
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling the semi-permeability of protein nanocapsules influences the cellular response to macromolecular payloads†
Marina
Machtakova
a,
Sebastian
Wirsching
b,
Stephan
Gehring
b,
Katharina
Landfester
 *a and
Héloïse
Thérien-Aubin
*ac
*a and
Héloïse
Thérien-Aubin
*ac
aMax Planck Institute for Polymer Research, Mainz, Germany. E-mail: landfester@mpip-mainz.mpg.de
bChildren's Hospital, University Medical Center, Johannes Gutenberg University, Mainz, Germany
cDepartment of Chemistry, Memorial University of Newfoundland, St. John's, NL, Canada. E-mail: htherienaubin@mun.ca
First published on 16th August 2021
Abstract
Nanocapsules are an excellent platform for the delivery of macromolecular payloads such as proteins, nucleic acids or polyprodrugs, since they can both protect the sensitive cargo and target its delivery to the desired site of action. However, the release of macromolecules from nanocapsules remains a challenge due to their restricted diffusion through the nanoshell compared to small molecule cargo. Here, we designed degradable protein nanocapsules with varying crosslinking densities of the nanoshell to control the release of model macromolecules. While the crosslinking did not influence the degradability of the capsules by natural proteases, it significantly affected the release profiles. Furthermore, the optimized protein nanocapsules were successfully used to deliver and effectively release a bioactive macromolecular vaccine adjuvant in vitro and, thus, can be used as an efficient platform for the design of potential nanovaccines.
Introduction
The delivery of macromolecular therapeutics is becoming increasingly important in the development of nanomedicines.1–4 From biomacromolecular therapeutic agents, such as peptides, proteins, or nucleic acids to high molecular weight polyprodrugs, new therapeutic strategies are emerging for the treatment of various diseases.2,5 However, the release of such large payloads from a nanocarrier matrix represents a major challenge, which is attributed to the restricted diffusion and large hydrodynamic radius of those molecules.6,7 Understanding the parameters influencing the release of such macromolecular payloads is the key to get insights into their release mechanism and to their successful application.6 We have designed a system based on degradable protein nanocapsules encapsulating macromolecular payloads to study the parameters influencing the release such as the size of the patload and the mesh size of the shell. The optimized system also provides a unique and practical platform for the delivery and release of bioactive macromolecular payloads in biological systems.The design of drug delivery systems for macromolecular payloads is more complex than for low molecular weight therapeutic agents. In addition to the typical challenges associated with drug delivery systems such as degradability, biocompatibility, extended circulation time, and targeting, the delivery of macromolecular payloads is also plagued by a complex release from the drug delivery system. The release usually occurs by a combination of degradation of the matrix and the diffusion of the payload through and from the nanocarrier.8 While the degradation is mainly controlled by the chemical composition of the delivery system, the diffusion process is dependent on the ratio of mesh size of the carrier to the radius of the payload. Consequently, macromolecular payloads with a large hydrodynamic radius display reduced diffusion and release kinetic. Furthermore, the mesh size of the matrix, in turn, is affected by the degree of crosslinking in the nanocarrier and the degradability of the system. Often, in smart polymer nanocarriers, the mesh size can be tuned by environmental cues, such as a change in the pH value of the environment or by the presence of a specific level of biomolecules, such as enzymes.9,10 These biochemical cues lead to an increase in the mesh size induced by the swelling or the partial degradation of the nanocarriers, promoting the release of the encapsulated payload.11 This strategy is efficient for the encapsulation and release of small molecules but is not necessarily well-suited for macromolecular payloads because their diffusion is heavily restricted due to their high molecular weight and large hydrodynamic radius.6,7 Therefore, the limited increase in the mesh size resulting from the swelling or partial degradation of the nanocarriers might no longer be sufficient to ensure the successful release.
Several nanosystems have shown potential for the encapsulation of (bio)macromolecules. For example, proteins and enzymes were successfully encapsulated in polymer nanosystems for the development of therapeutic or catalytic nanoreactors.12,13 Moreover, macromolecular genetic materials, such as DNA or RNA were efficiently encapsulated and delivered in vivo, which has led to the development of novel therapies.14–16 However, the challenge of a controlled release of the macromolecular payload remains. In some nanosystems, no release of such payloads can be observed. Rather, the selectivity of the carrier membrane is altered in order to ensure the influx of small molecule substrates to access the encapsulated (bio)macromolecule, while the target payload remains inside the carrier.17,18 In other systems, an uncontrolled release of encapsulated macromolecules can occur due to the limited stability of the nanocarrier system in complex biological environments.19,20
Crosslinked protein nanocapsules can provide a solution to both the controlled delivery with a stable carrier and the sufficient release of macromolecular payload. This class of delivery vehicle provides excellent structural stability and a high degree of biodegradability, which can be exploited to control the release of payload.21,22 The hollow nanocapsules are composed of crosslinked biopolymers and have a high loading capacity in their inner aqueous core.23 They are highly degradable by intracellular proteinases under natural conditions but preserve their structural integrity in biological media during their delivery to the site of action.24,25 The crosslinking degree of the proteins in the shell tunes the mesh size of the capsule and hence controls the release kinetic. Proteins from the albumin family, such as human serum albumin or ovalbumin, have been widely used to prepare protein nanosystems, and some of those have already been used in clinical therapy, demonstrating the potential of such nanocarriers.24,26,27 However, the release profile of payloads encapsulated in such nanocarriers is complex and is influenced by the semi-permeability of the nanocapsule shell and consequently affects the final biological response to the molecules vectorized with such nanocarriers.28
In this study, we aim at controlling the release of model macromolecular payload from crosslinked ovalbumin nanocapsules by tuning the crosslinking degree of the nanocapsule shell. Furthermore, to gain a fundamental understanding of the release mechanism of macromolecular payloads from protein NCs, the tunable biodegradability of the shell was correlated to the release kinetics of model payload of different molecular weights (Fig. 1). Then, we applied this fundamental understanding of the release of macromolecular payloads to tune the biological activity of macromolecular functional payload delivered to dendritic cells. Our results show that the appropriate biological response hinges on the adequate control of the release conditions.
