
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMembrane protein channels equipped with a cleavable linker for inducing catalysis inside nanocompartments†
Luisa
Zartner
a,
Viviana
Maffeis
ab,
Cora-Ann
Schoenenberger
 ab,
Ionel Adrian
Dinu
ab and
Cornelia G.
Palivan
*ab
ab,
Ionel Adrian
Dinu
ab and
Cornelia G.
Palivan
*ab
aDepartment of Chemistry, University of Basel, BPR1096, Mattenstrasse 24a, 4058 Basel, Switzerland. E-mail: cornelia.palivan@unibas.ch
bNCCR-Molecular Systems Engineering, BPR1095, Mattenstrasse 24a, 4058 Basel, Switzerland
First published on 4th October 2021
Abstract
Precisely timed initiation of reactions and stability of the catalysts are fundamental in catalysis. We introduce here an efficient closing–opening method for nanocompartments that contain sensitive catalysts and so achieve a controlled and extended catalytic activity. We developed a chemistry-oriented approach for modifying a pore-forming membrane protein which allows for a stimuli-responsive pore opening within the membrane of polymeric nanocompartments. We synthesized a diol-containing linker that selectively binds to the pores, blocking them completely. In the presence of an external stimulus (periodate), the linker is cleaved allowing the diffusion of substrate through the pores to the nanocompartment interior where it sets off the in situ enzymatic reaction. Besides the precise initiation of catalytic activity by opening of the pores, oxidation by periodate guarantees the cleavage of the linker under mild conditions. Accordingly, this kind of responsive nanocompartment lends itself to harboring a large variety of sensitive catalysts such as proteins and enzymes.
Introduction
Different nanocompartments are specifically engineered to protect their cargo while supporting a triggered release of the cargo or other functionalities in response to an environmental stimulus.1–6 The nanocompartments enable cargo release by a change of their architecture upon a stimulus, while they preserve their integrity when the aim is to trigger an in situ reaction.7–10 Such stimuli-responsive nanocompartments have an increasing potential for applications ranging from catalysis to therapeutics.11,12 Catalytic activity and selectivity are also greatly enhanced by confining catalytic sites in specific compartments.13 Moreover, there is a strong interest in the field of nanomedicine and other biological applications for the development of photoredox-active catalyst systems that can be operated in pure water. The ability to perform chemical reactions in aqueous media together with effective recycling strategies for catalysts represent keystones for sustainable and green chemistry. Polymer-derived nanocompartments, in particular polymersomes that support triggered reactions inside, are changing their bounding membrane upon the presence of a stimulus in their environment while preserving the architecture: the increased permeability favours the molecular flow across the compartment membrane, which will trigger an in situ functionality, for example by starting a specific reaction of the encapsulated active compounds. One strategy to generate stimuli-responsive polymersomes is to take advantage of stimuli-responsive copolymers to form nanocompartments that shrink or swell upon an external signal, e.g., pH,14 CO215,16 or a change in temperature.17,18 For example, polymersomes self-assembled from copolymers with tertiary, protonable amine groups show reversible swelling properties (breathing membrane) at distinct pH-values.14 Such pH-sensitive copolymers include e.g. poly[2-(diethylamino)ethyl methacrylate] (PDEAEMA), poly(2-[dimethylamino)ethyl methacrylate] (PDMAEMA), or poly[(N-amidino)dodecyl acrylamide] (PAD) as protonable blocks.14,19 Other polymers forming pH-responsive polymersomes are comprising polyacrylic acid (PAA)20,21 and poly(styrene sulfonate) (PSS).22 Another strategy, “bioinspired” by cellular membranes where numerous proteins mediate molecular transport across the membrane, hinges on incorporating proteins or small polypeptides into the polymersome membrane to achieve a desired permeability.23 For example, channel forming proteins such as the outer membrane protein F (OmpF), enable the diffusion of molecules with a cut-off largely corresponding to the pore diameter,24,25 while others, such as Ferrichrome outer membrane transporter (FhuA) or gramicidin, only allow diffusion of certain small molecules or ions.26,27 A step further in developing stimuli-responsive membrane permeabilization of polymersomes was achieved by turning inserted membrane proteins into “bio-valves” or “bio-locks” by corresponding modifications of the protein. For example, a pH-sensitive Gala3 peptide covalently bound to OmpF transformed the pores in the membrane of polymersomes into pH-triggerable valves that regulate the molecular flow to and from the compartment.28 In the random coil conformation (pH 7.4), Gala3 appeared not to perturb passage through the pore, while upon protonation (pH 5.5), peptides assume an α-helical structure that closed the pore, albeit only partially. Alternatively, permeability in response to a reducing environment was achieved in polymer-based artificial organelles by coupling a small molecular cap to cysteine residues of an OmpF double mutant: under reducing conditions, the cap was removed and the pores re-opened.29 Furthermore, polymersomes equipped with stimuli-responsive OmpF that encapsulate enzymes in their aqueous cavity served as catalytic nanocompartments (CNCs) whose activity was sensitive to open gates through which substrates could freely diffuse.28,29 Here we enlarge the concept of nanocompartments with stimuli-responsive in situ catalysis by developing a chemistry-based strategy for regulating substrate entry to the compartment. We rationally designed and synthesized a periodate sensitive linker to covalently bind to amino acids K89C/R270C of a double mutant of OmpF under mild conditions, thereby blocking the channel. We chose OmpF as a model membrane protein because it is a well characterized porin in terms of structural and functional characteristics.30 The linker was designed to crosslink two opposing cysteines located near the pore opening, and to be cleavable by periodate, an important oxidant for the selective oxidation of polysaccharides and glycopeptides.31,32 The advantages of periodate lie in its specificity, stability, and reactivity under neutral and mild conditions, and even more so in its compatibility with the preparation of a plethora of bioactive natural products and fine chemicals.33–36 Conditions used for the cleavage by periodate usually do not significantly perturb the structure of proteins37,38 although side reactions such as the cleavage of glycans, methionines, cysteines or the oxidation of N-terminal threonine and serine moieties may occur. In addition, there are different examples of enzymes with periodate tolerance, such as hydrolases39–41 or proteases.37,38 Considering the results published by several groups on chemical proteomics that reveal an efficient and selective cleavage of periodate-sensitive 1,2-diol linkers,37,38,42 we chose periodate for the oxidative cleavage of the 1,2-diol derived C–C bond in our linker because it presumably will not perturb OmpF function. 1,2-Diols are known to be oxidatively cleaved by sodium periodate via a cyclic transition state, in analogy with the oxidation by lead tetraacetate. The oxidation yields two carbonyl fragments, in this case two formyl-containing compounds.43 Besides, we were interested in synthesizing a water-soluble linker to enable applications without the use of organic solvents that are expected to harm membrane proteins.Results and discussion
OmpF monomer has the structure of a hollow beta barrel embedded in the membrane, with a diameter of around 30 Å (Fig. 1A), enabling diffusion of small (generally under 600 Da) water-soluble molecules16 in both directions. To achieve the chemical modification necessary to bind the linker, we used OmpF-M, a genetically modified porin that features two cysteine (Cys) residues opposing each other in the ‘eyelet’ region of the pore (OmpF double mutant K89C/R270C, OmpF-M).20 The thiol groups of these Cys moieties are exposed and can be readily modified (Fig. 1). To block OmpF pores by covalent binding of an organic compound and induce a controlled opening by periodate, different requirements have to be considered in terms of size and conformation of the linker, its accessibility and efficient cleavage. First, the steric size and conformation of the linker should efficiently block the pore, and second, both ends of the linker should be bound to OmpF to minimize the freedom of rotation. Thus, we opted to synthesize a small organic compound to provide a more rigid structure compared to coiled macromolecules where the spatial orientation and folding is more difficult to predict.44,45 Furthermore, the length of the linker was designed to span the diameter of the OmpF-M pore at the site of the two Cys moieties, i.e. approximately 30 Å, whereas a maleimide group at each linker end was inserted to favor the reaction with the thiol side chains of the two cysteines (Fig. 1C).46 Third, an important factor to consider in the linker design was the periodate-responsiveness that would result in its oxidative cleavage to unblock of the OmpF ‘gate’. Hence, we introduced a vicinal diol unit with a periodate-sensitive C–C bond in the middle of the symmetric linker (Fig. 1C, in blue). Periodate oxidizes the diol into the corresponding carbonyl groups under mild conditions, at pH = 7 and room temperature.25 Oxidation by periodate is predicted to produce two fragments able to rotate freely as they are bound only at one end to a Cys side chain. Hence, we anticipate that the periodate-triggered cleavage of the linker releases the OmpF pore obstruction. Finally, our linker design included two fluorophore (cyanine3 dye) side chains, both to create a bulkier structure for a more efficient pore blocking and to allow detection by fluorescence-based techniques. Synthesis of the periodate-responsive linker was performed in three consecutive steps (Scheme 1). We started from the vicinal diol element of a disuccinimidyl derivative of tartaric acid and symmetrically built up the linker on both sides. To achieve a final linker length of approximately 30 Å, we coupled two cysteine molecules to the respective ends of the reactive ester, to which we then covalently attached cyanine3 maleimides as fluorescent side chains via the thiol groups. The two functional maleimide end groups of the linker were integrated using a compound combining maleimide with a small hydrocarbon chain and hydrazide as the second end functional group. The hydrazide moiety reacted selectively with the carboxylate group of the cysteine fragments resulting in the final linker with a length of 30 Å (Fig. 1B). | ||
