
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTyrian purple: an ancient natural dye for cross-conjugated n-type charge transport†
Kealan J.
Fallon
 a,
Nilushi
Wijeyasinghe
b,
Anastasia
Leventis
a,
Jose M.
Marin-Beloqui
c,
Daniel T. W.
Toolan
d,
Mohammed
Al-Hashimi
e,
Tracey M.
Clarke
c,
Thomas D.
Anthopoulos
b and
Hugo
Bronstein
*a
a,
Nilushi
Wijeyasinghe
b,
Anastasia
Leventis
a,
Jose M.
Marin-Beloqui
c,
Daniel T. W.
Toolan
d,
Mohammed
Al-Hashimi
e,
Tracey M.
Clarke
c,
Thomas D.
Anthopoulos
b and
Hugo
Bronstein
*a
aDepartment of Chemistry & Physics, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: hab60@cam.ac.uk
bKing Abdullah University of Science and Technology, KAUST Solar Center, Thuwal 23955-6900, Saudi Arabia
cDepartment of Chemistry, University College London (UCL), London, WC1H 0AJ, UK
dDepartment of Chemistry, University of Sheffield, Dainton Building, Brook Hill, Sheffield S3 7HF, UK
eDepartment of Chemistry, Texas A&M University at Qatar, P. O. Box 23874, Doha, Qatar
First published on 3rd March 2021
Abstract
Herein, we present two novel organic semiconducting polymers synthesised from an ancient dye. By employing cross-conjugation within the polymer backbone as a synthetic strategy, we are able to engineer optical gaps such that the novel materials absorb over the entire visible spectrum. The cross-conjugated polymers exhibited relatively high n-type charge transport performance in organic field-effect transistors, a rare characteristic for this type of polymer. Quantum chemical calculations provide insight into this behaviour, suggesting that, whilst conjugation along the HOMO is indeed inhibited via molecular design, these materials possess highly delocalized LUMOs, facilitating high n-type charge transport.
Introduction
Organic semiconducting polymers have received large attention in the past two decades due to their low cost, mechanical flexibility, and ease of processing. Their effectiveness as donor materials in the bulk heterojunction of organic photovoltaics (OPVs) and in transporting charge carriers in organic field-effect transistors (OFETs) is reliant on the energetic tailoring of frontier molecular orbital (MO) energy levels and thus the band gap between them. The most widely employed strategy for band gap engineering is in alternating donor–acceptor (D–A) copolymers, where the energy of the highest occupied molecular orbital (HOMO) is typically dictated by the donor unit whilst the energy of the lowest unoccupied molecular orbital (LUMO) is dictated by the acceptor.1–3A design approach which has seldom been investigated is the interruption of conjugation along a polymer backbone. Due to their shorter effective conjugation lengths, cross-conjugated polymers typically have wider band gaps and increased ionisation potentials, making them more stable to oxidative doping when operated in air.4,5 Heeney et al. demonstrated the first successful use of a thieno[2,3-b]thiophene cross-conjugated polymer as a p-type OFET material, exhibiting hole transport up to 0.05 cm2 V−1 s−1.6 Recently, Zhang et al. reported high hole mobilities in diketopyrrolopyrrole (DPP) based cross-conjugated polymers.7 Other common building blocks have been exploited in cross-conjugated polymers including isoindigo,8,9 fluorene,10–13 carbazole,14 and some more atypical systems,15–20 however successful application in devices is rarely reported.
