
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fabrication of nanoparticles for bone regeneration: new insight into applications of nanoemulsion technology
Barbara
Kupikowska-Stobba
 * and
Mirosław
Kasprzak
* and
Mirosław
Kasprzak
ŁUKASIEWICZ Research Network – Institute of Ceramics and Building Materials, Ceramic and Concrete Division in Warsaw, Department of Biomaterials, Postępu 9, 02-677, Warsaw, Poland. E-mail: barbara.kupikowska-stobba@icimb.lukasiewicz.gov.pl
First published on 18th June 2021
Abstract
Introducing synthetic bone substitutes into the clinic was a major breakthrough in the regenerative medicine of bone. Despite many advantages of currently available bone implant materials such as biocompatiblity and osteoconductivity, they still suffer from relatively poor bioactivity, osteoinductivity and osteointegration. These properties can be effectively enhanced by functionalization of implant materials with nanoparticles such as osteoinductive hydroxyapatite nanocrystals, resembling inorganic part of the bone, or bioactive polymer nanoparticles providing sustained delivery of pro-osteogenic agents directly at implantation site. One of the most widespread techniques for fabrication of nanoparticles for bone regeneration applications is nanoemulsification. It allows manufacturing of nanoscale particles (<100 nm) that are injectable, 3D-printable, offer high surface-area-to-volume-ratio and minimal mass transport limitations. Nanoparticles obtained by this technique are of particular interest for biomedical engineering due to fabrication procedures requiring low surfactant concentrations, which translates into reduced risk of surfactant-related in vivo adverse effects and improved biocompatibility of the product. This review discusses nanoemulsion technology and its current uses in manufacturing of nanoparticles for bone regeneration applications. In the first section, we introduce basic concepts of nanoemulsification including nanoemulsion formation, properties and preparation methods. In the next sections, we focus on applications of nanoemulsions in fabrication of nanoparticles used for delivery of drugs/biomolecules facilitating osteogenesis and functionalization of bone implants with special emphasis on biomimetic hydroxyapatite nanoparticles, synthetic polymer nanoparticles loaded with bioactive compounds and bone-targeting nanoparticles. We also highlight key challenges in formulation of nanoparticles via nanoemulsification and outline potential further improvements in this field.
Barbara Kupikowska-Stobba, received her MSc degree in biotechnology from Warsaw University of Life Sciences (Poland) and PhD degree in biomedical engineering from Nalecz Institute of Biocybernetics and Biomedical Engineering PAS in Warsaw in 2019. She is currently a postdoctoral fellow in the Department of Biomaterials at LUKASIEWICZ Research Network – Institute of Ceramics and Building Materials in Warsaw. She has co-authored 16 scientific publications including 10 papers in scientific journals, 6 monograph chapters and 1 patent. Her current research interests include development of multifunctional bioactive hydroxyapatite/polymer composite implants for bone regeneration applications, synthesis and modification of hydroxyapatite with pro-osteogenic factors. |
Mirosław Kasprzak, received his MSc degree in Food Technology and Human Nutrition from University of Life Sciences in Lublin, Poland and PhD degree in animal science from Aarhus University, Denmark (2013). Currently, he is the leader of the research project “Multifunctional biologically active composites for applications in regenerative medicine of bone” conducted in the Department of Biomaterials at LUKASIEWICZ Research Network – Institute of Ceramics and Building Materials in Warsaw, Poland. He has co-authored 13 publications in scientific journals and 1 monograph. His research interest lies in the development of multifunctional bone implants composed of hydroxyapatite and biodegradable polymers. |