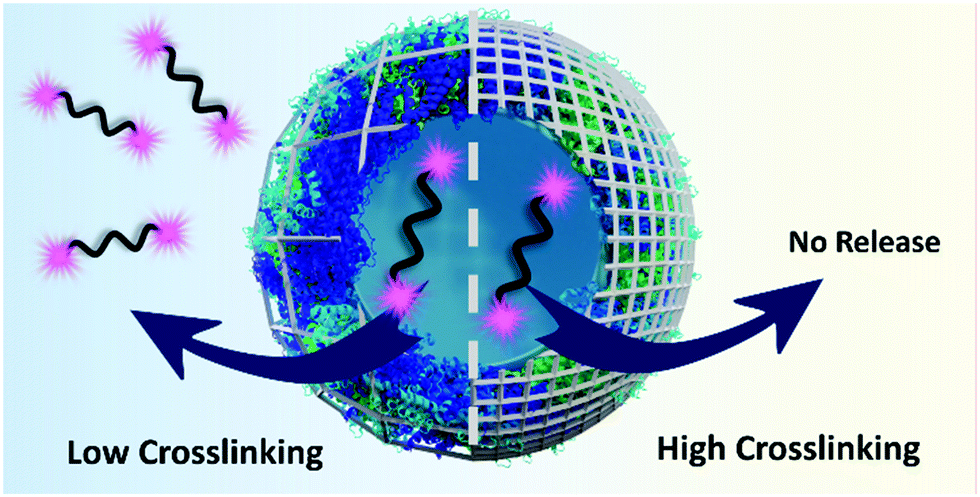 | ||
| Fig. 1 The crosslinking density of the shell governs the release of high molecular weight payload from protein nanocapsules induced by proteases. | ||
Results and discussion
Preparation of the nanocapsules
The density of the nanocapsule shell is of the utmost importance to control the release of encapsulated payload. The crosslinked protein shell hinders the diffusion of the macromolecules and prevents the leakage of the payload from the nanocapsules (NCs). Therefore, tuning the crosslinking density of the protein shell can influence the barrier properties of the NCs and the release of the cargo. Here, we synthesized protein NCs by crosslinking ovalbumin (OVA) with 2,4-toluene diisocyanate (TDI), resulting in NCs with varying shell densities.29,30 The reaction between OVA and TDI occurred at the interface of an inverse miniemulsion composed of aqueous droplets of OVA solution in toluene. Then, TDI was added to the toluene phase and was let to react with the nucleophilic amino groups of OVA. After the reaction, a solid protein shell surrounding an aqueous core was obtained (Fig. 2). Later, the NCs were transferred to water followed by the evaporation of the remaining toluene.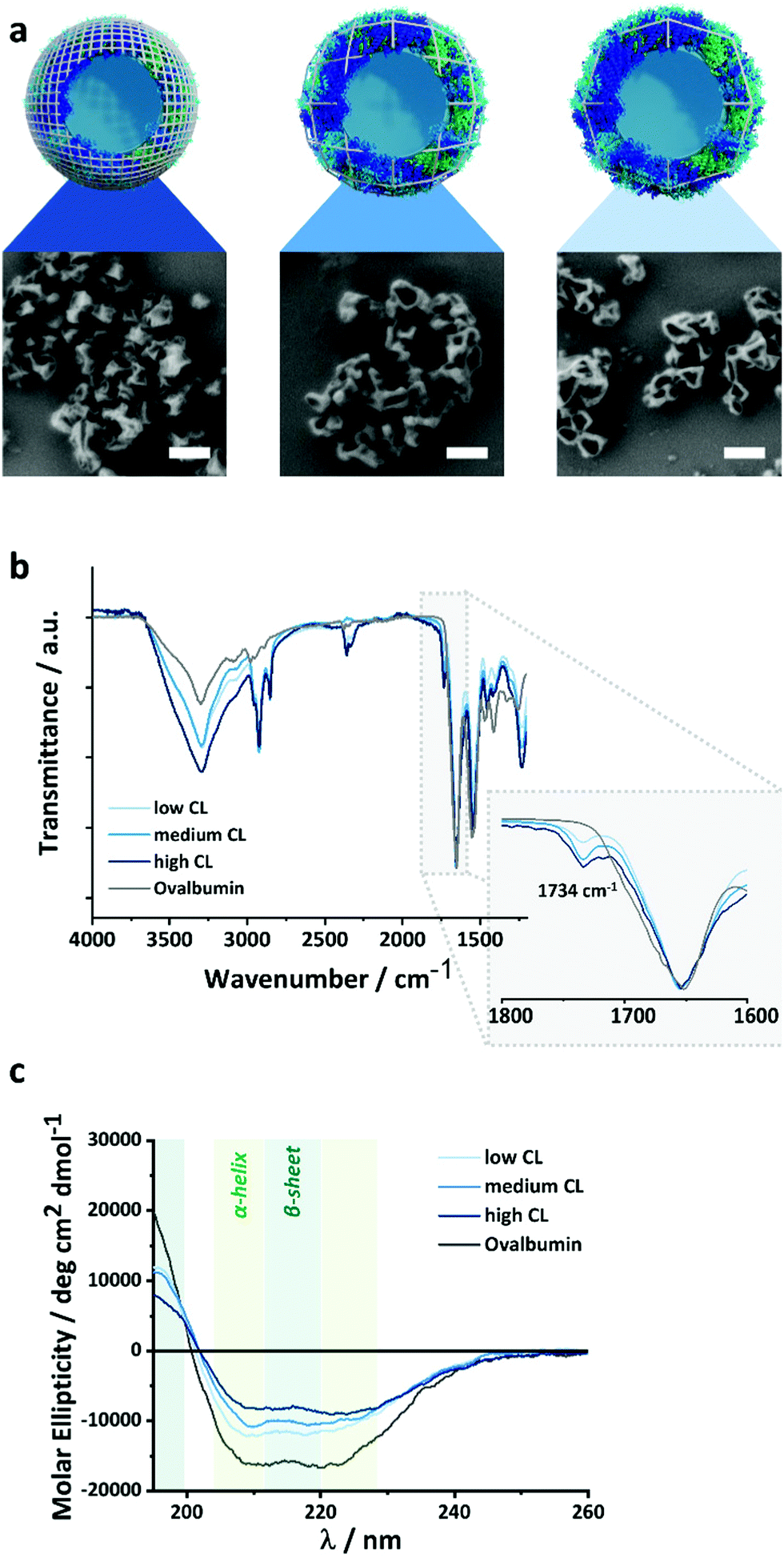 | ||
| Fig. 2 Effect of the CL on the structure and composition of the OVA NCs. (a) SEM images of the NCs with different CL, the scale bars are 200 nm, (b) FT-IR spectra of the NCs prepared with different crosslinker/protein ratio, (c) CD spectra of the NCs with different crosslinker/protein ratio. | ||
Tuning the molar ratio between the number of nucleophilic lysine residues in the OVA protein and the amount of TDI used allowed to control the crosslinking degree (CL) of the NCs shell. The CL directly affects the mesh size of crosslinked nanosystems, as the number of crosslinking points increases, the distance between two crosslinking points will decrease if the number of polymer chains present in the system remains constant. In our case, the polymer content was kept the same, and the number of crosslinking points was varied. An equimolar ratio of TDI/lysines resulted in OVA NCs with a medium CL. While increasing the molar ratio to 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 led to highly crosslinked NCs, decreasing the ratio to 1:3 results in NCs with a low CL. SEM and TEM (Fig. 2a and Fig. S1, ESI†) analysis show the formation of solid protein NCs with a clear hollow structure for every CL. The evaporation of the liquid core during the sample preparation for microscopy give rise to the expected crumpled structures of a dried hollow capsule. Consequently, if we consider that the extent of reaction between the TDI and the OVA remained constant with the different amounts of TDI used, the mesh size would decrease linearly with the molar concentration of TDI.
:1 led to highly crosslinked NCs, decreasing the ratio to 1:3 results in NCs with a low CL. SEM and TEM (Fig. 2a and Fig. S1, ESI†) analysis show the formation of solid protein NCs with a clear hollow structure for every CL. The evaporation of the liquid core during the sample preparation for microscopy give rise to the expected crumpled structures of a dried hollow capsule. Consequently, if we consider that the extent of reaction between the TDI and the OVA remained constant with the different amounts of TDI used, the mesh size would decrease linearly with the molar concentration of TDI.
The chemical microstructure of the NCs shell was studied for purified and NCs by TEM analysis, FT-IR spectroscopy and CD-spectroscopy (Fig. S1, ESI† and Fig. 2b, c). In TEM (Fig. S1, ESI†), the contrast of the nanocapsules increased with the increasing amount of crosslinker used. This can be ascribed to the increased inclusion of electron-dense TDI (in comparison to the protein) in the nanocapsule shell. Furthermore, during FT-IR analysis, all samples showed the peaks characteristic for the OVA protein, and the NCs samples also show an additional signal at 1734 cm−1 attributed to the vibration of the urea-groups created after the crosslinking reaction between the amino groups of the lysines and the TDI. The increase in the molar fraction of TDI added to the reaction resulted in an increase in the urea peak intensity, confirming that the shell of the nanocapsules prepared with increasing amounts of TDI (Fig. S2, ESI†) contained more urea crosslinking points. Furthermore, circular dichroism spectroscopy (CD) was used to evaluate the secondary structure of the OVA protein and to qualitatively monitor changes in the structure after the crosslinking with various amounts of TDI (Fig. 2c). The intensity of the peaks between 200 and 230 nm, typical of a mixture of α-helices and β-sheets, decreased accordingly with the increasing amount of TDI used. This loss of secondary structure of the protein was indicative of the stronger disruption of the protein secondary structure when the NCs were prepared with higher amount of TDI and can be correlated to the formation of more crosslinking points between the protein and the TDI, and, consequently, a denser NCs shell. Those results were in keeping with the loss of structure and function observed during the crosslinking of enzymes under similar conditions.31
While the ratio of TDI/lysines significantly influenced the composition and CL of the NCs shell, it had no impact on the size and size distribution of the capsules. The average size of all particles was determined by dynamic light scattering (DLS) (Table 1 and Fig. S5, ESI†). The average size of the NCs in toluene was between 240 and 250 nm, independently of the CL degree. The zeta-potentials of the redispersed NCs in PBS buffer were negative due to the residual amount of anionic surfactant (SDS) used to stabilize the NCs in water.
| CL density | Toluene | Water | |||
|---|---|---|---|---|---|
| Diameter/nm | PDI | Diameter/nm | PDI | ζ-Potential/mV | |
| High | 250 | 0.09 | 230 | 0.25 | −4.7 |
| Medium | 240 | 0.16 | 190 | 0.16 | −16.7 |
| Low | 240 | 0.22 | 160 | 0.13 | −21.8 |
Encapsulation of macromolecular payload
The interfacial crosslinking reaction of inverse-miniemulsion droplets is an ideal technique to encapsulate a variety of hydrophilic small and macromolecular payloads.32,33 Different macromolecular payloads with molecular weights ranging from 5000 to 600000 g mol−1 were prepared to study the effect of the payload size on the release. The model payloads were a series of different poly(ethylene glycol)s (PEGs) functionalized with rhodamine. Rhodamine-isothiocyanate was reacted with different PEGs to yield payloads with controlled molecular weight and size, highly water-soluble, and reliably quantifiable by fluorescence spectroscopy. The resulting PEG payloads had a specific hydrodynamic radius, determined by DLS, ranging from 2 to 22 nm for PEG 5 kDa and PEG 600 kDa, respectively (Fig. S6, ESI†). The encapsulation of the hydrophilic macromolecular payload did not influence the formation of stable protein nanocapsules (Fig. S7, ESI†).
