
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluating the impact of systematic hydrophobic modification of model drugs on the control, stability and loading of lipid-based nanoparticles†
Cameron
Hogarth
a,
Keith
Arnold
a,
Andrew
McLauchlin
a,
Steve P.
Rannard
 a,
Marco
Siccardi
b and
Tom O.
McDonald
*a
a,
Marco
Siccardi
b and
Tom O.
McDonald
*a
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: tomm@liverpool.ac.uk
bDepartment of Pharmacology and Therapeutics, Institute of Systems, Molecular and Integrative Biology, University of Liverpool, 70 Pembroke Place, Liverpool, L69 3GF, UK
First published on 30th November 2021
Abstract
A significant number of new chemical entities in the drug development pipeline are poorly soluble, therefore routes that facilitate effective administration is of considerable value. Lipid nanoparticles have proved an attractive approach for drug delivery; however, challenges that include optimising drug loading and understanding the impact of drug physiochemical parameters on nanoparticle properties have limited progression. In this work, we investigate the effect of modifying the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P of a model drug on the formation and stability of lipid-based nanoparticles. A range of model drug analogues with systematically varying alkyl chains were produced using a lamivudine (nucleoside analog reverse transcriptase inhibitor) scaffold and processed into lipid nanoparticles by nanoprecipitation. Characterisation included evaluation of particle diameter, size distribution, drug loading and nanoformulation stability. A distinct correlation with the LaMer model of nucleation was observed and logP appeared to strongly influence rates of nucleation. Model drugs with high logP were uniform in particle size and distribution and offered enhanced stability. In addition, various model drug/lipid blends were produced and their physical properties were investigated using dynamic light scattering (DLS) and differential scanning calorimetry (DSC). Complex mixtures of lipids were shown to influence formulation crystallinity and strategies to form uniform and stable lipid based nanoparticles of high drug loading- through manipulation of logP are discussed.
P of a model drug on the formation and stability of lipid-based nanoparticles. A range of model drug analogues with systematically varying alkyl chains were produced using a lamivudine (nucleoside analog reverse transcriptase inhibitor) scaffold and processed into lipid nanoparticles by nanoprecipitation. Characterisation included evaluation of particle diameter, size distribution, drug loading and nanoformulation stability. A distinct correlation with the LaMer model of nucleation was observed and logP appeared to strongly influence rates of nucleation. Model drugs with high logP were uniform in particle size and distribution and offered enhanced stability. In addition, various model drug/lipid blends were produced and their physical properties were investigated using dynamic light scattering (DLS) and differential scanning calorimetry (DSC). Complex mixtures of lipids were shown to influence formulation crystallinity and strategies to form uniform and stable lipid based nanoparticles of high drug loading- through manipulation of logP are discussed.
Introduction
For some years, reports have suggested that ≥40% of drugs within clinical use are hydrophobic,1 and ≥90% of new drug candidates within the developing pipeline may be described as either Class II (high permeability, low solubility) or Class IV (low permeability, low solubility) categories using the Biopharmaceutics Classification System.2 Although the pharmaceutical industry considers drug hydrophobicity as a negative physical property, new therapy development is increasingly reliant on poorly water soluble compounds. Drug delivery systems have therefore become more important and target the enhancement of administration to ensure the therapeutic behaviour of poorly soluble drugs. In particular, nanomedicines have shown clinical benefits for drugs with low aqueous solubility in a range of diseases and conditions such as various cancer treatments. Additionally, lipid-based nanomedicines have been deployed in the recently approved COVID-19 vaccines.3–5 Solid lipid nanoparticles (SLNs) are a type of lipid-based nanomedicine which can accommodate and offer benefits for hydrophobic drug administrations.SLNs can be produced by a range of methods including; high shear homogenisation, hot or cold homogenisation and ultrasonication. Flash nanoprecipitation is a particularly interesting route to SLN production due to its simplicity and fast process speed which also offers the potential for scale up via continuous processing.6–8 Indeed, this process has already been realised in the formulation of nanolipomers with the use of microfluidics.9 In this method the solid lipid and drug are dissolved in a water-miscible organic solvent (e.g. tetrahydrofuran) which is later mixed with a solution of aqueous surfactant. The organic solvent diffuses through the aqueous medium meanwhile the solid lipid and drug precipitate and are stabilised by the surfactant due to their poor aqueous solubility.8
Many materials used to form lipid nanoparticles have high degrees of crystallinity, and the formation of nanoparticles of crystalline materials can be followed by the LaMer model of crystal nucleation and growth,10 which applies to nanoparticles formed by a ‘bottom up’ method such as nanoprecipitation.11 The LaMer model states that upon mixing, the solute becomes supersaturated, thereby triggering nucleation. Nucleation and growth then proceed in a simultaneous fashion competing for the consumption of supersaturated solute, until the solute concentration falls below the critical nucleation threshold whereby new nucleation events can no longer occur. Meanwhile, nanoparticles continue to grow by diffusion of solute and are stabilised by surfactants.10 Dalvi and Dave suggested that according to the LaMer model, small nanoparticles with a narrow distribution (low polydispersity) can be produced if a high rate of nucleation can be achieved while also limiting any growth phase. On the other hand, a slower rate of nucleation results in a dominant growth phase and a formulation of large particle size and broad distribution (high polydispersity).12 Dalvi et al. continued by linking a higher rate of nucleation to a higher degree of supersaturation.12 Strategies that have been demonstrated to increase the degree of supersaturation are: increased concentration of solute; variation of temperature; and the selection and ratio of solvents and anti-solvents.13 Therefore, the solubility of a compound plays a critical role in controlling the size and polydispersity of the nanoparticles that are formed.
Unfortunately, SLNs have been reported to suffer from poor drug loading as well as poor stability primarily due to; aggregation, Ostwald ripening or polymorphic transitions of the solid core.14,15 Polymorphic transitions are linked to highly crystalline materials. Helgason et al., have previously shown that polymorphic transitions of SLNs lipid core can induce changes in nanoparticle morphology from spherical to a needle-like form, which can result in drug expulsion.16 As an approach to address polymorphic transitions in SLNs Jenning et al. demonstrated that blending liquid and solid lipids can disrupt the crystal structure of the solid lipid, therefore delaying recrystallisation and improve physical stability.17 It was found that increasing the concentration of oil in lipid carrier nanoparticles resulted in the depression and broadening of the solid lipid melting peak, thus indicating a reduction in crystal order. Additionally, a study by Bunjes et al. demonstrated how blends of various solid lipids at different ratios can cause imperfections and disrupt the crystal structure of lipid nanoparticles, showing that the greatest imperfections to the crystal structure of a lipid nanoparticle may be achieved using a complex mixture of lipids. The results highlighted how different ratios of lipids can change the melting temperature, enthalpy and rate of recrystallisation.18 An investigation by Salminen et al. highlighted how high melting lecithin facilitated physically stable SLNs, while low melting lecithin resulting in nanoparticle growth.19 Therefore, strategies that can be used to disrupt the ability of lipids and drug to crystallise are very useful in developing lipid-based nanocarriers of higher stability.
Ostwald ripening is another phenomenon that is often also responsible for colloidal instabilities in lipid nanoparticles. It is a thermodynamically driven process whereby smaller particles undergo dissolution into the continuous phase and deposit in the larger particles.14 The larger particles are favoured over smaller particles due to their lower chemical potential. Therefore, Ostwald ripening results in an increase in the overall particle size of the formulation. Some advances have been made to slow/prevent Ostwald ripening by employing various surfactant stabilisers to enhance steric and electrostatic stabilisation, however screening a diverse range of surfactants tends to be a lengthy process which lacks efficiency. Another key factor determining the rate of Ostwald ripening is the solubility of the hydrophobic material (e.g. the lipids) in the continuous phase. Early investigations by Wooster et al. have highlighted how the aqueous solubility of an oil had a major impact on the stability of nanoemulsions. Specifically, compounds with very low aqueous solubilities such as triglyceride oils possessed a profound resistance to Ostwald ripening due to their severe insolubility in water.20 As a result, it is likely that the solubility of components within SLNs are key factors in controlling Ostwald ripening.