Introduction
Introducing synthetic bone substitutes into the clinic was a major breakthrough in the regenerative medicine of bone.1 Advances in the field of biomaterials have allowed development of bone implants that are biocompatible, bioresorbable2,3 and osteoconductive4,5 (i.e. able to support bone growth on their surface). Despite these advantages, the vast majority of currently available implant materials suffer from poor bioactivity,4 osteoinductivity6 (ability to induce osteogenesis) and osteointegration.7 Over the past few decades, many efforts have been devoted to improving properties of implant materials by their functionalization with nanoparticles such as osteoinductive hydroxyapatite nanocrystals resembling inorganic part of the bone or bioactive nanoparticles loaded with different types of drugs/biomolecules such as factors facilitating osteogenesis (e.g. bone morphogenetic proteins, BMPs,8–10 osteopontin,11 dexamethasone12), antimicrobial (Ag,13 CuO,14 Se,15 curcumin,16–18 chlorhexidine19,20) or anti-inflammatory agents (e.g. curcumin,16–18,21 quercitrin22,23) and drugs inhibiting bone resorption (e.g. bisphosphonates such as alendronate24–26).One of the most widespread and versatile methods for fabrication of nanoparticles is nanoemulsification. It relies on formation of dispersions of nanosized droplets by mixing a polar phase with a non-polar phase in the presence of a surfactant27 and allows manufacturing of very small particles (<100 nm) that are injectable, 3D-printable, offer high surface-area-to-volume-ratio and minimal mass transport limitations. Nanoparticles obtained by nanoemulsion technology have received particular attention in biomedical engineering due to fabrication procedures requiring lower surfactant concentrations (compared to other emulsifications methods i.e. macro- and microemulsification28) or no surfactant at all, translating into reduced risk of in vivo adverse effects associated with high surfactant levels (e.g. cytotoxicity29,30 and mucosa irritation31) and improved biocompatibility of the final product. These qualities have contributed to the broad use of nanoparticles formulated via nanoemulsion technology in different fields of tissue engineering including bone,32–35 cartilage,36 neural37 and cardiac37,38 tissue engineering.39,40
Among nanoparticles (NPs) fabricated via nanoemulsion technology hydroxyapatite NPs and drug-loaded synthetic polymer NPs have been the most extensively studied in the context of bone regeneration applications. Hydroxyapatite nanoparticles have been widely explored as fillers and strengths enhancers of orthopaedic and dental implants41–44 due to their excellent biocompatibility45,46 and well documented osteoconductive47 and osteoinductive48–50 properties. When incorporated into implant material, they improve a number of properties crucial for its in vivo performance such as hydrophilicity,51 surface properties,52 mechanical resistance53–55 and osteoinductivity.56,57 Osteoinductivity of nano-hydroxyapatite is attributed to the size of its particles and their high resemblance to hydroxyapatite nanocrystals occurring in the natural bone.58–60 As a result, hydroxyapatite nanocrystals are better recognized by the host cells compared to their micron-sized counterparts61,62 and thus can more effectively stimulate cellular behaviors involved in osteogenesis such as osteogenic differentiation, cell adhesion, proliferation and migration.48