The NCs were used to encapsulate a model payload, PEG-Rhodamine derivative, of molecular weight of 5 or 600 kDa. The PEG was dissolved in the aqueous solution of protein used to prepare the NCs prior to the crosslinking reaction to encapsulate the payload. The payload was encapsulated in situ as the crosslinked network of the NCs was formed. The encapsulation efficiency was measured after the transfer of the NCs to water as the fraction of PEG remaining in the NCs after the separation of the NCs from the aqueous media by centrifugal filtration (Fig. S8, ESI†). The observed encapsulation efficiency was measured after the transfer and equilibration of the NCs in PBS buffer and accounted for both the unencapsulated payload molecules and those released following the transfer to water of the NCs (i.e. 20 h corresponding to the time needed to complete the water transfer process and the complete evaporation of the toluene) (Fig. 3a).
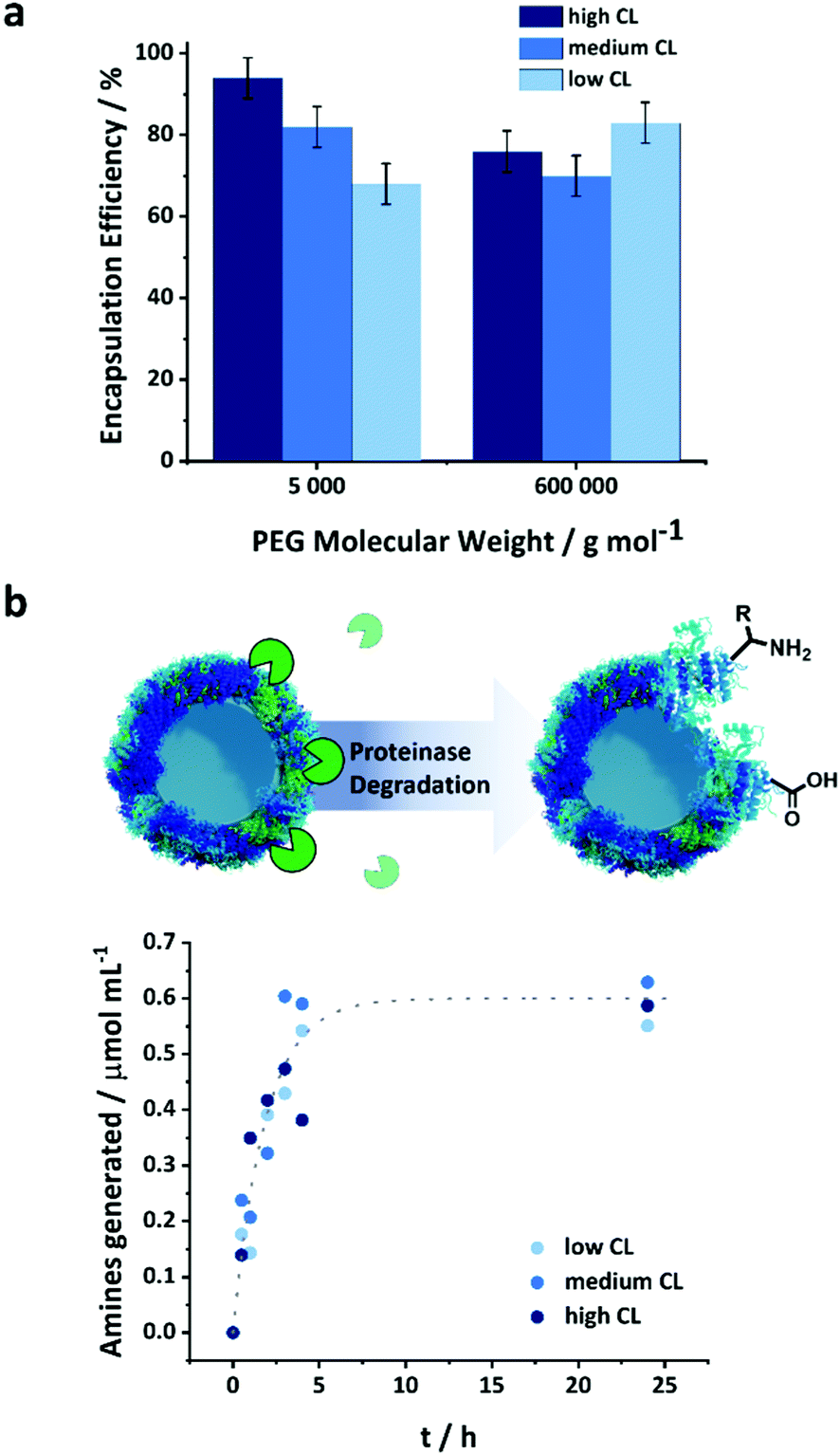 | ||
| Fig. 3 (a) Encapsulation efficiency of macromolecular payloads (rhodamine-labeled PEGs) in the NCs with varying crosslinking degrees, (b) generation of amines during the degradation of the OVA NCs by proteinase K (1 unit per mg of protein NCs). | ||
Furthermore, the size of the payload molecules affected their encapsulation efficiency. The largest PEG molecule, PEG 600 kDa, was encapsulated less efficiently than smaller payloads. A constant mass loading of the payloads was used for the different molecular weights; consequently, more molecules were present for smaller payloads, and the number of molecules washed away during the water transfer remained essentially the same for the different payloads. The CL also affected the encapsulation efficiency. This phenomenon was more pronounced for smaller payloads than large ones. With PEG 600 kDa, there were no statistically significant differences between the encapsulation efficiency measured for the NCs prepared with different CL densities. However, the encapsulation efficiency increased with the CL for PEG 5 kDa. At lower CL density, the looser mesh size of the NC shell allowed for the washing off of more molecules entrapped within the shell during the transfer to water. Once the NCs were transferred to water, and the initial washing of the loosely or non-encapsulated molecule occurred, there was no leakage of the encapsulated payload (Fig. S9, ESI†). The shell of the nanocapsule acted as a semi-permeable membrane and prevented the mass transport of the molecules trapped in the inner core of the nanocapsules.
