
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluencing prototropy by metal ion coordination: supramolecular transformation of a dynamer into a Zn-based toroidal species†‡
Miroslava
Čonková
 ab,
Wojciech
Drożdż
ab,
Zygmunt
Miłosz
c,
Piotr
Cecot
ab,
Jack
Harrowfield
d,
Mikołaj
Lewandowski
c and
Artur R.
Stefankiewicz
*ab
ab,
Wojciech
Drożdż
ab,
Zygmunt
Miłosz
c,
Piotr
Cecot
ab,
Jack
Harrowfield
d,
Mikołaj
Lewandowski
c and
Artur R.
Stefankiewicz
*ab
aFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, Poznań 61-614, Poland. E-mail: ars@amu.edu.pl
bCenter for Advanced Technologies, Adam Mickiewicz University, Uniwersytetu Poznańskiego 10, Poznań 61-614, Poland
cNanoBioMedical Centre, Adam Mickiewicz University, Wszechnicy Piastowskiej 3, Poznań 61-614, Poland
dInstitut de Science et d’Ingénierie Supramoléculaires (ISIS), Université de Strasbourg, 8 allée Gaspard Monge, Strasbourg 67083, France
First published on 3rd March 2021
Abstract
Disruption of a tautomeric equilibrium by coordination self-assembly depends upon a delicate interplay between supramolecular interactions but has not been studied for supramolecular polymers in two dimensions at the solid–liquid interface. Presented herein is the description of a functionalized molecular module which in DMSO solution generates a highly compact dynamic aggregate in its lactam (amide) form that undergoes reaction with Zn(II) ion to give a ML2-type complex in which one lactam unit on each ligand has been converted to its iminol form, enabling bidentate binding also involving pyrimidine-N. These solution measurements, as well as a study by scanning tunneling microscopy at the liquid/graphite interface, corroborated by density functional studies, have revealed how the tautomeric equilibrium can be modulated by the addition of a metal salt.
Supramolecular assemblies based on noncovalent bonds are reversible and sensitive to environment, entailing adaptivity and stimuli-responsiveness that can be employed to achieve desired topology and/or functionality.1–7 Of the numerous stimuli which have been used to displace dynamic equilibria,8,9 metal ions are amongst the most versatile, since their actual influence can vary greatly, depending upon the particular choice.10 Very simple, long-known examples of their varying preferences for interaction sites are provided by the complexes of transition metals with ambidentate ligands11–13 such as thiocyanate and nitrite ions, although more interesting cases arise with the coordination of primary amides, RCONHR′.14–17 In their neutral form, such amides behave as O-donor species but the enhancement of acidity due to their coordination results in their ready deprotonation followed by rearrangement to the N-bound anion, subsequent reacidification leading, at least in the case of kinetically inert Co(III),18 to a complex of the tautomeric iminol form of the neutral ligand.
Similar cases of metal ion binding leading to displacement of a tautomeric equilibrium have been seen in uranyl ion complexes of amidoximes, where the ligand is bound in its neutral but zwitterionic form obtained by proton transfer from O to oxime-N.19,20 Somewhat surprisingly therefore, these principles have not been explored for more complex dynamic aggregates in solution and in two dimensions on a flat surface, even though structurally distinct supramolecular assemblies can be efficiently studied in submolecular detail with scanning probe techniques.21,22
In seeking to explore enhancement of metal ion effects on a tautomeric equilibrium, we have examined the coordination behavior of the 2-pyridone-derived ligand H2L (Fig. 1), where the additional pyrimidine-N coordination sites adjacent to the amide unit were expected to favor action as a bis(bidentate) species with the amide units converted to their iminol form, thus making the bidentate unit a 2,2′-bipyridine derivative. Both theoretical and experimental studies of 2-pyridone itself have shown that its lactam and iminol forms do not differ greatly in energy, so that the position of the equilibrium between them is readily displaced.23,24 The ligand (H2L) was prepared in three steps, following a previously reported procedure.25 The molecule is insoluble in the most commonly used solvents except for dimethyl sulfoxide (DMSO). While it would appear possible that H2L could adopt a multitude of different forms (for other possible tautomeric forms for ligand H2L and coordination modes see Fig. S1 and S2 in the ESI†), previous study25 by 1H NMR spectroscopy of the molecule in DMSO solution and by STM when adsorbed (from 1,2,4-trichlorobenzene) on graphite, has shown only the form presented in Fig. 1 to be involved. Features of the ambient-temperature 1H NMR spectroscopy in DMSO (Fig. 2a), not previously commented on, are that the peaks are all rather broad and that the NH resonance appears at a rather low field (11.5 ppm) indicating its involvement in H-bonding interactions.