LogP also known as the n-octanol/water partition coefficient is a measure of a molecule's lipophilicity, by means of a balance between a molecules hydrophobic and hydrophilic features. Therefore, molecules of high lipophilicity can be correlated with extreme hydrophobicity. Additionally, the aqueous solubility of a compound is closely linked to logP; molecules with higher logP values are generally less water soluble. Calculated values for lipophilicity can be determined by a wide range of calculation descriptors, which typically are robust predictors of experimental logP values.21 Research by Zhu investigated the correlation between logP and stability of drug nanoparticles formulated by flash nanoprecipitation. The study investigated various hydrophobic drug molecules each ranging in value of logP calculated by the ACDlogP algorithm. They showed that nanoparticles made up of molecules with a logP > ∼12 showed good stability. Meanwhile, any nanoparticles formed from compounds with logP values between approximately 2 and 9 showed signs of fast Ostwald ripening and recrystallisation. Finally, compounds with a logP lower than 2 were generally unable to form nanoparticles due to their high solubility in water.22 Furthermore, the conditions surrounding logP and stability set by Zhu have been supported by further studies by Pustulka using the milogP algorithm which claim stable nanoparticles can be produced using compounds with a logP greater than ∼6.13 Although slight differences have been found between various algorithm models.23 It is also likely that the higher logP compounds also possessed lower aqueous solubilities which hindered any likelihood of Ostwald ripening. This work firstly supports how logP has potential to be used as a predictive tool with regards to nanoparticle stability, while there is a clear need for further research focused on the relationship between logP and nanoparticle stability. Therefore, it is likely that the solubility of a compound not only influences the stability of the nanoparticles but also the size and the polydispersity of the nanoparticles that are formed.
The systematic modification of a drug molecule to alter its logP would allow for a more detailed investigation into the factors controlling lipid nanoparticle formation and growth. A suitable approach for this modification has previously been demonstrated by Hobson et al. where chemical modification of the hydrophilic drug emtricitabine was conducted to increase its logP and enable formulation as drug nanoparticles which could be used as a long-acting injectable.24 Lamivudine (Fig. 1) is a very similar molecule to emtricitabine, however is structurally less complex due to the absence of fluorine. It has a logP < 2 (too hydrophilic to formulate efficiently within a hydrophobic nanoparticle) and possesses amine and alcohol groups to allow modification with various hydrophobic alkyl chloroformates to form stable drug analogues with reversible carbonate and carbamate bonds. We hypothesised that the same synthesis could be applied for lamivudine, with the alkyl chloroformates carefully selected based on logP values predicted for drug analogues.
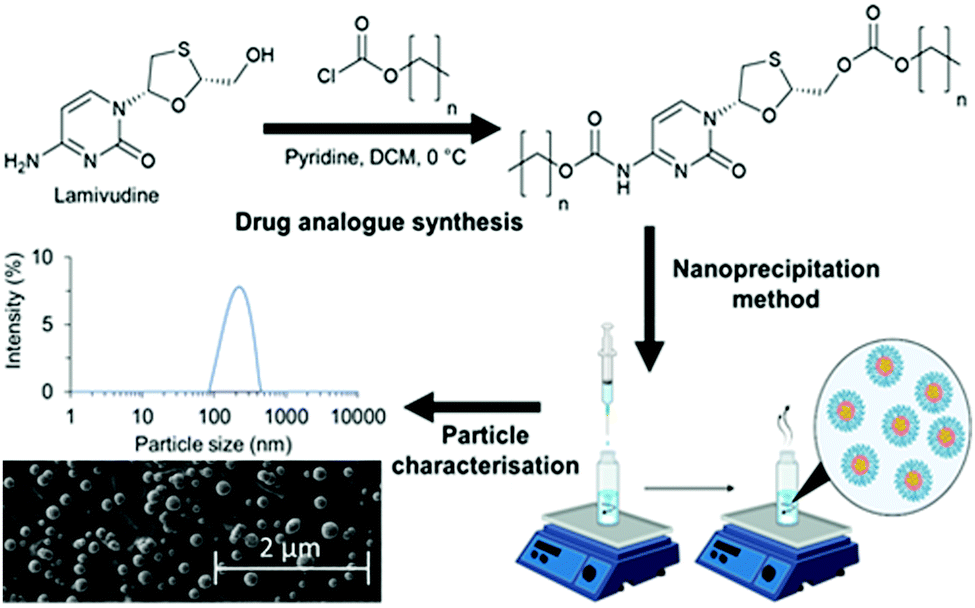 | ||
| Fig. 1 Schematic overview of a strategy to investigate the relationship between logP and nanoparticle characteristics such as particle size, polydispersity and morphology of lipid-based nanoparticles. Three drug analogues of lamivudine were synthesised by reacting with various length alkyl chloroformates to produce carbonate and carbamate bonds. The drug analogues were then nanoprecipitated into an aqueous solution of Brij 78 to achieve drug analogue nanoparticles for characterisation. | ||
In this work we undertake a systematic investigation of how logP of a set of drug analogues controls SLN formation and stability, (Fig. 1).
The approach also examines potential for solid drug analogue nanoparticles (which we refer to as SDANs from hereon) formed 100% drug analogue which have drug loadings much higher than the majority of SLNs reported in the literature. In order to investigate a broad range in logP, three drug analogues were synthesised from the hydrophilic drug lamivudine. We also investigated the effect of blending dodecyl drug analogue with the solid lipid Imwitor 900k. We investigated the degree of crystallinity of the drug analogue in various blends, while also documented the effect on drug loading, particle size, polydispersity and stability.
Results and discussion
Synthesis of drug analogues
Three alkyl modified drug analogues with carbamate and carbonate ester linkers were produced using the strategy previously demonstrated by Hobson et al. The reactions occurred at the 5′-hydroxyl and amino groups of lamivudine (Fig. 1). To better understand the relationship between logP and nanoparticle stability, we used chloroformates with specific alkyl chain lengths based on the calculated logP to produce drug analogues of lamivudine. The alkyl chloroformates were n-butyl, n-octyl and n-dodecyl chloroformates, while the corresponding calculated logP were 2.98, 7.21 and 11.44 respectively. Each drug analogue was a white solid and was characterised with the following proton and carbon nuclear magnetic resonance (NMR), Infra-red spectroscopy, mass spectrometry and CHNS elemental analysis (see ESI†) for methods and characterisation data, Fig. S1–S7 and Tables S1, S2, ESI†). The three hydrophobically modified drug analogues were successfully prepared; butyl drug analogue, octyl drug analogue and dodecyl drug analogue.
Solid drug analogue nanoparticles (SDANs)
The range of logP of drug analogues enabled us to investigate some of the key boundaries in logP previously reported by Zhu et al. Each of the drug analogues were nanoprecipitated into a fixed volume of the surfactant Brij 78 (polyoxyethylene 20 stearyl ether). Brij 78 is derived from stearic acid molecules conjugated to a polyethylene glycol (PEG) 1000 g mol−1 chain. PEG has been widely used to provide a steric stabilisation for lipid nanoparticles.25
The addition of the octyl and dodecyl drug analogue solutions in THF to the aqueous antisolvent resulted in the formation of a weakly turbid nanoparticle formulation as the nanoparticles were formed, Meanwhile, upon addition of the butyl drug analogue solution the formulation remained clear (Fig. S8A, ESI†), thus indicating potential solubility of the butyl drug analogue within the continuous phase and hindering nanoparticle formation. The samples were analysed by dynamic light scattering (DLS) 1 hour after formulation- while THF still present, the dodecyl SDAN formulation was on average 151 nm in diameter and 0.14 PDI. DLS measurements of the butyl SDAN formulations revealed a hydrodynamic diameter of 30 nm with a derived count rate approximately 1% of that of the dodecyl SDAN formulations, thus further suggesting solubility of the butyl drug analogue due to an extremely low concentration of nanoparticles of butyl drug analogue/micelles of Brij 78. Herein was concluded the butyl drug analogue was too soluble in the continuous phase mixture of both water and THF which was likely due it's low logP of 2.98 which in turn drastically hindered nanoparticle formation. The DLS data for the octyl drug analogue formulation was bimodal containing micron sized aggregates and therefore was deemed not suitable for DLS due to impacting the accuracy of the measurements. Soon after DLS measurements the octyl drug analogue formulation possessed a shimmering appearance that is associated with anisotropic crystals diffracting light at multiple angles, thus suggesting rapid particle size growth and crystallisation of the drug analogue likely due to Ostwald ripening (Fig. S8A, ESI†).26 The presence of anisotropic crystals within the octyl SDAN formulation was later confirmed by optical microscopy (Fig. S9A, ESI†).