Synthetic polymer nanoparticles are probably the largest category of NPs used for bone regeneration applications.63 This type of NPs is typically made of biodegradable polymers such as poly(lactide-co-glycolide) (PLGA), poly(lactic acid) (PLA), poly(ε-caprolactone) (PCL) or polyethylene glycol (PEG) and are predominantly applied as delivery vehicles for pro-osteogenic agents (e.g. bone morphogenetic proteins, BMPs8–10) and drugs preventing bone resorption,24–26 implant-related infections or inflammation.16–18,21 Drug-loaded nanoparticles can provide controlled sustained drug release over extended periods of time16,18,21,64 and can be easily introduced into bone defects (e.g. via injection), which may eliminate adverse side effects of drugs administered systemically and current therapeutic approaches to bone injuries based on supra-physiological doses of BMP-2.65
In this review, we discuss nanoemulsion technology and its current uses in manufacturing of nanoparticles for bone regeneration applications. In the first section, we introduce basic concepts of nanoemulsification including nanoemulsion formation, properties and preparation methods. In the next sections, we focus on applications of nanoemulsions in fabrication of nanoparticles used for delivery of drugs/biomolecules facilitating osteogenesis and functionalization of bone implants with special emphasis on biomimetic hydroxyapatite nanoparticles, synthetic polymer nanoparticles loaded with bioactive compounds and bone-targeting nanoparticles. We also highlight key challenges in formulation of nanoparticles via nanoemulsification and outline potential further improvements in this field.
Nanoemulsion technology
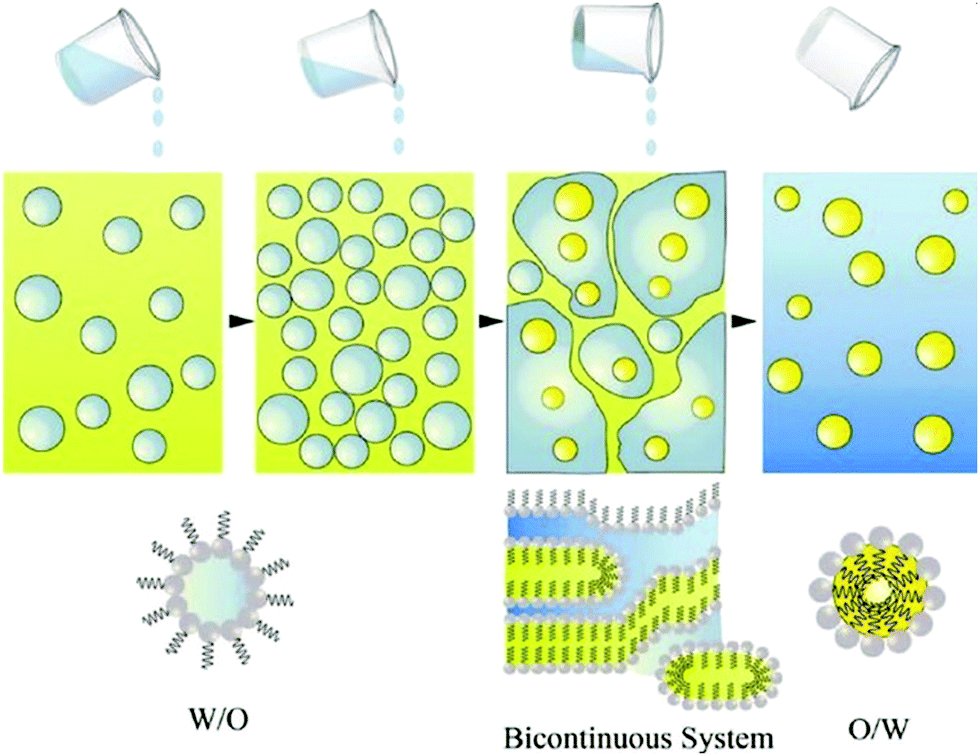
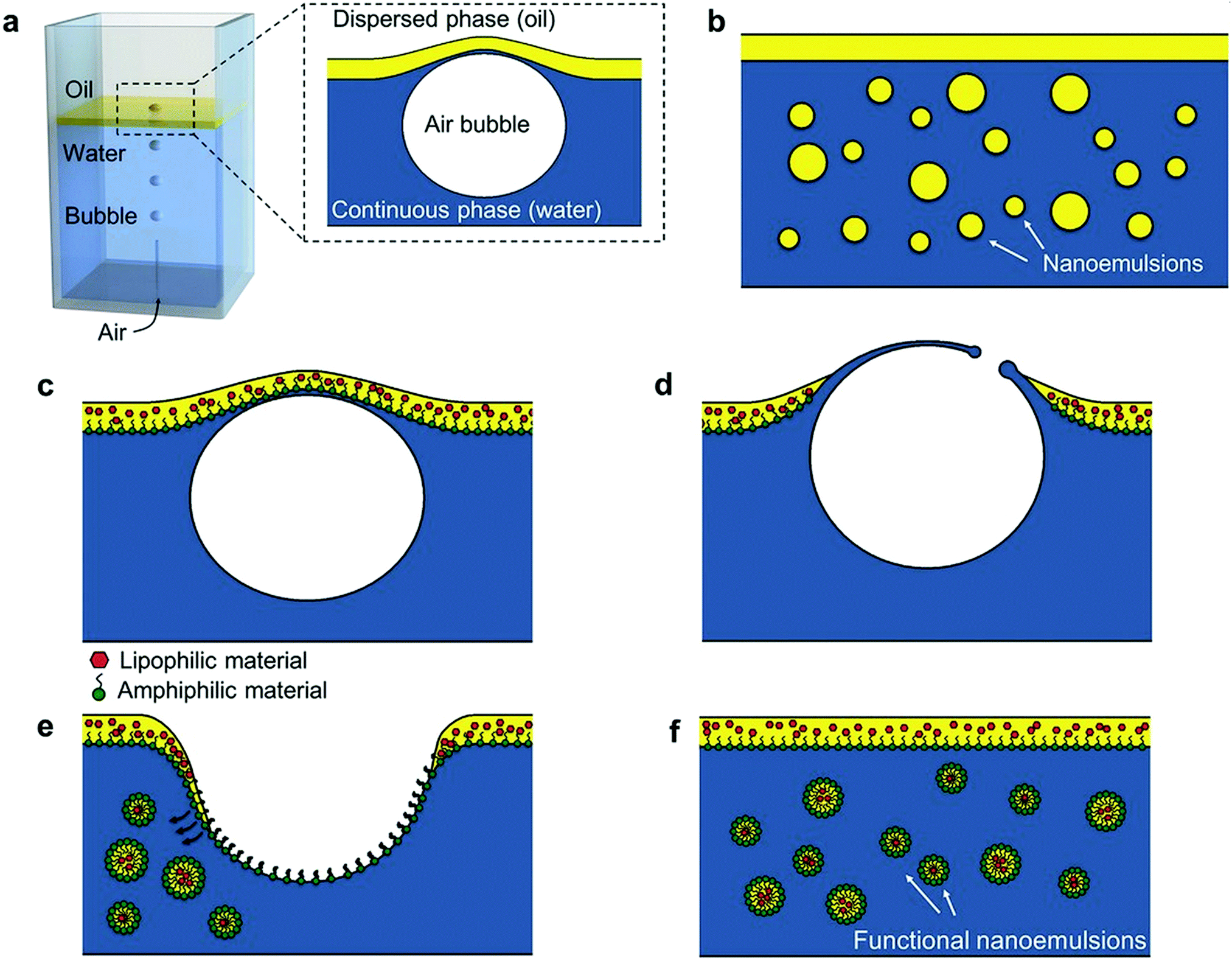
Nanoemulsions (also known as mini-emulsions) are heterogeneous systems, in which nanosized droplets (<100 nm27) of one immiscible liquid (the dispersed phase) are dispersed in another liquid (the continuous phase). Nanoemulsions are formed by mixing an oil (non-polar) phase with an aqueous (polar) phase in the presence of an emulsifier. First, the dispersed phase is broken up into larger droplets forming a macroemulsion (also called a coarse emulsion), which in the second step is broken up into nanodroplets.66 Deformation and breaking up of the droplets expands the interface between the phases, which significantly increases interfacial area and interfacial energy. For this reason, formation of nanoemulsions does not occur spontaneously and requires energy input.28 The role of emulsifier is to reduce the interfacial tension between the immiscible phases and lower the shear force needed to break up the droplets.67 At the same time, emulsifier molecules adsorbed to the surface of newly formed droplets impart a physical barrier between neighbouring droplets, which stabilizes them and prevents them from coalescing.28The ability of emulsifiers to stabilize the droplets results from their amphiphilic structure. Emulsifier molecules are composed of a hydrophilic group (the head) and a hydrophobic domain (the tail). When added to an oil-in-water (o/w) emulsion (dispersion of oil droplets in an aqueous phase), emulsifier molecules spontaneously surround the oil droplets with their non-polar tails extending into the droplet, while their heads direct outwards (towards the continuous phase) as depicted in Fig. 1. In the case of the reverse (water-in-oil, w/o) nanoemulsions (composed of water droplets dispersed in the oil phase), the orientation of emulsifier molecules is reversed: non-polar tails are directed outwards (facing the oil phase), whereas polar heads point to the center of water droplets (Fig. 1). Oil-in-water emulsions are referred to as regular emulsions,68 whereas water-in-oil emulsions are called reverse or invert emulsions. When emulsion is further dispersed into another continuous phase, it is called a multiple emulsion or emulsified emulsion. An example of multiple emulsion is oil-water-oil (o/w/o) emulsion, which consists of small oil droplets dispersed in larger water droplets suspended in the continuous oil phase.