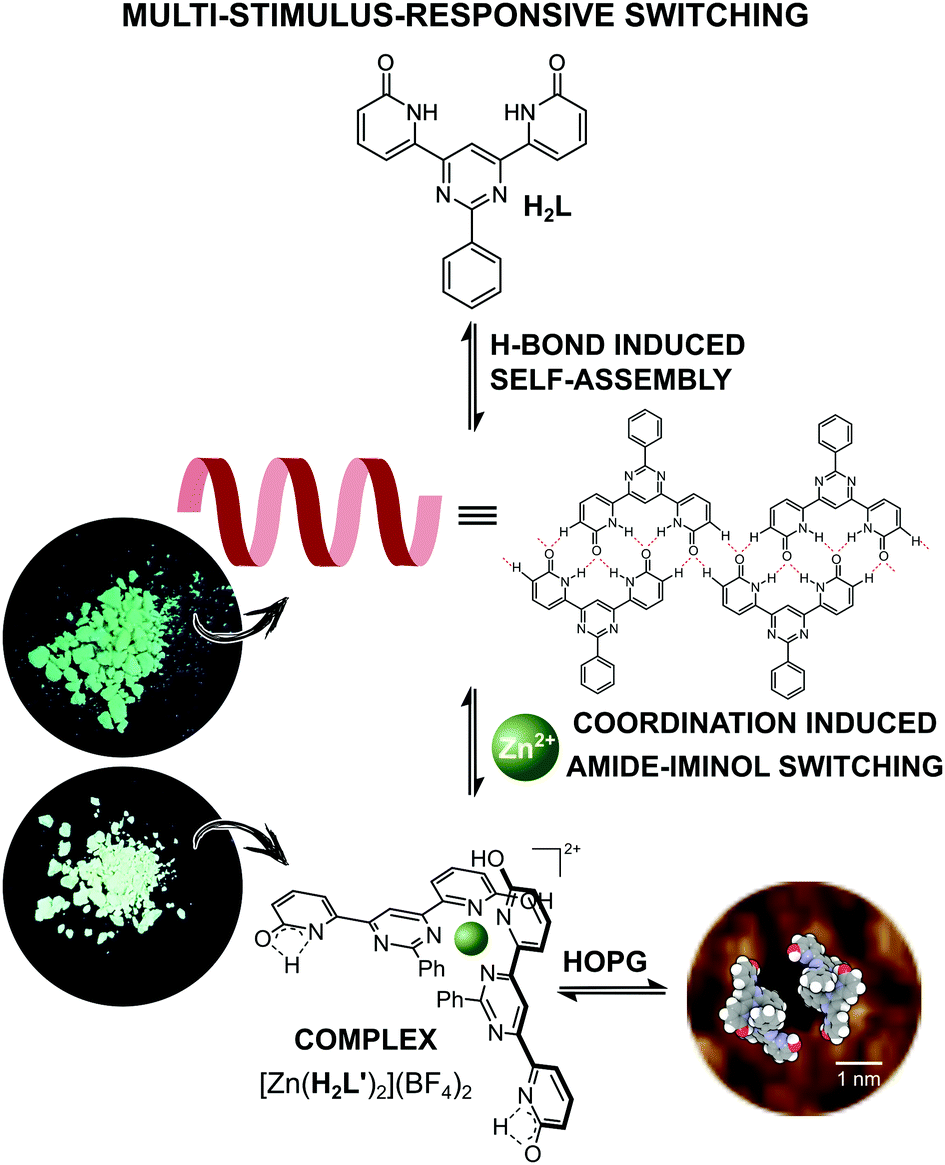 | ||
| Fig. 1 Schematic representation of the metal-ion coordination induced supramolecular transformation between organic dynamer and Zn-based ML2 type complex. | ||
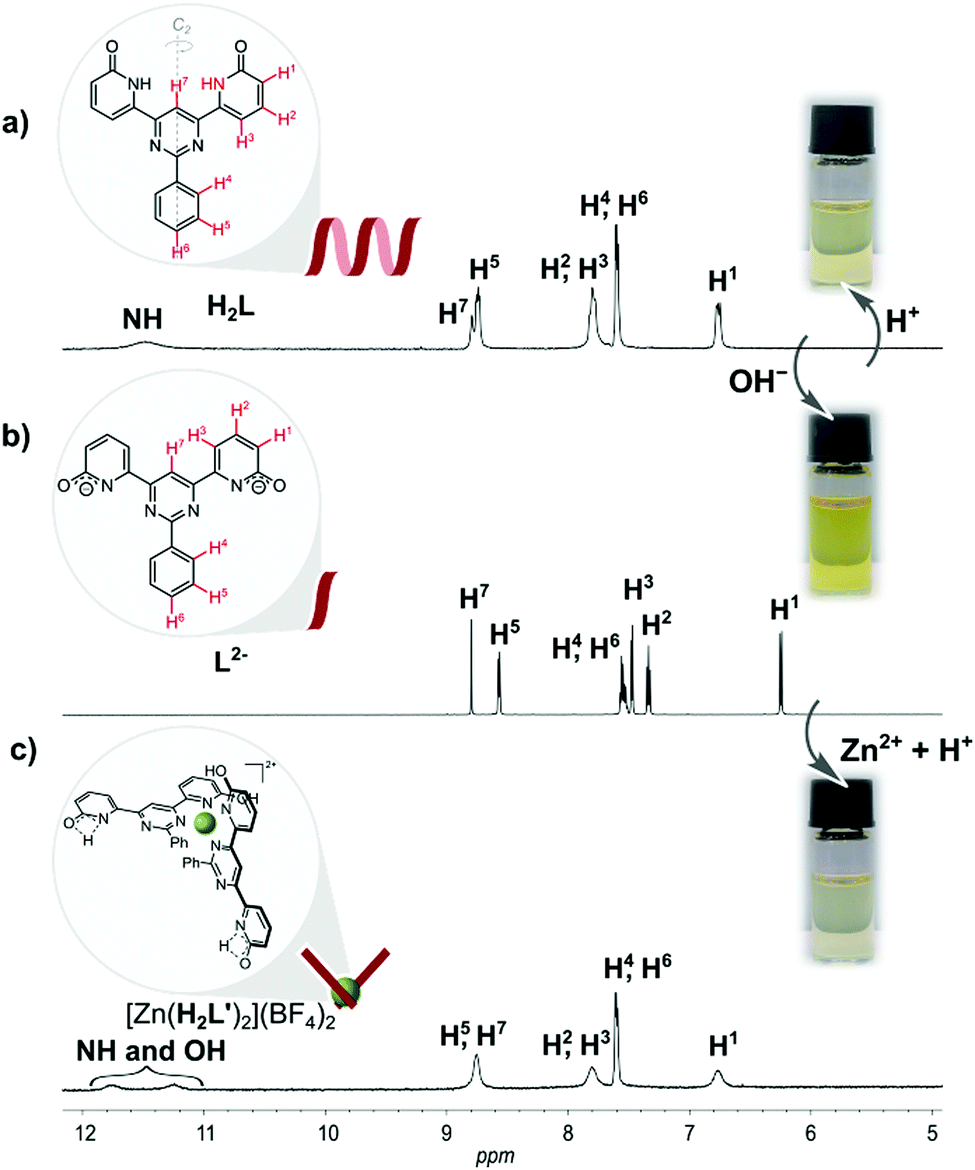 | ||
| Fig. 2 1H NMR (600 MHz) spectra at ambient temperature of: (a) fully organic dynamer H2L; (b) L2− in form of disodium salt; (c) [Zn(H2L′)2](BF4)2 in DMSO-d6. | ||
Since intramolecular H-bonding is not possible for the conformer shown in Fig. 1, this is taken as evidence that supramolecular aggregation due primarily to N–H⋯O type hydrogen bonding, to some extent analogous to that seen on graphite, must also occur in solution.
In contrast, the 1H NMR spectrum in DMSO-d6, of the O-methylated analogue of H2L, which cannot form hydrogen-bonded structures and can exists only in pseudo-iminol form, is markedly different and appears as sharp, well-resolved proton signals expected for the monomeric structures containing two methoxypyridine units (see Fig. S3 in the ESI†). Moreover, MALDI mass spectrometry provided further evidence for the presence of H2L-based supramolecular H-bonded aggregates (Fig. 3a), while only the monomer signal was observed for the reference O-methylated component (see Fig. S4 in the ESI†).