| Fig. 1 OmpF structure and linker design. (A) PyMol ribbon representation of OmpF-M secondary structure reveals a distance of about 30 Å between the side groups of the two amino acids replaced by Cys in OmpF-M. (B) The linker length corresponds to the distance between the two opposing Cys of OmpF-M, as estimated by PyMol modeling. (C) Chemical structure of the designed linker terminal maleimide groups, fluorophore side chains (pink) and the diol unit (blue). | ||
 | ||
| Scheme 1 Chemical synthesis of the periodate-responsive, fluorescent linker. | ||
In the first step, cysteine was coupled with disuccinimidyl tartrate (DST; a crosslinker with known periodate oxidation-induced cleavage properties)47 by the simple reaction of amine groups with the NHS-activated tartaric acid (Scheme 1). We assumed periodate to be the best choice of oxidant to selectively cleave the diol bond of the final linker as it reacts under mild conditions (i.e., no organic solvents or harsh additional substrates like acid or bleach required, but aqueous solutions, neutral pH, ambient temperature). Because oxidation of hydrazide bonds requires harsh conditions, we presume it unlikely that oxidative cleavage of the hydrazide groups occurs in our experimental system. In addition, compared to other oxidants, periodate is by itself a mild oxidant that presumably would not damage the sensitive structure of OmpF. This step was necessary to introduce the reactive thiol groups which were subsequently needed for coupling the fluorescent cyanine3 maleimide molecules. The first intermediate (compound 1) was only partially purified prior to being used for the synthesis of compound 2. The 1H-NMR spectrum of compound 1 showed the characteristic peaks of the coupled cysteine found in the range of 3.45–3.30 ppm and 4.65 ppm corresponding to the methylene (CH2–SH) and methine (NH–CH) groups, together with the two protons (CH–CH) of the vicinal diol unit at 4.7–4.6 pm. Additionally, the spectrum indicated the presence of monofunctional product, cysteine and free N-hydroxy succinimide. The 13C-NMR spectrum confirmed the presence of carbons around 180–170 ppm characteristic for carbonyl moieties from amides and carboxylic acids, the two carbons of the vicinal diol unit around 70 ppm, and the two carbon atoms of the cysteine connected to the tartaric acid by amide groups at around 50 ppm. Analysis by ESI-MS revealed a value of m/z = 179.05, which corresponds to double the protonated compound 1 [M + 2H]+ and could not be assigned to DST or cysteine. Compound 1 was used in the second reaction step aiming to induce its selective reaction with cyanine3 maleimide, resulting in compound 2. The 1H-NMR spectrum of compound 2 showed that possible impurities resulting from the synthesis of the first intermediate were efficiently removed during the purification step and did not lead to any other side products nor disrupt the reaction affording compound 2 (Fig. S1, ESI†). In addition to the protons of the vicinal diol element of tartaric acid and the cysteine fragments, we observed peaks in the 9.00–5.50 ppm range, characteristic for the heterocyclic and aromatic rings, as well as for the extended conjugation of the double bonds of the cyanine3 dye. The singlet peak characteristic for the two protons of the maleimide groups was not identified in the spectrum, indicating the complete coupling of cyanine3 maleimide and consequently the formation of compound 2. According to the 13C-NMR spectrum, the characteristic peaks of the carbon atoms from the carboxylic acids and amides were visible in the 180–170 ppm range. In addition, carbon peaks of cyanine3 dye were observed, including a peak at 168 ppm representing carboxamides, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups around 140 ppm, aromatic carbons around 120–110 ppm, aliphatic amines between 50–30 ppm, and the hydrocarbon chains in the 30–20 ppm range (Fig. S2, ESI†). Analysis of the product by MS (MALDI-TOF) revealed an m/z value of [M + 2H]+ = 379.16, corresponding exactly to compound 2. In the last synthesis step, the carboxylic acid groups of compound 2 were activated by EDC to form the amine-reactive O-acyl isourea intermediate. Subsequently, the reaction with N-ε-maleimidocaproic acid hydrazide (EMCH) led to the formation of a symmetric bismaleimide linker, as confirmed by the NMR spectra (Fig. S3 and S4, ESI†). In addition to the peaks that were already observed for compound 2, the 1H-NMR spectrum showed peaks between 2.0–1.0 ppm, characteristic for protons of the two hydrocarbon chains that were introduced by reacting compound 2 with EMCH. The hydrazide group of the crosslinker EMCH commonly generates a very broad peak between 4.5–3.5 ppm with an integral of 4, which was not found in the 1H-NMR spectrum of our final product. The 13C-NMR spectrum of the final linker indicated its large hydrocarbon backbone and showed the characteristic peaks of the two carboxamines carbons from the coupled maleimide groups around 165 ppm. Interestingly, the peaks of the aromatic carbons from the fluorophore were no longer detected. However, we concluded that this absence represents an effect of a decreased solubility in CDCl3 when the linker was dissolved in the NMR-tube (Fig. S4, ESI†). Additionally, MALDI-TOF revealed an m/z value of [M + H]+ = 643.68, which corresponds exactly to the bismaleimide linker.
C groups around 140 ppm, aromatic carbons around 120–110 ppm, aliphatic amines between 50–30 ppm, and the hydrocarbon chains in the 30–20 ppm range (Fig. S2, ESI†). Analysis of the product by MS (MALDI-TOF) revealed an m/z value of [M + 2H]+ = 379.16, corresponding exactly to compound 2. In the last synthesis step, the carboxylic acid groups of compound 2 were activated by EDC to form the amine-reactive O-acyl isourea intermediate. Subsequently, the reaction with N-ε-maleimidocaproic acid hydrazide (EMCH) led to the formation of a symmetric bismaleimide linker, as confirmed by the NMR spectra (Fig. S3 and S4, ESI†). In addition to the peaks that were already observed for compound 2, the 1H-NMR spectrum showed peaks between 2.0–1.0 ppm, characteristic for protons of the two hydrocarbon chains that were introduced by reacting compound 2 with EMCH. The hydrazide group of the crosslinker EMCH commonly generates a very broad peak between 4.5–3.5 ppm with an integral of 4, which was not found in the 1H-NMR spectrum of our final product. The 13C-NMR spectrum of the final linker indicated its large hydrocarbon backbone and showed the characteristic peaks of the two carboxamines carbons from the coupled maleimide groups around 165 ppm. Interestingly, the peaks of the aromatic carbons from the fluorophore were no longer detected. However, we concluded that this absence represents an effect of a decreased solubility in CDCl3 when the linker was dissolved in the NMR-tube (Fig. S4, ESI†). Additionally, MALDI-TOF revealed an m/z value of [M + H]+ = 643.68, which corresponds exactly to the bismaleimide linker.
Next, we mixed the bifunctional linker with purified recombinant OmpF-M in water to couple the maleimide groups of the linker to the thiol groups of the Cys residues in OmpF and close the OmpF pore. After the labeling reaction, an aliquot of the linker-OmpF-M solution was heated to 95 °C to denature all intramolecular interactions except covalent bonds, and then analyzed by SDS-PAGE (Fig. S5, ESI†). We observed a single fluorescent band running with the apparent molecular weight of monomeric OmpF-M (about 40 kDa,48 lanes 3, 4), which indicated a covalent binding of the fluorescent linker to the OmpF-M. As unspecific aggregation of the linker with the protein was not apparent, we concluded that covalent binding was successful.
We were interested to establish whether the linker attachment to the OmpF-M was able to (i) efficiently close the pore and (ii) control the molecular flow through the OmpF pore in a precise manner. Hence, we prepared several types of polymersomes encapsulating laccase in their aqueous cavity, henceforth termed catalytic nanocompartments (CNCs), as model for stimuli-responsive regulation of enzyme activity: (i) CNCs without OmpF-M pores (CNC-noOmpF-M), (ii) CNCs with inserted linker-modified OmpF-M (CNC-linker-OmpF-M), (iii) CNCs with inserted OmpF-M lacking the linker modification (CNC-OmpF-M), and (iv) CNCs with wild-type OmpF inserted in their membranes (CNC-OmpF-WT). We used the film rehydration method49 to form polymersomes and CNCs from poly(2-methyl-2-oxazoline)-block-poly(dimethyl-siloxane)-block-poly(2-methyl-2-oxazo-line), PMOXA11-b-PDMS104-b-PMOXA11. Such amphiphilic triblock copolymers (PMOXA-b-PDMS-b-PMOXA) are known to self-assemble into polymersomes with membranes that are sufficiently flexible to integrate OmpF despite the hydrophobic mismatch.50,51 The average number of MOXA and DMS repeating units in PMOXA11-b-PDMS104-b-PMOXA11 was assessed by proton nuclear magnetic resonance (1H-NMR) spectroscopy (Fig. S6, ESI†). This method also allowed for obtaining the number-average molecular weight of this copolymer (Mn = 9800 g mol−1), whereas a dispersity (D) of 1.23 was revealed by gel permeation chromatography (GPC) (Fig. S7, ESI†). The size and architecture of CNCs were characterized by transmission electron microscopy (TEM), fluorescence correlation spectroscopy (FCS), and light scattering (LS) (Fig. 2 and Table 1). All three techniques indicated a hollow sphere architecture with a diameter of around 200 nm with appropriate homogeneity (polydispersity index, PDI = 0.2).