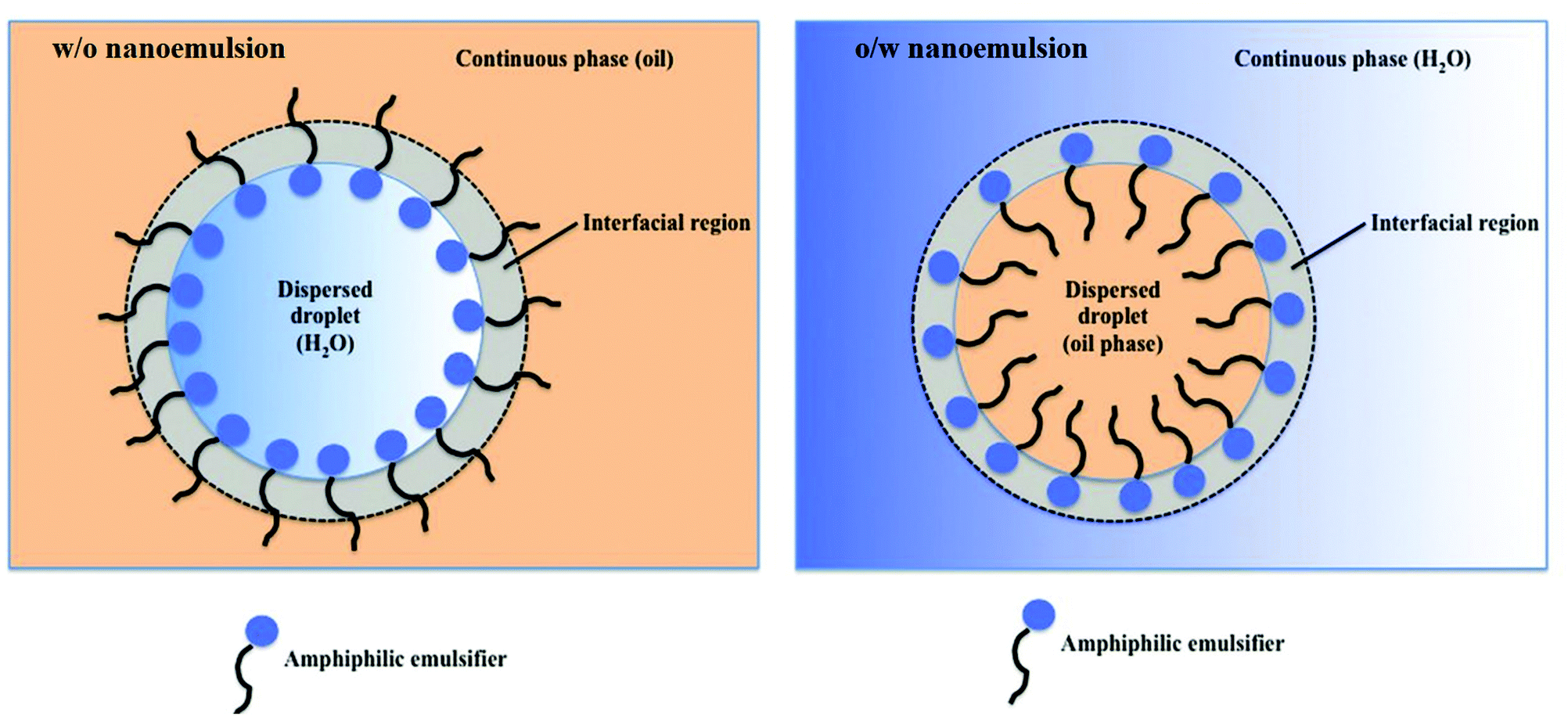 | ||
| Fig. 1 Illustration of a regular (oil-in-water, o/w) and reverse (water-in-oil, w/o) nanoemulsion droplets stabilized with an amphiphilic emulsifier. W/o nanoemulsion (left): non-polar tails are directed towards the oil phase, whereas polar heads point to the center of water droplets. O/w nanoemulsion (right): amphiphilic emulsifier molecules spontaneously surround the oil droplets with their non-polar tails extending into the droplet, while their heads direct outwards, towards the continuous polar phase. Copyright©2017 Scrivener Publishing LLC. Reproduced from ref. 69: https://doi.org/10.1002/9781119364221.ch6 with permission from Scrivener Publishing LLC. | ||
Properties of nanoemulsions
Nanoemulsions have several significant advantages over conventional emulsions (macroemulsions) containing droplets in the 0.1–5.0 μm range70 and microemulsions containing droplets ranging in size from 10 to 100 nm.71 First, nanoemulsions are much more kinetically stable than macroemulsions, i.e. they can resist phase separation for much longer periods of time.72 Increased kinetic stability of nanoemulsions is attributed to their very small droplet size, which prevents gravitation-driven phase separation processes such as droplet sedimentation and flocculation (a process where droplets group together, but do not merge, Fig. 2). Second, due to the droplet size significantly smaller than the wavelength of visible light (380–700 nm), nanoemulsion droplets scatter little light, making them appear transparent, as opposed to macroemulsions, which are usually opaque or turbid.73 This quality allows incorporation of nanoemulsions into products that need to be optically clear and enables their easy observation by light microscopy. Third, preparation of nanoemulsions requires much lower concentrations of surfactants compared to macro- and microemulsions28 or no surfactant at all,74 which substantially improves their biocompatibility. Although nanoemulsions have similar droplet size to microemulsions (10 to 100 nm71), they are much more resistant to physical and chemical changes than microemulsions,72 which tend to easily destabilize with changing composition or environmental conditions such as temperature and pH. Other advantages of nanoemulsions include low polydispersity of the droplets (<10–20% vs. >40% for macroemulsions72) and improved bioavailability of compounds encapsulated in the droplets.75 The fundamental difference between nanoemulsions and microemulsions is that nanoemulsions are thermodynamically unstable, i.e. they are not in chemical equilibrium and do not form/reform spontaneously (without energy input) when the emulsifier, oil and aqueous phase are brought together.27 However, they are kinetically stable, which means that they remain stable for extended periods of time (ranging from several days76 to several years77), but over time will eventually separate into two phases due to their natural tendency to lower their interfacial area and interfacial energy (which is lower in the two-phase state). The advantage of this metastability is that it removes the compositional limitation of microemulsions required for their thermodynamic stability. As a result, nanoemulsions can be functionalized with much wider variety of compounds.78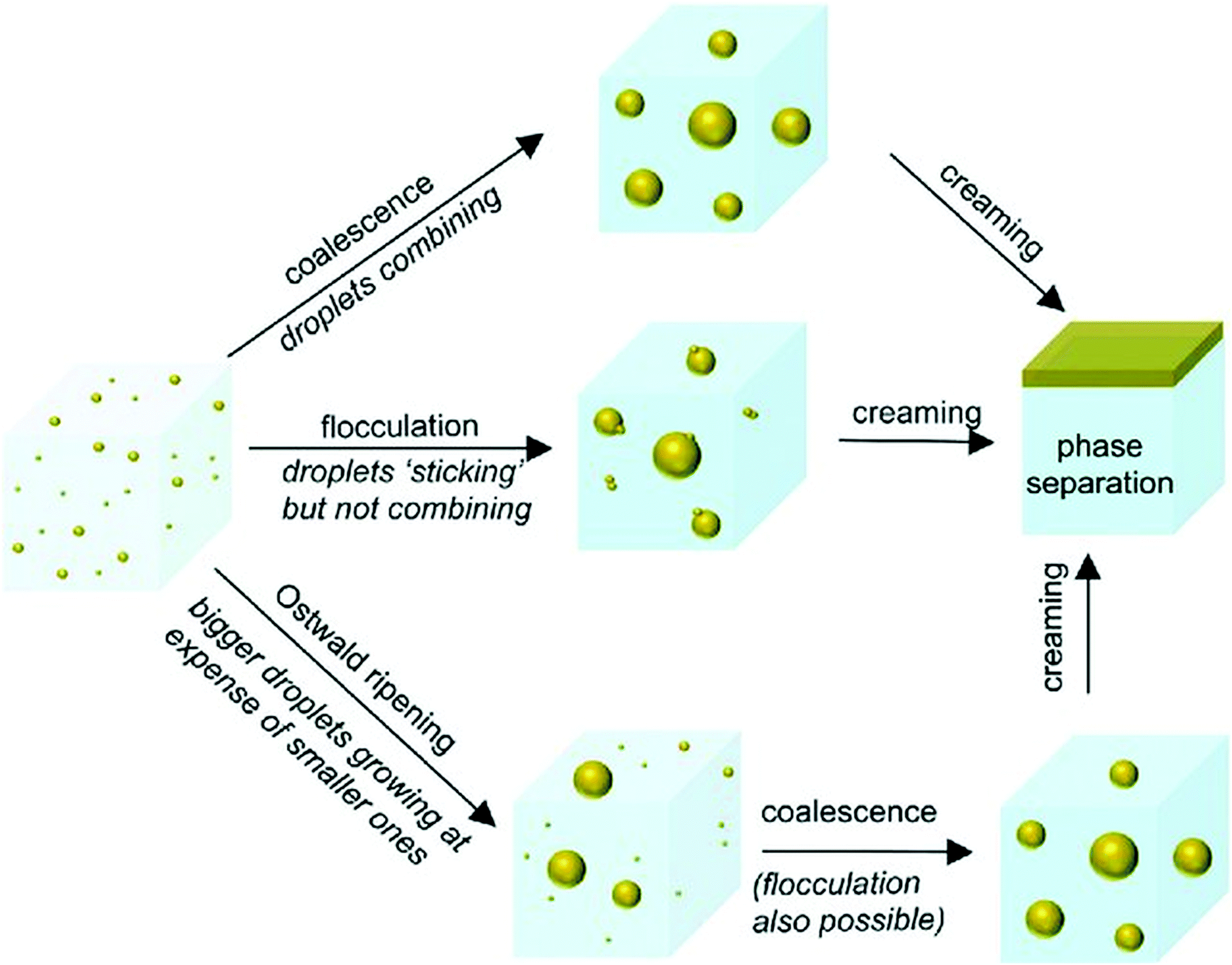 | ||