Biodegradability of the NCs
A major advantage of the protein NCs is their high potential to be biodegraded by naturally occurring enzymes. Various types of proteinases are commonly present under in vivo conditions and can induce the degradation of the proteins employed as building blocks for the NCs. However, the structural variations induced by the crosslinking of the protein could potentially change their conformation and hence their recognition and digestion by enzymes.34,35 Proteinase K is a natural protease with a high ability to cleave peptide bonds between the protein amino acids, more specifically where alanine residues are present.36 The OVA NCs can be enzymatically degraded by proteinase K leading to the generation of free amino groups on the surface of the nanocapsules, which can be quantified by a fluorescamine assay (Fig. S3, ESI† and Fig. 3b).37 The absolute number of amino groups found in the NCs samples with varying CL densities provides an insight into the crosslinking of the shell. Fig. S3 (ESI†) shows that the highest CL degree resulted in the lowest amount of remaining amino groups, while low CL led to more free amino groups in the crosslinked protein. This result shows that increasing the amount of crosslinker fed to the system led to the conversion of more amino groups on the native OVA. After the addition of 1 unit of proteinase K for each mg of NCs, an increased in the number of amino groups in the NCs suspension was observed (Fig. 3b and Fig. S4, ESI†). Because the initial number of amine groups in the NCs varied with the CL degree (Fig. S3, ESI†), the absolute amount of amine groups measured during the degradation (Fig. S4, ESI†) was corrected for the initial NH2 concentration, and we report (Fig. 3b) the number of amino groups generated by the addition of the proteinase K for the better and more direct comparison of the different samples. Interestingly, the number of amines formed by the degradation of the NCs prepared with different CL followed a similar kinetic. Consequently, the degradability of the capsules was independent of the CL density and the conformational changes observed in the protein structure (Fig. 2c). It is important to note that to monitor the degradation via the fluorescamine assay, a higher concentration of protease than what is normally found in serum was used.38 Under typical blood concentrations of proteases and in the presence of protease inhibitors, protein-based nanocarriers were shown to be stable over several days, which is a prerequisite for a successful in vivo application of those nanocarriers.39Release of macromolecular payload
As the nanocarriers were degraded by proteinase K, the mesh size of the NCs shell increased. The degradation influenced the semi-permeability of the nanocapsule shell, and led to the release of the encapsulated payload over the degradation period (Fig. 4). An obstruction model40 can be used to describe the release of the payloads from crosslinked NCs. Originally developed to explain the release from crosslinked polymer hydrogels, it can also be applied to describe the release from the protein nanocapsules used here. After the transfer to water, the crosslinked protein chains swell and behave similarly to a poorly swollen hydrogel. Consequently, the release of the payload from the NC is directly proportional to the diffusion of the payload through the crosslinked polymer shell. The effective diffusion coefficient (D) of the payload through the shell is governed by three major parameters, the diffusion coefficient of the payload in water (D0), the average mesh size of the network ξ, and the radius of the payload (R):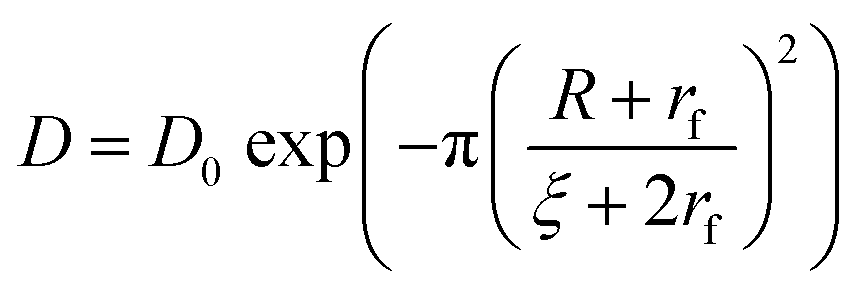 | (1) |
 | ||
| Fig. 4 (a) Release kinetics of PEG 600 kDa and PEG 5 kDa from OVA NCs with varying CL density (1 unit of proteinase K per mg of NCs), (b) release as a function of the degradation of PEG 600 kDa from NCs with different CL density (1 unit proteinase K per mg of nanocapsules), (c) release as a function of degradation of PEG of different molecular weight from NCs with a low CL density (0.025 unit proteinase K per mg of nanocapsules). | ||
In this model, the solute is considered to be a hard sphere not interacting significantly with the network. Furthermore, the network is considered as immobile compared to the mobility of the solute and there is a distribution of mesh sizes in the network resulting from the random distribution of fibers as proposed by Ogston.41 However, this model remains a suitable tool to qualitatively address the release of macromolecular payload from protein nanocapsules upon degradation.
According to eqn (1), the diffusion and the release of macromolecular payloads depend on their size and the mesh size of the protein network, which itself would vary with the initial CL degree and the degradation of that network. The protein nanocapsules with high, medium and low CL density all efficiently encapsulated the macromolecular payloads, and small molecules could even be efficiently encapsulated at a high CL degree. Without the degradation of the protein network, no release occurs for PEG 5 kDa, even from the low CL nanocapsules, showing that the final mesh size of the nanocapsule was smaller than the hydrodynamic radius of the payload (Fig. S6, ESI†). Given that the mesh size in eqn (1) represents the average of a distribution of mesh sizes in the network,41 the value obtained must be smaller than the hydrodynamic radius of the payload (eqn (S5), ESI†) to fully prevent its release.
As the OVA NCs were degraded by proteinase K, as evidenced by the generation of amine during the cleavage of the peptidic bonds, the release of the encapsulated payload occurred because the mesh size increased from the initial size ξ0 to a value superior to a threshold value varying on the hydrodynamic radius of the payload to be released (eqn (S6) and (S7), ESI†). The extend of the payload released was measured by fluorescence following centrifugal ultrafiltration, a mild technique to separate the media and the nanocarriers without subjecting the formulation to high shear forces.42Fig. 4a and b show the effect of the crosslinking density, i.e. the mesh size of the nanocapsules, on the release of encapsulated molecules. The release curves show that in the first 4 h, highly crosslinked NCs only released ca. 10% of their payload although degraded by 1 unit proteinase K per mg of nanocapsules, while NCs with lower CL degree release 20 and 80% of their payload for medium and low CL at identical conditions, respectively (Fig. 4a).
While the degradation kinetic of the crosslinked OVA NCs by proteinase was not affected by the crosslinking density (Fig. 3), the release kinetic of the payload encapsulated within those NCs was significantly affected by the CL degree (Fig. 4a). As the CL degree increased, the initial mesh size decreased. In every case, the proteinase K cleaved the same number of peptidic bonds. Statistically, the average increase in the mesh size generated by the cleavage of a peptidic bond would increase exponentially with the decreasing initial CL degree.
This phenomenon was evidenced by the rapid initiation of the release of macromolecular payload from the low CL NCs. For the release to occur from the low crosslinked nanocapsules, in the case of PEG 600 kDa, the threshold mesh size to observed release was ca. 26ξ0 (eqn (S7), ESI†) corresponding to the generation of ca. 0.5 μmol mL−1 of amine groups produced for a NC suspension of 0.1 wt% (eqn (S11), ESI†), which is in keeping with the results obtained. When increasing the CL density, the initial mesh size decreased, but not the mesh size needed to observe the release of the payload. Consequently, more peptidic segments need to be cleaved for the release to occur. For the highest CL density used, ca. 6 μmol mL−1 of amines should be generated before being able to observe the release of PEG 600 kDa. Hence, no release was observed from the high CL NCs even after 1 day of coincubation with the proteinase when the concentration of amine generated by the degradation process reached 0.8 μmol mL−1 (Fig. 4b). However, in the case of the NCs with the intermediate CL density, only ca. 0.9 μmol mL−1 of amine needs to be generated before the release to occur, which roughly corresponds to the degradation observed at the onset of the release (Fig. 4b).
The second parameter influencing the release of the payload is the molecular weight and hydrodynamic radius of the encapsulated cargo. Fig. 4a shows the release of two payloads (PEG 5 kDa and 600 kDa) in the presence of 1 unit of proteinase K per mg of NCs. In this case, there were no significant differences in the release kinetics. However, when the protease concentration was decreased to 0.025 μ mg−1 of NCs, the effect of the payload molecular weight on the release kinetics from OVA NCs can be observed (Fig. S4c and S10, ESI†). While the larger payload (600 kDa) showed limited release in the early stage of the degradation process, the smaller payload (5 kDa) was released up to 60–80%. For the release of the PEG 5 kDa to occur, the mesh size of the nanocapsules only needed to increase ca. 1.2ξ0, this translates to the generation of ca. 0.18 μmol mL−1 of amines, in comparison to the 26 time increase and the generation of 0.54 μmol mL−1 of amines needed in the case of PEG 600 kDa. Hence, for the release to occur 3 times more amines have to be generated in samples containing PEG 600 kDa compared to samples containing PEG 5 kDa, and a delayed release of PEG 600 kDa was observed when compared to PEG 5 kDa.
