low temperature, to a cubic crystal system in the plastic/paraelectric phase, above 370 K. Finally, the sample presents long-range magnetic ordering at ca. 4 K. We studied the magnetic behavior of this compound by the combination of magnetometry measurements and neutron diffraction. The structure was solved in the antiferromagnetic Ps1 Shubnikov space group. Table 1 summarizes the different phase transitions of the title compound with the temperature. Single crystal and powder diffraction using neutrons, laboratory X-ray and synchrotron diffraction techniques have been used to characterize the different structural and magnetic phases. In addition, macroscopic measurements were also carried out to follow the temperature dependence of the real part of the complex dielectric permittivity from RT to 430 K. Finally, the magnetic susceptibility was also investigated from RT to 2 K.
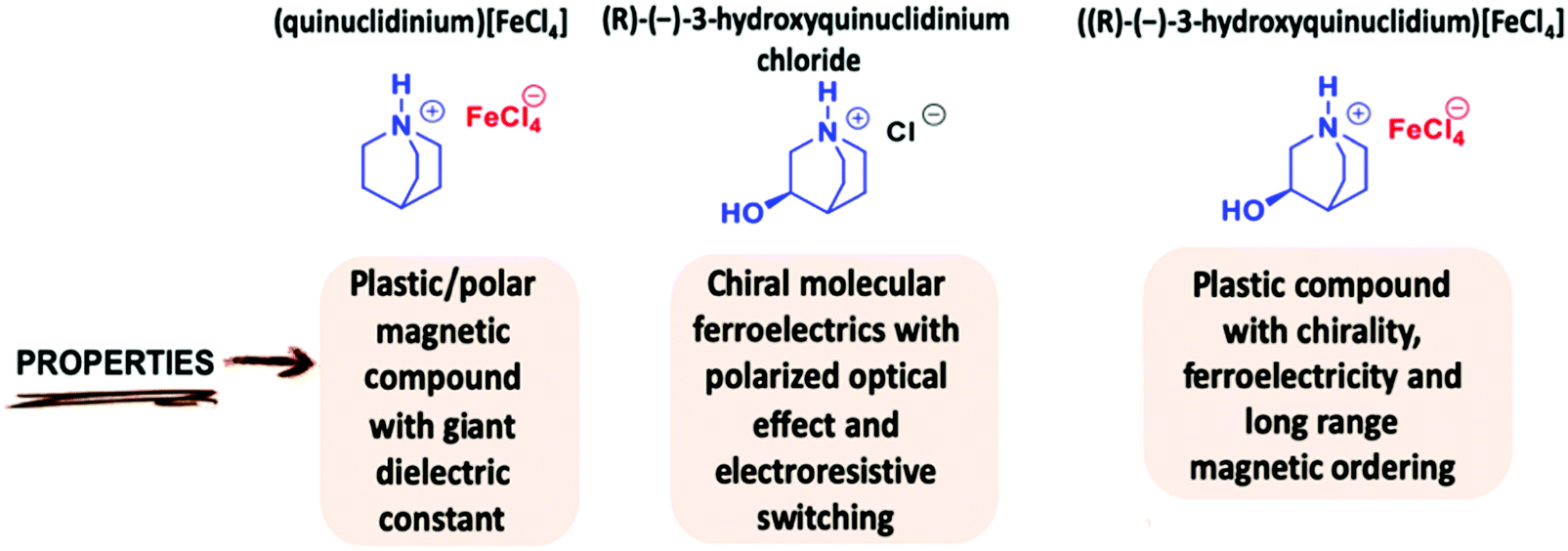 | ||
| Scheme 1 Main physical properties of some quinuclidinium-based compounds. | ||
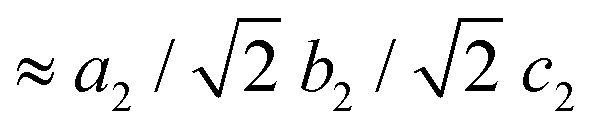

2. Experimental procedure
Synthesis of ((R)-(−)-3-hydroxyquinuclidium)[FeCl4]
Anhydrous FeCl3, (R)-(−)-3-quinuclidinol hydrochloride and 2-propanol were purchased from commercial sources and used without any further purification.1 equivalent of (R)-(−)-3-quinuclidinol hydrochloride (1 g, 6.11 mol) was placed in a round bottom flask with a stirring bar and dissolved in 20.0 mL of methanol. After addition of 1.0 equivalent of FeCl3 (0.99 g, 6.11 mol), the reaction mixture was heated at 30 °C for 18 h. Upon completion of the reaction, the solvent was removed in vacuo, obtaining a yellow powder. Yield: 98% (1.95 g) Single-crystals suitable for X-ray diffraction were grown by recrystallization of this compound in methanol. The solvent was allowed to slowly evaporate for 1 month. Elemental analysis: Found: C, 25.70; H, 4.08; N, 4.27; O 4.88; Fe, 17.10; Cl, 43.97%. Calcd. for C7H13NOFeCl4: C, 25.88; H, 4.03; N, 4.31; O 4.93; Fe, 17.19; Cl, 43.66%. Characteristic IR bands (cm−1): 3530 (O–H) 3184 (C–H), 2940 (C–H), 1459 (C–H sp2), 1405 (C–H sp2), 1120 (C–H sp3), 1020 (C–O), 961 (N–C3), 865 (N–C3), 613 (N–C2). Characteristic Raman bands (cm−1): 410 (N–C2), 332 (Fe–Cl), 133 (Fe–Cl), 107 (Fe–Cl) (see Fig. S1, ESI†). (R)-(−)-3-Quinuclidinol [α]25D = −34.4 (c = 5 mg/1 mL H2O). (R)-(−)-3-hydroxyquinuclidinium[FeCl4]. [α]25D = −22.1 (c = 5 mg/1 mL CH2Cl2) and [α]25D = −20.0 (c = 5 mg/1 mL H2O). UV-Vis [λmax/nm (ε/M−1 cm−1)]: 243 (7969), 314 (5856), 363 (5919) (Fig. S2, ESI†).
Fourier transform infrared spectroscopy (FT-IR)
FT-IR measurements were performed on a Bruker Alpha Series FT-IR spectrometer equipped with an attenuated total reflectance (ATR) module. The ATR FT-IR spectra were recorded by collecting 24 scans of a compound in the ATR module.Raman spectroscopy
The non-polarized Raman spectra of the sample were recorded in backscattering geometry with a Horiba T64000 triple spectrometer with the same protocol of ref. 34.UV-vis spectroscopy
UV-vis measurements were carried out on a Cary 500 SCAN UV-vis-NIR. Quartz cells of 1.0 cm path length were used. Samples were measured at 25 °C and with dichloromethane (CH2Cl2) as the solvent.Polarimetry
Optical rotations were measured in a 1.0 dm tube with a PerkinElmer spectropolarimeter (model 341) equipped with a Na lamp.Circular dichroism spectra
Electronic circular dichroism (CD) spectra were recorded using a Biologic Mos-450 spectropolarimeter (Barcelona, Spain). A standard quartz cell of 1.0 cm path length was used. Each spectrum was obtained from an average of 5 runs at 298.0 K.Thermal analysis
A Setaram calorimeter (DSC131) was used for the differential scanning calorimetry (DSC) analyses from 150 to 450 K under a nitrogen atmosphere at a heating rate of 10 K min−1. For this experiment, ca. 10 mg of sample were used and blank runs were performed. Thermogravimetric analyses (TGA) were carried out in a TGA-DTA Thermal Analysis SDT2960 equipment. Approximately 30 mg of powder was heated at a 10 K min−1 rate from 295 K to 1000 K.Variable temperature synchrotron X-Ray powder data collection
Synchrotron X-ray powder diffraction (SR-XRPD) measurements were performed at the high resolution station of the MSPD beamline (BL04) at ALBA synchrotron. The sample was introduced into a 0.7 mm glass capillary and measured in transmission at 22 keV energy (0.56376 Å wavelength determined from a Si NIST-640d reference) using the microstrip Mythen-II detector (six modules, 1280 channels per module, 50 μm per channel, sample-to-detector distance 550 mm). The temperature was controlled using an Oxford Cryosystems Series 700 Cryostream. Data from 1 to 43° (2θ) were collected during a 10 K min−1 ramp from 260 to 420 K every 30 s. The crystal structures were refined using the FullProf suite program.35Single-crystal X-ray diffraction (SCXRD)
A prismatic yellow single crystal of ((R)-(−)-3-hydroxyquinuclidium)[FeCl4] with approximate dimensions 0.1 mm × 0.06 mm × 0.05 mm was used for the single crystal X-ray crystallographic analysis on the dual-source D8 Venture equipped with a Photon III detector. The data were collected using Ag-Kα radiation (0.56086 Å) at 300 and 150 K. The data reduction was performed with the Bruker SAINT software package36 using a narrow-frame algorithm. Data were corrected for absorption effects using the Multi-Scan method (SADABS).37 All the structures were solved and refined using the Bruker SHELXTL Software Package,38 using the space groups C2 and P1, for RT and 150 K, respectively. At RT, the organic cation presents a notable disorder. Two different orientations of the ((R)-(−)-3-hydroxyquinuclidium)+ cation were included in the model equally weighted. To achieve a convergent model, 18 different distance restrains were used. Furthermore, the low data quality, due to the molecular thermal libration, precluded the anisotropic refinement of the ((R)-(−)-3-hydroxyquinuclidium)+ cation. The same single crystal was used for the 150 K measurement and the quality of the refinement was improved due to the reduction of the thermal effects. All the atoms, except the H, were refined anisotropically. For both models, the H atoms were included at calculated positions and treated as riding atoms with isotropic thermal motion related to that of its parent atom. The final structural parameters and figures of merit of the last refinements are summarized in Table S1 (ESI†) and the positional parameters are given in the corresponding CIF files.Neutron powder diffraction