Our recent work on conjugated organic polymers exploiting the relatively unexplored building block indolonaphthyridine (IND) was inspired by the chromophore of natural indigo dye. These materials with fully conjugated non-planar backbones exhibited narrow optical band gaps (∼1.2 eV), gave high power conversion efficiencies in OPVs (up to 4.1%), and gave high performance charge transport in OFETs for holes and electrons up to 1.0 and 3.1 cm2 V−1 s−1 respectively.21
INDs are formed through a dual condensation-annulation reaction of indigo and an aromatic acetyl chloride. The benzene rings of the indigo dye are not involved in the conjugation of the IND unit and offer the potential to introduce cross-conjugation. Tyrian Purple (6,6′-dibromoindigo, 5) is a naturally occurring dye of significant legacy. The precursors of the dye were historically extracted from the sea snail Murex brandaris, which underwent oxidative coupling in air to form the brilliantly purple solid. A ghastly 8500 snails were required to harvest 1 g of the dye, leading to the high wealth associated with the colour purple throughout history until cheaper synthetic alternatives were discovered in the late 1800s.22 Fatefully, it cannot be simply prepared by the bromination of indigo, which results in bromination of all positions except the 6-position.23 Whilst several preparations have been reported, none are particularly high yielding.24,25 Herein we report a facile, high-yielding four-step synthesis (Scheme 1), adapted from Imming et al.26
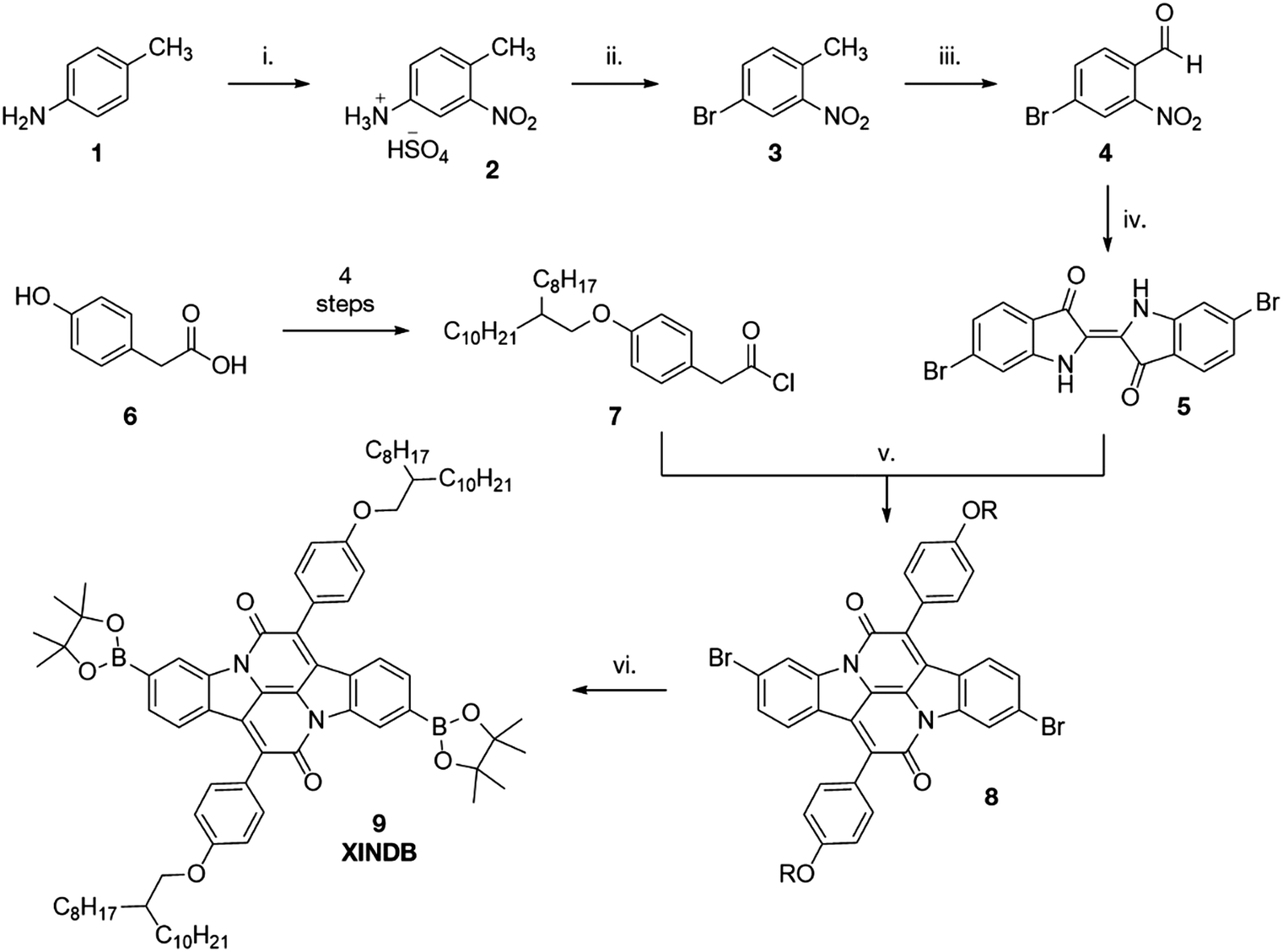 | ||
| Scheme 1 Synthesis of monomer (i) HNO3, H2SO4, 83% (ii) NaNO2, HBr, CuBr, 57% (iii) DMF·DMA then NaIO4, 67% (iv) NaOH, (CH3)2CO, 58% (v) xylenes, Δ, 6%, R = 2-octyldodecanyl (vi) PdCl2(dppf), B2pin2, dioxane, 87%. | ||
Results and discussion
Following nitration of 4-methylphenylammonium hydrosulfate, a Sandmeyer reaction yields the desired brominated nitrotoluene 3. Oxidation of the methyl group using DMF·DMA and NaIO4 provides the desired aldehyde 4 in high yield. Tyrian purple 5 was then synthesised from this aldehyde through a straightforward Baeyer–Drewson indigo synthesis in good yield. Condensation of 5 with acetyl chloride 7 gave the brominated indolonaphthyridine benzene 8, which underwent a high yielding Miyaura borylation to give the diborylated cross-conjugated monomer XINDB (9).This monomer subsequently underwent a high yielding Suzuki co-polymerisation of 9 with dibrominated DPPT27 and INDT monomers to give the cross-conjugated polymers XINDB–DPPT (P1) and XINDB–INDT (P2) in high yield (Fig. 1), which both exhibited excellent solubility in typical organic solvents.