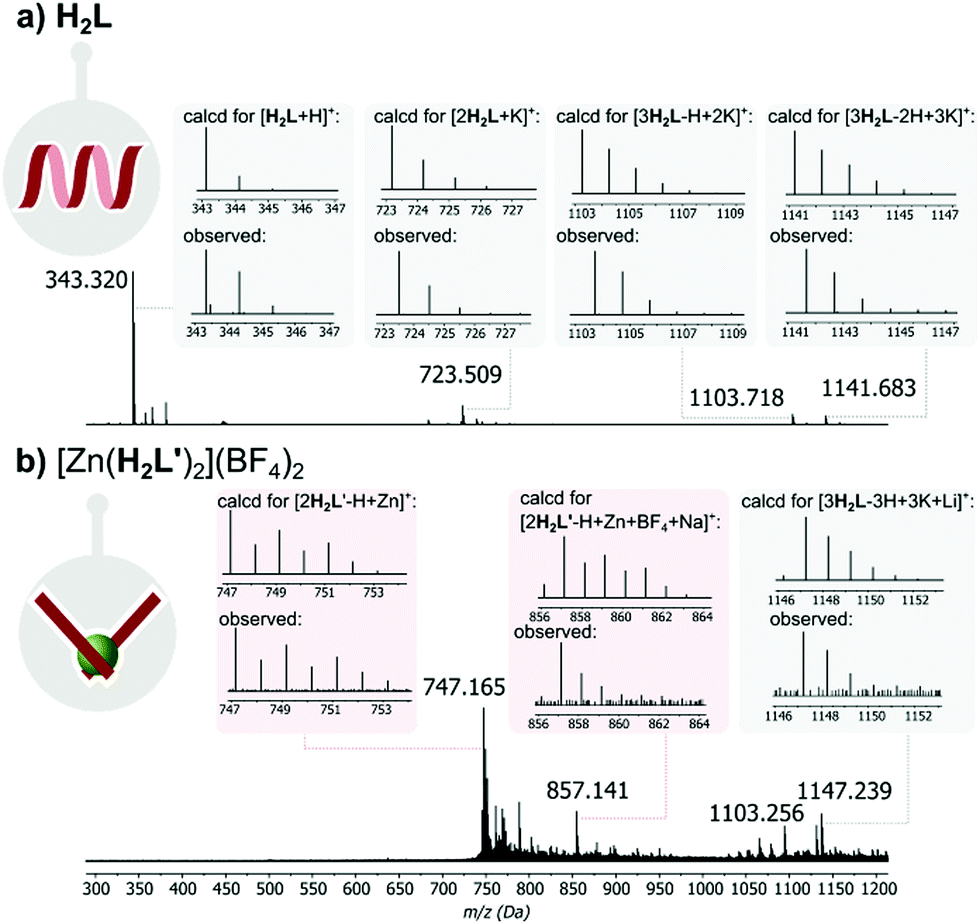 | ||
| Fig. 3 Simulated and experimental MALDI-MS signals of: (a) H2L and (b) complex [Zn(H2L′)2](BF4)2. | ||
Interestingly, when, as part of the present work, the DMSO solution was titrated with aqueous HCl to see if protonation might disrupt this aggregation, the spectrum remained essentially unchanged except for the disappearance of the NH signal (Fig. S5, in the ESI†). The lack of evidence of protonation is unsurprising given the weak basicity of both pyrimidine and amide units but the loss of the NH peak could be explained as a result of an acid-catalyzed exchange process passing through the iminol (hydroxypyridine) tautomer of H2L as an intermediate (Fig. S6 in the ESI†), thus being evidence that this form should be attainable and justifying efforts to explore the effects of metal ion coordination.
While it is known that different metal ions can favour binding of either tautomer of an amide ligand,17,26 Zn(II), in particular has been shown to favour the iminol form.
The diamagnetic nature of Zn(II) renders 1H NMR spectroscopy a particularly convenient way of following its complex formation. Addition of Zn(BF4)2 to a solution of H2L in DMSO-d6 produced no immediate change in the 1H NMR spectrum but on prolonged reaction (48 hours) at a high temperature, peaks, in particular two broad resonances at δ 11.3 and 11.8, attributable to a new species became evident (Fig. S7 in the ESI†). Assuming that the slowness of the reaction was due to the limited ability of the neutral, amide-form ligand to compete with DMSO for coordination to Zn(II), an alternative means of bringing the two species into contact was sought.