After 2 days the THF had evaporated and the dependence of stability on logP became highly noticeable as the butyl drug analogue formulation increased in turbidity. The butyl drug analogue formulation also contained visible aggregates which became even more visible on day 6 (Fig. S8B, C and S9B, ESI†), which likely formed due to loss of solubility within the continuous phase resulting in saturation of the butyl drug analogue as the THF evaporated. As a result, instigated the rapid and extensive growth of nanoparticles. Meanwhile, the octyl drug analogue formulation still possessed the shimmering appearance until day 6 where the formulation had sedimented (Fig. S8B–D, ESI†). The dodecyl SDAN formulation appeared unchanged (Fig. S8B and C, ESI†), however a slight increase in particle size and PDI was observed by DLS, Fig. 2. This was attributed to a small degree of Ostwald ripening during solvent evaporation. Nevertheless, the dodecyl SDAN formulations appeared to remain stable upon storage at 4 °C, as there was no further notable change on day 6 or 28, Fig. 2, likely due to steric stabilisation provided by Brij 78. In addition, the data from Fig. 2 also highlights the large degree of reproducibility between dodecyl analogue formulations – the relative standard deviation of particle size between three replicate samples was 7, 18, 23 and 9 for day 0, 2, 6 and 28 measurements. Measurement of zeta potential confirmed that the dodecyl nanoparticles were stabilised solely by steric stabilisation due to an average net charge of −1.9 mV.
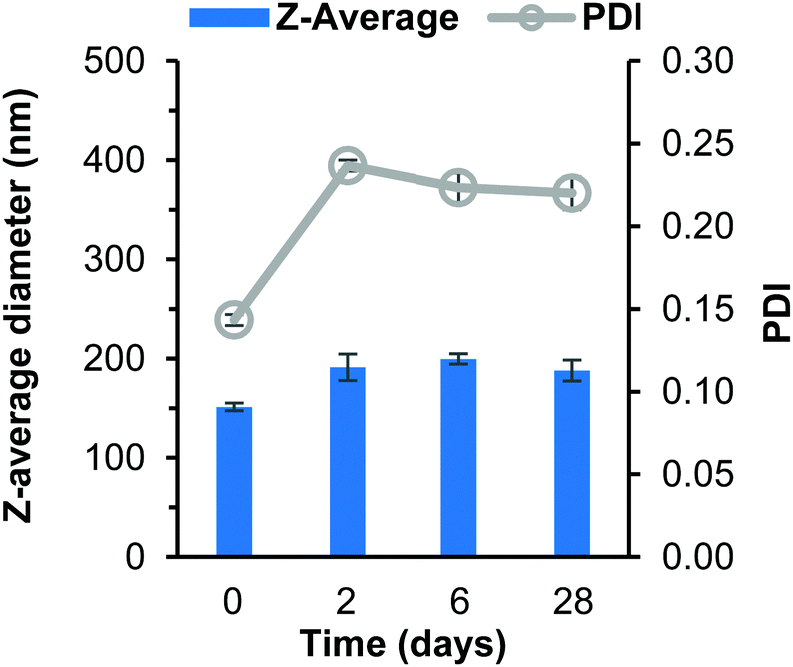 | ||
| Fig. 2 Data collected measuring both particle size and polydispersity of dodecyl SDAN formulations over 28 days. Samples prepared in triplicate and error bars calculated on standard deviation between measurements. Samples were stored at 4 °C after day 2 measurement. | ||
Attempts were made to characterise each of the SDAN formulations using cryogenic scanning electron microscopy (cryo-SEM) 2 days after formulation. Cryo-SEM was used in order to allow the visualisation of the particles without any significant drying effects that can occur in conventional scanning electron microscopy. Unfortunately, it was not possible to conclusively image either the butyl or octyl drug analogue formulations, however it was deemed with the dodecyl SDAN formulation, spherical nanoparticles were observed (Fig. 3) after comparing the cryoSEM images of both the dodecyl SDAN formulation and a brij 78 control (Fig. S10, ESI†) The measurement of 200 particles from the cryo-SEM images revealed an average particle diameter of 136 nm and a standard deviation of 44, thus in agreement with data obtained by DLS – after factoring in the solvent layer which adheres to the surface of nanoparticles in solution making particles appear bigger on DLS than in reality. The diameter distribution graph based on the cryo-SEM data can be found in the supplementary information (Fig. S11, ESI†).
 | ||
| Fig. 3 Depicts drug analogue structures and cryo-SEM images of the corresponding SDANs formulations. | ||
By combining data from various microscopy techniques alongside DLS and photographic data we hypothesised the relationships observed were evidence of the LaMer model and occur due to a difference in degree of supersaturation and rate of nucleation. Whereby, drug analogues of higher logP have a higher degree of supersaturation and thus nucleate at a faster rate which competes with the growth phase producing nanoparticles of smaller and uniform size. On the other hand, a low logP results in a lower degree of supersaturation therefore slow nucleation was resulting in a longer period of nucleation events, thus upon growth a broader particle distribution was established, as shown schematically in Fig. 4. In essence, the nanoparticle formulation properties were heavily dependent on the logP of the drug analogue and display evidence for the ability to tune these properties accordingly by applying LaMer model.
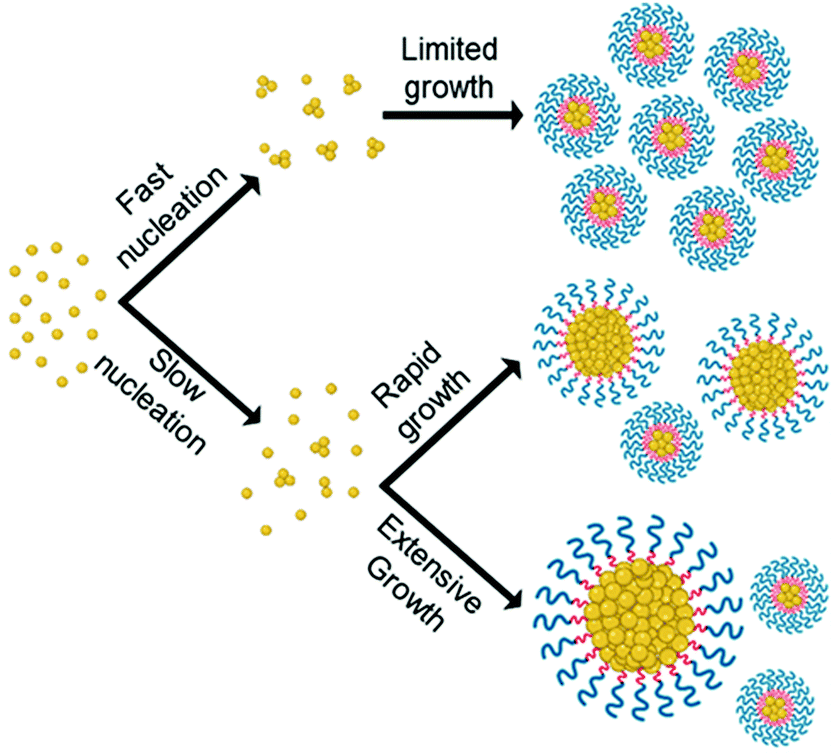 | ||
| Fig. 4 Illustrates how the differences in rate of nucleation as a result of differences in logP may affect particle size and size distribution of formulations according to LaMer model. | ||
Furthermore, the high logP of the dodecyl SDAN formulation appeared to resist Ostwald ripening over the analysis duration we have investigated. Therefore, it can be deduced that Ostwald ripening was greater in nanoparticles of lower logP, due to the greater solubility of the lower logP compounds in the continuous phase which enabled more rapid mass transport from the smaller nanoparticles to the larger nanoparticles/growing crystal. This data agrees with Zhu suggesting drugs with a logP between 9 and 12 are potentially stable. Given the favoured properties of the dodecyl drug analogue SDANs, further studies were carried out on this drug analogue.