| Fig. 2 Nanoemulsion destabilization mechanisms: Ostwald ripening, flocculation and coalescence. Though nanoemulsions can be destabilized through any of the possible routes, initial growth of droplets generally occurs through Ostwald ripening. Coalescence and flocculation become more prevalent as the droplet size increases. Copyright©The Royal Society of Chemistry 2016. Reproduced from ref. 72: https://doi.org/10.1039/C5SM02958A available via license: CC BY-NC 3.0. | ||
Factors affecting nanoemulsion droplet size
Droplet size is a key factor that determines nanoemulsion stability and physicochemical properties including its transparency, density and rheological properties.79,80 Nanoemulsion droplet size depends primarily on applied emulsification method (e.g. high or low energy method, see section “Nanoemulsion preparation methods”), process parameters (e.g. shear force, feed pressure of dispersed phase, emulsification time, temperature, etc.80), phase mixture composition (e.g. type of emulsifier, oil and aqueous phase, and their relative concentrations), and the physicochemical properties of the immiscible phases (e.g. interfacial tension, solubility and viscosity).80,81 The final size of nanoemulsion droplet is the resultant of droplet break-up and coalescence processes.28 Break-up of the droplets can be induced by shear stress applied to the dispersed phase. Increasing shear force or emulsification time usually leads to formation of smaller droplets.72,80,82 The resistance of the droplets to deformation is determined by the Laplace pressure (the difference in the pressure inside and outside the droplet), which can be reduced by adding a surfactant into the oil/water mixture.28,83 In general, an increase in the surfactant concentration lowers the interfacial tension between the dispersed phase and continuous phase84 leading to reduction in droplet diameter.80,85,86Droplet size correlates also with surfactant surface activity and molecule size, which both influence the rate of surfactant adsorption to the surface of newly formed droplets.28,83 Small-molecule surfactants (such as sodium dodecyl sulfate, SDS and Tween) adsorb more rapidly to the droplet surface and thus tend to produce smaller droplets than larger surfactants (e.g. caseinate and β-lactoglobulin) under the same emulsification conditions.80,81,87 Nanoemulsion droplet size can be also controlled by altering relative viscosity of the dispersed phase to the continuous phase. As the viscosity of the disperse phase increases, it becomes more difficult to break up, which increases the size of the droplets.88 Increasing viscosity of the continuous phase has the opposite effect – it reduces the size of the droplets due to higher shear stress generated during emulsification.80,83,89 Another parameter affecting final droplet size is temperature. Temperature strongly influences viscosity of the dispersed and continuous phase, as well as solubility of the emulsifier in the oil and water mixture.84 Nanoemulsions produced at higher temperatures contain smaller droplets of broader size distribution, which results from enhanced rate of coarsening of the droplets via Ostwald ripening and coalescence.71,84
Nanoemulsion stability