Neutron powder diffraction measurements were performed on D1B and D2B powder diffractometers at the Institute Laue-Langevin (ILL, Grenoble, France). ca. 2 g of ((R)-(−)-3-hydroxyquinuclidium)[FeCl4] were used for the experiments, which were placed in a cylindrical vanadium container and held in a liquid helium cryostat. High flux and medium resolution diffractometer D1B, operated at λ = 2.525 Å, was used to study the evolution of the sample in the low temperature range. Besides, high counting time neutron diffraction patterns were measured between 6 and 1.4 K in the angular range 1 ≤ 2θ ≤ 128°. The collected data allowed us to solve and refine the magnetic structure. The high resolution diffractometer D2B (λ = 1.595 Å) was used to refine the structure at low temperature (10 K) in the angular range 0 ≤ 2θ ≤ 160°. In particular, we focused on the position of the hydrogen atom bonded to the oxygen. In order to do this, we ran one refinement cycle without the hydrogen to generate a Fourier density map. Then, we used the coordinates of the lowest density peaks to place the hydrogen and we refined the structure again. In this Rietveld refinement, the atomic coordinates were refined with distance restrictions (range 2.6 Å, sigma 0.05 Å) and the Debye Waller factors were refined isotropically by groups of atoms (Fe and Cl; C, N and O; H).Magnetization measurements
DC magnetic susceptibility measurements were performed using a Quantum Design PPMS magnetometer whilst heating from 2 to 300 K in zero field cool and field cool mode at 1 kOe. Magnetization as a function of field (H) was measured using the same magnetometer in the −50 ≤ H/kOe ≤ 50 at 2 K after cooling the sample in zero field.Dielectric measurements
The complex dielectric permittivity (εr = ε’r′ − iεr′′) of cold-press pelletized samples was measured as a function of frequency and temperature, building a parallel-plate capacitor coupled to a Solartron1260A Impedance/Gain-Phase Analyzer, capable of measuring in the frequency range from 10 Hz up to 1 MHz using an amplitude of 1 V. The capacitor was mounted in a Janis SVT200T cryostat refrigerated, and with a Lakeshore 332 incorporated to control the temperature from 295 K up to 390 K. The data were collected on heating and, before carrying out the measurement, the pellets were maintained at each temperature for two minutes to achieve thermal equilibrium.Pelletized samples, with an area of approximately 13 mm2 and a thickness of approximately 1 mm, were prepared by cold-press to fit into the capacitor. Gold was sputtered on the surfaces of the pelletized samples to ensure a good electrical contact.
All the dielectric measurements were carried out in a nitrogen atmosphere, performing several purging cycles with nitrogen gas to ensure that the sample chamber is completely free of atmospheric moisture.
Piezoresponse force microscopy (PFM)
The ferroelectric response of the obtained compound was analysed by piezoresponse force microscopy (PFM), which is a powerful technique for electromechanical characterization of materials. The PFM measurement was carried out on a commercial atomic force microscope (AFM), model MFP-3D (Asylum Research), with a high-voltage package. Images were obtained by using a Dual AC Resonance Tracking (DART) PFM mode using Pt/Ir-coated cantilevers with a spring constant of ∼2 N m−1 and a free resonance frequency of ∼75 kHz. PFM was carried out on a thin-film, which was prepared by spin-coating. For this purpose, the compound was dissolved in methanol and 100 μl of the obtained solution was deposited on a cleaned indium-tin-oxide coated glass substrate, which spins at 4000 rpm for 30 s. After that, the sample was heated at 350 K to favor the twinning of the material, with the phase transition, and to better observe the ferroelectric domains.3. Results and discussion
Optical analysis
The presence of the [FeCl4]− anion was confirmed by UV-vis spectroscopy. The spectrum of ((R)-(−)-3-hydroxyquinuclidium)[FeCl4] shows three peaks around 243, 314 and 363 nm, which are characteristics for charge-transfer absorptions of the [FeCl4]− anion (Fig. S2, ESI†).39–41The chirality of the species was studied by circular dichroism (CD) spectroscopy in the UV-vis absorption range. A solution of the title compound in CH2Cl2 was used in the CD measurement, sensing CD signals around 243, 314 and 363 nm, which correspond to the UV-vis absorption bands of ((R)-(−)-3-hydroxyquinuclidium)[FeCl4] (Fig. S3 and S4, ESI†). Notwithstanding the low CD spectrum resolution, these CD data can give a clue about the enantiomeric purity of the iron-based species. In order to gain more insight into the enantiomeric purity of the chiral compound, measurements of the optical rotation were performed. The iron-based molecule exhibits similar optical activity to its chiral precursor ([α]25D = −34.4 for (R)-(−)-3-Quinuclidinol and [α]25D = −20.0 for ((R)-(−)-3-hydroxyquinuclidium)[FeCl4], thus confirming the chirality of the new complex.
Thermal analysis
Differential scanning calorimetry curves display a sharp endothermic peak upon heating from 150 K at 305 K (Fig. 1 left), suggesting a first-order phase transition, which is attributed to the change from the ordered structure to a disordered one. Likewise, an endothermic peak upon heating from RT was observed at the offset temperature of 370 K (Fig. 1 right), associated with another disordered phase transition to a high-temperature plastic phase. Finally, TGA curves show that the title compound starts to decompose at 450 K (see Fig. S5, ESI†)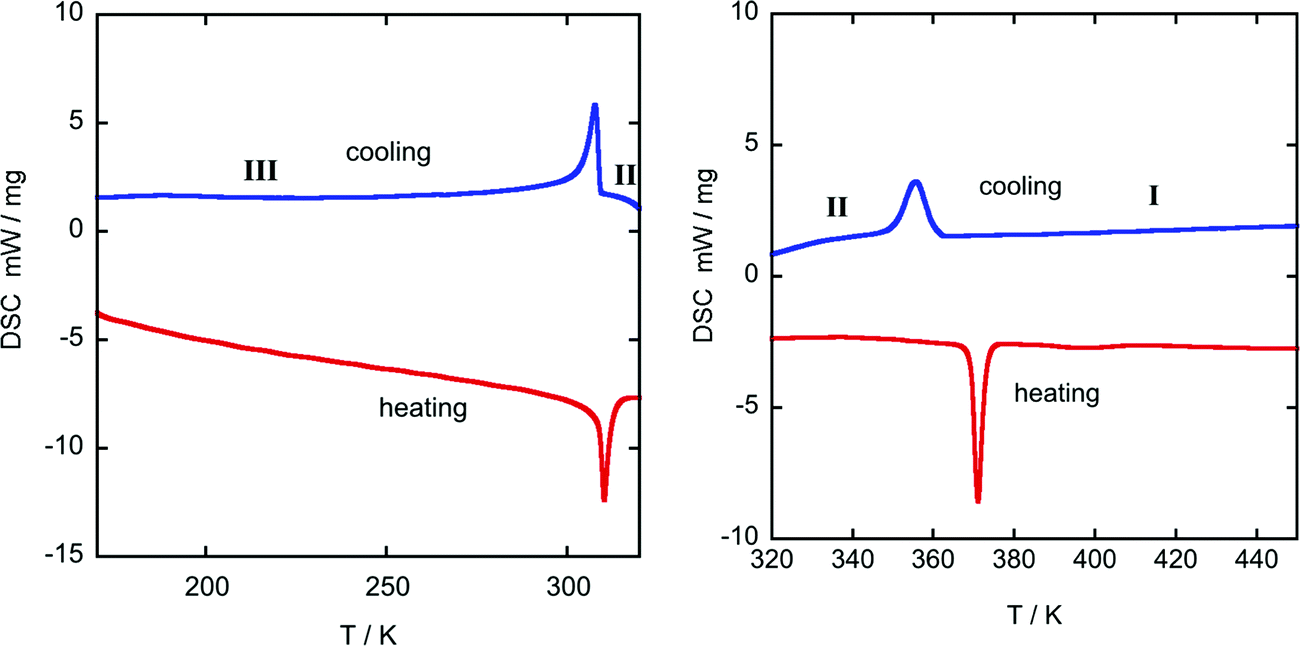 | ||
| Fig. 1 DSC-thermogram from 175-320 K (left) and 320-450 K (right) of ((R)-(−)-3-hydroxyquinuclidium)[FeCl4]. Blue: 1st cooling cycle; red: 2nd heating cycle; heating rate 10 K min−1. | ||
Table 2 shows the values of the associated latent heat (ΔH) and entropy change (ΔS), obtained from the peak integration of the heat flow curve. It is worth noting that the obtained values of latent heat and entropy change are slightly lower than those reported for similar plastic crystals with [FeCl4]− anions.42,43
| Parameters | Heating | Cooling | ||
|---|---|---|---|---|
| T III→II | T III→II | T III←II | T II←I | |
| T t (K) | 310.3 | 371.0 | 307.8 | 355.7 |
| |ΔH| (kJ kg−1) | 12.4 | 9.2 | 11.6 | 8.2 |
| |ΔS| (kJ kg−1 K−1) | 0.0399 | 0.0248 | 0.0377 | 0.0230 |
Crystal structures from phase I to III
Along this section, we will describe the different phases observed in the title compound with the temperature. As defined by convention, the highest temperature solid phase is denoted as phase I, with subsequent lower temperature phases denoted as phase II, III and IV, respectively.Above 370 K, phase I is observed in the SR-XRPD data at ALBA synchrotron44 (see Fig. 2, right). The crystal structure can be described in the cubic crystal system, with cell parameters a = b = c = 7.0940(1) Å and a unit cell volume of 357.01(1) Å3. The indexing of the synchrotron powder diffraction data suggests Pmm as a possible space group. The orientation of both cations and anions is highly disordered, which is in agreement with the isotropic character of the rotations described in plastic phases.32,45,46 The free rotation of the ((R)-(−)-3-hydroxyquinuclidium) cation results in a centrosymmetric crystal structure. This behavior is similar to the previously reported results for other quinuclidine-based compounds,32,45,46 suggesting that this type of complex is a promising material to obtain plastic crystal phases.