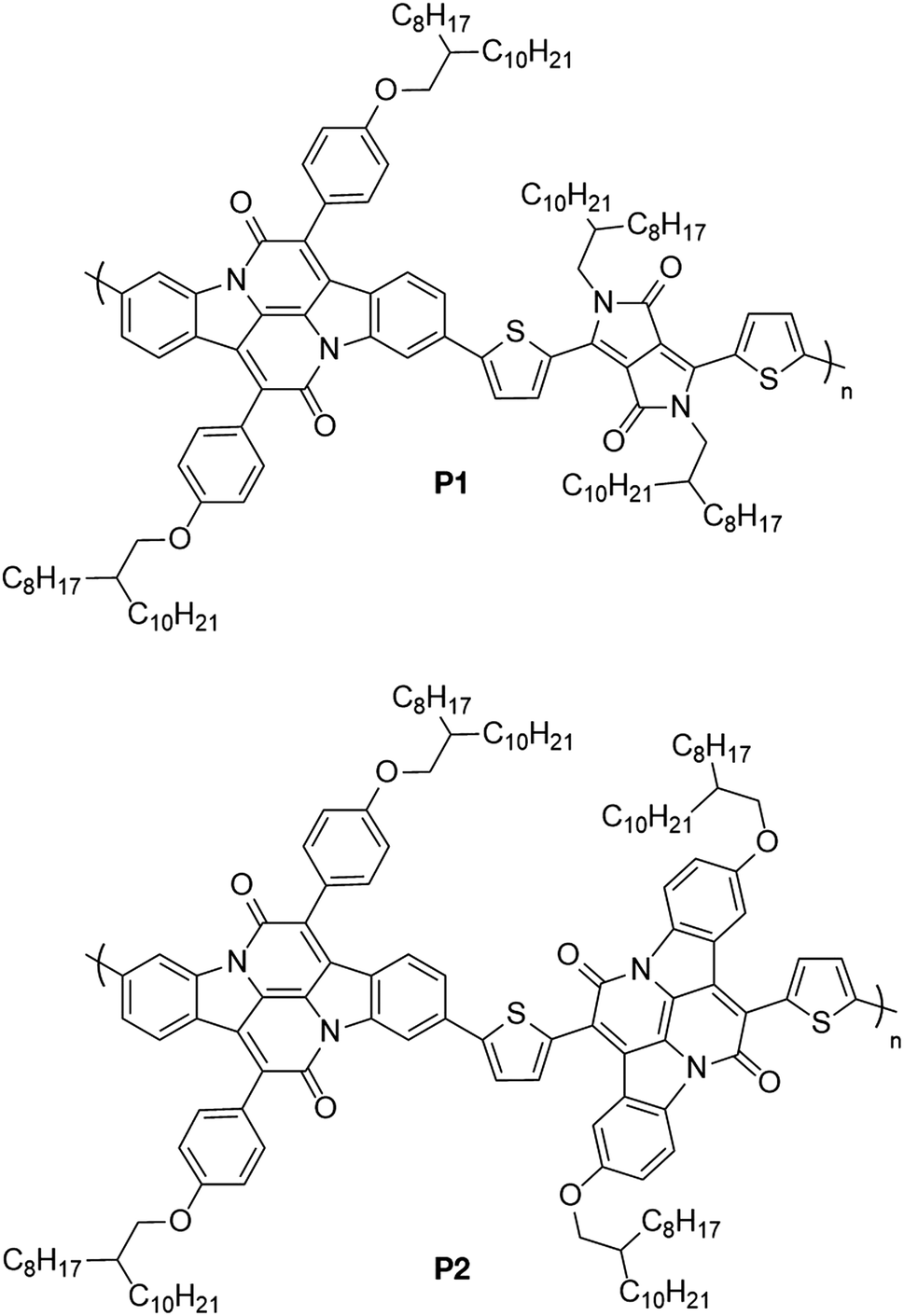 | ||
| Fig. 1 Structures of the two polymers in this study. | ||
The polymers were purified by Soxhlet extraction first with acetone then hexane, to remove impurities and low molecular weight oligomers, then finally chloroform. The physical properties of the polymers are shown in Table 1. Molecular weights were determined by size exclusion chromatography (SEC) using polystyrene (PS) standards eluting with chlorobenzene at 80 °C. The DPP-containing polymer P1 exhibited very high molecular weight, whilst the INDT-containing polymer P2 exhibited relatively lower molecular weight averages.
| Polymer | M n (kDa) | M w (kDa) | Đ |
λ
solnmax![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (nm) b (nm) |
λ
filmmaxc (nm) |
HOMOd (eV) | LUMOe (eV) | E g (eV)b |
E
calcgf (eV) |
|---|---|---|---|---|---|---|---|---|---|
| a Determined by SEC(PS) using PhCl as eluent. b PhCl solution. c Spin-coated from PhCl 5 mg mL−1. d Determined by PESA. e HOMO + optical band gap. f Determined by TD-DFT using B3LYP/6-31g*. | |||||||||
| P1 | 84 | 551 | 6.6 | 720 | 727 | −5.29 | −3.75 | 1.54 | 1.61 |
| P2 | 11 | 53 | 5.0 | 743 | 750 | −5.21 | −3.78 | 1.43 | 1.70 |
Fig. 2 shows the UV-vis absorption spectrum of the brominated monomer and the two polymers. Both cross-conjugated polymers exhibited broad absorption in the visible and near-IR regions. The optical band gaps (Eg) of the polymers were estimated from the absorption onsets of 805 nm for P1 and 865 nm for P2, indicating Eg of 1.54 and 1.43 eV respectively. These band gaps are notably wider than typical DPP and INDT conjugated polymers, ratifying the strategy of cross-conjugated design to widen the optical gap. Both absorptions exhibited a bathochromic shift in thin-film, attributed to solid-state packing effects. The ionisation potential (EHOMO) of the two polymers were measured using photoelectron spectroscopy in air (PESA) and the electron affinities (ELUMO) were estimated by the addition of the respective optical band gap to the measured ionisation potential. In our previous work, all conjugated IND-based polymers exhibited isoenergetic electron affinities of approximately −3.75 eV, irrespective of co-monomer (benzene, thiophene, selenophone, benzothiadiazole). Interestingly, ELUMO of P1 and P2 were −3.75 and −3.78 eV respectively, fitting this trend, demonstrating the remarkable ability of the electron deficient indolonaphthyridine building block at controlling ELUMO. The widening of the optical band gap seen in these cross-conjugated polymers results from a deeper EHOMO of −5.29 and −5.21 eV for P1 and P2 respectively.