Nanoemulsion stability is defined as its ability to resist changes in the physicochemical properties and droplet size over time.90 As mentioned before, nanoemulsions are thermodynamically unstable, but kinetically stable systems in nonequilibrium state. The lower the droplet size, the higher the stability of nanoemulsion.90 This effect results from lower attraction between smaller droplets and their reduced coalescence. Initial growth of droplets leading to nanoemulsion separation occurs predominantly via Ostwald ripening.28 Ostwald ripening is based on incorporation of smaller droplets into larger droplets (Fig. 2) driven by differences in the pressure between the inside of the droplets and the continuous phase, as well as differences in solubility of non-uniformly sized droplets in the continuous phase. Since smaller droplets display higher solubility in the continuous phase,70 Brownian motion and random collisions between the droplets of different sizes causes inclusion of smaller droplets into the larger ones. Ostwald ripening phenomena is largely dependent on the solubility of the dispersed phase in the continuous phase.91 The ripening process can be minimized by selecting oil and water phases that are highly immiscible, i.e. the dispersed phase solubility in the continuous phase is very low.92 As the droplets increase in size, coalescence and flocculation become more prevalent mechanisms of nanoemulsion separation.The period of time after which nanoemulsion will separate is highly variable and can be controlled by a number of factors including nanoemulsion composition (emulsifier type and concentration, water-to-oil volume ratio, presence of additives such as co-surfactants, etc.), droplet polydispersity index, physicochemical properties of mixture components (e.g. viscosity, density, interfacial tension, polarity, ionic strength) and temperature.72 One of the most important factors affecting nanoemulsion stability is choice of the emulsifier. The key properties that need to be considered during emulsifier selection is its size and charge. Nanoemulsions can be stabilized using ionic (cationic or anionic) or non-ionic emulsifiers. Ionic emulsifiers contain a charged polar group (positively charged in cationic emulsifiers or negatively charged in anionic emulsifiers) and work by forming a charged shell around the droplets causing electrostatic repulsion between them. The degree of repulsion between the droplets correlates with the surface charge and ionization degree of emulsifier molecules, which in turn depend on nanoemulsion pH and presence of other charged species (e.g. electrolytes).70 As a result, stabilizing effect of ionic emulsifiers is highly sensitive to nanoemulsion composition. This restricts application possibilities of ionic emulsifiers to only certain types of emulsions. For example, cationic emulsifiers are effective in low pH and neutral solutions, but not in alkaline solutions, while anionic emulsifiers are more suitable for alkaline emulsions. Non-ionic emulsifiers are considered more effective in stabilization of different kinds of nanoemulsions.70 The molecules of non-ionic emulsifiers contain large, bulky, non-charged polar groups that sterically hinder the droplets from coalescing. This stabilization mechanism is not sensitive to medium pH and composition. Thus, most nanoemulsions are prepared using emulsifiers from this group such as Pluronics,70 Tween-80, Span 80 or Brij 30.76