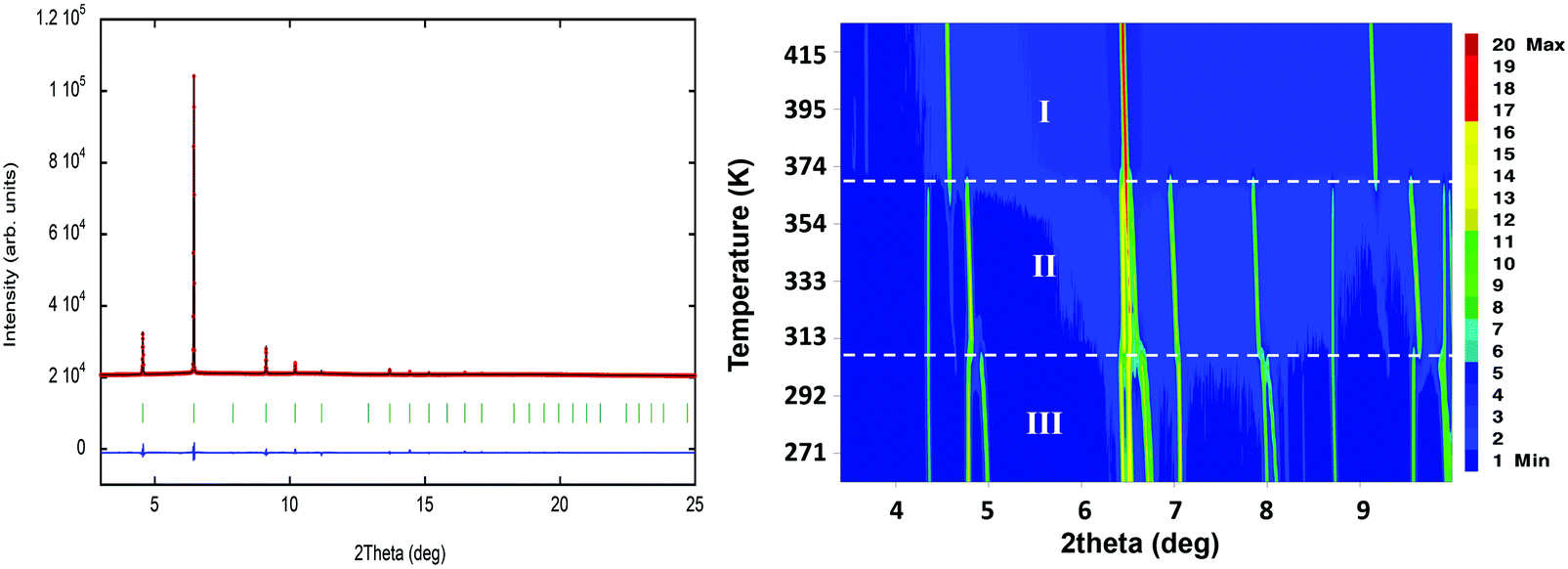 | ||
| Fig. 2 (left) Fit of the powder synchrotron X-ray data to the plastic phase (ca. 420 K, phase I), using the spherical shell refinement method. The experimental data are the red circles and the calculation is the black solid line. The difference between the experimental data and the fit is represented as a solid blue line; the markers (green vertical lines) indicate the position of the Bragg reflections in the Pmm S. G. (right) Temperature evolution of the synchrotron powder diffraction data of ((R)-(−)-3-hydroxyquinuclidium)[FeCl4] from 260 to 420 K. | ||
The plastic phase was modelled using a series of spherical shells centred on the 1a and 1b Wyckoff positions. The (0, 0, 0) and the (0.5, 0.5, 0.5) positions were filled by the [FeCl4] unit and the organic cation, respectively. The refinement at 420 K was carried out using the adapted spherical harmonics restrained to be spherical shells (see Fig. 2, left). The large value obtained for the thickness of the shell suggests a more complex rotation behavior on the plastic phase for both anions and cations.
The observed density map obtained through the Fourier transform is very similar to that previously observed for the (quinuclidinium)[FeCl4] compound.32 The shape difference in the Fourier map between the 1a and 1b Wyckoff positions, with a predominance of octahedral shape in the [FeCl4]− site and spherical shape in the (quinuclidinium)+ position, should be a symmetry artefact, as the 1a Wyckoff position has octahedral (Oh) symmetry.
Below the plastic phase, between 370 and 300 K, the crystal structure can be described in the non-centrosymmetric monoclinic space group C2, with cell parameters a = 9.257(3) Å, b = 9.852(3) Å, c = 7.466(3) Å and β = 90.704(9)°, obtained from the SCXRD data at 300 K. The nuclear phase transition from I to II is compatible with the signal observed on the DSC measurements at ca. 371 K (Fig. 1), as well as with the dielectric permittivity measurements, as it will be discussed below. The asymmetric unit consists of half [FeCl4]− anion and half ((R)-(−)-3-hydroxyquinuclidium)+ cation, therefore achieving electroneutrality. The other half of the inorganic and organic counterparts are generated by the 2-fold rotation axis along the b-axis. This induces a structural disorder in the organic part, with two positions of the O1 and N1/C1 atoms, sharing a 0.5 occupation value. The asymmetric unit can be seen in Fig. 3. For the sake of clarity, the symmetry operators have been applied to complete the fragments.
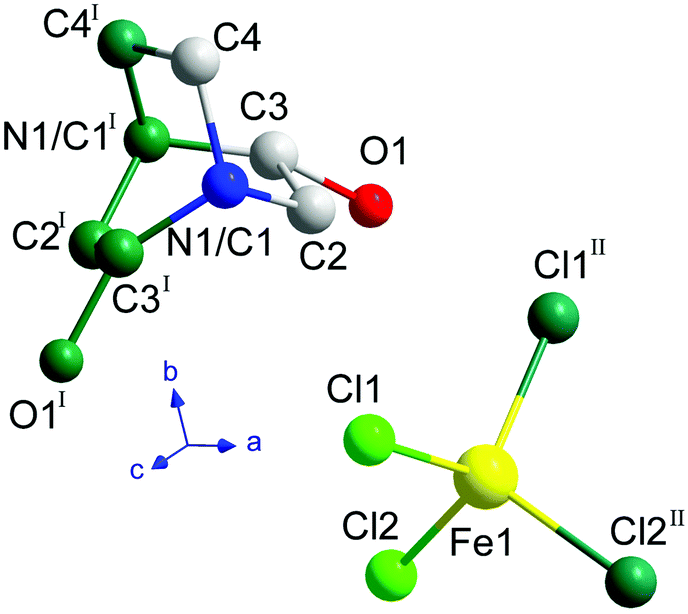 | ||
| Fig. 3 Asymmetric unit of ((R)-(−)-3-hydroxyquinuclidium)[FeCl4] at 300 K. The atoms generated by the symmetry operations are depicted in dark green (I: 1 − x, y, 1 − z; II: 1 − x, y, −z). The hydrogen atoms have been omitted for the sake of clarity. | ||
The three-dimensional assembly can be described as a stacking of organic ((R)-(−)-3-hydroxyquinuclidium)+ and inorganic [FeCl4]− layers along the crystallographic c-axis (see Fig. 4, left). The layers are pillared following an ABAB stacking sequence. The crystal packing is sustained by an intricate network of electrostatic and non-covalent interactions that include hydrogen bonds and van der Waals forces. Based on the crystal structure, a net polarization is possible along the b-direction, which is compatible with the electric polarization tensor Pi = (0, Py, 0).