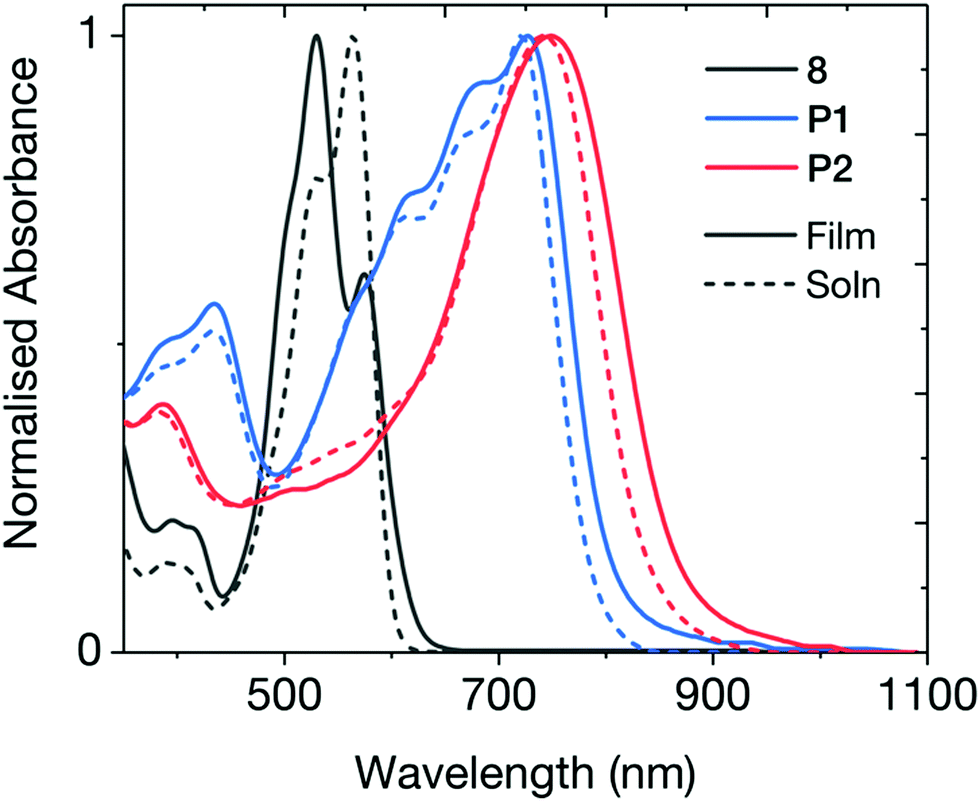 | ||
| Fig. 2 Normalized UV-vis absorption spectra of the two cross-conjugated polymers and the dibrominated indolonaphthyridine. Solution spectra were recorded in chlorobenzene, and thin-films were spun from 5 mg mL−1 solutions in chlorobenzene. | ||
Theoretical analysis of the electronic structure of the two polymers was carried out on model trimers using the B3LYP hybrid functional and the 6-31G* basis set.28 The calculated frontier molecular orbital distributions are shown in Fig. 3. Despite the cross-conjugation, the calculations indicate the LUMO of both polymers are delocalised along the polymer backbones. In contrast, the HOMO of both polymers is localised strongly on the DPPT and INDT units. This is in agreement with previous theoretical studies on the electronic structure of indigo-containing polymers.29
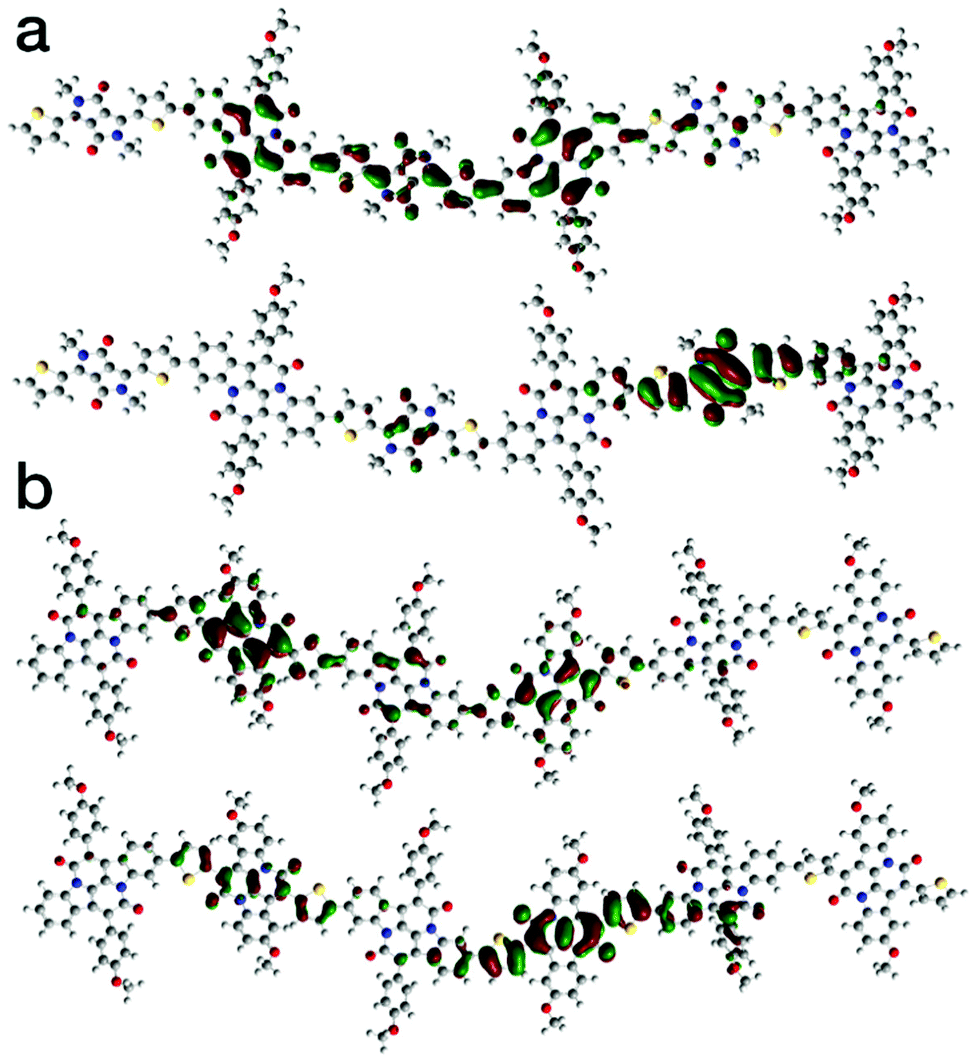 | ||
| Fig. 3 Theoretical distributions of the LUMO (top) and HOMO (bottom) of (a) P1 and (b) P2 calculated at B3LYP/6-31G*. | ||
To investigate the charge transport properties of the two cross-conjugated polymers, unipolar top gate bottom contact (TG-BC) OFET devices were fabricated on glass substrates using thermally evaporated source/drain (SD) electrodes. For n-type operation, Al SD electrodes were treated with ethoxylated polyethylenimine (PEIE) to facilitate electron injection by reducing the work function of the electrodes to better match the electron affinities of the polymers. For p-type operation, the treatment of Au electrodes with UV/O3 removes surface contaminants and is expected to improve the hole injection into polymers with HOMO energies of −5.29 eV (P1) and −5.21 eV (P2). Kelvin probe contact potential difference measurements verified that the work function of the treated Au surface (5.3 eV) was an excellent match to the HOMO energies of the two polymers. The semiconductor layer was spin-cast onto the SD electrodes from 10 mg mL−1 solutions in chlorobenzene, followed by annealing at 190 °C for 15 min. Finally, an amorphous fluoropolymer dielectric (CYTOP) was spin-cast onto the semiconductor and the device further annealed for 1 h at 100 °C. Each device was completed with a thermally evaporated Al gate electrode.