Another important parameter taken into account when choosing optimal emulsifier is its hydrophilic–lipophilic balance (HLB). HLB is a measure of emulsifier partitioning tendency between oil and water phase. It is used to predict emulsifier's ability to form a particular type of emulsion. HLB correlates with emulsifier solubility in water and oil, as well as size and strength of its hydrophilic and lipophilic moieties.93 It is calculated from the weight percentage of the hydrophilic groups to the hydrophobic groups in a molecule.94 The HLB scale ranges from 0 to 20. HLB equal to 10 means that the emulsifier is equally attracted to the water and oil phase (i.e. is equally hydrophilic and hydrophobic). Emulsifiers with HLB above 10 (e.g. phospholipids, proteins, potassium/sodium salts) are more hydrophilic and therefore tend to form oil-in-water emulsions.94 Emulsifiers with HLB below 10 (e.g. glycerol esters, sorbitol fatty acid esters, propylene glycol fatty acid esters) are more hydrophobic and promote formation of water-in-oil emulsions.93 To achieve maximum nanoemulsion stability, HLB value should be matched to the HLB value of oil used as the non-polar phase.95 For instance, vegetable oil emulsions typically require emulsifiers with an HLB in the 7–8 range, while in the case of castor oil emulsions HLB should be around 14. However, the HLB concept has certain limitations such as low accuracy of predicting type of emulsions formed by some emulsifiers.96,97 For example, depending on emulsification process variables such as temperature or emulsifier concentration, the same emulsifier can form both w/o and o/w emulsions.95 In addition, o/w emulsions can be produced using emulsifiers across the entire HLB range and the theoretical HLB calculation does not apply to blends of different emulsifiers.
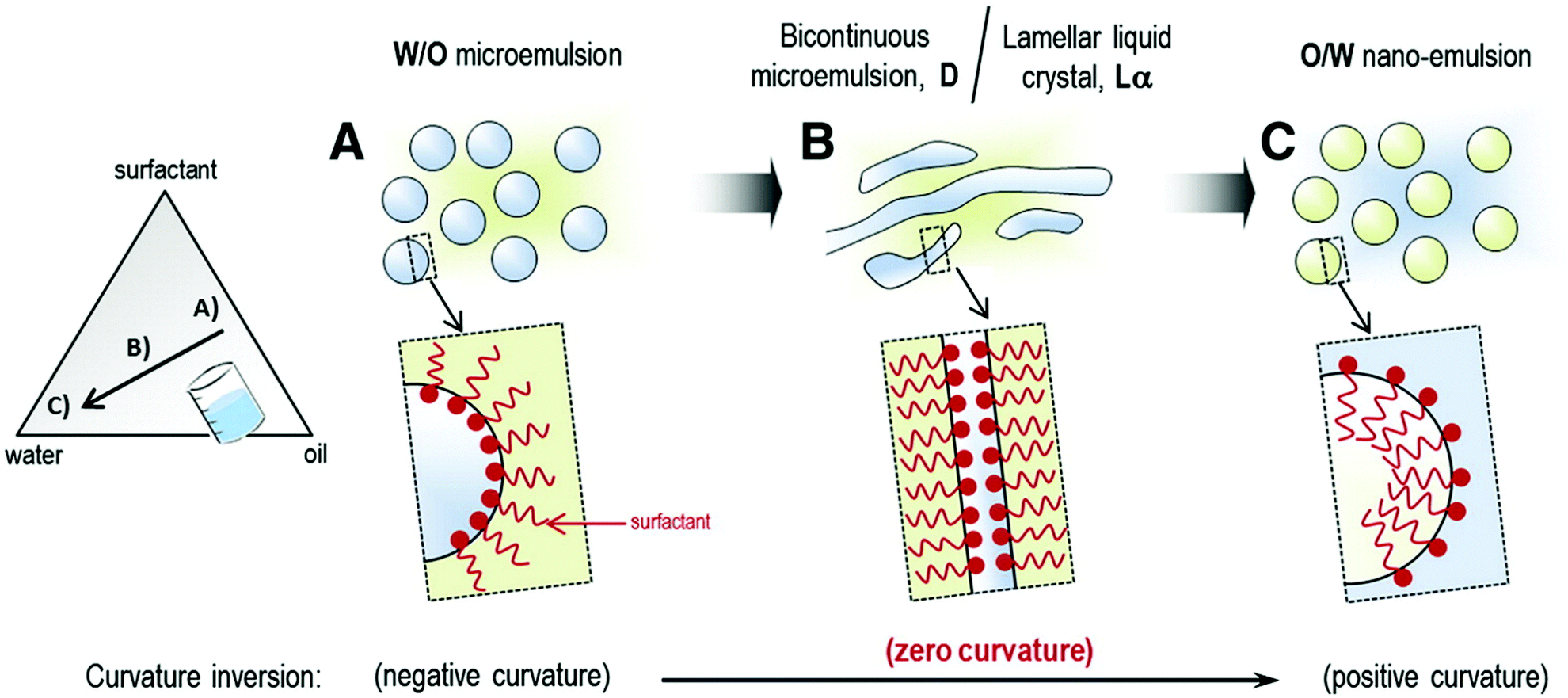
To allow more accurate characterization of emulsifiers, hydrophilic–lipophilic difference (or hydrophilic–lipophilic deviation, HLD) concept has been introduced.98,99 HLD takes into account numerous factors characterizing the emulsion such as surfactant concentration and type, hydrophobicity of the oil phase, salinity, temperature and the characteristic curvature (Cc) of the emulsifier. The Cc value reflects whether the emulsifier has a tendency to curve around an oil droplet in water (negative Cc) or a water droplet in oil (positive Cc). Hydrophilic emulsifiers tend to have negative Cc (e.g. sodium laurel sulphate, SLS has a Cc = −2.3), while hydrophobic emulsifiers have positive Cc (e.g. dioctyl sodium sulfosuccinate has a Cc of 2.6). HLD equal to 0 corresponds to the optimal emulsifier with equal affinity to both phases. A negative HLD value implies that the emulsifier is preferentially soluble in water and tends to produce o/w emulsions,100 whereas positive HLD value means that the emulsifier is more hydrophobic and tends to generate w/o emulsions.