 | ||
| Fig. 2 Insertion of linker-modified OmpF-M in CNC membranes. Ultrastructural morphology of (A) CNCs without OmpF, (B) CNCs with linker-OmpF-M inserted in the membrane, (C) CNCs with OmpF-M, and (D) OmpF wild-type. Scale bars: 200 nm. (E) FCS autocorrelation curves (solid line) and raw data (dots) of PBS solutions of the linker (black), standalone linker-OmpF-M in 1% OG (blue), and CNCs with linker-OmpF-M inserted in the membrane (red). | ||
| CNC-noOmpF | CNC-linker-OmpF-M | CNC-OmpF-M | CNC-OmpF-WT | |
|---|---|---|---|---|
| PDI | 0.2 | 0.2 | 0.2 | 0.2 |
| R h (nm) | 138 | 118 | 119 | 121 |
| R g (nm) | 134 | 113 | 101 | 101 |
| Ratio | 1.0 | 0.9 | 0.8 | 0.8 |
| Plot | Guinier | MIE | MIE | Guinier |
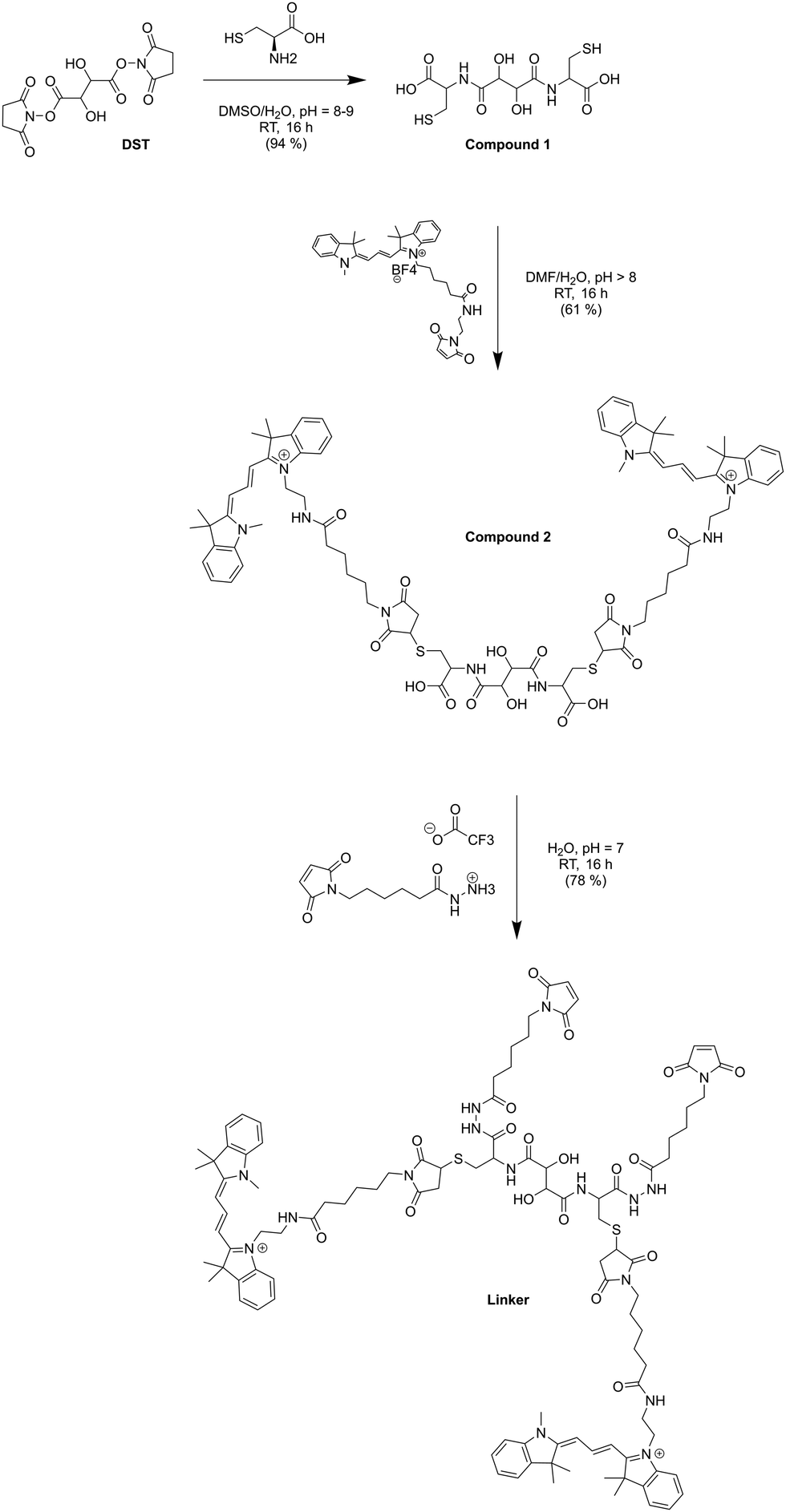
TEM micrographs revealed 200 nm structures characteristic for collapsed vesicles (Fig. 2A–D). We used static (SLS) and dynamic light scattering (DLS) to obtain the radius of gyration (Rg) and radius of hydration (Rh) for all CNCs (Table 1). CNC-linker-OmpF-M, CNC-OmpF-M and CNC-OmpF-WT were in a similar range of size with Rh of around 120 nm and Rg of 100–110 nm, while CNC-noOmpF were slightly bigger (Rh = 138, Rg = 134 nm). The calculated ratios of Rh/Rg ranged between 0.8–1.0, presenting typical values for hollow sphere architectures linker to OmpF-M was assessed by FCS (Fig. 2E). The autocorrelation curves revealed significant shifts in the diffusion time values of standalone linker-OmpF-M relative to free linker and linker-OmpF-M permeabilized CNCs relative to standalone linker-OmpF-M (Table S1, ESI†). These characteristic increases in diffusion time values indicate a successful coupling of the linker to OmpF-M and a successful insertion of the linker-OmpF-M in the membrane of polymersomes. In addition, we used brightness measurements to evaluate the number of linker molecules/OmpF pore. With the counts per molecules given in Table S1 (ESI†), we calculated that one linker molecule is bound per OmpF-M monomer and obtained an average number of 9 reconstituted linker-OmpF-M molecules per CNC. We next assayed the activity of encapsulated laccase in CNCs after treatment with NaIO4, quenching with glucose and SEC purification. To determine laccase activity, we used 2,6-dimethoxyphenol (DMP; MW 154.16), a substrate that is small enough to diffuse through OmpF pores. Laccase catalyses the oxidation of DMP to the corresponding radical species. The resulting orange radical can be detected spectrophotometrically at 470 nm.52,53 As CNCs self-assembled from PMOXA–PDMS–PMOXA triblock co-polymers show a characteristic absorption at 280 nm,54 and the fluorescent linker at 550 nm, their interference with oxidized DMP (470 nm) could be largely excluded. The periodate-triggered unblocking of OmpF-M pores in CNCs was evaluated by measuring the absorbance related to DMP oxidation for several hours (Fig. 3). When DMP was added to CNC-linker-OmpF-M in the absence of NaIO4, where OmpF pores should be blocked, the enzyme activity was similarly low as for CNCs lacking OmpF. This data confirms that the pores are blocked by the linker and thus the substrate is unable diffuse through the pores to reach the enzyme. NaIO4 treatment of CNC-linker-OmpF-M led to a significantly higher absorbance at 470 nm as cleavage of the linker unblocked the pores and DMP was able to diffuse into the cavity where it was oxidized to the coloured product (Fig. 3, green curve). Periodate treatment of CNC-noOmpF-M did not yield notable absorbance, indicating that NaIO4 by itself did not damage the CNCs (Fig. 3, red curve) which would have otherwise led to a release of laccase and to an enzyme-catalysed oxidation of DMP. In all laccase activity assays, periodate treatment was followed by quenching with glucose to avoid false positive signals from periodate-induced oxidation reactions. Our spectrophotometric results clearly show that while the linker closes OmpF pores efficiently, a stimuli-responsive opening is achieved by periodate-triggered linker cleavage. Furthermore, the increased absorbance over several hours demonstrated that periodate did not affect the activity of encapsulated laccase.