The NH acidity of primary amides is well-known, the pKa of 2-pyridone itself in DMSO being 17,27 and an anionic ligand should of course interact more strongly with a cation than it would with its neutral parent. Even more significantly, there is an extensive known coordination chemistry of amide-derived anions which shows that the form of their complexes is usually closer to what would be expected for an iminolate than an amidate.27,28 Reaction of H2L with 2 molar equivalents of NaOH in DMSO-d6 provided a deep yellow solution giving a 1H NMR spectrum with very well resolved peaks consistent with the formation of the presumably largely dissociated disodium complex (Fig. 2b). Addition of 0.5 molar equivalent of Zn(BF4)2 to this solution resulted in drastic broadening of all peaks, indicating rather strong interaction, and careful addition of 2 molar equivalents of HCl then led to the emergence of two broad resonances as seen in the first experiment (Fig. 2c). Assignment of one of these resonances as due to the NH of the uncoordinated amide unit and the other as OH of the bound iminol unit is completely consistent with the structure shown in Fig. 2, although we have not been able to establish which is which.
Repetition of the second experiment on a preparative scale and addition of methanol and diethyl ether to the final solution led to the isolation of [Zn(H2L′)2](BF4)2 in the form of yellow powder.
Since a 2,2′-bipyridine-like ligand should be weakly basic and Zn(II) complexes are generally labile, we reasoned that release of the ligand from [Zn(H2L′)2](BF4)2 should occur on further addition of acid. Consistent with this expectation, an 1H NMR titration of [Zn(H2L′)2](BF4)2 with acid (0.06 M HCl, in H2O/DMSO-d6) revealed, after addition of 1 equivalent, that the spectrum of dynamer H2L was regenerated (see ESI,† Fig. S13). Since chloride ion is poorly solvated in DMSO, formation of chloro complexes may have assisted this reaction also.
Since in DMSO solution, [Zn(H2L′)2](BF4)2 has a rather dynamic nature, we decided to examine fluorescence of both H2L and [Zn(H2L′)2](BF4)2 in solid state. These measurements show that described system is photo-responsive, which opens several avenues towards their application in various technological fields. As shown in Fig. 4, after excitation at 468 nm, the emission spectrum of H2L shows a broad band with its maximum at 555 nm, while in the emission spectrum of [Zn(H2L′)2](BF4)2 there is a notable shift in the band maximum = 578 nm (for absorbance spectra see ESI,† Fig. S12).
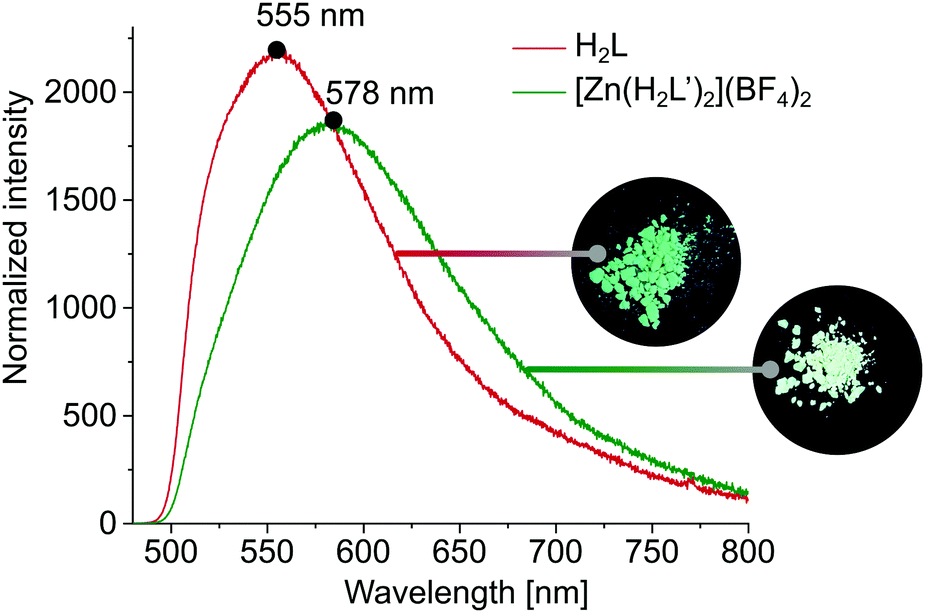 | ||
| Fig. 4 Emission spectra of H2L and [Zn(H2L′)2](BF4)2 in the solid state, together with photographs made under UV light (254 nm). | ||
The difference in fluorescence under excitation at 254 nm is also visible to the naked eye as a slight difference in colour (Fig. 4). The observed significant Stokes shifts of 87 nm (H2L) and 110 nm ([Zn(H2L′)2](BF4)2) predispose both materials to application as dynamic fluorescence dyes.