Blends of drug analogues
The conclusion from earlier data was the octyl drug analogue was slower to nucleate than the dodecyl drug analogue, therefore we hypothesised that a blend may result in the dodecyl drug analogue aiding nanoparticle formation by providing nuclei from which the octyl drug analogue could grow. Another advantage of the blending approach is the potential to achieve higher drug loadings due to a higher proportion of the octyl drug analogue is made up of the model active lamivudine. Fig. 5 shows data obtained by DLS for various blends of dodecyl and octyl drug analogue.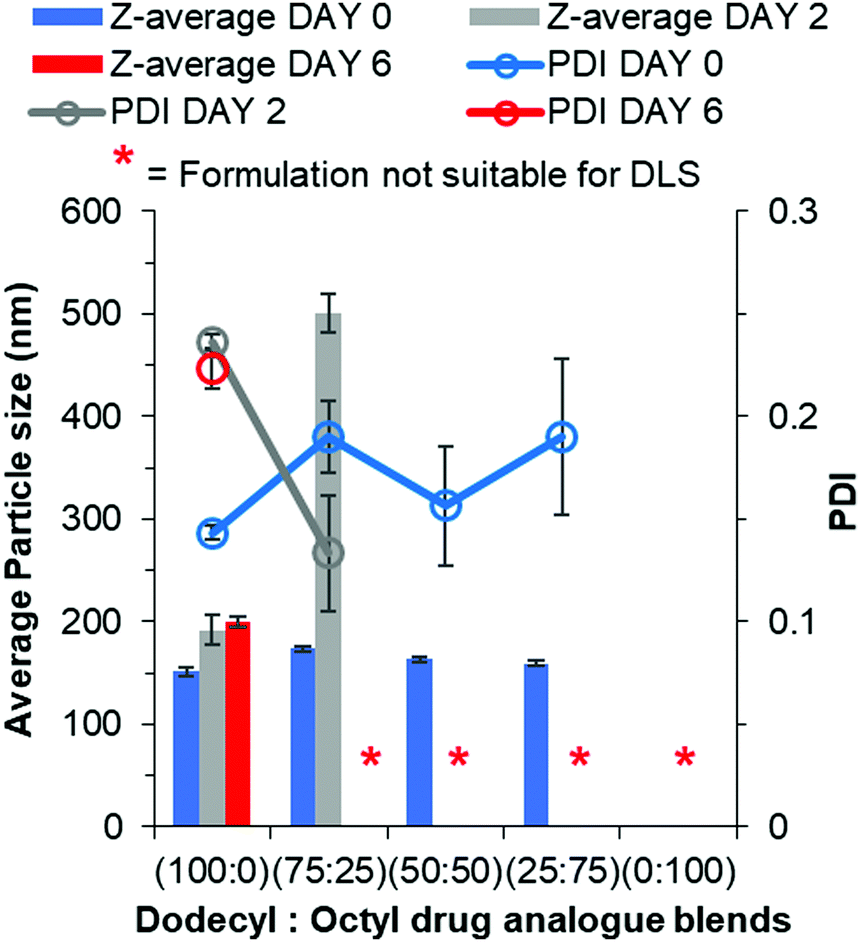 | ||
| Fig. 5 Particle size and size distribution data for blends of dodecyl and octyl drug analogue formulations obtained by DLS. | ||
For each blend, particles were detected and measured by DLS, all of which were of comparable average particle size to that of 100% dodecyl drug analogue formulation. Thus, we concluded the dodecyl drug analogue had indeed aided in nanoparticle formation. Furthermore, as the % of octyl drug analogue was increased in the blends the PDI generally increased along with broader margins for error thus suggesting a loss of control over nanoparticle formation. However, shortly after the DLS measurement the blend of 25% dodecyl/75% octyl drug analogue displayed signs of instability in a similar manner to the 100% octyl drug analogue formulation by means of a shimmering effect again suggesting the presence of anisotropic crystals. On the other hand, the blends of 75% dodecyl/25% octyl drug analogue and 50% dodecyl/50% octyl drug analogue appeared stable on day 0 due to no notable changes in appearance. Nevertheless, during the 2 day period of removal of THF by evaporation, the blends of dodecyl and octyl drug analogue quickly increased in particle size as the 75% dodecyl/25% octyl formulation grew on average by >300 nm after 2 days. While the 50% dodecyl/50% octyl drug analogue formulation also possessed a shimmering effect again suggesting a large degree of crystal particle growth to form anisotropic crystals and was no longer suitable for particle size measurement by DLS. This suggests despite aiding particle formation the continuous phase of water/THF was still too soluble for the octyl drug analogue thus enabling Ostwald ripening.
Lipid/drug analogue nanoparticle blends
We further investigated the effect of blending with the dodecyl drug analogue and a complex mixture of lipids in the form of Imwitor 900k, a mixture of mono-, di- and triglycerides of both palmitic and stearic acid.An important consideration in terms of blending the drug analogue with lipid is that it will in turn reduce the active drug loading. Fig. 6 shows the relationship between the drug loading in the formulation and the composition of the formulation. It is clear that formulations composed of 100% drug analogue offer excellent active drug loadings (32% w/w) in the core of the nanoparticles. Furthermore, Fig. 6 also shows how a good level of drug loading is maintained as the dodecyl drug analogue is blended with Imwitor 900k at various ratios.
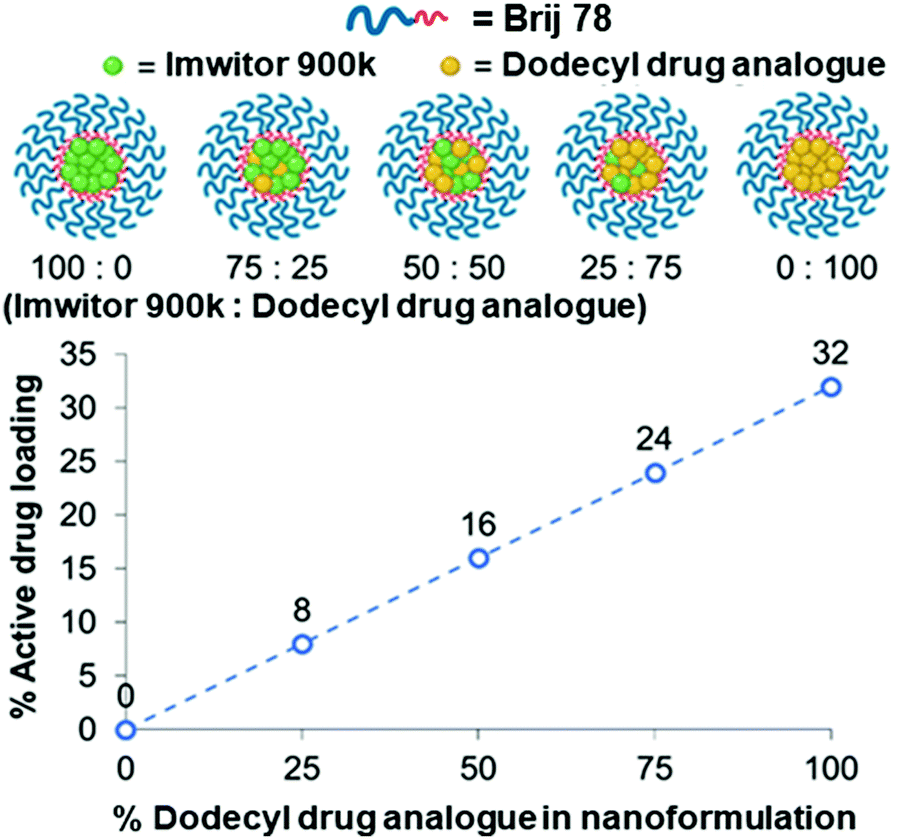 | ||
| Fig. 6 The relationship between drug analogue loading and active drug loading for each of the various lipid/drug analogue blend formulations. | ||
Lipid-based nanoparticles were then formed using blends of drug analogue with Imwitor 900k to investigate the effect of composition on particle size, distribution and stability. Fig. 7 displays particle size and polydispersity data obtained for each of the lipid/drug analogue blends over a 4 week period where samples were stored at 4 °C once all THF had been removed. The measurement on day 0 reflects particle properties immediately after preparation, day 2 provides a timepoint for when no more THF remained in the continuous phase due to the disappearance of peaks at 1.85 and 3.7 ppm on a 1H NMR spectrum (Fig. S12, ESI†).27 While, day 6 reflected on short term stability upon storage at 4 °C and day 28 provides a measure of the longer-term dispersion stability at 4 °C. On Day 0, the effect of increasing the drug analogue concentration in the nanoparticles resulted in a slight decrease in the average particle size for each of the blends. Additionally, there was a more pronounced decrease in polydispersity with increasing drug analogue concentration in the blend suggesting the formulations become much narrower. A possible explanation for the decrease in dispersity and particle size is the differences in logP. Imwitor 900k consists of 40–55% monoglycerides,28 both glyceryl monostearate and glyceryl monopalmitate have a moderate logP of 6.3 and 7.4. logP values in this range will result in an overall decrease in rate of nucleation as the percentage of Imwitor 900k was increased. Furthermore, low logP may also offer an explanation as to why the 100% Imwitor nanoparticles suffer from growth between day 6 and day 28 resulting in a shimmering effect similar to that seen with the octyl drug analogues (Fig. S13, ESI†). Despite this, the 100% Imwitor 900k nanoparticles appear to be stable and resist Ostwald's ripening up to a point between day 2 and 6, which was likely due to the high logP of the remaining di- and triglycerides of Imwitor 900k following a fast degree of nucleation. All the lipid/drug analogue blend formulations showed very little change in particle properties at the time points, any changes were likely within error. This finding suggests that each of the blend formulations of lipid and dodecyl drug analogue were stable for up to 4 weeks.