Intracellular uptake and cytotoxicity
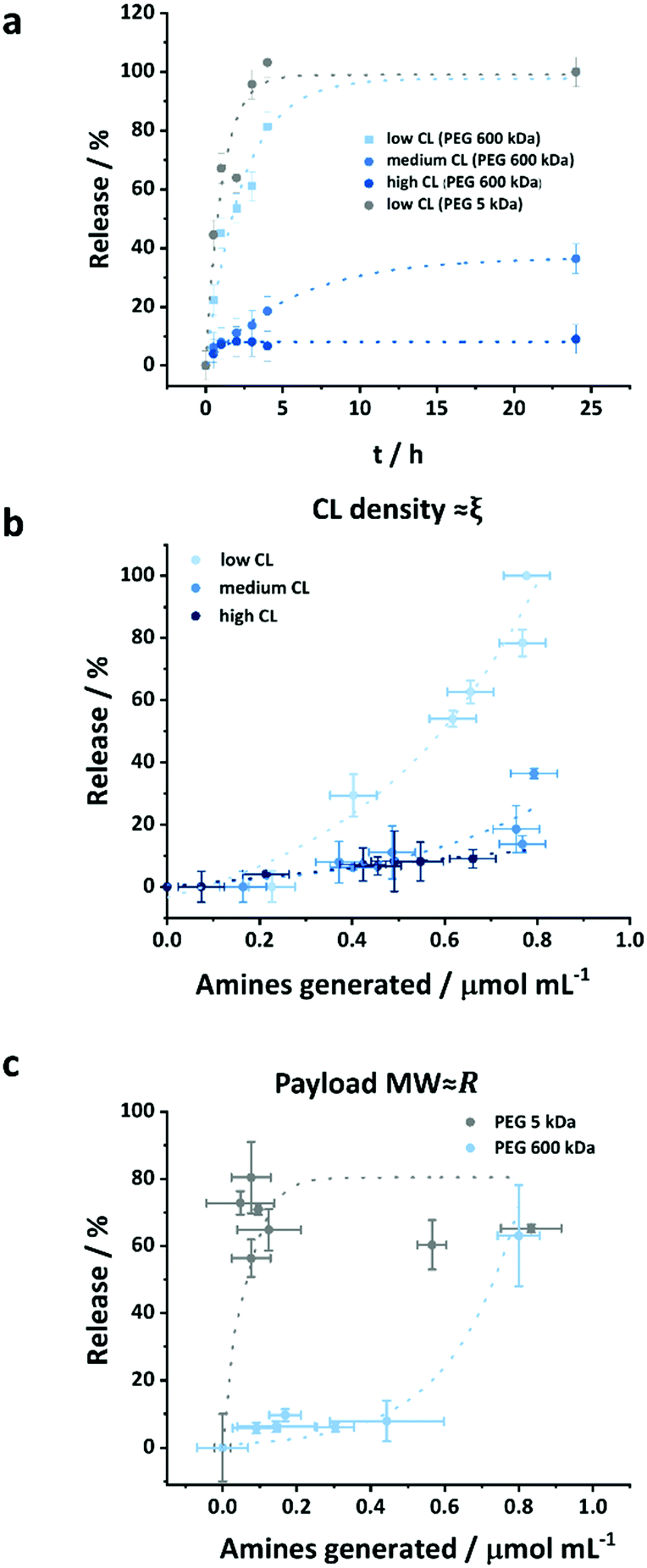
The use of OVA NCs is particularly appealing to target peripheral blood dendritic cells (DCs) because it is a model antigen, which, when co-delivered with immune-stimulating adjuvants, can be used to elicit a robust immune response. The intracellular uptake of the OVA NCs prepared with different CL densities was analyzed in vitro in human DCs. For those cellular uptake experiments, OVA labeled with cyanine-5 (Cy-5), a fluorescent molecule, was used in the synthesis of the NCs. First, the DCs were incubated with different concentrations of NCs for 20 h, and the amount of DCs containing the NCs, the Cy-5+ cells, was determined by flow cytometry. Additionally, the fraction of dead cells was quantified by incubating the cells with the live/dead stain 7-AAD. The uptake of NCs moderately increased when increasing the concentration of the NCs in the cell medium from 25 to 100 μg mL−1 (Fig. S11, ESI†). Simultaneously, the toxicity only marginally increased in the same conditions. Furthermore, the CL degree also influenced the cellular uptake and cytotoxicity. Between 40 and 50% of the cells were Cy-5+ indicating the uptake of the NCs. Interestingly, as the CL of the NCs decreased, the fraction of dead cells observed decreased; 15% of the DCs coincubated with the highly CL NCs were 7-AAD positive, whereas only 11 and 10% of the DCs were dead when coincubated with medium and low CL NCs, respectively (Fig. 5b). The results observed here were similar to those observed for other NCs made from OVA or other proteins albeit the uptake reported here was moderately lower at comparable NC concentrations.43–45 However, those studies were not performed using primary dendritic cells, but rather monocyte-derived dendritic cells, DC cell lines or macrophages. Another study using primary plasmacytoid DCs (pDC) and targeting different receptors using functionalized PLGA NCs reported cellular uptake ranging from 5–45%, depending on the utilized targeting moiety.46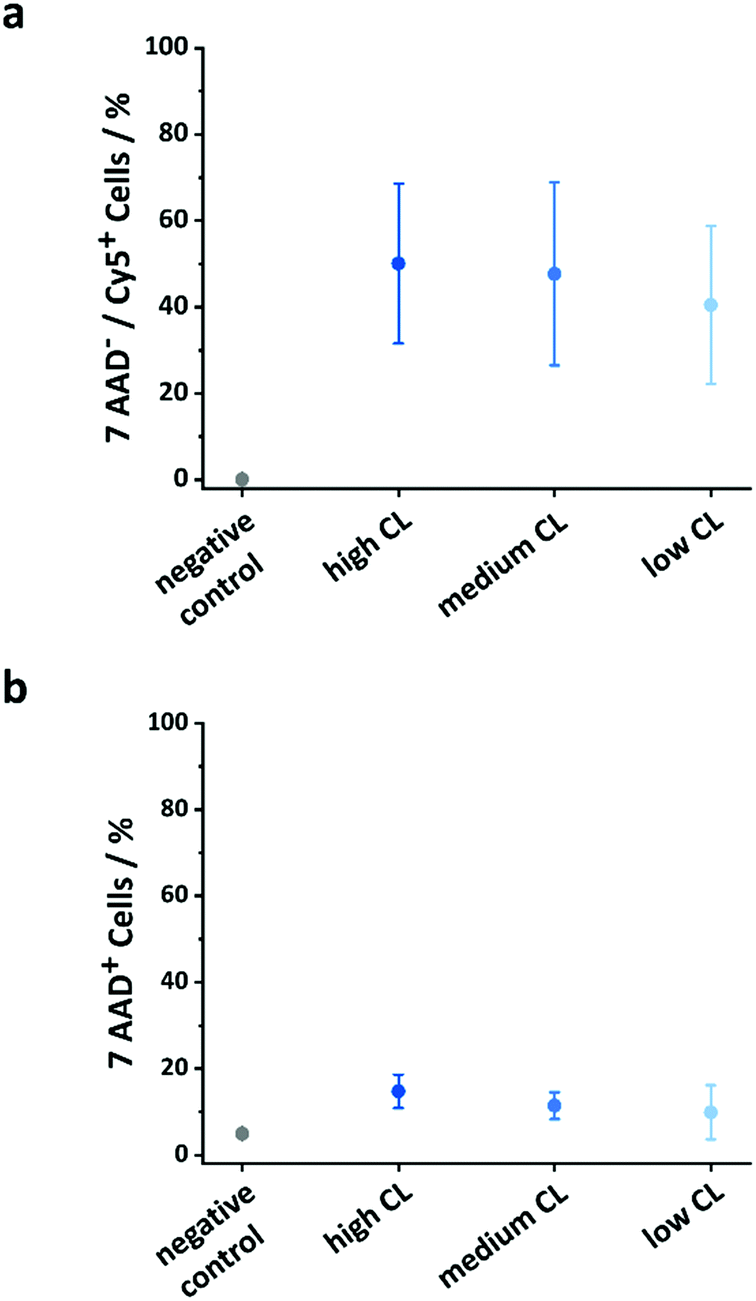 | ||
| Fig. 5 Comparison of uptake and toxicity of OVA-Cy-5 NCs with different CL degrees. (a) Percentage of Cy-5 positive and 7-AAD negative cells, (b) percentage of 7-AAD negative cells (dead cells) after the incubation with 50 μg mL−1 OVA NCs with different CL degrees for 20 h. Data represents mean ± SD (n = 3). | ||
In vitro release of a macromolecular adjuvant and cell activation
The design and the resulting properties of the protein NCs are essential for their use as nanodelivery system in vitro and potentially in vivo. In this regard, the structure–function relationship of the OVA NCs with varying CL degrees was tested with a model macromolecular payload in peripheral blood dendritic cells aiming at the design of a nanovaccine. For the development of nanovaccines, OVA NCs are potent model antigens, which can be used to co-deliver immune-stimulating adjuvants.24 The immune-stimulant resquimod (R848) served as a relevant model adjuvant cargo due to its potent anti-viral and anti-tumor properties.47 A successful delivery and release of R848 in the immune system cells, like the dendritic cells, leads to the upregulation of specific surface proteins on the DC.48 In order to develop this stimulatory effect, the immune-stimulant has to reach specific receptors, which in the case of R848, are localized in the endosomes of the DCs. Hence, the controlled intracellular delivery and release of R848 are essential.However, R848 is a poorly water-soluble small molecule, and its use as a vaccine adjuvant is hampered by the rapid diffusion of the small R848 molecule and its rapid dissociation from the antigen upon injection.49,50 A possible approach to solve those two pitfalls includes the co-encapsulation of an antigen and R848 in liposomal formulation,51 or polymer nanoparticles.46 However, this solution comes with the risk of also generating immunity against the carrier compounds.52 Here, the strategy proposed, to solve both of those issues, was to attach the hydrophobic R848 to the water-soluble, non-immunogenic polymer PEG and to encapsulate the macromolecular adjuvant in NCs entirely composed of the antigen protein (OVA).