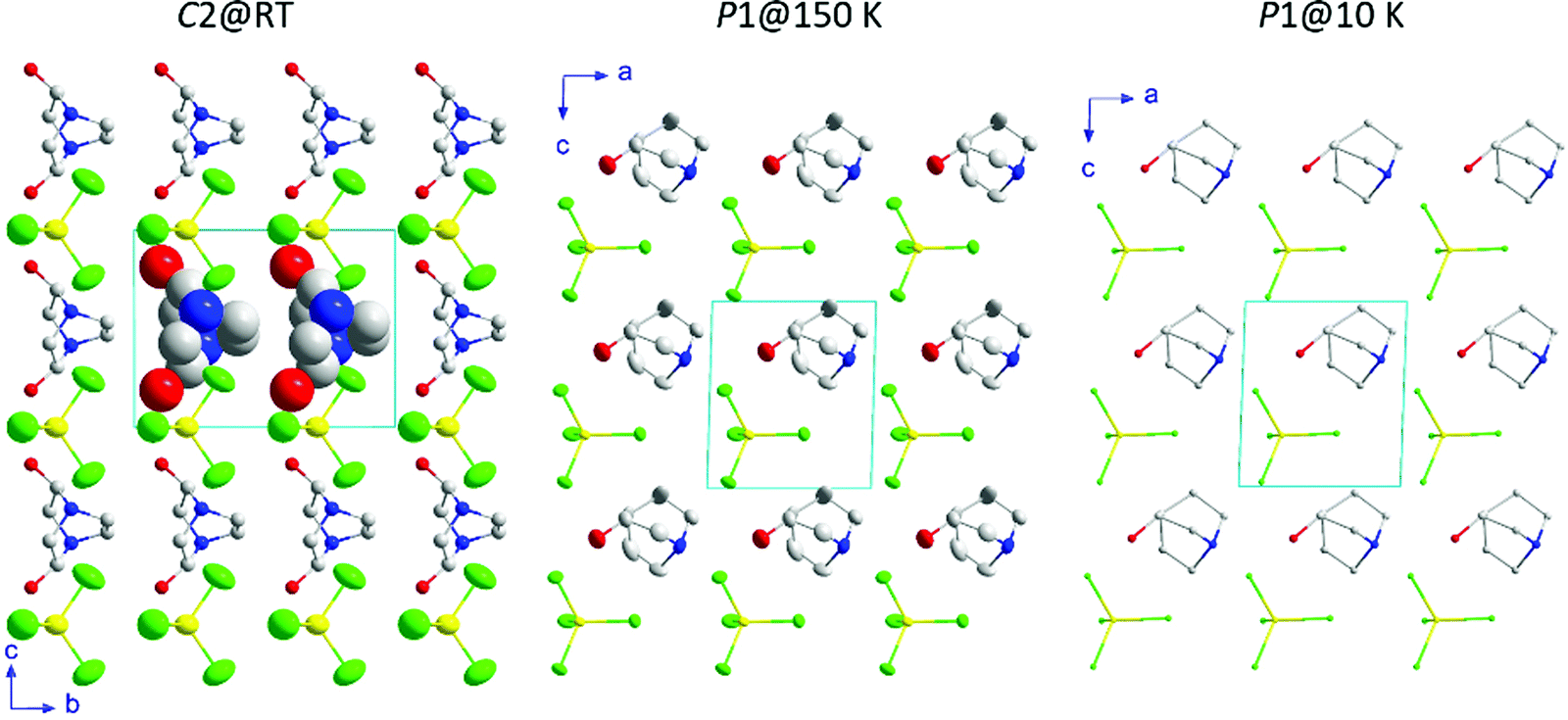 | ||
| Fig. 4 View of the crystal structures at 300, 150 and 10 K. The displacement ellipsoids are shown with 50% probability. Only the isotropic displacement spheres of two organic molecules are shown at 300 K for clarity. Likewise, the hydrogen atoms for all temperatures have been omitted for the sake of clarity. Color codes: yellow, iron; green, chloride; grey, carbon; blue, nitrogen; red, oxygen. | ||
Phase III is stable from 300 to 4 K. From SR-XRPD and DSC data, the nuclear phase transition from II to III is detected at ca. 310 K, in agreement with the dielectric permittivity measurements. There is a slight difference between the temperature obtained on the SR-XRPD and DSC with respect to the SCXRD. This difference can be attributed to the thermal inertia, as SR-XRPD and DSC measurements were performed following a ramp of 10 K min−1. The structure of phase III was solved by SCXRD at 150 K and the obtained model was refined against the X-ray (Fig. S6, ESI†) and neutron powder diffraction data (D2B) (Fig. S7, ESI†) at 100 and 10 K, respectively (see Fig. 4, right). The model corresponds to the triclinic P1 space group, with cell parameters: a = 6.4375(16) Å, b = 6.7708(16) Å, c = 7.3462(19) Å, α = 90.147(7)°, β = 91.589(7)° and γ = 92.021(7)°. The previously observed disorder in phase II is now well resolved. In this case, due to the absence of symmetry operations, all the atoms in the unit cell are unique. Therefore, the asymmetric unit consists of one ((R)-(−)-3-hydroxyquinuclidium)+ and one [FeCl4]− unit (Fig. 5). Regarding the 3D-assembly, phase III is structured on the same way as phase II, with stacking layers of organic and inorganic ions along the c-axis in an ABAB sequence. Based on the crystal structure, a net polarization could be possible in all directions, described by the electric polarization tensor: Pi = (Px, Py, Pz).
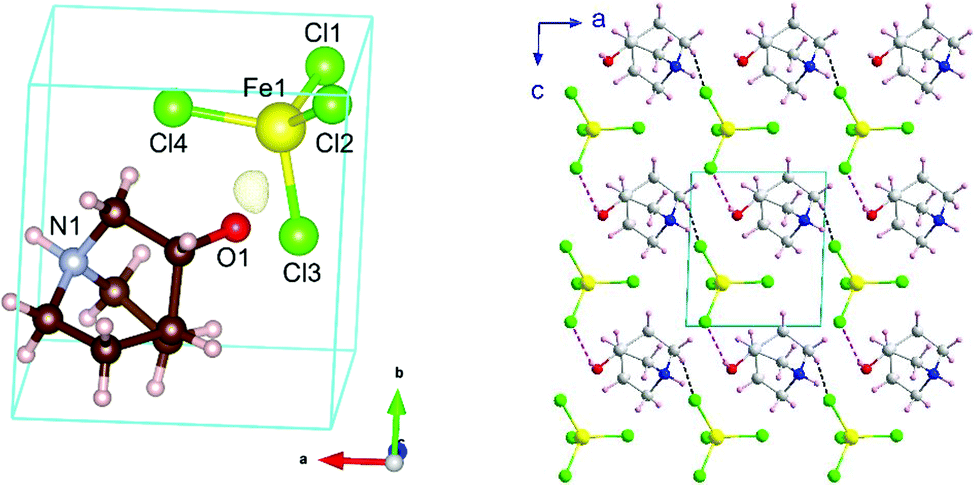 | ||
| Fig. 5 (left) Unit cell/asymmetric unit of the title compound at 10 K resulting from the refinement to the NPD data from D2B. The negative Fourier density isosurface is shown in yellow, corresponding to the most probable position of the hydrogen atom of the OH group. (right) View of the crystal packing on phase III together with the most probable H-bonds, O–H⋯Cl and C–H⋯Cl, are highlighted as red and blue dashed lines, respectively. | ||
Neutron diffraction data collected on the high resolution powder diffractometer D2B was employed to localize and refine the position of the hydrogen atoms. The location of all H atoms allows us to know all possible H-bonds within the structure, which play a crucial role in building up the crystal structure in this type of complex. These possible H-bonds were selected taking into account the IUPAC recommendations (H⋯Cl distance shorter than the sum of the van der Waals radii and X–H⋯Cl angle greater than 110°, where X refers to the donor atom).47 The strongest most probable H-bond corresponds to the one bonded to the oxygen, with a H⋯Cl distance of 2.58(4) Å and an O–H⋯Cl angle of 150(3)° (marked as pink in Fig. 5). Among all the possible ones, this bond presents the shortest H⋯Cl length and one of the greatest X–H⋯Cl angles (Table S2, ESI†). Furthermore, the oxygen is more electronegative than nitrogen or carbon, which makes the bond stronger.47 Regarding the Fe⋯Fe distances, which are relevant in the magnetic ordering, we can see a trend of decreasing lengths with lowering temperature, which ranges from 6.759(7) Å at 300 K to 6.323(18) Å at 10 K. These data can be seen in Table S2–S4 (ESI†) which list the shortest intralayer Fe⋯Fe distances for 10, 150 and 300 K, respectively, together with the bond lengths within the ((R)-(−)-3-hydroxyquinuclidium)+ and the [FeCl4]− ions.
SR-XRPD data from 260 to 365 K were refined using the pattern matching method with the FullProf suite program.33 The evolution of the crystal parameters and cell volume of phase II and III SR-XRPD is shown in Fig. 6. As it can be seen in the figure, all parameters follow an almost linear tendency. However, the study of the thermal expansion process in this temperature range displays anisotropically evolving cell parameters with an axial negative thermal expansion. This trend is maintained after the crystal phase transition detected at 310 K from II to III and it is attributed to the translational and reorientational dynamic displacements of the ((R)-(−)-3-hydroxyquinuclidium)+ cation. The changes in the lengths of the principal orthogonal axis (Fig. S8, ESI†), the principal thermal expansion (TE) coefficients, α (Table S5, ESI†), and their indicatrices (Fig. S9, ESI†) were obtained via linear fits using the PASCal program,48 using orthogonal lattice parameter evolution. On one hand, the volume thermal expansion coefficient, αV, is positive in both phases at all temperatures and almost four times larger than that in ice,49 [392(20) and 383(10) M K−1 for II and III]. On the other hand, the strong anisotropic positive TE values, αx3 = 356(27) and 199(3) M K−1 for II and III, respectively, are larger than the well-known “colossal” (|αx| ≥ 100) mechanical responses observed in cyanide-based inorganic material Ag3[Co(CN)6],50 and comparable to those recently reported in some imidazolium salts.51 Finally, the value of principal axis X2 and the uniaxial negative TE coefficient αx1 increase from 78(2) to 199(4) M K−1 and −48(6) to −10(1) M K−1, respectively, after the phase transition.