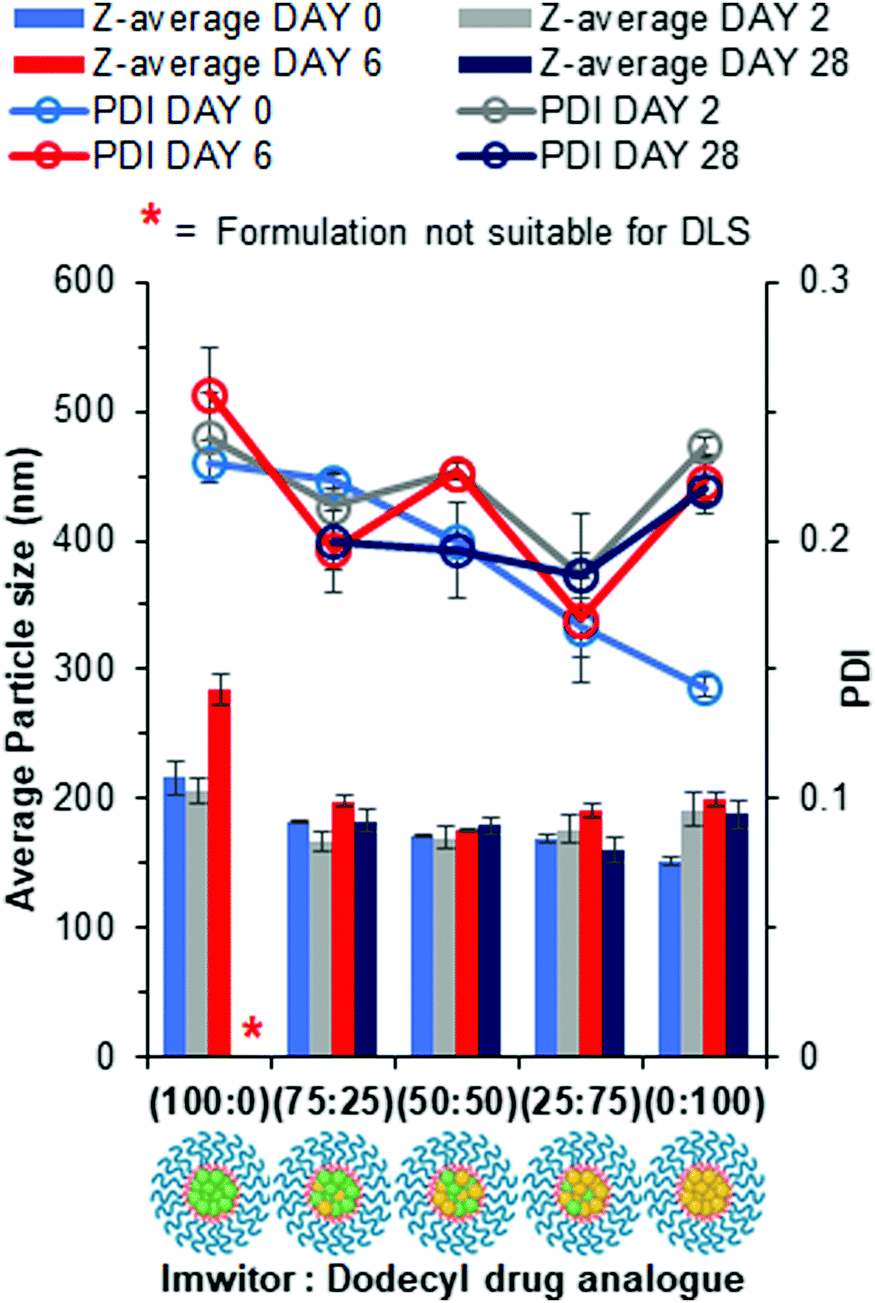 | ||
| Fig. 7 Particle size and size distribution data for each of the lipid/dodecyl drug analogue blends obtained by DLS over a 28 day period. Samples prepared in triplicate and error bars calculated on standard deviation between measurements. Samples were stored at 4 °C after day 2 measurement. | ||
In order to better understand the effect of blending on the crystallinity of both the lipid and dodecyl drug analogue DSC experiments were carried out. Firstly, the starting materials for the nanoparticle formulations were investigated individually, (Fig. 7). Brij 78 showed crystalline behaviour with a melting peak at approximately 40 °C. Imwitor 900k showed a broad melting endotherm with a melting temperature of approximately 60 °C. The slightly broader melting behaviour was likely due to Imwitor 900k existing as a mixture of lipids rather than a single pure molecule. The endotherm for the dodecyl drug analogue was sharp, with a peak at high temperature at approximately 89 °C. In order to assess the compatibility of the lipid and the drug analogue, a 50:50 binary mixture of dodecyl drug analogue and Imwitor 900k was prepared by melting the two compounds together allowing to solidify followed by thermo-analysis by DSC. The thermogram for this binary mixture possessed an intense peak at a lower temperature than that of both Imwitor 900k and dodecyl drug analogue which suggests the presence of a eutectic mixture.29 On the other hand, an additional weaker transition was also observed in the range of 55–62 °C which may indicate the co-existence of a small amount of crystalline Imwitor 900k suggesting the dodecyl drug analogue may have instead solubilised within Imwitor 900k (Fig. 8). Nevertheless, the absence of a dodecyl drug analogue transition indicates the dodecyl drug analogue is of good compatibility with Imwitor 900k, meanwhile introduced a degree disorder of the Imwitor 900k crystal structure.30 The onset temperatures and peak temperatures for each of the samples is summarised in Table 1.
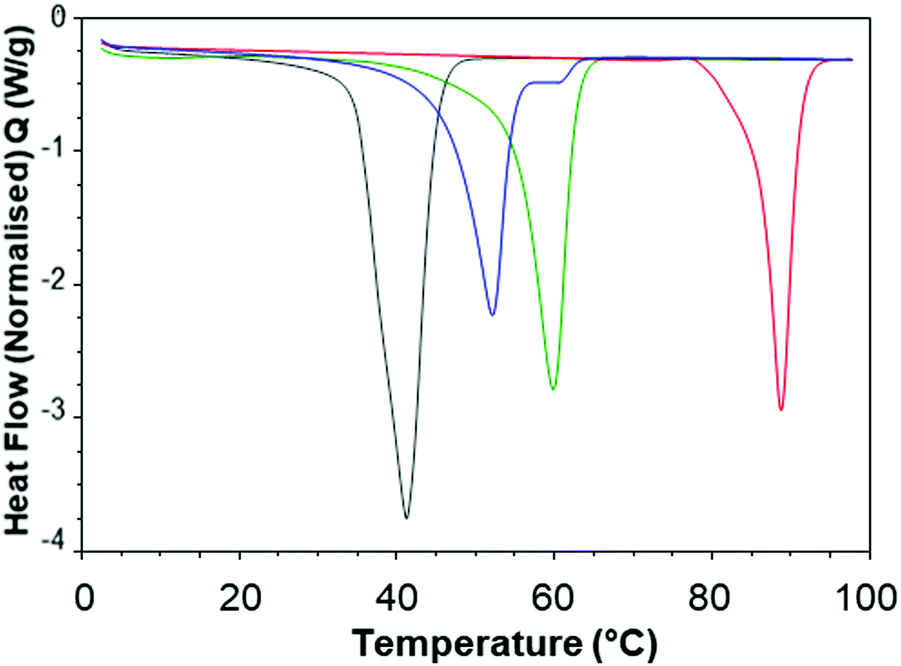 | ||
| Fig. 8 Overlay of 2nd heat DSC thermograms for each bulk material nanoparticle component as non-formulated materials Brij 78 (grey), Imwitor 900k (green), dodecyl drug analogue (red). Overlay includes a 50:50 binary mixture between Imwitor 900k and the dodecyl drug analogue (blue). | ||
| Onset temperature (°C) | Peak temperature (°C) | |
|---|---|---|
| Brij 78 | 32.9 | 39.7 |
| Imwitor 900k | 54.6 | 59.9 |
| Dodecyl drug analogue | 85.6 | 88.8 |
| Binary mixture (Imwitor 900k/Dodecyl drug analogue at 50:50) |
46.2 | 52.1 |
The thermal properties of the lipid-based nanoparticle blends were then investigated. Fig. 9 displays an overlay of thermograms for each of the nanoparticle formulations. The most pronounced melting transition that was observed for all formulations was Brij 78 peak at approximately 35–45 °C, this was due to Brij 78 comprising 86% of the total mass of each sample while the core comprised 14%. The endotherms ocurring in the region 45–65 °C corresponded to the nanoparticle core crystallinity, with endotherms in the range ∼45–55 °C for the Imwitor 900k and ∼55–75 °C for the dodecyl drug analogue. For the formulation of 100% Imwitor 900k the average peak temperature for Imwitor 900k was 51.1 °C. It was clear that the ratio of Imwitor 900k and dodecyl drug analogue determined the crystallinity of the two compounds. Firstly, as expected for 100% Imwitor 900k nanoparticles only one peak was present in this region (see Fig. 9, inset). As the percentage of dodecyl drug analogue was increased to 25% the magnitude of the Imwitor 900k endotherm decreased, whilst also shifting left to a lower melting temperature. This change in peak behaviour was described by Jenning et al. as peak depressions.17 No endotherm for the drug analogue was observed in the region of 60–75 °C. The presence of a single peak at this ratio suggested the Imwitor 900k and dodecyl drug analogue exists as a solid solution with the dodecyl drug analogue completely solvated within the solid lipid core of the nanoparticles. This behaviour in the nanoparticles was that same as seen for the binary mixture of only lipid and drug analogue shown in Fig. 8. As the ratio of dodecyl drug analogue was increased further to ratio of 50:50 the Imwitor 900k endotherm again experienced peak depression, however an additional peak was also present at a higher temperature of approximately 54 °C, thus indicating the presence of two different crystalline forms.18 As the amount of drug analogue in the composition was further increased to a ratio of 25:75 the endotherm corresponding to the drug analogue became more pronounced and shifted to higher temperatures of approximately 60 °C and 65 °C as the ratio of dodecyl drug analogue was increased to 75 and then 100%. Additionally, the peak for Imwitor 900k completely disappeared at 75% drug analogue. Consequently, we hypothesise that at 75% drug analogue, the core existed as Imwitor 900k solubilised within the drug analogue. Comparison between the unformulated material and nanoparticle formulations showed peak broadening and a shift of endotherms to lower melting temperature for both the core nanoparticle materials i.e. Imwitor 900k or dodecyl drug analogue (see Table 2) compared to the corresponding bulk material (Table 1). Similar findings have been reported by Siekmann and Westesen.31 This suggests a decrease in crystal perfection which can be attributed to small size of the nanoparticles and an interaction between the surfactant and the core.