Due to the limited solubility of both H2L and [Zn(H2L′)2](BF4)2, we were not able to obtain a crystal suitable for an X-ray diffraction measurement, even after multiple trials. However, computational methods are a powerful tool for compound characterization. Therefore the B3LYP/6-31+G(d,p) molecular geometry optimization of H2L together with H2L′ and [Zn(H2L′)2](BF4)2 was performed. Additionally, in order to capture the effects of dispersion interactions, the empirical Grimme GD3 dispersion parameters were used, supplemented by Becke–Johnson (BJ) damping. The accuracy of the method was tested by first optimizing H2L, until results from the literature25 were reproduced (Table S3 in the ESI†). Zn(H2L′)22+ was optimized stepwise using the same method. First H2L′ was optimized and based on these results, the structure of the cation Zn(H2L′)22+ was obtained. Ligand molecules coordinate the Zn2+ cation in their pyrimidine–pyridine pocket, the Zn center having the symmetry of a distorted tetrahedron with angles N(pyr)–Zn–N(pyr) 153°, N(py)–Zn–N(py) 105° and N(pyr)–Zn–N(py) 81.5° and Zn–N(py) and Zn–N(pyr) bond lengths of 2.08 Å and 2.01 Å, respectively (see ESI,† Section II for details).
Intrinsic properties of a molecule can be predicted from the energy levels of the HOMO and LUMO orbitals. The calculations showed a symmetrical pattern for the H2L MO, while the introduction of a metal ion caused a shift of the electron density in the HOMO towards the metal centre and a displacement of the LUMO to the outer region of the ligand (Table S2 in the ESI†). It has been previously shown, that H2L forms both 2D arrays and helical polymer chains at a solid–liquid interface.25Fig. 5a presents the STM image of a helical dynamer H2L drop-casted onto HOPG, heated up and imaged at room temperature. The measured period of the model structure is ∼25 nm – in agreement with the experimental value. In addition to helical structures, we also observed 2D molecular arrays shown in Fig. 5d, which are built from the same molecule H2L but with a different H-bonding motif (Fig. 5e). Following similar drop-casting of a solution of complex [Zn(H2L′)2](BF4)2, the STM image revealed a well-ordered molecular array, visible as a 2D array of dark spots surrounded by bright rings (Fig. 5f). The measured in-plane lattice parameters of this structure are ∼3.6 nm. STM topography fits very well with the calculated LUMO orbitals of Zn(H2L′)22+ (Fig. 5i). The two pyridone moieties not involved in coordination should still be able to form H-bonds, regardless of their tautomeric form. This we consider to be the reason why two molecules of the complex assemble into toroidal dimer units (Fig. 5), which then form more extended arrays.