 | ||
| Fig. 3 Linker-based control of laccase activity in CNCs in response to NaIO4. (A) Schematic representation of periodate-induced linker cleavage, activating in situ catalysis. Addition of NaIO4 cleaves the linker and thereby unblocks the pore. In the open state, substrate enters the compartment where it is oxidized to a detectable product by the confined laccase. (B) An increase in absorbance at 470 nm reflects laccase-catalyzed DMP conversion in NaIO4-treated CNC-linker-OmpF-M. Laccase activity was minimal in CNCs lacking OmpF or in CNC-linker OmpF-M in the absence of NaIO4 (closed pores). Measurements were carried out at pH 7.4 for 9 hours at RT. | ||
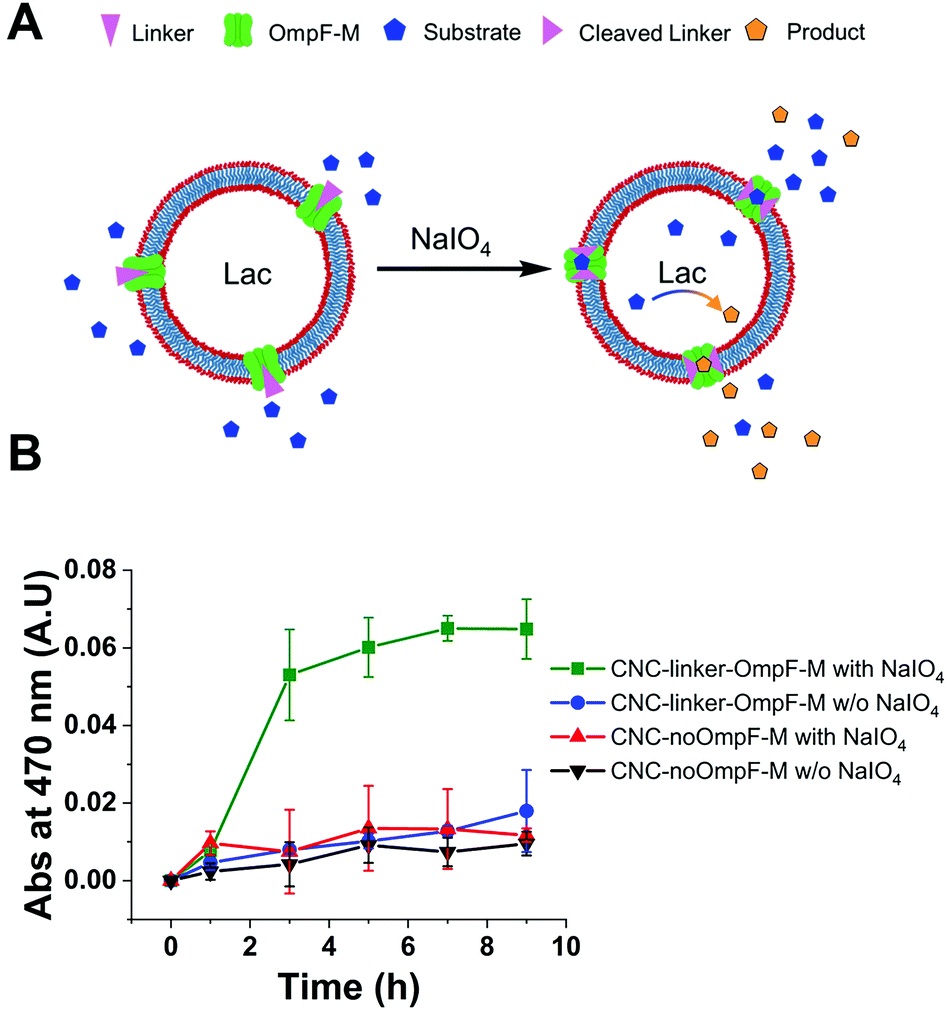
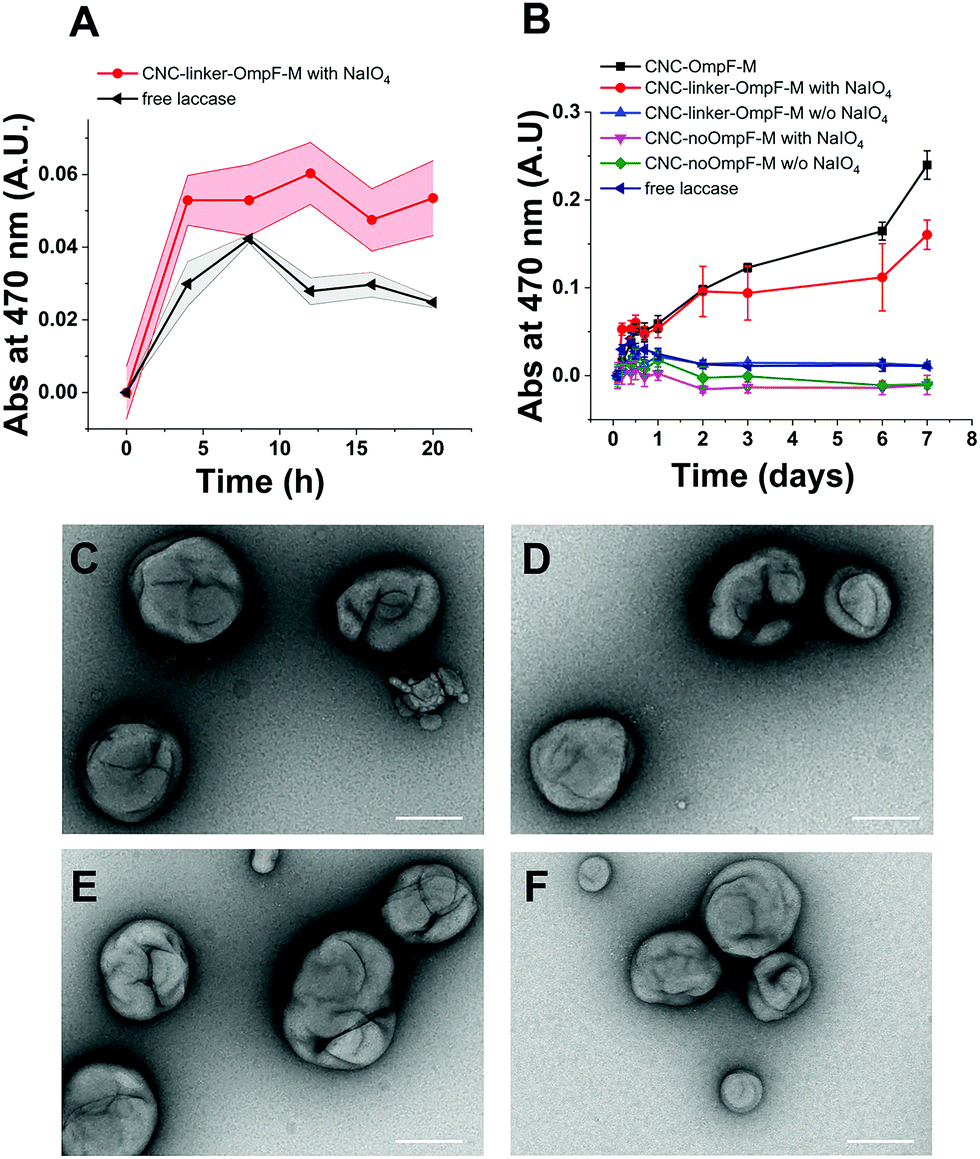
To test how long triggered laccase activity persists when protected by the polymersome membrane, we measured DMP of free laccase reached a maximum after 8 h and then continuously decreased over the following 12 h (Fig. 4A). In contrast, enzymatic activity of CNCs whose membrane was permeable to DMP either by unmodified OmpF-M pores (CNC-OmpF-M) or by periodate-induced opening of linker-OmpF-M pores (CNC-linker-OmpF-M), increased for seven days (Fig. 4B). Again, without OmpF pores in the membrane, NaIO4 treatment (CNC-no-OmpF with NaIO4) did not lead to the oxidation of DMP, nor did it affect CNC morphology, as shown by TEM micrographs (Fig. 4C–F). For comparison and to rule out that the increase in absorbance was related to an unspecific effect of NaIO4 on DMP, we also tested laccase activity of corresponding CNCs using 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid diammonium salt) (ABTS) as substrate (Fig. S8, ESI†). These enzyme assays were performed over several months, indicating that CNCs were active over long time periods if stored at 4 °C. Because DMP exhibited notably lower auto-oxidation42 over 9 h at pH 7.4, it was more suitable for long-term measurements of NaIO4-induced CNC catalysis (Fig. 4). ABTS on the other hand, as non-phenolic substrate is reported to be a more efficient substrate under acidic conditions.55 Further evidence of the longevity of CNCs encapsulating laccase was provided by TEM micrographs after eleven months which revealed an unchanged morphology (Fig. 5). Long-term stability of CNCs represents a key aspect for future applications of this system.
 | ||
| Fig. 4 Long-term laccase activity of CNCs in response to NaIO4. Absorbance intensity of the laccase product was measured at 470 nm. Measurements were carried out in triplicate (±SD) at pH 7.4 and RT. (A) Activity of free laccase in PBS compared to laccase encapsulated in CNC-linker-OmpF in the presence of NaIO4 over 20 h. (B) Activity measured for one week of free laccase in PBS compared to CNCs with and without periodate-cleaved pores or CNCs lacking pores. Periodate treatment had no effect on the ultrastructural morphology of (C) CNC-linker-OmpF-M compared to (D) CNC-no OmpF after 1 week nor after 3 weeks (E and F, respectively). Scale bars: 200 nm. | ||
 | ||
| Fig. 5 Longevity of CNC morphology. TEM micrographs of CNCs encapsulating laccase with (A) unmodified OmpF-M inserted in the compartment membrane, and (B) linker-OmpF-M reveal an intact morphology after 11 months at 4 °C. Scale bars: 500 nm. | ||
Materials and methods
Disuccinimidyl tartrate (DST) and 3,3′-N-[ε-maleimido caproic acid] hydrazide, trifluoroacetic acid salt (EMCH) were purchased from Thermo Fisher, cyanine3 maleimide from Lumiprobe. All other materials were purchased from Sigma Aldrich. All reagents were of the highest commercially available grade and were used without any further purification, unless stated otherwise.Synthesis of amphiphilic triblock copolymer
The amphiphilic block copolymer PMOXA11-b-PDMS104-b-PMOXA11 was previously synthesized in our group according to reported protocols.56,57 Briefly, a hydroxyl-terminated bifunctional PDMS was reacted below −10 °C with trifluoromethanesulfonic anhydride in dry hexane, leading to the activated PDMS macroinitiator. After filtration under argon and removal of hexane under vacuum, dry chloroform was added as reaction solvent, in which dry 2-methyl-2-oxazoline (MOXA) reacted with the macroinitiator via symmetric cationic ring-opening polymerization. The hydroxyl-terminated triblock copolymer was obtained after quenching the polymerization reaction with a water/ethanol mixture. A number-average molecular weight (Mn) of 8700 g mol−1 and a chemical composition of PMOXA12-b-PDMS87-b-PMOXA12 was obtained from the 1H-NMR spectrum of this triblock copolymer, whereas its dispersity (Đ) of 1.6 was revealed by gel permeation chromatography (GPC) in tetrahydrofuran (THF).57 To decrease the dispersity and thereby better control the shape morphology of the self-assembled structures, the copolymer was extracted with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) hexane/methanol mixture and the copolymer fraction with a longer PDMS block was recovered from the hexane phase after solvent removal and characterized by 1H NMR (Fig. S6, ESI†) and SEC (Fig. S7, ESI†). Chemical structure determination yielded PMOXA11-b-PDMS104-b-PMOXA11 with Mn = 9800 g mol−1 (from 1H-NMR) and Đ = 1.23 (from SEC).