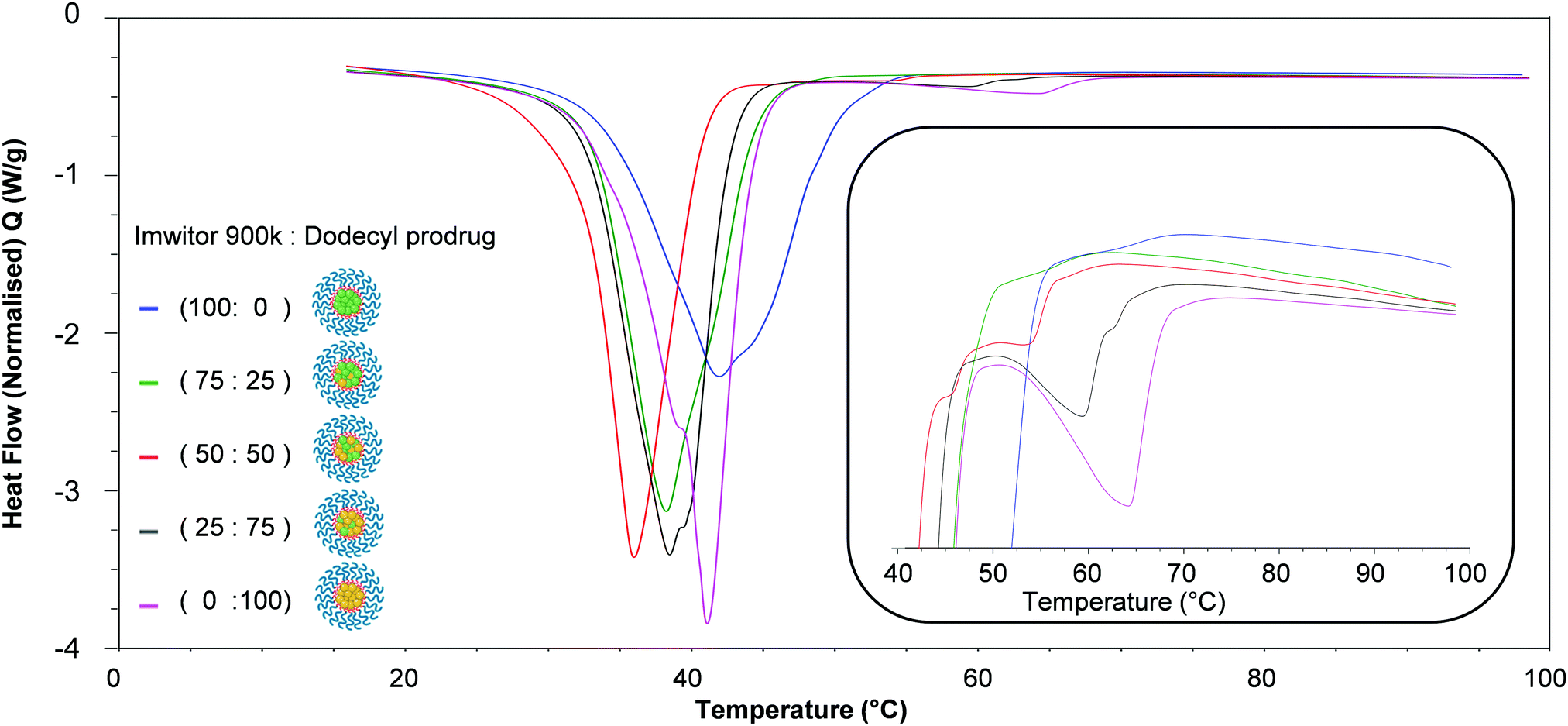 | ||
| Fig. 9 Overlay of DSC traces for each of the nanoparticle formulations. The inset shows the region 40–100 °C which indicates the effect of core composition on the crystallinity of the lipid and drug analogue within the core of nanoparticles. | ||
| Average onset temperature (°C) | Difference (±) to unformulated onset temperature (°C) | Average peak temperature (°C) | Difference (±) to unformulated peak temperature (°C) | |
|---|---|---|---|---|
| Imwitor 900k | 55.2 | +0.6 | 58.9 | −1.0 |
| Dodecyl drug analogue | 53.1 | −32.5 | 64.9 | −23.9 |
In addition, a notable trend between formulation ratio of Imwitor 900k to drug analogue and shifts in the melting temperatures and enthalpies for both the dodecyl drug analogue and Imwitor 900k. For the drug analogue, the melting temperature and crystallinity decreased as the percentage of Imwitor increased in the formulation (Fig. 10). Indeed, the drug analogue was entirely amorphous when the drug analogue represented 25% of the core mass formulation. It was not possible to carry out a similar analysis for the Imwitor 900k due to some overlap of the melting enthalpies with those from Brij 78, however it was clear that the melting temperature decreased and the enthalpy for the melted also decreased with increasing drug analogue concentration. These results provide strong evidence of the Imwitor and the dodecyl drug analogue were contained in the same particles (rather than separate nanoparticle populations), additionally the blending reduced the crystallinity of both compounds relative to that of the bulk material. This work demonstrates how the crystallinity of a nanoparticle core can be tuned by blending with various components.