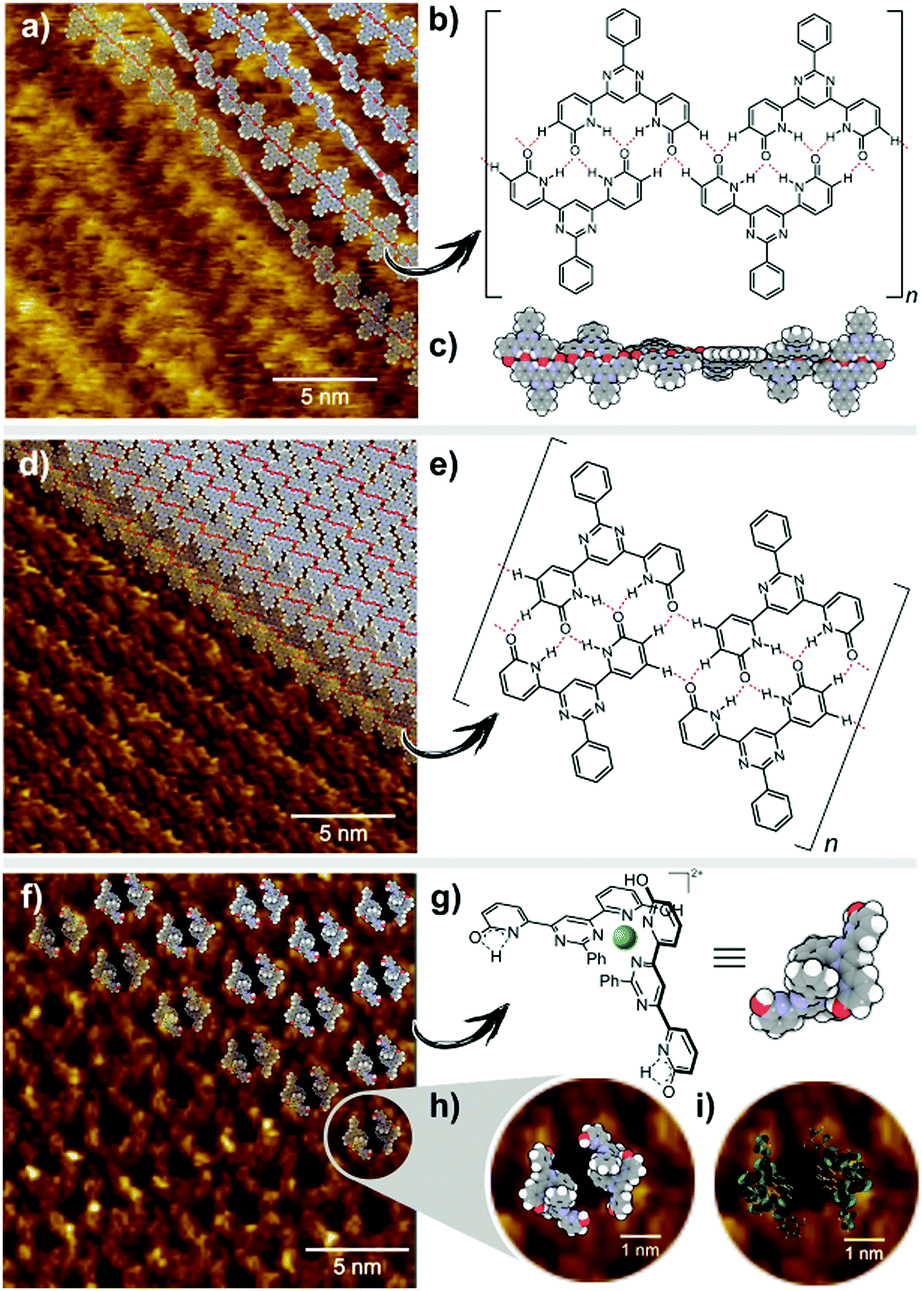 | ||
| Fig. 5 (a) STM image of a helical dynamer with model arranged on top of it. Image size: 20 × 20 nm2; Vsample = +0.6 V; IT = 0.1 nA; (b) chemical structure of H2L with presented H-bond motif; (c) model of helical dynamer H2L; (d) STM image of a 2D array of dynamer with model an arranged on top of it. Image size: 20 × 20 nm2; Vsample = +0.6 V; IT = 0.1 nA; (e) chemical structure of H2L with presented H-bond motif; (f) STM image of 2D an array formed by complex [Zn(H2L′)2](BF4)2 with a model arranged on top of it. Image size: 20 × 20 nm2; Vsample = +0.6 V; IT = 0.1 nA; (g) chemical structure of the cation Zn(H2L′)22+ and its model; (h) zoomed area of STM image shown in (f). Image size: 5 × 5 nm2; Vsample = +0.6 V; IT = 0.1 nA; (i) zoomed area of STM image with model of LUMO orbitals on top of it. Image size: 5 × 5 nm2; Vsample = +0.6 V; IT = 0.1 nA. | ||
The present work provides a previously unknown example of the desymmetrisation of a ligand resulting from the metal-ion-induced tautomerisation of one binding site within an initially symmetrical organic species. This mode of coordination produces two different H-bonding sites within the metallosupramolecular species, which are both used to provide different forms of association in the solid state and in solution. Retention of the amide form of the unbound coordination site is significant factor in leading to the H-bonding which appears to control the adsorbed form of the complex on the surface and which reveals its multifunctionality. Reversible interconversion between organic dynamer and metallosupramolecular toroidal species is readily achieved by simple control of metal ion concentration, base and acid additions and results in the generation of dynamic materials of distinct fluorescent properties. Thus, we have demonstrated that simple 2-pyridone derivatives are well suited for the generation of distinct forms of fluorescent adaptive materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Science Centre of Poland grants: SONATA BIS 2018/30/E/ST5/00032 (A.R.S.), PRELUDIUM UMO-2016/21/N/ST5/00849 (W. D.) and OPUS 2014/15/B/ST3/02927 (Z. M. and M. L.). The work of P. C. was also co-financed by European Social Fund under the Operational Program Knowledge Education Development and Poznan Supercomputing and Networking Centre computing grant no. 401. The work of M. C. was co-financed by the National Centre for Research and Development grant POWR.03.02.00-00-I032/16. We thank Prof. Jean-Marie Lehn, Dr Monika Wałęsa-Chorab and Jakub Lewandowski for their contribution at the initial stage of this project.Notes and references
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 16024 CrossRef CAS
.