Despite the outlined efforts, the cross-conjugated polymers only exhibited n-type charge transport – no hole transport was observed in the devices designed for p-type operation. These results correspond well with the theoretical molecular orbital distributions and we attribute the lack of hole current primarily to the strongly localized HOMOs on the DPPT and INDT units, which is expected to hinder hole transport, although we note that further work is required to confirm this. In contrast, the clear delocalization of the LUMOs along the polymer backbone correlates well with the good electron transport in the n-type optimized OFETs.
The transfer and output characteristics of the n-type devices are shown in Fig. 4 and the performance data summarised in Table 2. The maximum drain currents measured in OFETs based on P1 (≈10 μA) were two orders of magnitude higher than the maximum drain currents in OFETs based on P2 when similar voltages were applied, indicating clearly that P1 exhibits superior electron transport in comparison to P2. Interestingly, considering the almost isoenergetic LUMO energies of the two polymers, this result is supported by the molecular orbital distributions in Fig. 3, which indicate that the LUMO of P1 exhibits a greater degree of delocalization along the polymer backbone relative to P2. The on–off ratios in the linear and saturated regimes for P1 (103 to 104) were up to two orders of magnitude higher than P2 (102), again demonstrating the better performance of the DPP-based polymer.
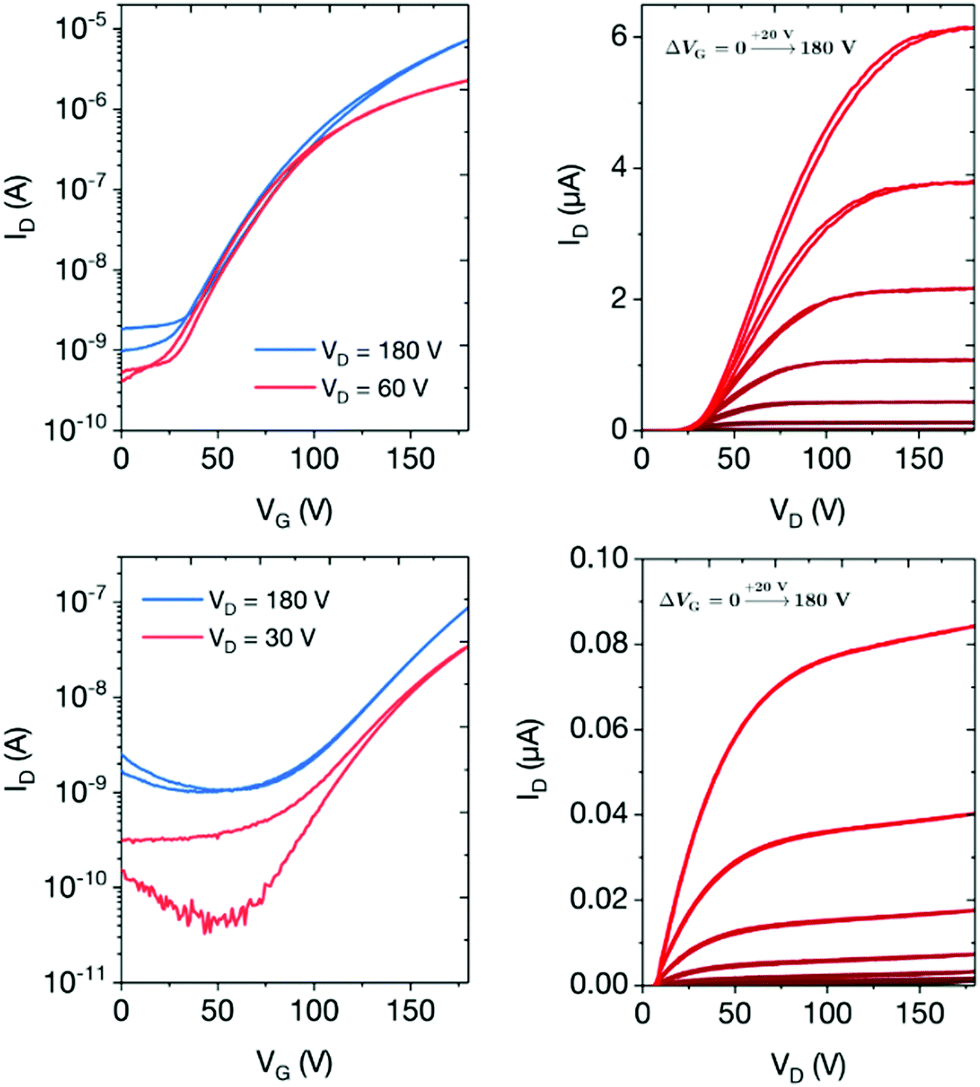 | ||
| Fig. 4 Transfer curves (left) and output characteristics (right) of n-type TG-BC OFET devices based on P1 (top) and P2 (bottom) semiconductor layers annealed at 190 °C, with channel dimensions of L = 40 μm and W = 1000 μm. | ||
| Polymer | I on/Ioff (lin) | I on/Ioff (sat) | V linon (V) | V satth (V) | μ lin (cm2 V−1 s−1) | μ sat (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|
| a Field-effect electron mobility extracted from smoothed data, indicating the best of three devices measured and the mean value given in brackets. | ||||||
| P1 | 103 to 104 | 103 to 104 | +23 | +91 | 0.012 (0.010) | 0.10 (0.09) |
| P2 | 102 | 102 | +41 | +123 | 0.0007 (0.0007) | 0.0019 (0.0017) |
Field-effect electron mobilities for the two polymers were calculated from the transfer characteristics using the standard gradual channel approximation. Due to poor device performance at low VD, electron mobilities calculated in the linear regime were 0.012 and 0.0007 cm2 V−1 s−1 for P1 and P2, respectively. In the saturation regime, the electron mobilities were 0.10 and 0.002 cm2 V−1 s−1 for P1 and P2 respectively, representing respectable values for these types of materials. Notably, the mobility of P1 is 17× higher than the most comparable previously reported polymer.30 Whilst conjugated indolonaphthyridine polymers have exhibited ambipolar charge transport, our results showcase the ability to switch to unipolar characteristics by exploiting cross-conjugation and MO localization as a synthetic design strategy.