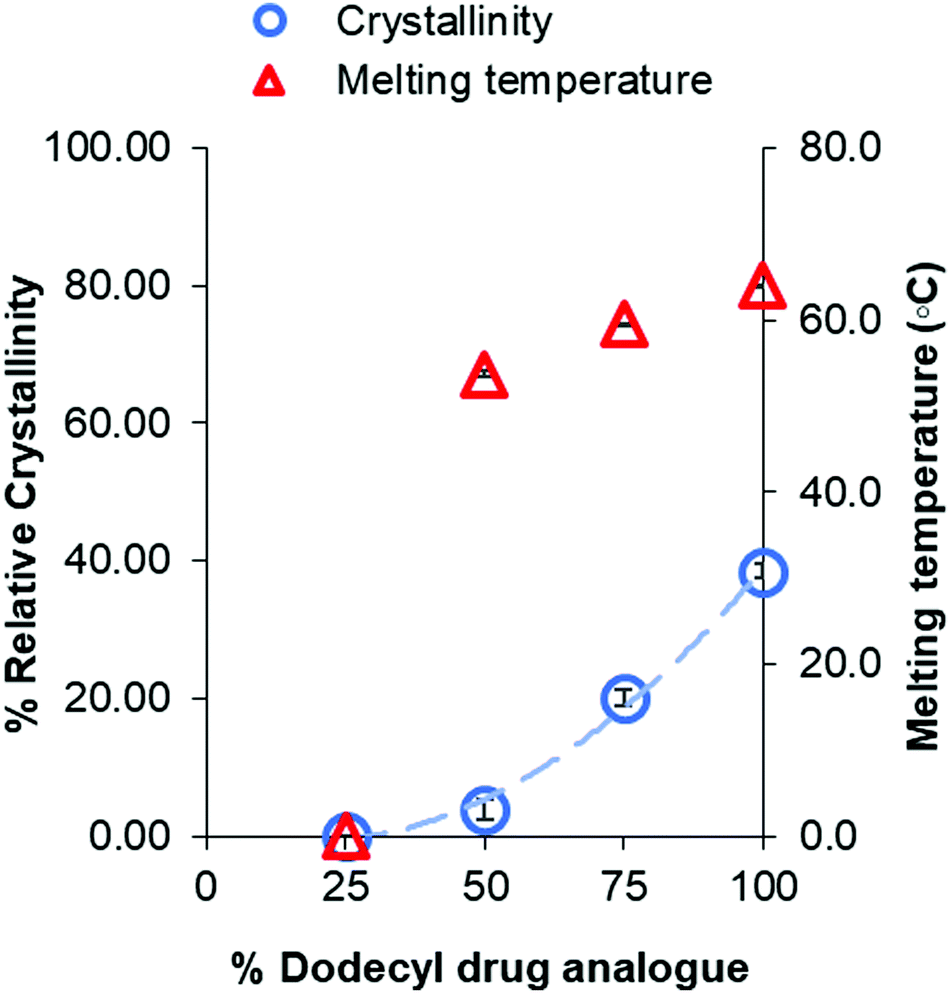 | ||
| Fig. 10 Graph showing the relationship between percentage of dodecyl drug analogue within the core and the crystallinity of dodecyl drug analogue relative to that of the bulk. | ||
Conclusions
In this study the effects of logP of carbamate and carbonate-based drug analogues on the formation and stability of SLN and SDAN were investigated. As per the LaMer model, with increasing logP (and the likely higher degree of supersaturation and faster rate of nucleation) smaller nanoparticles with reduced standard deviation were produced. Furthermore, the drug analogue with the highest logP (11.44) resulted in highly stable formulations which maintained size and dispersity for up to at least 4 weeks. Meanwhile drug analogues of lower logP experienced considerable growth resulting in micron sized aggregates as a result of Ostwald ripening. This work shows how logP can be used as a tool to tune nanoparticle formulations in the future. Additionally, the approach to produce SDANs (that included no carrier material offered the opportunity to produce nanoparticles with an active drug loading of up to 32%. Additionally, we also demonstrated how using a drug analogue of high logP and a complex mixture of lipids such as Imwitor 900k, SLN formulations of high drug loading and good immediate stability may be achieved. By blending with a complex mixture of lipids such as Imwitor 900k we also managed to control the crystallinity of the drug analogue within the core of the SLN formulations due to a strong correlation between % drug analogue loading and % crystallinity. DSC analysis was highly suggestive of a solid solution at both 25 and 75% drug analogue loadings, whereby the drug analogue or lipid would dissolve into the higher concentrated material effectively becoming amorphous due to the absence of an endothermic melting peak.
The ability to produce lipid-based nanoparticles of originally hydrophilic drugs is attractive in terms of its ability to obtain high drug loading formulations. The strategy of producing drug analogues and then forming SDANs could also be employed for hydrophobic drugs of relatively low logP with high drug loading. While also potentially allow the formation of combination nanoparticles with hydrophilic and hydrophobic drugs.
Experimental
Materials
Brij 78, tetrahydrofuran, anhydrous materials (pyridine, dichloromethane) and deuterated solvents (CDCl3) were all purchased from Sigma Aldrich. Each were used as received apart from CDCl3 where 0.1% tetramethylsilane was added. All other solvents were reagent grade and purchased from Fischer Scientific and used as received. Alkyl chloroformates were purchased from Tokyo chemical industry and used as received. Magnesium sulphate (MgSO4) and concentrated hydrochloric acid (HCl) were also purchased from Fischer Scientific. HCl was diluted down to a 1M solution, meanwhile MgSO4 was used as received. Imwitor® 900K (glyceryl monostearate) was kindly gifted from IOI Oleochemical, Hamburg. Lamivudine was purchased from Top Well Medipharma Group.Methods
:50 ethyl acetate) to obtain a white powder which was dried in a vacuum oven at 20 °C.
P values were calculated using ChemDrawProfessional version 18.1 software.
Characterisation data of drug analogues
Butyl drug analogue (butyl(1-((2R,5S)-2-((((butyloxy)carbonyl)oxy)methyl)-1,3-oxathiolan-5-yl)-2-oxo-1,2-dihydropyrimidin-4-yl)carbamate): yield 0.7 g (37%); m.p. 82–84 °C; elemental analysis (calcd for C18H27N3O7S: C, 50.34; H, 6.34; N, 9.78; S, 7.46; found: C, 50.36; H, 6.35; N, 9.9; S, 7.22%); Vmax/cm−1 3455w (NH), 3295s, 3114w, 2960s, 2936w and 2903w (CH); 1750s, 1727s and 1677s (CO), 1501s (CC), 1260s (CO); 1H NMR (400 MHz, CD3OD) δ ppm; 0.9 (6H, t, CH3), 1.3–1.4 (4H, m, CH2), 1.6–1.75 (4H, m, CH2), 3.4 (2H, dd, CH2), 4.2 (4H, t, CH2), 4.6 (2H, m, CH2), 5.4 (1H, s, CH), 6.3 (1H, s, CH), 7.25 (1H, d, CH), 7.4–8.1 (1H, broad s, NH), 8.2 (1H, d, CH); 13C NMR NMR (100 MHz, CD3OD) δ ppm; 13.60 (s, 1C), 13.63 (s, 1C), 18.86 (s, 1C), 18.92 (s, 1C), 30.56 (s, 1C), 30.59 (s, 1C), 38.83 (s, 1C), 66.25 (s, 1C), 66.58 (s, 1C), 68.62 (s, 1C), 84.37 (s, 1C), 87.98 (s, 1C), 94.68 (s, 1C), 144.24 (s, 1C), 152.49 (s, 1C), 154.60 (s, 1C), 154.89 (s, 1C), 162.65 (s, 1C); HRMS (ESI) m/z: calcd for C18H27N3O7S, 430.16 [M + H]+, 452.15 [M + Na]+, found 430.16, 452.15.Octyl drug analogue (octyl(1-((2R,5S)-2-((((octyloxy)carbonyl)oxy)methyl)-1,3-oxathiolan-5-yl)-2-oxo-1,2-dihydropyrimidin-4-yl)carbamate): yield 0.88 g (37%); m.p. 72–74 °C; elemental analysis (calcd for C26H43N3O7S: C, 57.65; H, 8.00; N, 7.76; S, 5.92; found: C, 57.74; H, 8.04; N, 7.94; S, 5.97%); Vmax/cm−1 3457w (NH), 3295s, 3114w, 2955s, 2920w and 2852w (CH); 1747s, 1729s and 1673s (CO), 1501s (CC), 1278s (CO); 1H NMR (400 MHz, CD3OD) δ ppm; 0.9 (6H, t, CH3), 1.3–1.4 (20H, m, CH2), 1.6–1.75 (4H, m, CH2), 3.4 (2H, dd, CH2), 4.2 (4H, t, CH2), 4.6 (2H, m, CH2), 5.4 (1H, s, CH), 6.3 (1H, s, CH), 7.25 (1H, d, CH), 7.4–8.1 (1H, broad s, NH), 8.2 (1H, d, CH); 13C NMR NMR (100 MHz, CD3OD) δ ppm; 14.06 (s, 2C), 22.60 (s, 2C), 25.62 (s, 1C), 25.69 (s, 1C), 28.59 (s, 1C), 29.11 (s, 1C), 29.12 (s, 2C), 29.14 (s, 1C), 31.73 (s, 1C), 31.75 (s, 1C), 38.85 (s, 1C), 66.58 (s, 2C), 68.94 (s, 1C), 84.39 (s, 1C), 88.00 (s, 1C), 94.64 (s, 1C), 144.28 (s, 1C), 152.42 (s, 1C), 154.61 (s, 1C), 154.90 (s, 1C), 162.60 (s, 1C); HRMS (ESI) m/z: calcd for C26H43N3O7S, 542.29 [M + H]+, 564.27 [M + Na]+, found 542.29, 564.27.