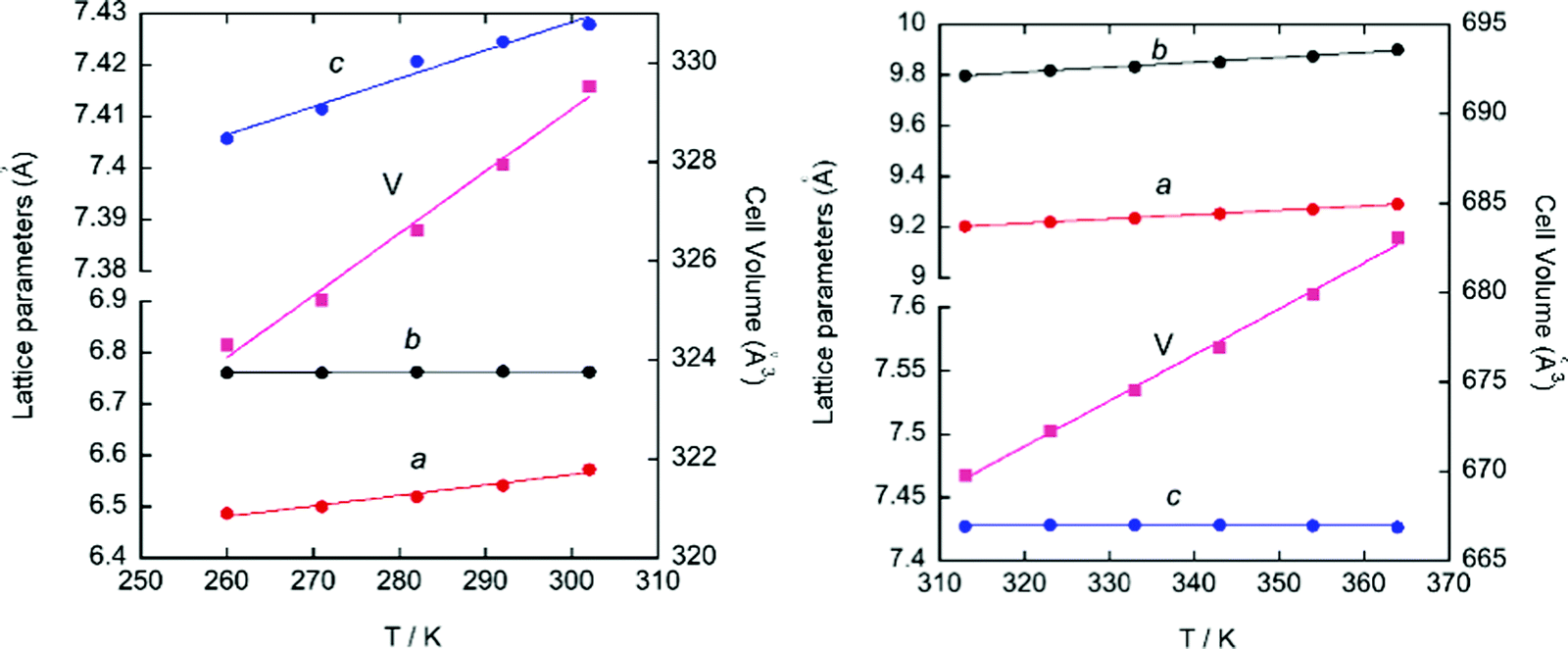 | ||
| Fig. 6 Evolution of the cell parameters and cell volume obtained from the pattern matching of the SR-XRPD data for phase III (left) and II (right). | ||
Magnetometry measurements
The magnetic properties of the presented compound were determined over the temperature range 2–300 K at 1 kOe in zero field cool (ZFC) and field cool (FC) modes. The magnetic susceptibility (χm) and the χmT versus T are shown in Fig. 7 left. χm displays paramagnetic behavior above 10 K. The magnetic susceptibility data can be fitted to a Curie-Weiss law (Fig. S10, ESI†) with a Weiss constant, θp, close to −3.5 K and an effective paramagnetic moment μeff = 6.07(1) μB per Fe ion, which agrees with the expected one for high spin d5 Fe(III) ions (μeff = 5.92 μB per Fe ion) and is in good agreement with those found in other hybrid halometallate compounds based on [FeCl4]− ions.34,52–54 At RT, the χmT value of 4.54 emu K mol−1 Oe−1 is also consistent with the expected one (4.38 emu K mol−1 Oe−1 for Fe(III) ions with a magnetic spin S = 5/2). This value remains almost constant down to 30 K, where it begins to fall due to the evolution of antiferromagnetic interactions between Fe(III) ions. Moreover, below 10 K, the ZFC-FC molar susceptibility measured under 1 kOe displays a broad peak near 4 K (see inset of Fig. 7, left), suggesting the existence of long-range magnetic ordering. The negative θp value together with the absence of splitting in the ZFC-FC molar susceptibility is characteristic of a predominant antiferromagnetic behavior. | ||
| Fig. 7 (left) Magnetic susceptibility (χm) and the χmT versus T at 1 kOe. The inset shows the low temperature detail of ZFC-FC measurements at 1 kOe. (Right): magnetization measurements at 2 K. The inset shows the derivative of the magnetization with respect to the applied field. | ||
In order to study the response of the magnetization to the applied magnetic field, we measured the M(H) curves within ±50 kOe at 2 K (Fig. 7, right). The M(H) curve displays a maximum value of ±4.35 μB per Fe ion at ±50 kOe, which is near the expected fully-saturated value of 5 μB per Fe(III) ion. In addition, the curve shows an inflexion point near 15 kOe (see the derivative with respect to the magnetic field in the inset of Fig. 7 right), suggesting the presence of a metamagnetic transition. Overall, we can conclude that ((R)-(−)-3-hydroxyquinuclidium)[FeCl4] shows an antiferromagnetic behavior in the absence of a ferromagnetic component.
Magnetic structure (phase IV)
Neutron powder diffraction is the most suitable technique to evidence the apparition of long-range magnetic order. To confirm the occurrence of magnetic ordering, high-flux patterns were collected from 1.6 to 6 K using a high flux medium resolution D1B diffractometer (ILL, Grenoble), operated with a wavelength of λ = 2.52 Å (Fig. S11, ESI†). A comparative view of both patterns collected at 6 K and 1.6 K reveals the presence of a new sharp Bragg reflection in the low temperature pattern, which is not overlapped with the structural peaks observed at 6 K. The new reflections are coincident in temperature with the signal observed in the magnetometry measurements, which proves that they have a magnetic origin and, therefore, providing direct evidence for the existence of long magnetic ordering.The magnetic reflections were indexed using the k-search program included in the FullProf suite.35 The best solution provides a magnetic unit cell with a doubled c-axis with respect to the paramagnetic unit cell. This corresponds to a magnetic propagation vector k = (0, 0, 0.5). In order to work only with the magnetic contribution, the nuclear contribution from the paramagnetic pattern collected at 6 K has been subtracted from the pattern at 1.6 K (see Fig. 8). Both diffraction patterns were normalized to the same monitor in order to obtain the best difference pattern and the difference was shifted to positive intensities to avoid negative values. The difference pattern isolates the magnetic contribution and, therefore, a more accurate refinement of the magnetic structure can be undertaken.
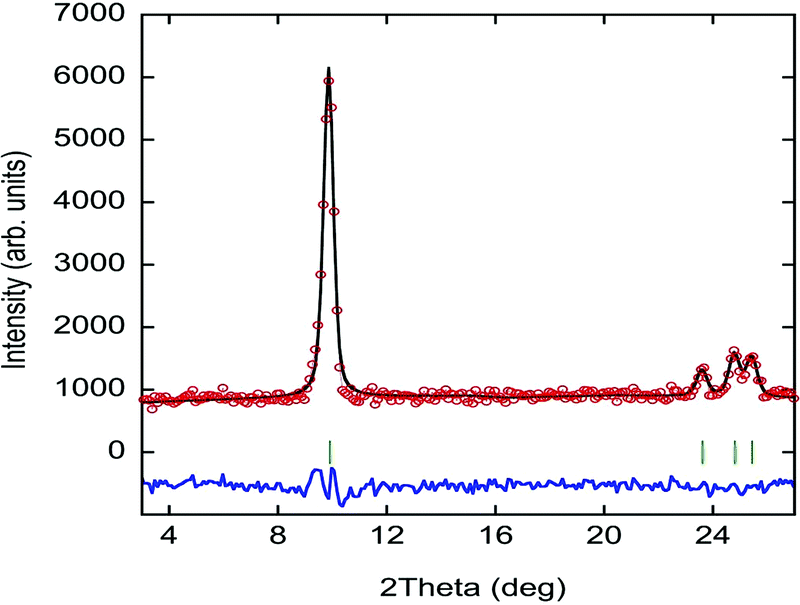 | ||
| Fig. 8 Experimental neutron diffraction data (red circles) and fit (black solid line) to the difference pattern between 1.6 and 10 K collected at the D1B diffractometer. The difference between the experimental data and the fit is represented as a solid blue line; the markers (green vertical lines) indicate the position of the magnetic Bragg reflections. | ||
Symmetry analysis using the Bertaut's symmetry analysis method,55 employing the BCS k-subgroupsmag utility56 and BasIreps program in FullProf,35 shows a unique potential maximal magnetic space group, corresponding to the Ps1 Shubnikov space group. This Shubnikov space group is a subgroup of the P11′ paramagnetic space group. The doubling of the unit cell due to the application of a k = (0, 0, 0.5) propagation vector gives rise to two magnetic sites within the magnetic unit cell, which are symmetry-related by an anti-translation symmetry operator. Therefore, the magnetic structure is strictly antiferromagnetic.