To examine the solid state structures of the two polymers, we performed grazing incidence X-ray scattering (Fig. 5). For both the as-cast polymers, only scattering from the glass substrate is observed (q = 0.45 Å−1). Thermal annealing results in the development of perpendicular peaks parallel to the substrate at q = 0.20 and 0.217 Å−1, corresponding to domain spacings of 31.4 and 29.0 Å for P2 and P1, respectively. The observed perpendicular alignment of the scattering features indicates preferential molecular ordering perpendicular to the substrate. Additionally AFM was carried out on the two polymers (see ESI†) which showed very similar surface morphologies. The similarities in solid state packing between both P2 and P1 suggest that the difference in charge-carrier motilities are due to our proposed electronic effects as opposed to differences in polymer microstructure.
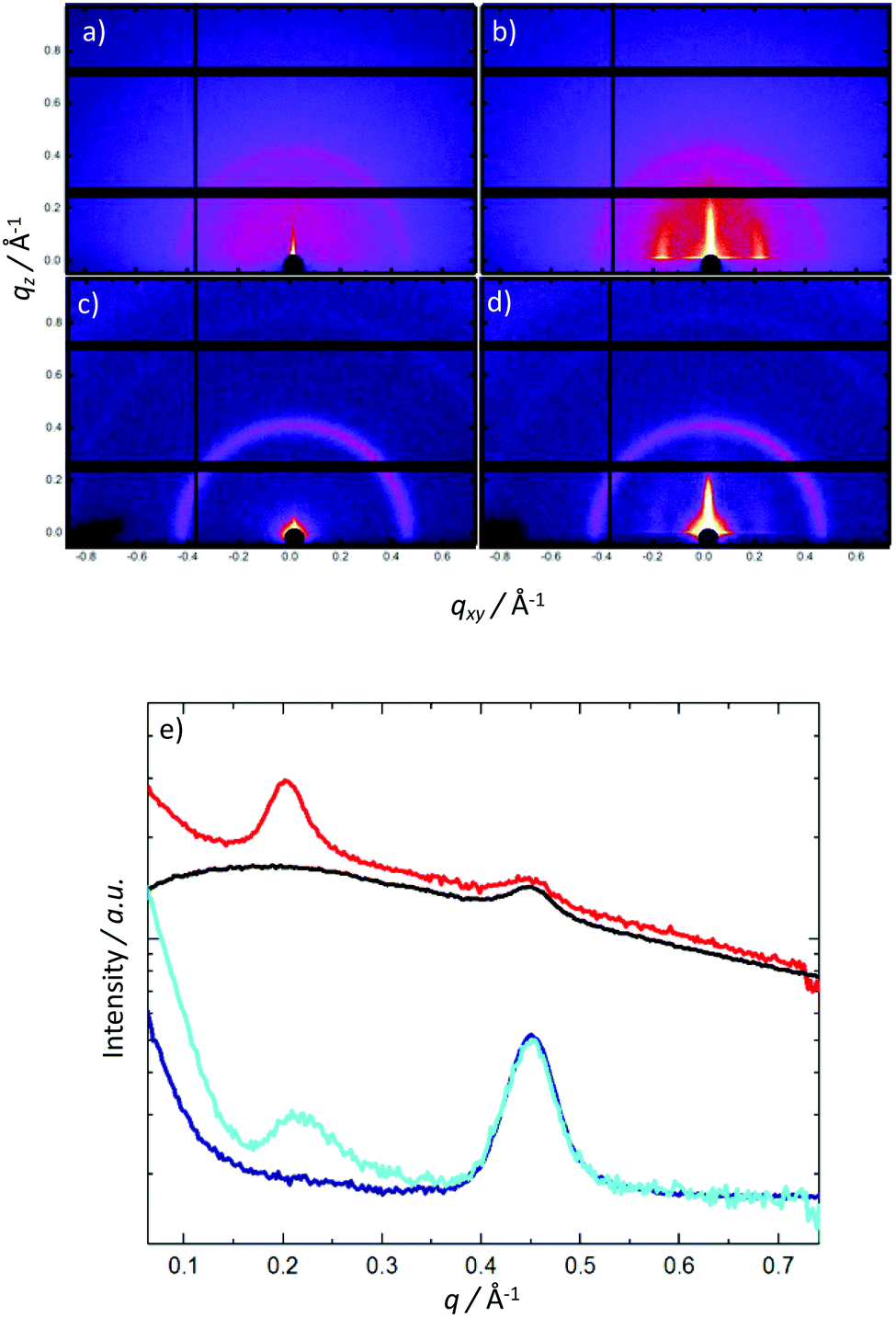 | ||
| Fig. 5 2D X-ray grazing incidence scattering data, for P2 [as-cast (a) & annealed (b)] and P1 [as-cast(c) & annealed (d)], and horizontally integrated X-ray scattering data (e) to highlight the development of in plane scattering features for P2 [as-cast(black), annealed (red)] and P1 [as-cast(blue) & annealed (cyan)]. | ||
Conclusions
In conclusion, we report the synthesis and characterization of two cross-conjugated polymers derived from Tyrian Purple dye. Using this relatively unexplored design strategy, the polymers exhibited notably wider optical gaps compared to previously reported conjugated polymers. Quantum chemical calculations revealed that, despite their cross-conjugated electronic nature, the polymers remarkably exhibited delocalization of their LUMO wavefunction. This characteristic resulted in electron mobilities in field-effect transistors up to 0.1 cm2 V−1 s−1. To the best of our knowledge this is the highest efficiency n-type OFET transport to date in indigo-based cross-conjugated organic polymers. These findings confirm the effectiveness of cross-conjugation as a tool for widening the optical band gap of organic semiconductors and demonstrate the ability of indolonaphthyridine to furnish high performance n-type materials with predictable electron affinities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support for this work from the Qatar National Research Fund project number: NPRP 12S-0304-190227, EU project 679789 – 455 CONTREX and the EPSRC grant EP/S003126/1.Notes and references
- K. Müllen and W. Pisula, Donor-Acceptor Polymers, J. Am. Chem. Soc., 2015, 137(30), 9503–9505 CrossRef PubMed
.
- R. S. Kularatne, H. D. Magurudeniya, P. Sista, M. C. Biewer and M. C. Stefan, Donor-Acceptor Semiconducting Polymers for Organic Solar Cells, J. Polym. Sci., Part A: Polym. Chem., 2013, 51(4), 743–768 CrossRef CAS
- Y. Zhao, Y. Guo and Y. Liu, 25th Anniversary Article: Recent Advances in n-Type and Ambipolar Organic Field-Effect Transistors, Adv. Mater., 2013, 5372–5391 CrossRef CAS PubMed