- S. Chen, R. Costil, F. K. Leung and B. L. Feringa, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202007693
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, e1905966 CrossRef PubMed
- A. Brzechwa-Chodzyńska, M. Zieliński, M. Gilski, J. M. Harrowfield and A. R. Stefankiewicz, Inorg. Chem., 2020, 59, 8552–8561 CrossRef PubMed
- A. Bocian, W. Drożdż, M. Szymańska, J. Lewandowski, M. Fik-Jaskółka, A. Gorczyński, V. Patroniak and A. R. Stefankiewicz, Nanoscale, 2020, 12, 4743–4750 RSC
- G. Markiewicz, M. M. J. Smulders and A. R. Stefankiewicz, Adv. Sci., 2019, 6, 1900577 CrossRef PubMed
- W. Drożdż, A. Walczak, Y. Bessin, V. Gervais, X.-Y. Cao, J.-M. Lehn, S. Ulrich and A. R. Stefankiewicz, Chem. – Eur. J., 2018, 24, 10802–10811 CrossRef PubMed
- J. M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC
- S. Sobczak, W. Drożdż, G. I. Lampronti, A. M. Belenguer, A. Katrusiak and A. R. Stefankiewicz, Chem. – Eur. J., 2018, 24, 8769–8773 CrossRef CAS
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS
- R. A. A. Abdine, G. Kurpik, A. Walczak, S. A. A. Aeash, A. R. Stefankiewicz, F. Monnier and M. Taillefer, J. Catal., 2019, 376, 119–122 CrossRef CAS
- A. Walczak and A. R. Stefankiewicz, Inorg. Chem., 2018, 57, 471–477 CrossRef CAS PubMed
- A. Walczak, H. Stachowiak, G. Kurpik, J. Kaźmierczak, G. Hreczycho and A. R. Stefankiewicz, J. Catal., 2019, 373, 139–146 CrossRef CAS
- R. C. R. H. E. Toma, Croat. Chem. Acta, 2001, 499–528 Search PubMed
-
W. G. Jackson and A. M. Sargeson, Essay 11 in Rearrangements in Ground and Excited States, in Organic Chemistry: A Series of Monographs, ed. P. de Mayo, Academic Press, New York, 1980, vol. 42-3, pp. 273–378 Search PubMed
- D. B. Brown, M. B. Robin and R. D. Burbank, J. Am. Chem. Soc., 1968, 90, 5621–5622 CrossRef CAS
- D. P. Fairlie, T. C. Woon, W. A. Wickramasinghe and A. C. Willis, Inorg. Chem., 1994, 33, 6425–6428 CrossRef CAS
- D. P. Fairlie, P. M. Angus, M. D. Fenn and W. G. Jackson, Inorg. Chem., 1991, 30, 1564–1569 CrossRef CAS
- D. A. Decato and O. B. Berryman, Org. Chem. Front., 2019, 6, 1038–1043 RSC
- E. G. Witte, K. S. Schwochau, G. Henkel and B. Krebs, Inorg. Chim. Acta, 1984, 94, 323–331 CrossRef CAS
- D. B. Amabilino and P. A. Gale, Chem. Soc. Rev., 2017, 46, 2376–2377 RSC
- T. Kudernac, S. Lei, J. A. Elemans and S. De Feyter, Chem. Soc. Rev., 2009, 38, 402–421 RSC
- M. W. Wong, K. B. Wiberg and M. J. Frisch, J. Am. Chem. Soc., 1992, 114, 1645–1652 CrossRef CAS
- J. Seliger and V. Zagar, J. Phys. Chem. A, 2013, 117, 1651–1658 CrossRef CAS PubMed
- A. Ciesielski, A. R. Stefankiewicz, F. Hanke, M. Persson, J.-M. Lehn and P. Samorì, Small, 2011, 7, 342–350 CrossRef CAS PubMed
- Z. Xu, K. H. Baek, H. N. Kim, J. Cui, X. Qian, D. R. Spring, I. Shin and J. Yoon, J. Am. Chem. Soc., 2010, 132, 601–610 CrossRef CAS PubMed
- C. G. Swain and J. F. Brown, J. Am. Chem. Soc., 1952, 74, 2538–2543 CrossRef CAS
- P. J. Stang and B. Olenyuk, Acc. Chem. Res., 1997, 30, 502–518 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc05598k |
| ‡ Dedicated to Prof. Bogdan Marciniec on the occasion of his 80th birthday. |
| This journal is © The Royal Society of Chemistry 2021 |