Hence, we modified the small molecule R848 at the exocyclic –NH2 by attaching it to a PEG (5 kDa) via NHS-Ester chemistry. The PEG was further labeled with rhodamine for its efficient quantification (Fig. S12a and b, ESI†). This modification did not alter the therapeutic efficacy of the adjuvant, as shown by comparison of the activation of DCs with R848 and the modified R848 (Fig. S13, ESI†). The results were in keeping with the preserved activity of R848 on the stimulation of TLR-7 receptors after derivatization at the NH2 with a low molecular weight PEG-linker and RNA.53
After the synthesis, we encapsulated the macromolecular stimulant, PEG-R848 in OVA NCs with varying CL, to demonstrate how the delivery and release of macromolecular payloads can be controlled even in vitro by the rational design of the NCs.
Due to the high molecular weight of the modified R848, the macromolecular payload was encapsulated with high encapsulation efficiencies (>80%) in the NCs with different CL densities. Furthermore, the release of the resulting macromolecular adjuvant from the OVA NCs is essential to the upregulation of specific cell markers in the DC cells. To study the release from the NCs, the NCs loaded with the modified R848 were coincubated with DCs at an effective R848 concentration of 500 ng mL−1 for 20 h. Afterwards, the cells were harvested, and the expression of the two activation markers, CD80 and CD83, was measured by flow cytometry. Fig. 6 shows that only the OVA NCs with low CL density induced a significant upregulation of the cell markers CD80 (∼80%) and CD83 (∼70%). The activation was comparable to the positive control with soluble PEG-R848 in an equimolar dose. However, the OVA NCs with high and medium CL density showed no cell marker upregulation, illustrating that those two formulations could not successfully release their macromolecular payload in vitro (Fig. 6a and b) in keeping with the release kinetics measured with the model payload (Fig. 4a). This key finding shows that the OVA NCs with a low CL degree provided both a high encapsulation of macromolecular payloads and a controlled release of those molecules in vitro.
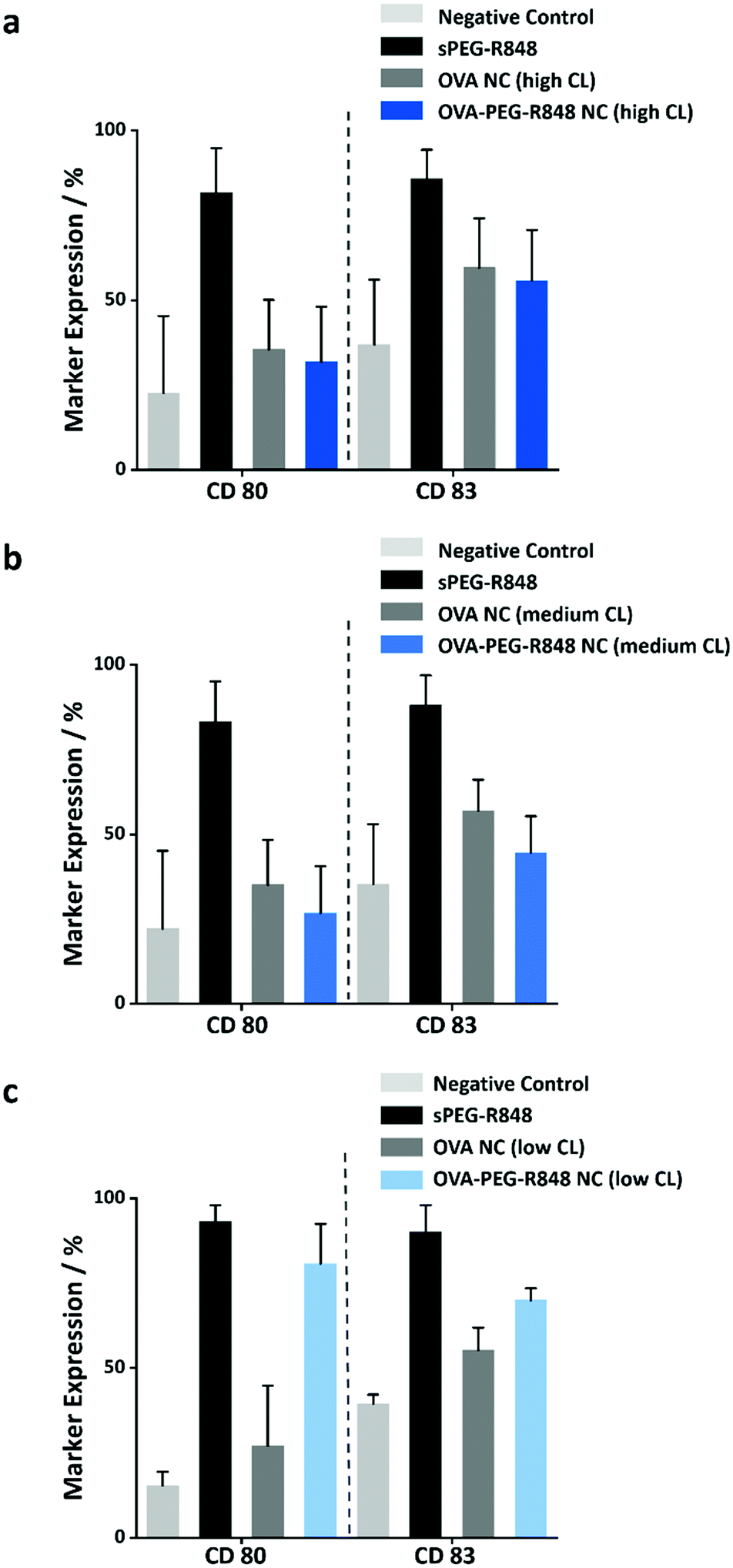 | ||
| Fig. 6 Activation of DCs after incubation with different OVA-PEG-R848 NC dispersions. The DCs were incubated for 20 h with OVA-PEG-R848 NCs with (a) high, (b) medium or (c) low CL densities. Activation of cells was assessed by measuring CD80 and CD83 expression levels via flow cytometry. Data was compared to negative control (no stimulation) and significance was given with p < 0.01 (**), p < 0.0001 (****) (two-way ANOVA). Data represents mean ± SD (n = 3). | ||
Conclusions
We synthesized degradable protein nanocapsules with varying crosslinking degrees to understand the parameters influencing the release of macromolecular payloads from protein nanocapsules. Protein nanocapsules were prepared by the interfacial crosslinking of nanodroplets containing a solution of proteins and payload. The use of different amounts of crosslinking molecules during the interfacial reaction yielded nanocapsules with different crosslinking density, but with a comparable morphology and encapsulation efficiency. The degradability of the nanocapsules by a natural protease was not affected by the crosslinking density, but the resulting release profiles significantly differed. The initial semi-permeability of the nanocapsule and its response to degradation was influenced by the initial crosslinking density. Submitting the nanocapsules with the highest crosslinking density to enzymatic degradation resulted in no release of the encapsulated payload. However, release occurred from nanocapsules with medium and low crosslinking density. Even though the number of peptidic bonds cleaved in the three samples was similar, the release kinetic was different because the same amount of protein degradation did not lead to similar variation in the effective mesh size in the crosslinked polymer shell.Those nanocapsules were than used to encapsulate an adjuvant (PEG-R848) able to activate specific markers in dendritic cells. The in vitro studies showed that the cellular uptake and toxicity of the nanocapsules were only marginally influenced by the crosslinking density, whereas the release of the macromolecular adjuvant only occurred from nanocapsules with the lowest crosslinking density. The results show that such protein nanocapsules can act as an efficient delivery system for macromolecules in a complex biological environment, but only when design with the understanding of the release kinetic. The resulting protein nanocapsules represent a versatile nanocarrier system that can be employed with different small molecule or macromolecular payloads if the initial crosslinking density and mesh size are controlled accordingly. Thus, it provides an adaptable platform for more complex biomedical applications, such as the delivery and release of genetic materials or polyprodrugs.