:1 (v/v) hexane/methanol mixture and the copolymer fraction with a longer PDMS block was recovered from the hexane phase after solvent removal and characterized by 1H NMR (Fig. S6, ESI†) and SEC (Fig. S7, ESI†). Chemical structure determination yielded PMOXA11-b-PDMS104-b-PMOXA11 with Mn = 9800 g mol−1 (from 1H-NMR) and Đ = 1.23 (from SEC).
1H NMR (500 MHz, CDCl3) δ/ppm = 3.84–3.15 (bm, 8H), 2.30–1.96 (bm, 6H), 1.64–1.53 (bm, 4H), 1.38–1.26 (bm, 4H), 0.50–0.60 (m, 4H), 0.20–0.10 (bs, 6H).
Synthesis and characterization of the bismaleimide linker
To obtain the thiol-reactive linker able to covalently bind to both thiol groups of the OmpF double mutant (K89C/R270C), the disuccinimidyl-activated derivative of tartaric acid was used as diol-containing starting compound. For synthesis of the first intermediate (compound 1), an established procedure for crosslinking proteins was followed with small modifications.47 Briefly, DST (10.7 mg, 31 mmol, 1.0 eq.) was dissolved in 100 μL DMSO in an Eppendorf tube (1.5 mL) and then a 0.24 M aqueous solution containing L-cysteine was added (284 μL, 68.2 mmol, 2.2 eq.). After the pH was adjusted to a value between 8 and 9 with 1 M NaOH (aqueous solution), the reaction mixture was shaken in the dark for 16 h at RT. The resulting suspension was centrifuged (13.400 rpm, 5 min) and the supernatant was collected. The solvent was removed using a rotary evaporator and the resulting white solid was used for the synthesis of the second intermediate (compound 2).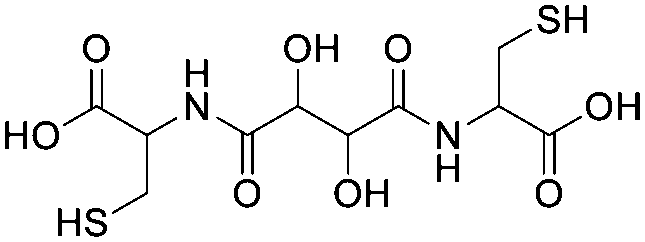
MF: C10H16N2O8S2, MW: 356.36, ESI-MS m/z calculated for C10H16N2O8S2 [M + 2H]+: 179.02; found: 179.05.
1H-NMR (D2O, 500 MHz): δ/ppm = 4.68 (d), 4.65 ppm (d), 3.45–3.30 (dd, 4H).
13C-NMR (D2O, 126 MHz): δ/ppm = 176.49, 173.51, 162.97 (d, J = 35.5 Hz), 72.45, 72.35 (d, J = 7.0 Hz), 54.87, 53.49, 38.72, 37.55, 25.17.
The synthesis of compound 2 was adapted and modified from a published study.58 2.8 mg compound 1 were dissolved in 200 μL of a 1/1 mixture of H2O/DMF, pH > 8, and 10.7 mg cyanine3 maleimide (16.0 mmol) dissolved in 400 μL DMF were added. After adjusting the pH value back to 8 with 1 M NaOH, the reaction mixture was shaken in the dark for 16 h at RT. The solvent was removed by adding an excess of toluene and subsequent rotary evaporation. To the crude product, 10 μL H2O with a pH > 8 (adjusted with 1 M NaOH) was added. The aqueous layer was extracted 3 times with CH2Cl2. The organic phases were collected, and the solvent was evaporated to give compound 2 as pink solid (7.64 mg, 4.9 mmol, 61%).
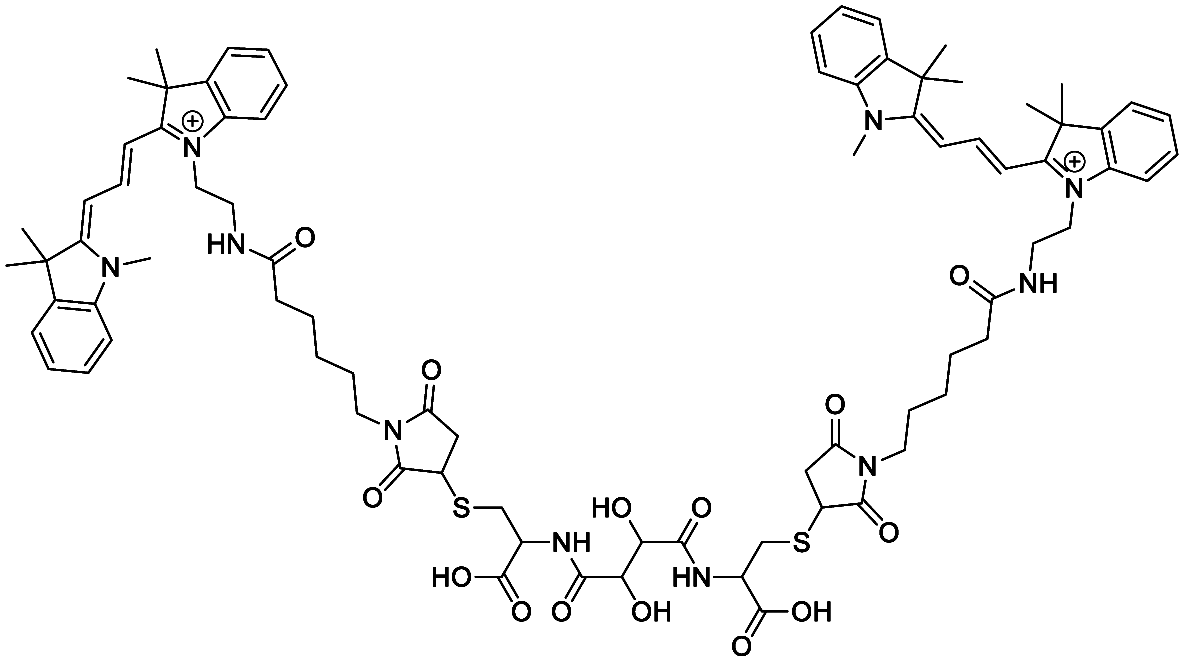
MF (cation): C82H102N10O14S2, MW: 1515.87 (cation), ESI-MS m/z calculated for C82H102N10O14S2 [M + 2H]+: 379.18; found: 379.16.
1H-NMR (500 MHz, CDCl3): δ/ppm = 11.35 (s, 2H), 8.38 (t, J = 13.4 Hz, 2H), 7.45–7.32 (m, 9H), 7.17 (d, J = 8.0 Hz, 2H), 7.12 (d, J = 8.0 Hz, 2H), 6.70 (t, J = 13.9 Hz, 4H), 6.41 (d, J = 13.1 Hz, 2H), 5.82 (d, J = 13.1 Hz, 2H), 4.17 (t, J = 7.6 Hz, 4H), 3.69 (s, 6H), 3.52–3.39 (m, 7H), 2.30 (t, J = 6.8 Hz, 4H), 2.23 (d, J = 10.4 Hz, 8H), 1.84 (p, J = 7.8 Hz, 4H), 1.80–1.75 (m, 4H), 1.72 (d, J = 5.5 Hz, 22H), 1.56–1.41 (m, 4H).
13C-NMR (126 MHz, CDCl3): δ/ppm = 174.73, 174.57, 174.04, 171.47, 168.05, 151.03, 143.03, 142.15, 140.97, 140.69, 138.71, 129.47, 129.34, 128.15, 126.04, 125.79, 122.43, 122.40, 111.80, 111.10, 104.46, 104.18, 77.67, 77.41, 77.16, 49.61, 49.32, 44.84, 39.84, 39.47, 36.74, 31.88, 28.50, 27.47, 26.05, 25.53.
The bismaleimide linker was synthetized according to the general procedure used for chemical labelling with small modifications.59 Briefly, 3,3′-N-[ε-maleimido caproic acid] hydrazide, trifluoroacetic acid salt (EMCH, 4.2 mg, 12.3 mmol, 2.5 eq.) was dissolved in 300 μL H2O, and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC·HCl, 4.7 mg, 24.5 mmol, 5 eq.) in 100 μL H2O. A solution of compound 2 (7.6 mg, 4.9 mmol, 1 eq.) was prepared in H2O/DMF (1:1000, 3 mL). All three solutions were combined, and then the pH was adjusted to approximately 7.0 with 1 M NaOH. The reaction mixture was stirred for 16 h in the dark at RT. The crude product was purified by extraction with dichloromethane (DCM, 3×). The organic layers were combined, and the solvent was evaporated to yield the bifunctional linker as pink solid (8.0 mg, 3.8 mmol, 78%).