- E. J. Meijer, C. Detcheverry, P. J. Baesjou, E. Van Veenendaal, D. M. De Leeuw and T. M. Klapwijk, Dopant Density Determination in Disordered Organic Field-Effect Transistors, J. Appl. Phys., 2003, 93(8), 4831–4835 CrossRef CAS
- T. M. Swager and R. H. Grubbs, Synthesis and Properties of a Novel Cross-Conjugated Conductive Polymer Precursor: Poly(3,4-Diisopropylidenecyclobutene), J. Am. Chem. Soc., 1987, 109(3), 894–896 CrossRef CAS
- M. Heeney, C. Bailey, K. Genevicius, M. Shkunov, D. Sparrowe, S. Tierney and I. McCulloch, Stable Polythiophene Semiconductors Incorporating Thieno[2,3-6]Thiophene, J. Am. Chem. Soc., 2005, 127(4), 1078–1079 CrossRef CAS PubMed
- W. Zhang, Z. Mao, N. Zheng, J. Zou, L. Wang, C. Wei, J. Huang, D. Gao and G. Yu, Highly Planar Cross-Conjugated Alternating Polymers with Multiple Conformational Locks: Synthesis, Characterization and Their Field-Effect Properties, J. Mater. Chem. C, 2016, 4(39), 9266–9275 RSC
- G. W. P. Van Pruissen, J. Brebels, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Effects of Cross-Conjugation on the Optical Absorption and Frontier Orbital Levels of Donor-Acceptor Polymers, Macromolecules, 2015, 48(8), 2435–2443 CrossRef CAS
- A. Ganguly, J. Zhu and T. L. Kelly, Effect of Cross-Conjugation on Derivatives of Benzoisoindigo, an Isoindigo Analogue with an Extended π-System, J. Phys. Chem. C, 2017, 121(17), 9110–9119 CrossRef CAS
- T. P. Voortman, D. Bartesaghi, L. J. A. Koster and R. C. Chiechi, Cross-Conjugated n-Dopable Aromatic Polyketone, Macromolecules, 2015, 48(19), 7007–7014 CrossRef CAS
- S. Romero-Servin, L. A. Lozano-Hernández, J. L. Maldonado, R. Carriles, G. Ramos-Ortíz, E. Pérez-Gutiérrez, U. Scherf and M. G. Zolotukhin, Light Emission Properties of a Cross-Conjugated Fluorene Polymer: Demonstration of Its Use in Electro-Luminescence and Lasing Devices, Polymers, 2016, 8(2), 43 CrossRef PubMed
- L. Aparicio-Ixta, G. Ramos-Ortiz, J. L. Pichardo-Molina, J. L. Maldonado, M. Rodríguez, V. M. Tellez-Lopez, D. Martinez-Fong, M. G. Zolotukhin, S. Fomine and M. A. Meneses-Nava,
et al., Two-Photon Excited Fluorescence of Silica Nanoparticles Loaded with a Fluorene-Based Monomer and Its Cross-Conjugated Polymer: Their Application to Cell Imaging, Nanoscale, 2012, 4(24), 7751–7759 RSC
- T. P. Voortman, H. D. De Gier, R. W. A. Havenith and R. C. Chiechi, Stabilizing Cations in the Backbones of Conjugated Polymers, J. Mater. Chem. C, 2014, 2(17), 3407–3415 RSC
- X. Li, X. Zhang, W. Li, Y. Wang, T. Liu, B. Zhang and W. Yang, Synthesis and Enhanced Two-Photon Absorption Properties of Tetradonor-Containing Anthracene-Centered 2-D Cross-Conjugated Polymers, J. Mater. Chem., 2011, 21(11), 3916–3924 RSC
- T. Maeda, T. Tsukamoto, A. Seto, S. Yagi and H. Nakazumi, Synthesis and Characterization of Squaraine- Based Conjugated Polymers with Phenylene Linkers for Bulk Heterojunction Solar Cells, Macromol. Chem. Phys., 2012, 213(24), 2590–2597 CrossRef CAS
- L. V. Kayser, E. M. Hartigan and B. A. Arndtsen, Multicomponent Coupling Approach to Cross-Conjugated Polymers from Vanillin-Based Monomers, ACS Sustainable Chem. Eng., 2016, 4(12), 6263–6267 CrossRef CAS