The iron site has no symmetry constraints and, consequently, the magnetic moment can present components in any direction [m = (mx, my, mz)]. During the first cycles of refinements, the three components of the magnetic moment were refined without restrictions. However, based on the results of the refinement, the component along the c-axis was zero within the experimental error. Therefore, in the last refinement cycle, the mz component was forced to be null (see Fig. 8).
The value of the magnetic moment obtained from the D1B data was 4.42(3) μB, corresponding to the modulus of the vector m = (−1.26(2), 4.20(7), 0.0), which is not far away from the expected value for a Fe3+ (S = 5/2).
The magnetic structure can be described as ferromagnetic layers extended along the ab-plane, which are antiferromagnetically coupled along the c-axis. The main component of the magnetic moment is along the b-axis, with a tilt within the ab-plane, due to the magnetic moment component along the a-axis (see Fig. 9).
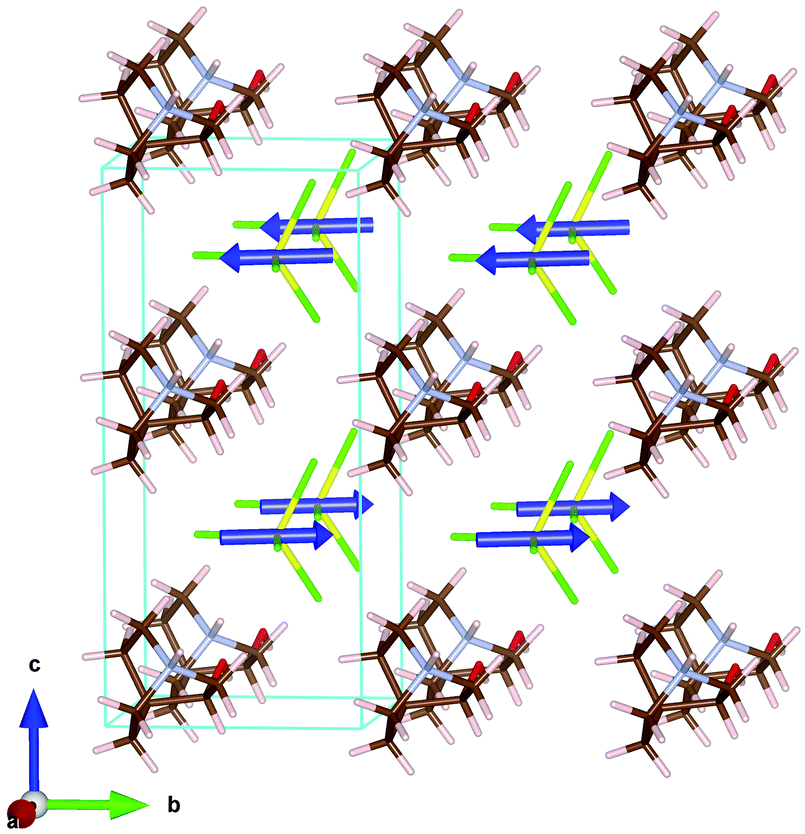 | ||
| Fig. 9 Magnetic structure of the ((R)-(−)-3-hydroxyquinuclidium)[FeCl4] compound at 1.6 K. Color codes: yellow, iron; green, chloride; blue, magnetic spin. The organic cations have been included in the model with the positions obtained from the refinement of the high resolution neutron diffraction data at 10 K. | ||
The shortest Fe⋯Fe distance (6.4375 Å) is along the a-axis. While the distance along the b-axis is slightly larger (6.7708 Å), both present a sigma-hole crystal packing. These two intralayer interactions exhibit ferromagnetic character, while for the interlayer interaction along the c-axis, the shortest Fe⋯Fe distance is notably longer (7.3462 Å), presenting antiferromagnetic interactions. Moreover, the shortest Cl⋯Cl distance is also along the c-direction (3.7446 Å) with a Fe–Cl⋯Cl–Fe torsion angle of ca. 12.7°. However, the Cl⋯Cl distances are longer than the sum of the standard van der Waals radii, thus it is not certain that the Cl⋯Cl interaction plays a principal role in the antiferromagnetic ordering along the c-axis. Nevertheless, dipolar interaction can couple the ferromagnetic planes (which are extended along the ab-plane) antiferromagnetically along the c-direction. Similar magnetic structures, with ferromagnetic layers antiferromagnetically coupled in the stacking direction, have been reported on 1,3-dimethylimidazolim tetrabromoferrate, Dimim[FeBr4].52 Although in the case of this halometallate complex the magnetic propagation vector was k = (0, 0, 0), and the volume of the paramagnetic unit cell was ca. twice the unit cell of the title compound at 10 K. Therefore, the magnetic unit cells of both compounds are quite similar a = 6.745(3) Å, b = 14.364(3) Å, c = 6.759(3) Å, and β = 90.80(2)° for Dimim[FeBr4] and a = 6.4375(16) Å, b = 6.7708(16) Å, c = 14.4596(19) Å, α = 90.147(7)°, β = 91.589(7)° and γ = 92.021(7)° for ((R)-(−)-3-hydroxyquinuclidium)[FeCl4] compound. Although both compounds crystallize in a different crystalline system, the topology of the crystal structure is similar and the Fe⋯Fe distances are comparable.
Dielectric properties and piezoresponse force microscopy
The complex dielectric permittivity of a polycrystalline pellet of ((R)-(−)-3-hydroxyquinuclidium)[FeCl4] was measured as a function of temperature and frequency. Fig. 10 shows the temperature dependence of the real part of the complex dielectric permittivity (εr′) at different frequencies. Very interestingly, this compound shows two dielectric anomalies at the εr′ vs. T data associated with the observed phase transitions by DSC and SR-XRPD patterns. A sharp step-like dielectric transition around T ∼ 315 K is associated with the phase transition between two different non-centrosymmetric structures: phase III (S.G.:P1) and phase II (S.G.:C2). This phase transition is a chiral to chiral transition described as 2F1 according to Aizu notation,57 and thus this belongs to one of the ferroelectric and ferroelastic phase transitions. Additionally, the εr′ vs. T data exhibit a narrow peak around T ∼ 370 K, corresponding to the phase transition between the non-centrosymmetric phase II (S.G.:C2) and centrosymmetric phase I (S.G.: Pmm). It is worth noting that the observed dielectric transitions show some resemblance to those reported for different plastic crystals.29–31,49,50,58
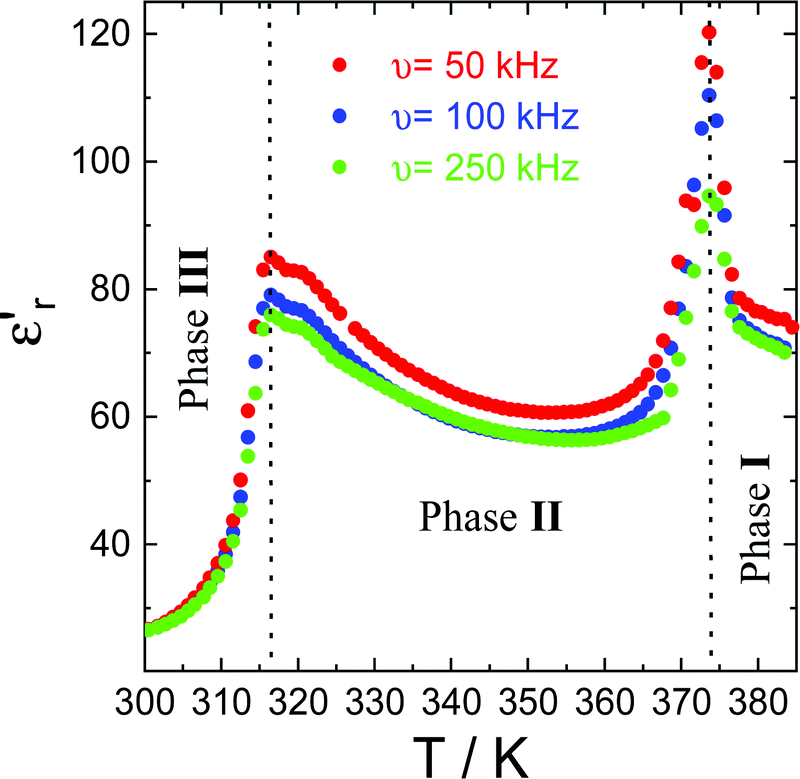 | ||
| Fig. 10 The real part of the complex dielectric permittivity (εr′) at different frequencies in the temperature range of 300–390 K. | ||
The change in amplitude of the real part of the complex dielectric permittivity with the frequency suggests the emergence of a ferroelelctric behaviour, related to an order–disorder transition. The high mobility of the constituents in the plastic phase produces a paraelectric state, where the dipoles are statistically disordered with respect to position and time. The decrease in temperature freezes the rotation movements of the constituents, creating an alignment of the dipoles, which should be responsible for the emergence of a polar ordering.