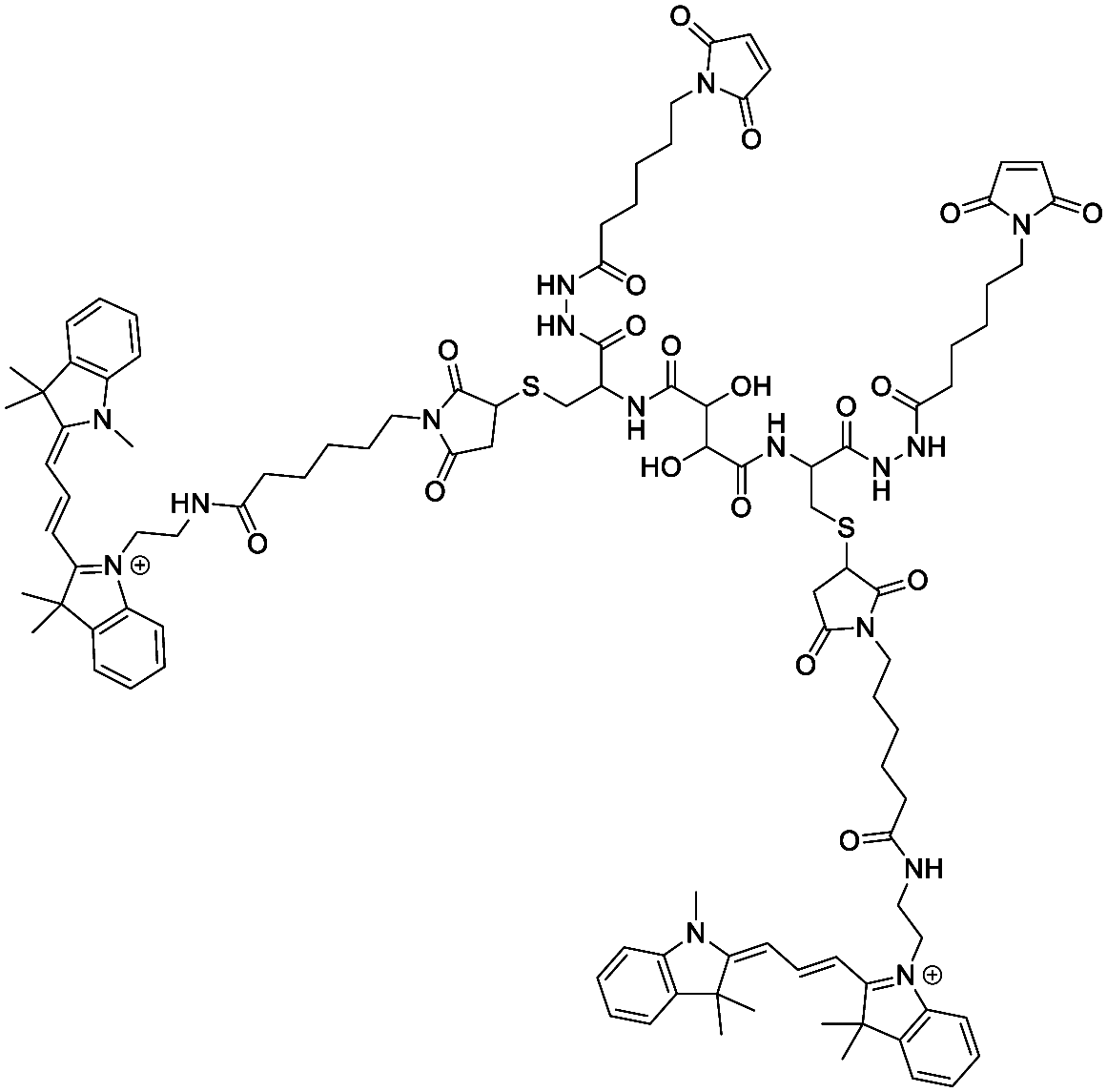
MF (cation): C102H128N16O18S2, MW (cation): 1930.36, ESI-MS m/z calculated for C102H128N16O18S2 [M + H]+: 643.64; found: 643.68.
1H-NMR (500 MHz, CDCl3): δ/ppm = 7.71 (d, J = 16.0 Hz, 1H), 7.58–7.49 (m, 3H), 7.46–7.30 (m, 9H), 6.87–6.67 (m, 2H), 6.46 (d, J = 16.0 Hz, 2H), 6.15 (t, J = 1.3 Hz, 2H), 5.62–5.53 (m, 1H), 5.29 (s, 9H), 4.51–4.39 (m, 6H), 4.22–3.96 (m, 1H), 3.75–3.58 (m, 4H), 3.52–3.29 (m, 3H), 2.95 (s, 1H), 2.88 (s, 1H), 2.41–2.10 (m, 4H), 1.99–1.85 (m, 15H), 1.72 (d, J = 5.9 Hz, 16H), 1.41 (s, 3H), 1.32–1.17 (m, 13H), 1.10 (s, 8H), 0.90–0.79 (m, 3H), 0.07 (s, 1H).
13C-NMR (126 MHz, CDCl3): δ/ppm = 167.20, 166.71, 145.43, 135.96, 134.29, 130.45, 128.94, 128.93, 128.16, 126.11, 117.54, 77.24, 62.52, 62.21, 59.53, 53.44, 38.15, 31.24, 29.71, 28.13, 18.29, 14.13, 1.02.
Recombinant OmpF production
Both OmpF wild type and the OmpF-M double mutant (OmpF K89C/R270C) were expressed and purified following a published procedure.28 Protein purity was evaluated on a 12% SDS-PAGE gel, and the concentration measured spectrophotometrically at λ = 280 nm with Nanodrop (Witec Ag). Wild type and mutant OmpF were stored at 4 °C in 3% n-octyl-β-D-glucopyranoside (OG) at a concentration of around 3 mg mL−1 for several months. Before use, OmpF was dialysed against 1 L of 0.05% OG in PBS for 16 h, then twice against phosphate-buffered saline (PBS) for 2 h. After the dialysis, the protein concentration was adjusted to 2.0 mg mL−1 with PBS.Chemical modification of OmpF-M
OmpF-M was diluted with PBS containing 3% OG to a concentration of 1 mg mL−1 (500 μL, 25 mM, 12.5 nmol). Next, a solution of the linker in MilliQ-H2O (7.5 mM) was added (2.5 μL, 19 nmol). The mixture was stirred overnight in the dark at RT. Sample and detergent concentration of the OmpF-M labeling mix was reduced by centrifuging 12 times with 1% OG in PBS through an Amicon Ultra-0.5 mL centrifugal filter for protein purification and concentration, molecular cut-off 10 kDa (Millipore). Subsequently, the volume of linker-OmpF-M suspension was adjusted to 450 μL with 1% OG in PBS, transferred to a dialysis tubing with a 14 kDa molecular cut-off (Carl Roth), and dialyzed against 1% OG solution in PBS for 16 h, followed by 2 × 2 h dialysis against PBS. After dialysis, the protein concentration of linker-OmpF-M was measured spectrophotometrically at λ = 280 nm with Nanodrop (Witec Ag) and adjusted to 2.0 mg mL−1.Characterization of linker-OmpF-M
2.5–5 μL of OmpF WT, OmpF-M and linker-OmpF-M were each mixed with 4× Laemmli loading buffer (2.5 μL) containing mercaptoethanol (0.7 mM). The volume was adjusted to 12.5 μL with 1% OG solution and samples denatured at 95 °C for 5 min. Samples (10 μL) were loaded on a 4–20% precast polyacrylamide gel (Mini-PROTEAN® TGX™) and ran at 200 V for 45 min (Fig. S5, ESI†). Before Coomassie staining, the gel was imaged by Bio-Rad GelDoc to observe the fluorescence emitted by the linker (fluorescence of cyanine3 maleimide: λex = 550 nm/λem = 580 nm).Preparation of laccase containing CNCs permeabilized by OmpF-M membrane insertion
5 mg PMOXA11-b-PDMS104-b-PMOXA11 triblock copolymer were dissolved in 186 μL EtOH. 154 μL of dialyzed OmpF (2.0 mg mL−1) or dialyzed OG solution (3%) as control, were added. After evaporating the solvents by a rotary evaporator (155 rpm, 40 °C, 175 mbar for 10 min, then vacuum for 15 min), the polymer films were rehydrated with 1 mL laccase from Agaricus bisporus dissolved in PBS (4.6 μg mL−1). For self-assembly of nanocompartments, the mixture was stirred overnight at RT. After extrusion (200 nm polycarbonate filters), CNCs were purified by size exclusion chromatography (SEC) and subsequently incubated with proteinase K (1 mg mL−1, 1–2 μL per 100 μL) at 37 °C for 3 hours to remove non-encapsulated laccase.Characterization of CNCs
The size and morphology of polymersomes were evaluated by a combination of static and dynamic light scattering (SLS, DLS), transmission electron microscopy (TEM) and fluorescence correlation spectroscopy (FCS). Quantification of reconstituted linker-OmpF-M was carried out by FCS measurements. All three techniques revealed a vesicular, hollow sphere structure of the CNCs with a size of around 200 nm. Additionally, we calculated the value for Rh based on the diffusion times of BODIPY stained vesicles measured by FCS (Table S1, ESI†). With a diffusion time of around 6000 μs, we received an Rh of 92.1 nm, resulting in a polymersome diameter of 184.2 nm, which is in agreement with the dimensions obtained by TEM and LS (Fig. 2). Cryo-TEM images of vesicles formed by PMOXA11-b-PDMS104-b-PMOXA11 revealed a membrane thickness of around 15 nm (Fig. S10, ESI†). To exclude unspecific binding of linker-OmpF-M to the CNC membrane, FCS measurements were performed with linker-OmpF-M added to preformed CNC-noOmpF (Fig. S11 and Table S1, ESI†). Although FCS measurements revealed an increased diffusion time compared to the free linker, the shift of fitted autocorrelation curves was less pronounced than that observed for linker-OmpF-M reconstituted into CNCs. In addition, to confirm that the detergent (1% OG) used to solubilize linker-OmpF-M did not disrupt CNCs, we added hydrophobic BODIPY 630/650-X to stain the nanocompartment membrane and measure the vesicle diffusion time by FCS.DLS/SLS
Light scattering experiments were performed using an ALV goniometer (ALV GmbH, Germany) equipped with an ALV He–Ne laser (JDS Uniphase, wavelength λ = 632.8 nm). Following film rehydration, samples (10 μL of self-assembled nanocompartments added to 990 μL PBS) were measured in a 10 mm cylindrical quartz cell at angles ranging from 35° to 135° at 293 K with angular steps of 5°. ALV/static and dynamic FIT and PLOT program version 4.31 10/10 were used for data analysis. Static light scattering (SLS) data were processed according to the MIE and Guinier plotting models.TEM
Nanocompartment solutions were diluted in a ratio of 1:3 with PBS. 5 μL aliquots were deposited on a carbon-coated copper grid and negatively stained with 2% uranyl acetate solution. The samples were examined with a transmission electron microscope (Philips CM-100) operated at 80 kV.