- S. Barik and S. Valiyaveettil, Synthesis and Self-Assembly of Polyhydroxylated and Electropolymerizable Block Copolymers, J. Polym. Sci., Part A: Polym. Chem., 2014, 52(15), 2217–2227 CrossRef CAS
- E. Preis and U. Scherf, Poly(Diindenonaphthalene) and Poly(Indenofluorene) – Tuning the Absorption Properties of Low Bandgap Cross-Conjugated Polymers, Macromol. Rapid Commun., 2006, 27(14), 1105–1109 CrossRef CAS
- H. Wang, R. Helgeson, B. Ma and F. Wudl, Synthesis and Optical Properties of Cross-Conjugated Bis(Dimethylaminophenyl)Pyridylvinylene Derivatives, J. Org. Chem., 2000, 65(18), 5862–5867 CrossRef CAS PubMed
- Q. Fang and T. Yamamoto, Preparation of a New Polymer Containing Photoluminescent Pyrazoline Unit in the Main Chain, J. Polym. Sci., Part A: Polym. Chem., 2004, 42(11), 2686–2697 CrossRef CAS
- K. J. Fallon, N. Wijeyasinghe, E. F. Manley, S. D. Dimitrov, S. A. Yousaf, R. S. Ashraf, W. Duffy, A. A. Y. Guilbert, D. M. E. Freeman and M. Al-Hashimi,
et al., Indolo-Naphthyridine-6,13-Dione Thiophene Building Block for Conjugated Polymer Electronics: Molecular Origin of Ultrahigh n-Type Mobility, Chem. Mater., 2016, 28(22), 8366–8378 CrossRef CAS
- P. Friedländer, Über Den Farbstoff Des Antiken Purpurs Aus Murex Brandaris, Ber. Dtsch. Chem. Ges., 1909, 42(1), 765–770 CrossRef
- P. F. Schatz, Indigo and Tyrian Purple—In Nature and in the Lab, J. Chem. Educ., 2009, 78(11), 1442 CrossRef
- J. L. Wolk and A. A. Frimer, A Simple, Safe and Efficient Synthesis of Tyrian Purple (6,6′-Dibromoindigo), Molecules, 2010, 15(8), 5561–5580 CrossRef CAS PubMed
- C. J. Cooksey, Tyrian Purple: 6,6′-Dibromoindigo and Related Compounds, Molecules, 2001, 736–769 CrossRef CAS
- P. Imming, I. Imhof and M. Zentgraf, An Improved Synthetic Procedure for 6,6′-Dibromoindigo (Tyrian Purple), Synth. Commun., 2001, 31(23), 3721–3727 CrossRef CAS
- A. Leventis, J. Royakkers, A. G. Rapidis, N. Goodeal, M. K. Corpinot, J. M. Frost, D. K. Bučar, M. O. Blunt, F. Cacialli and H. Bronstein, Highly Luminescent Encapsulated Narrow Bandgap Polymers Based on Diketopyrrolopyrrole, J. Am. Chem. Soc., 2018, 140(5), 1622–1626 CrossRef CAS PubMed
- D. M. E. Freeman, A. J. Musser, J. M. Frost, H. L. Stern, A. K. Forster, K. J. Fallon, A. G. Rapidis, F. Cacialli, I. McCulloch and T. M. Clarke,
et al., Synthesis and Exciton Dynamics of Donor-Orthogonal Acceptor Conjugated Polymers: Reducing the Singlet-Triplet Energy Gap, J. Am. Chem. Soc., 2017, 139(32), 11073–11080 CrossRef CAS PubMed
- C. Guo, J. Quinn, B. Sun and Y. Li, Regioisomeric Control of Charge Transport Polarity for Indigo-Based Polymers, Polym. Chem., 2015, 6(39), 6998–7004 RSC
- C. Guo, J. Quinn, B. Sun and Y. Li, An Indigo-Based Polymer Bearing Thermocleavable Side Chains for n-Type Organic Thin Film Transistors, J. Mater. Chem. C, 2015, 3(20), 5226–5232 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc05553k |
| This journal is © The Royal Society of Chemistry 2021 |