Dodecyl drug analogue (dodecyl (1-((2R,5S)-2-((((dodecyloxy)carbonyl)oxy)methyl)-1,3-oxathiolan-5-yl)-2-oxo-1,2-dihydropyrimidin-4-yl) carbamate): yield 0.75 g (26%); m.p. 78–80 °C; elemental analysis (calcd for C34H59N3O7S: C, 62.45; H, 9.09; N, 6.43; S, 4.90; found: C, 62.37; H, 8.98; N, 6.26; S, 5.00%); Vmax/cm−1 3459w (NH), 3279s, 3146w, 2955s, 2916w and 2849w (CH); 1747s, 1728s and 1676s (CO), 1500s (CC), 1276s (CO); 1H NMR (400 MHz, CD3OD) δ ppm; 0.9 (6H, t, CH3), 1.3–1.4 (32H, m, CH2), 1.6–1.75 (8H, m, CH2), 3.4 (2H, dd, CH2), 4.2 (4H, t, CH2), 4.6 (2H, m, CH2), 5.4 (1H, s, CH), 6.3 (1H, s, CH), 7.25 (1H, d, CH), 7.4–8.1 (1H, broad s, NH), 8.2 (1H, d, CH); 13C NMR NMR (100 MHz, CD3OD) δ ppm; 14.10 (s, 2C), 22.67 (s, 2C), 25.63 (s, 1C), 25.70 (s, 1C), 28.60 (s, 1C), 28.68 (s, 1C), 28.88 (s, 2C), 29.19 (s, 1C), 29.22 (s, 1C), 29.33 (s, 2C), 29.48 (s, 2C), 29.54 (s, 1C), 29.57 (s, 1C), 29.60 (s, 1C), 29.63 (s, 1C), 31.90 (s, 1C), 32.65 (s, 1C), 38.87 (s, 1C), 66.55 (s, 1C), 66.62 (s, 1C), 68.96 (s, 1C), 84.44 (s, 1C), 88.01 (s, 1C), 94.68 (s, 1C), 144.31 (s, 1C), 152.41 (s, 1C), 154.89 (s, 1C), 155.43 (s, 1C), 162.55 (s, 1C); HRMS (ESI) m/z: calcd for C34H59N3O7S, 654.41 [M + H]+, 676.40 [M + Na]+, found 654.41, 676.40.
General nanoparticle preparation
Method adopted for SDAN and SLN formulation was nanoprecipitation. For the aqueous phase, the surfactant Brij 78 was dissolved to prepare a 500 ml stock solution in distilled water (1 mg ml−1) and left overnight at 21 degrees Celsius under mechanical stirring (300 rpm). Regardless of the composition a stock solution of the lipid/drug analogue phase (2 mg ml−1) was prepared in tetrahydrofuran which was sealed and left at room temperature under mechanical stirring (300 rpm) for 30 minutes. Portions of the stock solution were then diluted down ready for injection (2 mg ml−1, 2 ml).The 2 ml lipid/drug analogue solution was charged dropwise into the vortex of Brij 78 aqueous solution (24 ml) in a 40 ml vial while mechanically stirring (800 rpm). To ensure consistency in time of injection the shot was charged by removing the plunger of a clamped syringe resulting in a steady flow through the hypodermic needle. The combined mixture was left stirring to allow evaporation of tetrahydrofuran over 2 days at a room temperature (∼21 °C) in a fume cupboard with an average air velocity of 0.35 m s−1. Samples were then stored at 4 °C.
Nanoparticle characterisation methods
:1 and measured in a Malvern zetasizer nano series disposable folded capillary cell. All measurements were carried out at 25 °C using automated setting of a maximum of 1000 processed run.
Conflicts of interest
There are no conflicts to be declared.Acknowledgements
The research leading to these resulted has been supported by a DTP provided by the Department of Chemistry. We also acknowledge the EPSRC for the grant EP/S012265/1, as equipment from this grant was used in this research. Also, thanks to IOI Oleo Gmbh for kindly gifting various lipids including Imwitor®900K which was used in these results.References
- A. Fahr and X. Liu, Expert Opin. Drug Delivery, 2007, 4, 403–416 CrossRef CAS PubMed.
- J. M. Custodio, C.-Y. Wu and L. Z. Benet, Adv. Drug Delivery Rev., 2008, 60(6), 717–733 CrossRef CAS PubMed.
- B. García-Pinel, C. Porras-Alcalá, A. Ortega-Rodríguez, F. Sarabia, J. Prados, C. Melguizo and J. M. López-Romero, Nanomaterials, 2019, 9, 638 CrossRef PubMed.
- Nat. Nanotechnol., 2020, 15, 963, DOI:10.1038/s41565-020-00820-0.
- J. Prados, C. Melguizo, R. Ortiz, C. Velez, P. J. Alvarez, J. L. Arias, M. A. Ruiz, V. Gallardo and A. Aranega, Anticancer. Agents Med. Chem., 2012, 12, 1058–1070 CrossRef CAS PubMed.
- M. Schubert, Eur. J. Pharm. Biopharm., 2003, 55, 125–131 CrossRef CAS PubMed.
- Y. Dong, W. K. Ng, S. Shen, S. Kim and R. B. H. Tan, Colloids Surf., B, 2012, 94, 68–72 CrossRef CAS PubMed.
- J. M. Taylor, K. Scale, S. Arrowsmith, A. Sharp, S. Flynn, S. Rannard and T. O. McDonald, Nanoscale Adv., 2020, 2, 5572–5577 RSC.
- A. Gdowski, K. Johnson, S. Shah, I. Gryczynski, J. Vishwanatha and A. Ranjan, J. Nanobiotechnol., 2018, 16, 12 CrossRef PubMed.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- J. Tao, S. F. Chow and Y. Zheng, Acta Pharm. Sin. B, 2019, 9, 4–18 CrossRef PubMed.
- S. V. Dalvi and R. N. Dave, Ind. Eng. Chem. Res., 2009, 48, 7581–7593 CrossRef CAS.
- K. M. Pustulka, A. R. Wohl, H. S. Lee, A. R. Michel, J. Han, T. R. Hoye, A. V. McCormick, J. Panyam and C. W. Macosko, Mol. Pharmaceutics, 2013, 10, 4367–4377 CrossRef CAS PubMed.
- A. Deshpande, M. Mohamed, S. B. Daftardar, M. Patel, S. H. S. Boddu and J. Nesamony, Emerging Nanotechnologies for Diagnostics, Drug Delivery and Medical Devices, Elsevier, 2017, pp. 291–330 Search PubMed.
- V. Mishra, K. K. Bansal, A. Verma, N. Yadav and S. Thakur, Pharmaceutics, 2018, 1–21 Search PubMed.
- T. Helgason, T. S. Awad, K. Kristbergsson, D. J. McClements and J. Weiss, J. Am. Oil Chem. Soc., 2008, 85, 501–511 CrossRef CAS.
- V. Jenning, A. F. Thünemann and S. H. Gohla, Int. J. Pharm., 2000, 199, 167–177 CrossRef CAS PubMed.
- H. Bunjes, K. Westesen and M. H. J. Koch, Int. J. Pharm., 1996, 129, 159–173 CrossRef CAS.
- H. Salminen, A.-S. Stübler and J. Weiss, Eur. Food Res. Technol., 2020, 246, 599–608 CrossRef CAS.
- T. J. Wooster, M. Golding and P. Sanguansri, Langmuir, 2008, 24, 12758–12765 CrossRef CAS PubMed.
- R. Mannhold, G. Cruciani, K. Dross and R. Rekker, J. Comput. Aided. Mol. Des., 1998, 12, 573–581 CrossRef CAS PubMed.
- Z. Zhu, Mol. Pharmaceutics, 2014, 11, 776–786 CrossRef CAS PubMed.
- A. Pyka, M. Babuska and M. Zachariasz, Acta Pol. Pharm., 2006, 63, 159–167 Search PubMed.
- J. J. Hobson, A. Al-khouja, P. Curley, D. Meyers, C. Flexner, M. Siccardi, A. Owen, C. F. Meyers and S. P. Rannard, Nat. Commun., 2019, 10, 1413 CrossRef PubMed.
- J. Q. Zhang, J. Liu, X. L. Li and B. R. Jasti, Drug Delivery, 2007, 14, 381–387 CrossRef CAS PubMed.
- S. Wada, S. Kudo and H. Takiyama, J. Cryst. Growth, 2016, 435, 37–41 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- Imwitor 900K, https://www.ioioleo.de/produkte/pharma/imwitor-900-k/.
- P. Stott, J. Controlled Release, 1998, 50, 297–308 CrossRef CAS PubMed.
- A. Tatke, N. Dudhipala, K. Janga, S. Balguri, B. Avula, M. Jablonski and S. Majumdar, Nanomaterials, 2018, 9, 33 CrossRef PubMed.
- B. Siekmann and K. Westesen, Colloids Surf., B, 1994, 3, 159–175 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb02297k |
| This journal is © The Royal Society of Chemistry 2021 |