Cryo-TEM
Aliquots of the samples (4 μL) were deposited on a holey carbon-coated grid (Lacey, Tedpella, USA), and vitrified into liquid ethane at −178 °C using a Leica GP plunger (Leica, Austria). The frozen samples were examined with a Talos electron microscope (FEI, USA) operated at 200 kV.NTA
Nanoparticle tracking analyses (NTA) were performed using a NanoSight NS3000 equipped with a blue (488 nm) and green laser (532 nm). The samples were diluted at 1:1000 in PBS to avoid concentration-related interference. The CNCs were excited with the blue laser and their emission was detected by using appropriate filters. The average particle concentration was calculated using the Stokes–Einstein equation for spherical particle suspensions. The hydrodynamic radius distribution was analysed by NanoSight's single particle tracking system.
FCS
All FCS measurements were carried out using a confocal laser scanning microscope (ZEISS LSM 880, Carl Zeiss, Germany) equipped with a water immersion objective (C-Apochromate 40×/1.2 W korr FCS M27) using the Zen Black software. A HeNe laser (561 nm or 633 nm) was used to excite the samples. The measurements were carried out at room temperature using a sample volume of 20 μL. Measurements were recorded over 10 s, and each measurement was repeated 30 times. The diffusion times of the free dye (100 nM cyanine3 maleimide), the linker and the different types of CNCs were measured independently. The autocorrelation function was calculated using a software correlator and fitted with a one component fit. Less than 10% of the correlation curves could not be fitted and were excluded from the analysis.Enzyme encapsulation efficiency
To assess laccase encapsulation efficiency, we first determined the initial concentration of the laccase (MW: 65000 g mol−1,60 confirmed by SDS PAGE, Fig. S12, ESI†) in solution (4.60 μg mL−1) by BCA according to the supplier's protocol (Thermo Fisher Scientific, USA, Fig. S12, ESI†) and subsequently the concentration of the non-encapsulated in the eluate from SEC purification of CNC-noOmpF. The difference between the amount of laccase added for the formation of the CNCs and the amount of enzyme that was not encapsulated (3.70 μg), corresponded to the amount of laccase encapsulated in our CNCs-noOmpF. Dividing the total number of encapsulated laccase molecules by the number of vesicles (total volume 0.85 mL, 2.6 × 1011 ± 1.6 × 1010 vesicles per mL, measured by NTA; see Table S2, ESI†) yielded a value of 8–9 encapsulated laccase molecules per vesicle.
Opening of linker-OmpF-M
NaIO4 dissolved in MilliQ-H2O (0.3 M, 10 μL, 1 eq.) was added to 100 μL of CNCs and incubated at 37 °C. Assuming approximately 2 × 1010 CNCs in the periodate/CNC mixture (110 μL), we calculated 1.6 × 10−7 mmol or 3.5 × 10−5 mg periodate per vesicle. After 24 h, an excess of glucose dihydrate dissolved in MilliQ-H2O (0.6 M, 25 μL, 5 eq.) was added to quench NaIO4, and incubation at 37 °C was continued for 24 h. Subsequently, CNCs were purified by SEC and incubated with proteinase K (1 mg mL−1, 2 μL per 100 μL CNCs) to remove residual laccase attached to the outer surface of CNCs.Enzymatic assay with DMP
Aliquots of free laccase (1 pg in 5 μL, corresponding to the amount of laccase in 1 × 109 CNCs), CNCs without NaIO4-treatment (5 μL), and CNCs incubated with NaIO4 and glucose (20 μL, accounting for the dilution after SEC purification of treated CNCs) were added to 2,6-dimethoxyphenol (DMP) in PBS (20 mM, 10 μL) at pH 7.4 and adjusted to a final volume of 210 μL per well with PBS in a 96-well plate. The absorbance at 470 nm was measured with a Spectramax M5e microplate reader (Molecular Devices). The concentration of all CNC samples in PBS was determined by NTA (Table S2, ESI†) to perform the assays at comparable enzyme concentrations.Enzymatic assay with ABTS
Corresponding laccase activity measurements were carried out using 2,2′-azinobis[3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt (ABTS) as substrate. The oxidation product was monitored by determining the increase of absorbance at 420 nm using a Spectramax M5e microplate reader (Molecular Devices).Conclusions
By means of a specifically designed, periodate sensitive linker we achieved exquisite spatiotemporal control of catalysis in polymeric nanosized compartments with OmpF channels inserted in their membranes. Covalent coupling of the linker to OmpF pores efficiently blocked the entry of substrates to the compartment, preventing the catalytic activity of the enzyme confined inside. We have previously reported a related strategy to control OmpF pore function via a molecular cap that is cleaved off in response to reductive agents, leading to the opening of OmpF,30 whereas here, we developed a complementary approach based on a linker with an entirely different chemistry. Periodate-induced linker cleavage opened the ‘locks’ for the passage of substrates and catalysis occurred. Importantly, the catalytic reactions are induced under mild conditions (no organic solvents, at pH 7, RT) where sensitive biomolecules remain intact. Moreover, the periodate cleavage of the linker did not affect the overall CNC structure, ensuring the full functionality of our system. Another attractive feature of this well controllable system is that the catalytic activity is preserved over months. Our means of switching on confined catalysis pave the way to new molecular technology approaches which might be extremely useful in multifunctional cluster activity when specific substrates or products need to be made available at a specific site and with precise timing to a downstream catalyst. We believe that our linker-based, periodate responsive OmpF modification conferring controllable functionality and improved properties upon CNCs could advance the development of sensitive catalysts based on CNCs for a broad range of applications including catalysis, food science and nanotechnology.Author contributions
Conceptualization: L. Zartner, C. G. Palivan, formal analysis: L. Zartner, V. Maffeis, I. A. Dinu, investigation: L. Zartner, V. Maffeis, methodology: L. Zartner, supervision: C.-A. Schoenenberger, C. P. Palivan, validation: L. Zartner, visualization: L. Zartner, V. Maffeis, C.-A. Schoenenberger, writing – original draft: L. Zartner, V. Maffeis, C. A. Schoenenberger, writing – review & editing: I. A. Dinu, C.-A. Schoenenberger, C. G. Palivan.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support provided by the Swiss National Science Foundation (SNSF), the National Centre of Competence in Research Molecular Systems Engineering, and the University of Basel. Authors thank Prof. Dr Timm Maier (Biozentrum, University of Basel, Switzerland) for his advice on the linker design and Dr Voichita Mihali (Department of Chemistry, University of Basel, Switzerland) for TEM micrographs.Notes and references
- T. M. Reineke, ACS Macro Lett., 2016, 5, 14–18 CrossRef CAS.
- A. Abdollahi, H. Roghani-Mamaqani, B. Razavi and M. Salami-Kalajahi, Polym. Chem., 2019, 10, 5686–5720 RSC.
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu, B. Phillips, M. B. Carter, N. J. Carroll, X. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. Chackerian, W. Wharton, D. S. Peabody and C. J. Brinker, Nat. Mater., 2011, 10, 389–397 CrossRef CAS PubMed.
- T. Anajafi and S. Mallik, Ther. Delivery, 2015, 6, 521–534 CrossRef CAS PubMed.
- J. Leong, J. Y. Teo, V. K. Aakalu, Y. Y. Yang and H. Kong, Adv. Healthcare Mater., 2018, 7, 1701276 CrossRef PubMed.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Adv. Mater., 2009, 21, 2787–2791 CrossRef CAS.
- M. Lomora, G. Gunkel-Grabole, S. Mantri and C. G. Palivan, Chem. Commun., 2017, 53, 10148–10151 RSC.
- C.-Y. Xing, Y.-C. Fan, X. Chen, J.-S. Guo, Y. Shen, P. Yan, F. Fang and Y.-P. Chen, Sci. Total Environ., 2020, 717, 137030 CrossRef CAS PubMed.