Experimentals
Synthesis of the protein nanocapsules
The nanocapsules were prepared by a polyaddition reaction in an inverse miniemulsion, as described previously.22 In a typical experiment, the aqueous phase was prepared by dissolving 20 mg of OVA in 0.2 mL PBS-buffer (pH 7.0). When the nanocapsules were used to encapsulate PEG-derivatives, 1 mg of PEG-derivative was added to the aqueous solution. The organic continuous phase was prepared by dissolving 75 mg of PGPR in 3 mL toluene. The aqueous and organic phases were combined and pre-emulsified with an ultraturrax (20 000 rpm) for 60 seconds and homogenized by one cycle through a microfluidizer (LV1, microfluidic corporation) at an operating pressure of 10000 MPa in a 75 μm Y-shape emulsion chamber. A solution of 25 mg PGPR and 4 μL, 1 μL or 0.1 μL of crosslinking agent (TDI) in 0.5 mL of toluene was added dropwise to the miniemulsion over a period of 2 min. The reaction was stirred over 20 h at room temperature. The resulting nanocapsules were purified by three cycles of centrifugation (30 min, 1200 RCF) followed by redispersion in fresh toluene for the removal of excess surfactant and unreacted crosslinker. To transfer the nanocapsules to an aqueous solution, 500 μL of the dispersion in toluene were added dropwise to a solution of Lutensol AT50 (0.1 wt%) in PBS buffer under sonication in an ultrasonic bath. The resulting dispersion was stirred in an open vial for 3 h to evaporate the toluene. The dispersion was purified using vivaspin centrifugal filter (MWCO 300000, 2 times, 30 min) and redispersed in fresh PBS buffer to remove the unreacted protein, excess of surfactant, and unencapsulated payload.
Biodegradability of protein NCs
The biodegradability of the protein nanocapsules was conducted with proteinase K. A concentration of 1 unit proteinase K was added to 1 mg of nanocapsules dispersed in 1 mL of PBS buffer, pH 7.4.Fluorescamine assay
The quantification of amine groups was determined by the fluorescamine assay. First, 0.5 mg of fluorescamine was dissolved in 1 mL of DMSO and 0.1 M borate buffer (pH 7.4) was prepared. Then, 363 μL of the borate buffer were mixed with 13 μL of nanocapsule dispersion or with hexylamine as standard and 125 μL of fluorescamine solution in DMSO were added. Immediately after vortexing the mixture, the fluorescence (λexc = 410 nm, λem = 470 nm) was recorded, and the amine content was determined by comparison to the standard curve prepared with hexylamine.Release experiments
After the transfer of the NCs to PBS buffer, 10 mL of the suspension was filtered by centrifugal ultrafiltration for 30 minutes at 1770 RCF using Vivaspin 1000 K centrifugal concentrators. The NCs collected on the filter were redispersed in 10 mL of phosphate buffer pH = 7.4.The concentration of the protein nanocapsules was determined using the bicinchoninic acid (BCA) protein assay. First, 100 mg of BCA, 200 mg of sodium carbonate, 16 mg of sodium tartrate and 95 mg of sodium hydrogen carbonate were dissolved in 10 mL of deionized water, and the pH was adjusted to 11.3 by using 3.0 M NaOH. To this solution, 200 μL of 50 mg of CuSO4·5H2O in 1 mL of deionized water were added. Then, 200 μL of this solution were mixed with 10 μL of protein standard (ovalbumin) or the protein nanocapsule dispersion of unknown concentration and incubated at 60 °C for 30 min. The absorbance at 565 nm was recorded and the enzyme concentration was determined by comparison to the standard curve prepared with native OVA. Then, the protein nanocapsule dispersion was diluted to a concentration of 1 mg mL−1.
The release of the macromolecular payload was measured by fluorescence spectroscopy after the incubation of the nanocapsules with proteinase K (1 unit of proteinase K for 1 mg of protein NCs) for different periods of time in buffer solution. After appropriate time intervals (0.5 h, 1, 2 h, 3 h, 4 h and 24 h), 500 μL of the suspension was taken out and filtered by centrifugal ultrafiltration at 1770 RCF for 30 min using a spin filter (vivaspin 500 μL 1000 K), and the fluorescence of the filtrate was measured at λex = 553 nm and λem = 576 nm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Else Kröner-Fresenius-Stiftung, the Max Planck Society and the MaxSynBio consortium are acknowledged for financial support. This project was also funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 213555243 – SFB1066. The authors thank Gunnar Glasser for recording the SEM images, and Stefan Schuhmacher for helping with graphical design. Open Access funding provided by the Max Planck Society.References
- J. Zhou, Z. Shao, J. Liu, Q. Duan, X. Wang, J. Li and H. Yang, ACS Appl. Bio Mater., 2020, 3, 2686–2701 CrossRef CAS.
- S. Mitragotri, P. A. Burke and R. Langer, Nat. Rev. Drug Discovery, 2014, 13, 655–672 CrossRef CAS PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- R. van der Meel, E. Sulheim, Y. Shi, F. Kiessling, W. J. M. Mulder and T. Lammers, Nat. Nanotechnol., 2019, 14, 1007–1017 CrossRef CAS PubMed.
- S. Wang, Z. Wang, G. Yu, Z. Zhou, O. Jacobson, Y. Liu, Y. Ma, F. Zhang, Z.-Y. Chen and X. Chen, Adv. Sci., 2019, 6, 1801986 CrossRef PubMed.
- M. S. Rehmann, K. M. Skeens, P. M. Kharkar, E. M. Ford, E. Maverakis, K. H. Lee and A. M. Kloxin, Biomacromolecules, 2017, 18, 3131–3142 CrossRef CAS PubMed.
- D. Sandrin, D. Wagner, C. E. Sitta, R. Thoma, S. Felekyan, H. E. Hermes, C. Janiak, N. de Sousa Amadeu, R. Kühnemuth, H. Löwen, S. U. Egelhaaf and C. A. M. Seidel, Phys. Chem. Chem. Phys., 2016, 18, 12860–12876 RSC.
- J. H. Lee and Y. Yeo, Chem. Eng. Sci., 2015, 125, 75–84 CrossRef CAS PubMed.
- J. Mu, J. Lin, P. Huang and X. Chen, Chem. Soc. Rev., 2018, 47, 5554–5573 RSC.
- Y. Qiao, J. Wan, L. Zhou, W. Ma, Y. Yang, W. Luo, Z. Yu and H. Wang, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 11, e1527 Search PubMed.
- N. Deirram, C. Zhang, S. S. Kermaniyan, A. P. R. Johnston and G. K. Such, Macromol. Rapid Commun., 2019, 40, 1800917 CrossRef PubMed.
- G. Yang, L. Xu, J. Xu, R. Zhang, G. Song, Y. Chao, L. Feng, F. Han, Z. Dong, B. Li and Z. Liu, Nano Lett., 2018, 18, 2475–2484 CrossRef CAS PubMed.
- J. Li, Y. Li, Y. Wang, W. Ke, W. Chen, W. Wang and Z. Ge, Nano Lett., 2017, 17, 6983–6990 CrossRef CAS PubMed.
- S. Grabbe, H. Haas, M. Diken, L. M. Kranz, P. Langguth and U. Sahin, Nanomedicine, 2016, 11, 2723–2734 CrossRef CAS PubMed.
- J. D. Finn, A. R. Smith, M. C. Patel, L. Shaw, M. R. Youniss, J. van Heteren, T. Dirstine, C. Ciullo, R. Lescarbeau, J. Seitzer, R. R. Shah, A. Shah, D. Ling, J. Growe, M. Pink, E. Rohde, K. M. Wood, W. E. Salomon, W. F. Harrington, C. Dombrowski, W. R. Strapps, Y. Chang and D. V. Morrissey, Cell Rep., 2018, 22, 2227–2235 CrossRef CAS PubMed.