At ca. 375 K, well-pronounced peaks with values of εr′ above 120 are clearly revealed. Interestingly, the temperature of the maximum peak remains almost constant for the studied frequencies, while the amplitude strongly decreases (up to 25% decrease) with increasing frequencies. Similar behaviour is observed at ca. 315 K, corresponding to the phase transition between phases II and III. This behaviour has been previously observed in other order–disorder-type ferroelectrics.59,60
To confirm the ferroelectric behavior (stable domain structure and switchable polarization) of phase III, we have used piezoresponse force microscopy (PFM), which is an effective tool to provide non-destructive visualization and manipulation of ferroelectric domains at the nanoscale.16,27,61Fig. 11a–c show the topography, the vertical amplitude and vertical phase images, respectively. The phase image (Fig. 11c) exhibits several antiparallel domains with a clear contrast. The amplitude image (Fig. 11b) shows that the domains are separated by the domain walls, which are irrelevant to the topography (Fig. 11a). The obtained butterfly curve and hysteresis loop exhibit a low coercive voltage of ∼10 V (see Fig. 11d and e), which gets very close to the goal of application in ferroelectric random access memories (FeRAMs).62
 | ||
| Fig. 11 The images show the local ferroelectric domain. (a) The topography images, (b) the vertical PFM amplitude image and (c) the vertical PFM phase image. Amplitude (d) and phase (e) signal as functions of the tip voltage for a selected point, showing the typical butterfly curve and hysteresis loop of a FE material. | ||
4. Conclusions
((R)-(−)-3-hydroxyquinuclidium)[FeCl4] is a plastic/chiral magnetic halometallate compound with ferroelectric and ferroelastic behavior in a wide range of temperatures. We applied the approach described in the quasi-spherical theory in order to decrease the symmetry of the molecular constituents, by adding modifications on the globular molecules to the quinuclidinium counter ions, to obtain ((R)-(−)-3-hydroxyquinuclidium), which, in combination with tetrachloroferrate ions, crystallize in low symmetry monoclinic or triclinic space groups below the plastic phase. Moreover, this cation helps in maintaining the plastic/polar behavior at similar temperatures to the quinuclidinium cation. The replacement of this cation for the homochiral enantiomer ((R)-(−)-3-hydroxyquinuclidium)+ forces the emergence of non-centrosymmetric space groups below the rotational phase, favoring the development of ferroelectricity. The complete phase diagram involves a phase transition between paraelectric-to-polar/chiral phase at 370 K, from polar/chiral-to-ferroelectric/chiral phase around 300 K and from paramagnetic-to-antiferromagnetic at 4 K. Above RT, the increase of the molecular libration produces a significant modification of the assembly of the electrical dipoles in the structure, giving rise to an increase of the real part of the dielectric permittivity. When the thermal energy is increased, the constituents develop rotational motions, giving rise to the plastic phase. Although the organic cation is chiral, the plastic phase can be described in a centrosymmetric space group due to the high molecular disorder. Therefore, a paraelectric state is achieved above the plastic temperature. Consequently, the real part of the complex dielectric permittivity shows a significant decrease due to the almost random distribution of the electric dipoles in the paraelectric phase.A structural phase transition close to RT with a hysteresis lower than 3 degrees is detected. However, the most interesting result is the direct evidence of the electric polarization switching and local ferroelectric behavior at RT, with a low coercive voltage of ∼10 V, which is in the limit for application in ferroelectric random access memories (FeRAMs). Unfortunately, the presence of a non-negligible sample conductivity precludes the characterization of the macroscopic ferroelectricity through the P–E hysteresis loops. Moreover, below RT, the system crystallizes in the P1 space group, therefore, the polarization tensor is not constrained by symmetry and it is thus free to rotate, which could be of interest to develop new devices based on thin films, as polarization could be switched more easily between multiple ferroelectric axes. This finding opens an avenue to construct quinuclidinine-based homochiral ferroelectrics and it will inspire the exploration of more eminent enantiomeric molecular ferroelectrics.
Author contributions
P. G. I. and O. F and I. P contributed equally. They designed and executed experiments, prepared figures, and wrote the original draft. L. C.-D., G. B., O. V. and J. R.-F. assisted with the experiments and data interpretation of single-crystal, synchrotron and neutron powder X-ray diffraction. J. S.-B. and M. S.-A. performed the dielectric and piezoresponse force microscopy measurements and analysis. C. M. B. and M. S.-A. executed the optical measurements and analysis. J. E. G. executed the P–E measurements and analysis. M. T. F.-D. led and guided the project. The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from Universidad de Cantabria (Proyecto Puente convocatoria 2018 funded by SODERCAN_FEDER), Universidad del País Vasco/Euskal Herriko Unibertsitatea (GIU17/50 and PPG17/37) and Ministerio de Economia y Competividad (MAT2017-89239-C2-(1,2)-P, MAT2017-83631-C3-3-R, MAT2017-86453-R, PGC2018-097520-A-100 and PID2019-104050RA-I00) is acknowledged. The authors gratefully acknowledge the technical and human support provided by SGIKer (UPV/EHU, MINECO, GV/EJ, ERDF, and ESF). Carmen Martín is grateful to VI PPIT-2018 from Universidad de Sevilla. The paper is (partly) based on the results of experiments carried out at the ALBA Synchrotron Light Source in Barcelona (proposal 2019083666) and Institute Laue-Langevin (ILL) of Grenoble (Proposals 5-31-2580 and 5-31-2460).References
- D. Gelman, B. Shvartsev, I. Wallwater, S. Kozokaro, V. Fidelsky, A. Sagy, A. Oz, S. Baltianski, Y. Tsur and Y. Ein-Eli, J. Power Sources, 2017, 364, 110–120 CrossRef CAS.
- M. Kar, O. Tutusaus, D. R. MacFarlane and R. Mohtadi, Energy Environ. Sci., 2019, 12, 566–571 RSC.
- K. Y. Chan, B. Jia, H. Lin, N. Hameed, J. H. Lee and K. T. Lau, Compos. Struct., 2018, 188, 126–142 CrossRef.
- X. Huang, G. Alva, Y. Jia and G. Fang, Renewable Sustainable Energy Rev., 2017, 72, 128–145 CrossRef CAS.
- J. Salgado-Beceiro, J. M. Bermúdez-García, A. L. Llamas-Saiz, S. Castro-Garcia, M. A. Señarís-Rodríguez, F. Rivadulla and M. Sánchez-Andújar, J. Mater. Chem. C, 2020, 8, 13686–13694 RSC.
- R. Gupta, M. Yadav, R. Gaur, G. Arora, P. Yadav and R. K. Sharma, Mater. Horiz., 2020, 7, 3097–3130 RSC.
- M. K. Leu, I. Vicente, J. A. Fernandes, I. de Pedro, J. Dupont, V. Sans, P. Licence, A. Gual and I. Cano, Appl. Catal., B, 2019, 245, 240–250 CrossRef CAS.
- J. Estager, J. D. Holbrey and M. Swadźba-Kwaśny, Chem. Soc. Rev., 2014, 43, 847–886 RSC.
- F. Scé, I. Cano, C. Martin, G. Beobide, O. Castillo and I. de Pedro, New J. Chem., 2019, 43, 3476–3485 RSC.
- R. L. Vekariya, J. Mol. Liq., 2017, 227, 44–60 CrossRef CAS.
- R. Kore, P. Berton, S. P. Kelley, P. Aduri, S. S. Katti and R. D. Rogers, ACS Catal., 2017, 7, 7014–7028 CrossRef CAS.
- I. Cano, C. Martin, J. A. Fernandes, R. W. Lodge, J. Dupont, F. A. Casado-Carmona, W. Lucena, S. Cardenas, V. Sans and I. de Pedro, Appl. Catal., B, 2020, 260, 118110 CrossRef CAS.
- J. Kurley, H. Zhang, D. V. Talapin, J. Russell and M. H. Hudson, Halometallate Ligand-capped Semiconductor Nanocrystals, US Patent No. 10541134, US Patent and Trademark Office, Washington, DC, 2020 Search PubMed.
- J. Timmermans, J. Phys. Chem. Solids, 1961, 18, 1–8 CrossRef CAS.
- H. Y. Zhang, Y. Y. Tang, P. P. Shi and R. G. Xiong, Acc. Chem. Res., 2019, 52, 1928–1938 CrossRef CAS PubMed.
- Y.-M. You, W.-Q. Liao, D. Zhao, H.-Y. Ye, Y. Zhang, Q. Zhou, X. Niu, J. Wang, P.-F. Li, D.-W. Fu, Z. Wang, S. Gao, K. Yang, J.-M. Liu, J. Li, Y. Yan and R.-G. Xiong, Science, 2017, 357, 306–309 CrossRef CAS PubMed.
- W.-Q. Liao, Y.-Y. Tang, P.-F. Li, Y.-M. You and R.-G. Xiong, J. Am. Chem. Soc., 2018, 140, 3975–3980 CrossRef CAS PubMed.