- J. Song, W. He, H. Shen, Z. Zhou, M. Li, P. Su and Y. Yang, Chem. Eng. J., 2020, 390, 124488 CrossRef CAS.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, J. Am. Chem. Soc., 2009, 131, 13908–13909 CrossRef CAS PubMed.
- J. F. Mukerabigwi, Z. Ge and K. Kataoka, Chem. – Eur. J., 2018, 24, 15706–15724 CrossRef CAS PubMed.
- F. Eisenreich, E. W. Meijer and A. R. A. Palmans, Chem. – Eur. J., 2020, 26, 10355–10361 CrossRef CAS PubMed.
- I. J. Minten, V. I. Claessen, K. Blank, A. E. Rowan, R. J. M. Nolte and J. J. L. M. Cornelissen, Chem. Sci., 2011, 2, 358–362 RSC.
- S. Yu, T. Azzam, I. Rouiller and A. Eisenberg, J. Am. Chem. Soc., 2009, 131, 10557–10566 CrossRef CAS PubMed.
- L. Xu, N. Ren, J. Pang, H. Deng, X. Zhu, M. Sun and D. Yan, Polym. Chem., 2017, 8, 6283–6288 RSC.
- Q. Yan, J. Wang, Y. Yin and J. Yuan, Angew. Chem., Int. Ed., 2013, 52, 5070–5073 CrossRef CAS PubMed.
- H. Xu, F. Meng and Z. Zhong, J. Mater. Chem., 2009, 19, 4183–4190 RSC.
- F. Liu, V. Kozlovskaya, S. Medipelli, B. Xue, F. Ahmad, M. Saeed, D. Cropek and E. Kharlampieva, Chem. Mater., 2015, 27, 7945–7956 CrossRef CAS.
- B. Iyisan, J. Kluge, P. Formanek, B. Voit and D. Appelhans, Chem. Mater., 2016, 28, 1513–1525 CrossRef CAS.
- J. Qin, Q. Liu, J. Zhang, J. Chen, S. Chen, Y. Zhao and J. Du, ACS Appl. Mater. Interfaces, 2015, 7, 14043–14052 CrossRef CAS PubMed.
- H. C. Chiu, Y. W. Lin, Y. F. Huang, C. K. Chuang and C. S. Chern, Angew. Chem., Int. Ed., 2008, 47, 1875–1878 CrossRef CAS PubMed.
- J. Gaitzsch, D. Appelhans, L. Wang, G. Battaglia and B. Voit, Angew. Chem., Int. Ed., 2012, 51, 4448–4451 CrossRef CAS PubMed.
- M. Garni, S. Thamboo, C.-A. Schoenenberger and C. G. Palivan, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 619–638 CrossRef CAS PubMed.
- S. Lee, N. G. Housden, S. A. Ionescu, M. H. Zimmer, R. Kaminska, C. Kleanthous and H. Bayley, J. Am. Chem. Soc., 2020, 142, 12157–12166 CrossRef CAS PubMed.
- A. Belluati, I. Craciun, J. Liu and C. G. Palivan, Biomacromolecules, 2018, 19, 4023–4033 CrossRef CAS PubMed.
- M. Lomora, M. Garni, F. Itel, P. Tanner, M. Spulber and C. G. Palivan, Biomaterials, 2015, 53, 406–414 CrossRef CAS PubMed.
- O. Onaca, P. Sarkar, D. Roccatano, T. Friedrich, B. Hauer, M. Grzelakowski, A. Güven, M. Fioroni and U. Schwaneberg, Angew. Chem., Int. Ed., 2008, 47, 7029–7031 CrossRef CAS PubMed.
- C. Edlinger, T. Einfalt, M. Spulber, A. Car, W. Meier and C. G. Palivan, Nano Lett., 2017, 17, 5790–5798 CrossRef CAS PubMed.
- T. Einfalt, D. Witzigmann, C. Edlinger, S. Sieber, R. Goers, A. Najer, M. Spulber, O. Onaca-Fischer, J. Huwyler and C. G. Palivan, Nat. Commun., 2018, 9, 1127 CrossRef CAS PubMed.
- E. Yamashita, M. V. Zhalnina, S. D. Zakharov, O. Sharma and W. A. Cramer, EMBO J., 2008, 27, 2171–2180 CrossRef CAS PubMed.
- K. A. Kristiansen, A. Potthast and B. E. Christensen, Carbohydr. Res., 2010, 345, 1264–1271 CrossRef CAS PubMed.
- A. S. Perlin, in Advances in Carbohydrate Chemistry and Biochemistry, ed. D. Horton, Academic Press, 2006, vol. 60, pp. 183–250 Search PubMed.
- A. Sudalai, A. Khenkin and R. Neumann, Org. Biomol. Chem., 2015, 13, 4374–4394 RSC.
- W. Wang, L. Lu, K.-J. Wu, J. Liu, C.-H. Leung, C.-Y. Wong and D.-L. Ma, Sens. Actuators, B, 2019, 288, 392–398 CrossRef CAS.
- S. Sakamoto, G. Yusakul, Y. Tsuneura, W. Putalun, K. Usui, T. Miyamoto, H. Tanaka and S. Morimoto, Analyst, 2017, 142, 1140–1148 RSC.
- N. K. Dunlap, W. Mergo, J. M. Jones and J. D. Carrick, Tetrahedron Lett., 2002, 43, 3923–3925 CrossRef CAS.
- Y. Yang, H. Hahne, B. Kuster and S. H. Verhelst, Mol. Cell. Proteomics, 2013, 12, 237–244 CrossRef PubMed.
- A. Maurer, C. Zeyher, B. Amin and H. Kalbacher, J. Proteome Res., 2013, 12, 199–207 CrossRef CAS PubMed.
- E. Nyfeler, J. Grognux, D. Wahler and J.-L. Reymond, Helv. Chim. Acta, 2003, 86, 2919–2927 CrossRef CAS.
- Q. Zhu, M. Uttamchandani, D. Li, M. L. Lesaicherre and S. Q. Yao, Org. Lett., 2003, 5, 1257–1260 CrossRef CAS PubMed.
- K. L. Larsen, R. J. Andersen and J. Brask, Enzyme Microb. Technol., 2016, 91, 66–71 CrossRef CAS PubMed.
- Y. Yang and S. H. L. Verhelst, Chem. Commun., 2013, 49, 5366–5368 RSC.
- B. Sklarz, Q. Rev., Chem. Soc., 1967, 21, 3–28 RSC.
- D. Y. Shroder, L. G. Lippert and Y. E. Goldman, Methods Appl. Fluoresc., 2016, 4, 042004 CrossRef PubMed.
- S. Tyagi and E. A. Lemke, in Methods in Cell Biology, ed. P. M. Conn, Academic Press, 2013, vol. 113, pp. 169–187 Search PubMed.
- T. E. Gartner and A. Jayaraman, Macromolecules, 2019, 52, 755–786 CrossRef.
- N. Geisler, FEBS Lett., 1993, 323, 63–67 CrossRef CAS PubMed.
- X. Wang, D. Teng, Q. Guan, R. Mao, Y. Hao, X. Wang, J. Yao and J. Wang, AMB Express, 2017, 7, 155 CrossRef PubMed.
- D. Wu, S. Rigo, S. Di Leone, A. Belluati, E. C. Constable, C. E. Housecroft and C. G. Palivan, Nanoscale, 2020, 12, 1551–1562 RSC.
- F. Itel, A. Najer, C. G. Palivan and W. Meier, Nano Lett., 2015, 15, 3871–3878 CrossRef CAS PubMed.
- S. T. Poschenrieder, S. K. Schiebel and K. Castiglione, Eng. Life Sci., 2017, 18, 101–113 CrossRef PubMed.
- E. Breslmayr, M. Hanžek, A. Hanrahan, C. Leitner, R. Kittl, B. Šantek, C. Oostenbrink and R. Ludwig, Biotechnol. Biofuels, 2018, 11, 79 CrossRef PubMed.
- C. Draghici, V. Mikhalevich, G. Gunkel-Grabole, J. Kowal, W. Meier and C. G. Palivan, Langmuir, 2018, 34, 9015–9024 CrossRef CAS PubMed.
- A. Moquin, J. Ji, K. Neibert, F. M. Winnik and D. Maysinger, ACS Omega, 2018, 3, 13882–13893 CrossRef CAS PubMed.
- L. I. Ramírez-Cavazos, C. Junghanns, N. Ornelas-Soto, D. L. Cárdenas-Chávez, C. Hernández-Luna, P. Demarche, E. Enaud, R. García-Morales, S. N. Agathos and R. Parra, J. Mol. Catal. B: Enzym., 2014, 108, 32–42 CrossRef.
- P. Baumann, M. Spulber, I. A. Dinu and C. G. Palivan, J. Phys. Chem. B, 2014, 118, 9361–9370 CrossRef CAS PubMed.
- F. Itel, M. Chami, A. Najer, S. Lörcher, D. Wu, I. A. Dinu and W. Meier, Macromolecules, 2014, 47, 7588–7596 CrossRef CAS.
- C. W. Scales, A. J. Convertine and C. L. McCormick, Biomacromolecules, 2006, 7, 1389–1392 CrossRef CAS PubMed.
- C.-C. Lin and K. S. Anseth, Adv. Funct. Mater., 2009, 19, 2325–2331 CrossRef CAS PubMed.
- C. R. Perry, S. E. Matcham, D. A. Wood and C. F. Thurston, J. Gen. Microbiol., 1993, 139, 171–178 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb01463c |
| This journal is © The Royal Society of Chemistry 2021 |