- J. Li, H. Liang, J. Liu and Z. Wang, Int. J. Pharm., 2018, 546, 215–225 CrossRef CAS PubMed.
- O. Rifaie-Graham, S. Ulrich, N. F. B. Galensowske, S. Balog, M. Chami, D. Rentsch, J. R. Hemmer, J. Read de Alaniz, L. F. Boesel and N. Bruns, J. Am. Chem. Soc., 2018, 140, 8027–8036 CrossRef CAS PubMed.
- L. D. Blackman, S. Varlas, M. C. Arno, A. Fayter, M. I. Gibson and R. K. O’Reilly, ACS Macro Lett., 2017, 6, 1263–1267 CrossRef CAS PubMed.
- R. Münter, K. Kristensen, D. Pedersbæk, J. B. Larsen, J. B. Simonsen and T. L. Andresen, Nanoscale, 2018, 10, 22720–22724 RSC.
- C. Tan, M. Arshadi, M. C. Lee, M. Godec, M. Azizi, B. Yan, H. Eskandarloo, T. W. Deisenroth, R. H. Darji, T. V. Pho and A. Abbaspourrad, ACS Nano, 2019, 13, 9016–9027 CrossRef CAS PubMed.
- M.-L. Frey, J. Simon, M. Brückner, V. Mailänder, S. Morsbach and K. Landfester, Polym. Chem., 2020, 11, 3821–3830 RSC.
- K. Piradashvili, M. Fichter, K. Mohr, S. Gehring, F. R. Wurm and K. Landfester, Biomacromolecules, 2015, 16, 815–821 CrossRef CAS PubMed.
- A. K. Yamala, V. Nadella, Y. Mastai, H. Prakash and P. Paik, Nanoscale, 2017, 9, 14006–14014 RSC.
- D. Paßlick, K. Piradashvili, D. Bamberger, M. Li, S. Jiang, D. Strand, P. R. Wich, K. Landfester, M. Bros, S. Grabbe and V. Mailänder, J. Controlled Release, 2018, 289, 23–34 CrossRef PubMed.
- K. Piradashvili, J. Simon, D. Paßlick, J. R. Höhner, V. Mailänder, F. R. Wurm and K. Landfester, Nanoscale Horiz., 2017, 2, 297–302 RSC.
- T. Lin, P. Zhao, Y. Jiang, Y. Tang, H. Jin, Z. Pan, H. He, V. C. Yang and Y. Huang, ACS Nano, 2016, 10, 9999–10012 CrossRef CAS PubMed.
- C. Zhao, M. Zhu, Y. Fang, X. Liu, L. Wang, D. Chen and X. Huang, Mater. Horiz., 2020, 7, 157–163 RSC.
- S. K. Pramanik, S. Seneca, M. Peters, L. D'Olieslaeger, G. Reekmans, D. Vanderzande, P. Adriaensens and A. Ethirajan, RSC Adv., 2018, 8, 36869–36878 RSC.
- G. Baier, A. Cavallaro, K. Vasilev, V. Mailänder, A. Musyanovych and K. Landfester, Biomacromolecules, 2013, 14, 1103–1112 CrossRef CAS PubMed.
- I. Schlegel, R. Muñoz-Espí, P. Renz, I. Lieberwirth, G. Floudas, Y. Suzuki, D. Crespy and K. Landfester, Macromolecules, 2017, 50, 4725–4732 CrossRef CAS.
- M. Machtakova, S. Han, Y. Yangazoglu, I. Lieberwirth, H. Thérien-Aubin and K. Landfester, Nanoscale, 2021, 13, 4051–4059 RSC.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS PubMed.
- A. Musyanovych and K. Landfester, Macromol. Biosci., 2014, 14, 458–477 CrossRef CAS PubMed.
- E. Monogioudi, G. Faccio, M. Lille, K. Poutanen, J. Buchert and M.-L. Mattinen, Food Hydrocolloids, 2011, 25, 71–81 CrossRef CAS.
- E. V. Petrotchenko, J. J. Serpa, D. B. Hardie, M. Berjanskii, B. P. Suriyamongkol, D. S. Wishart and C. H. Borchers, Mol. Cell. Proteomics, 2012, 11, M111.013524 CrossRef PubMed.
- W. Ebeling, N. Hennrich, M. Klockow, H. Metz, H. D. Orth and H. Lang, Eur. J. Biochem., 1974, 47, 91–97 CrossRef CAS PubMed.
- S. Udenfriend, S. Stein, P. Böhlen, W. Dairman, W. Leimgruber and M. Weigele, Science, 1972, 178, 871 CrossRef CAS PubMed.
- J. M. Artigas, M. E. Garcia, M. R. Faure and A. M. Gimeno, Postgrad. Med. J., 1981, 57, 219 CrossRef CAS PubMed.
- A. O. Elzoghby, W. M. Samy and N. A. Elgindy, J. Controlled Release, 2012, 161, 38–49 CrossRef CAS PubMed.
- N. A. Hadjiev and B. G. Amsden, J. Controlled Release, 2015, 199, 10–16 CrossRef CAS PubMed.
- A. G. Ogston, Trans. Faraday Soc., 1958, 54, 1754–1757 RSC.
- L. Nothnagel and M. G. Wacker, Eur. J. Pharm. Sci., 2018, 120, 199–211 CrossRef CAS PubMed.
- S. Gou, Q. Chen, Y. Liu, L. Zeng, H. Song, Z. Xu, Y. Kang, C. Li and B. Xiao, ACS Sustainable Chem. Eng., 2018, 6, 12658–12667 CrossRef CAS.
- T. Z. Chang, S. S. Stadmiller, E. Staskevicius and J. A. Champion, Biomater. Sci., 2017, 5, 223–233 RSC.
- A. Pietrzak-Nguyen, K. Piradashvili, M. Fichter, L. Pretsch, F. Zepp, F. R. Wurm, K. Landfester and S. Gehring, Nanomedicine, 2016, 12, 2383–2394 CrossRef CAS PubMed.
- J. Tel, S. P. Sittig, R. A. M. Blom, L. J. Cruz, G. Schreibelt, C. G. Figdor and I. J. M. de Vries, J. Immunol., 2013, 191, 5005 CrossRef CAS PubMed.
- H. Hemmi, T. Kaisho, O. Takeuchi, S. Sato, H. Sanjo, K. Hoshino, T. Horiuchi, H. Tomizawa, K. Takeda and S. Akira, Nat. Immunol., 2002, 3, 196–200 CrossRef CAS PubMed.
- M. M. Alam, D. Yang, A. Trivett, T. J. Meyer and J. J. Oppenheim, Front. Immunol., 2018, 9, 2982 CrossRef CAS PubMed.
- A. K. Andrianov, A. Marin, R. Wang, H. Karauzum, A. Chowdhury, P. Agnihotri, A. S. Yunus, R. A. Mariuzza and T. R. Fuerst, ACS Appl. Bio Mater., 2020, 3, 3187–3195 CrossRef CAS PubMed.
- N. Van Hoeven, C. B. Fox, B. Granger, T. Evers, S. W. Joshi, G. I. Nana, S. C. Evans, S. Lin, H. Liang, L. Liang, R. Nakajima, P. L. Felgner, R. A. Bowen, N. Marlenee, A. Hartwig, S. L. Baldwin, R. N. Coler, M. Tomai, J. Elvecrog, S. G. Reed and D. Carter, Sci. Rep., 2017, 7, 46426 CrossRef PubMed.
- K. J. Peine, G. Gupta, D. J. Brackman, T. L. Papenfuss, K. M. Ainslie, A. R. Satoskar and E. M. Bachelder, J. Antimicrob. Chemother., 2014, 69, 168–175 CrossRef CAS PubMed.
- N. Marasini, M. Skwarczynski and I. Toth, Expert Rev. Vaccines, 2014, 13, 1361–1376 CrossRef CAS PubMed.
- I. Hellmuth, I. Freund, J. Schlöder, S. Seidu-Larry, K. Thüring, K. Slama, J. Langhanki, S. Kaloyanova, T. Eigenbrod, M. Krumb, S. Röhm, K. Peneva, T. Opatz, H. Jonuleit, A. H. Dalpke and M. Helm, Front. Immunol., 2017, 8, 312 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb01368h |
| This journal is © The Royal Society of Chemistry 2021 |