- W.-Q. Liao, Y.-Y. Tang, P.-F. Li, Y.-M. You and R.-G. Xiong, J. Am. Chem. Soc., 2017, 139, 18071–18077 CrossRef CAS PubMed.
- Y. Zhang, X.-J. Song, Z.-X. Zhang, D.-W. Fu and R.-G. Xiong, Matter, 2020, 2, 697–710 CrossRef.
- P.-F. Li, Y.-Y. Tang, W.-Q. Liao, P.-P. Shi, X.-N. Hua, Y. Zhang, Z. Wei, H. Cai and R.-G. Xiong, Angew. Chem., Int. Ed., 2018, 130, 11939–11942 CrossRef PubMed.
- Z. Wei, W.-Q. Liao, Y.-Y. Tang, P.-F. Li, P.-P. Shi, H. Cai and R.-G. Xiong, J. Am. Chem. Soc., 2018, 140, 8110–8113 CrossRef CAS PubMed.
- Z.-H. Wei, Z.-T. Jiang, X.-X. Zhang, M.-L. Li, Y.-Y. Tang, X.-G. Chen, H. Cai and R.-G. Xiong, J. Am. Chem. Soc., 2020, 142, 1995–2000 CrossRef CAS PubMed.
- J.-C. Liu, W.-Q. Liao, P.-F. Li, Y.-Y. Tang, X.-G. Chen, X.-J. Song, H.-Y. Zhang, Y. Zhang, Y.-M. You and R.-G. Xiong, Angew. Chem., Int. Ed., 2020, 59, 3495–3499 CrossRef CAS PubMed.
- H. Liu, H. Y. Zhang, X. G. Chen and R.-G. Xiong, J. Am. Chem. Soc., 2020, 142, 15205–15218 CrossRef CAS PubMed.
- C.-K. Yang, W.-N. Chen, Y.-T. Ding, J. Wang, Y. Rao, W.-Q. Liao, Y. Xie, W. Zou and R.-G. Xiong, J. Am. Chem. Soc., 2019, 141, 1781–1787 CrossRef CAS PubMed.
- W. J. Xu, Y. Zeng, W. Yuan, R. G. Qiu, W. X. Zhang and X. M. Chen, Chem. Commun., 2018, 54, 3347–3350 RSC.
- Q. Pan, Z.-B. Liu, H.-Y. Zhang, W.-Y. Zhang, Y.-Y. Tang, Y.-M. You, P.-F. Li, W.-Q. Liao, P.-P. Shi, R.-W. Ma, R.-Y. Wei and R.-G. Xiong, Adv. Mater., 2017, 29, 1700831 CrossRef PubMed.
- H.-Y. Zhang, Y.-Y. Tang, P.-P. Shi and R.-G. Xiong, Acc. Chem. Res., 2019, 52, 1928–1938 CrossRef CAS PubMed.
- C.-K. Yang, W.-N. Chen, Y.-T. Ding, J. Wang, Y. Rao, W.-Q. Liao, Y.-Y. Tang, P.-F. Li, Z.-X. Wang and R.-G. Xiong, Adv. Mater., 2019, 31, 1808088 CrossRef PubMed.
- R.-G. Xiong, S.-Q. Lu, Z.-X. Zhang, H. Cheng, P.-F. Li and W.-Q. Liao, Angew. Chem., Int. Ed., 2020, 59, 9574–9578 CrossRef CAS PubMed.
- P.-F. Li, Y.-Y. Tang, Z.-X. Wang, H.-Y. Ye, Y.-M. You and R.-G. Xiong, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- P. Gonzalez-Izquierdo, O. Fabelo, L. Canadillas-Delgado, G. Beobide, O. Vallcorba, M. Sánchez-Andújar, M. T. Fernández-Díaz and I. de Pedro, J. Mater. Chem. C, 2020, 8, 11389–11398 RSC.
- X.-M. Zhao, D. Li, H.-X. Zhao, Y.-P. Ren, L.-S. Long and L.-S. Zheng, Inorg. Chem., 2020, 59, 5475–5482 CrossRef CAS PubMed.
- A. García-Saiz, I. de Pedro, O. Vallcorba, P. Migowski, I. Hernández, L. F. Barquin, I. Abrahams, M. Motevalli, J. Dupont, J. A. Gonzalez and J. R. Fernández, RSC Adv., 2015, 5, 60835–60848 RSC.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- Bruker, SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- Bruker, SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2001 Search PubMed.
- G. M. Sheldrick, SHELXS version-2018/3 and SHELXL version-2018/3: programs for crystal structure solution and refinement, University of Gottingen, Germany Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- K. B. Yoon and J. K. Kochi, Inorg. Chem., 1990, 29, 869–874 CrossRef CAS.
- A. Abedi, N. Safari, V. Amani and H. R. Khavasi, Dalton Trans., 2011, 40, 6877–6885 RSC.
- C. Martin, I. Cano, F. Scé, R. Pérez-Aguirre, C. Gimbert-Suriñach, P. Lopez-Cornejo and I. de Pedro, New J. Chem., 2020, 44, 6375–6383 RSC.
- J. Harada, Y. Kawamura, Y. Takahashi, Y. Uemura, T. Hasegawa, H. Taniguchi and K. Maruyama, J. Am. Chem. Soc., 2019, 141, 9349–9357 CrossRef PubMed.
- F. Fauth, I. Peral, C. Popescu and M. Knapp, Powder Diffr., 2013, 28, S360–S370 CrossRef CAS.
- J. Harada, T. Shimojo, H. Oyamaguchi, H. Hasegawa, Y. Takahashi, K. Satomi, Y. Suzuki, J. Kawamata and T. Inabe, Nat. Chem., 2016, 8, 946–952 CrossRef CAS PubMed.
- S. Horiuchi and Y. Tokura, Nat. Mater., 2008, 7, 357–366 CrossRef CAS PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CAS.
- M. J. Cliffe and A. L. Goodwin, J. Appl. Crystallogr., 2012, 45, 1321–1329 CrossRef CAS.
- S. J. La Placa and B. Post, Acta Crystallogr., 1960, 13, 503–505 CrossRef CAS.
- A. L. Goodwin, M. Calleja, M. J. Conterio, M. T. Dove, J. S. Evans, D. A. Keen, L. Peters and M. G. Tucker, Science, 2008, 319, 794–797 CrossRef CAS PubMed.
- I. de Pedro, A. García-Saiz, J. Dupont, P. Migowski, O. Vallcorba, J. Junquera, J. Rius and J. R. Rodríguez Fernández, Cryst. Growth Des., 2015, 15, 5207–5212 CrossRef CAS.
- A. García-Saiz, P. Migowski, O. Vallcorba, J. Junquera, J. A. Blanco, J. A. González, M. T. Fernández-Díaz, J. Rius, J. Dupont, J. R. Fernández and I. de Pedro, Chem. – Eur. J., 2014, 20, 72–76 CrossRef PubMed.
- P. González-Izquierdo, O. Fabelo, G. Beobide, I. Cano, I. R. de Larramendi, O. Vallcorba, J. R. Fernández, M. T. Fernández-Díaz and I. de Pedro, RSC Adv., 2020, 10, 11200–11209 RSC.
- P. González-Izquierdo, O. Fabelo, I. Cano, O. Vallcorba, J. R. Rodríguez Fernández, M. T. Fernandez-Diaz and I. de Pedro, J. Mol. Liq., 2021, 325, 114570 CrossRef.
- E. Bertaut, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1968, 24, 217–231 CrossRef CAS.
- M. I. Aroyo, J. M. Perez-Mato, C. Capillas, E. Kroumova, S. Ivantchev, G. Madariaga, A. Kirov and H. Wondratschek, Z. Kristallogr., 2006, 221, 15–27 CAS.
- K. Aizu, Phys. Rev. B: Solid State, 1970, 2, 754 CrossRef.
- J. Harada, N. Yoneyama, S. Yokokura, Y. Takahashi, A. Miura, N. Kitamura and T. Inabe, J. Am. Chem. Soc., 2018, 140, 346–354 CrossRef CAS PubMed.
- M. E. Lines and A. M. Glass, Principles and applications of ferroelectrics and related materials, Clarendon, Oxford, 1977 Search PubMed.
- P. Lunkenheimer, J. Müller, S. Krohns, F. Schrettle, A. Loidl, B. Hartmann, R. Rommel, M. de Souza, C. Hotta, J. A. Schlueter and M. Lang, Nat. Mater., 2012, 11, 755–758 CrossRef CAS PubMed.
- P.-P. Shi, S. Lu, S.-Q. X.-J. Song, X.-G. Chen, W.-Q. Liao, P.-F. Li, Y.-Y. Tang and R.-G. Xiong, J. Am. Chem. Soc., 2019, 141, 18334–18340 CrossRef CAS PubMed.
- P.-P. Shi, Y.-Y. Tang, P.-F. Li, H.-Y. Ye and R.-G. Xiong, J. Am. Chem. Soc., 2017, 139, 1319–1324 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV/Vis and CD results, crystallographic information, CIF data, details on the thermal analysis, neutron diffraction data and fit, single-crystal X-ray diffraction analysis, variable-temperature and synchrotron X-ray powder diffraction analysis. CCDC 2045021–2045023. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0tc05800a |
| This journal is © The Royal Society of Chemistry 2021 |