Another factor strongly affecting nanoemulsion stability is the emulsifier concentration. In most cases, applying higher emulsifier concentrations produces more stable nanoemulsions.101 However, in some systems the opposite effect can occur due to various mechanisms of inter-droplet interactions in different nanoemulsions.101 Other variables influencing nanoemulsion destabilization rate is temperature, ionic strength, pH and viscosity of the continuous phase.72,102 Ionic strength and pH of the continuous phase have a major impact on electrostatic interactions and the repulsive barrier between the droplets102,103 and therefore most strongly affect emulsions stabilized with ionic emulsifiers. The higher the ionic strength of the continuous phase, the lower the repulsive barrier between the droplets and the higher probability of their flocculation/coalescence. Temperature alters viscosity of the phases, interfacial tension, adsorption of the surfactant to the droplets, dispersed phase solubility and diffusivity in the continuous phase,104 which has indirect effect on nanoemulsion stability. Since both interfacial tension and viscosity decrease with an increase in temperature, elevating the temperature facilitates nanoemulsion formation. However, emulsion stability decreases as the temperature rises due to higher solubility of the dispersed phase in the continuous phase and accelerated Ostwald ripening.72,105,106 Sharp changes in temperature are particularly harmful for the stability of nanoemulsions.104
Nanoemulsion stability can be improved by increasing viscosity of the continuous phase. More viscous continuous phase slows down gravitational separation (flocculation and sedimentation) and reduces the frequency of droplet collisions and coalescence, which contributes to increased resistance of the emulsion to separation.102,107 Nanoemulsion stability may be also increased by lowering dispersed to continuous phase volume ratio104 and droplet polydispersity index. Lower stability is generally displayed by polydisperse nanoemulsions containing a range of different droplet sizes.28,90 As mentioned before, in polydisperse systems smaller droplets display larger solubility in the continuous phase than the large ones.70 As the polydispersity of nanoemulsion increases, the differences in solubility between droplets become higher, which in turn accelerates the Ostwald ripening. Over the recent years, many efforts have been devoted to improving control over nanoemulsion stability by using emulsifiers responsive to specific stimuli such as light (e.g. azobenzene modified emulsifiers),108,109 pH (e.g. poly(methacrylic acid-co-methyl methacrylate-co-7-(4-vinylbenzyloxyl)-4-methylcoumarin emulsifier110) or salts (e.g. zwitterionic emulsifiers). Stimuli-responsive emulsifiers allow reversible stabilization/destabilization of nanoemulsions in response to physical or chemical changes, which can prove very useful in on-demand site-specific drug delivery.
Nanoemulsion preparation: high and low energy methods
Nanoemulsion preparation methods can be categorized into two main types: high energy methods and low energy methods. High energy methods employ high shear forces to disrupt the dispersed phase into the nanodroplets.111 In low energy methods, the energy required for nanodroplet formation originates from changes in chemical potential of emulsion components, driven by changes in temperature or composition of the oil–water mixture, which allows production of nanoemulsions under gentle mixing conditions. The devices used in high energy methods to generate shear stress include high pressure homogenizers, microfluidizers and ultrasonicators.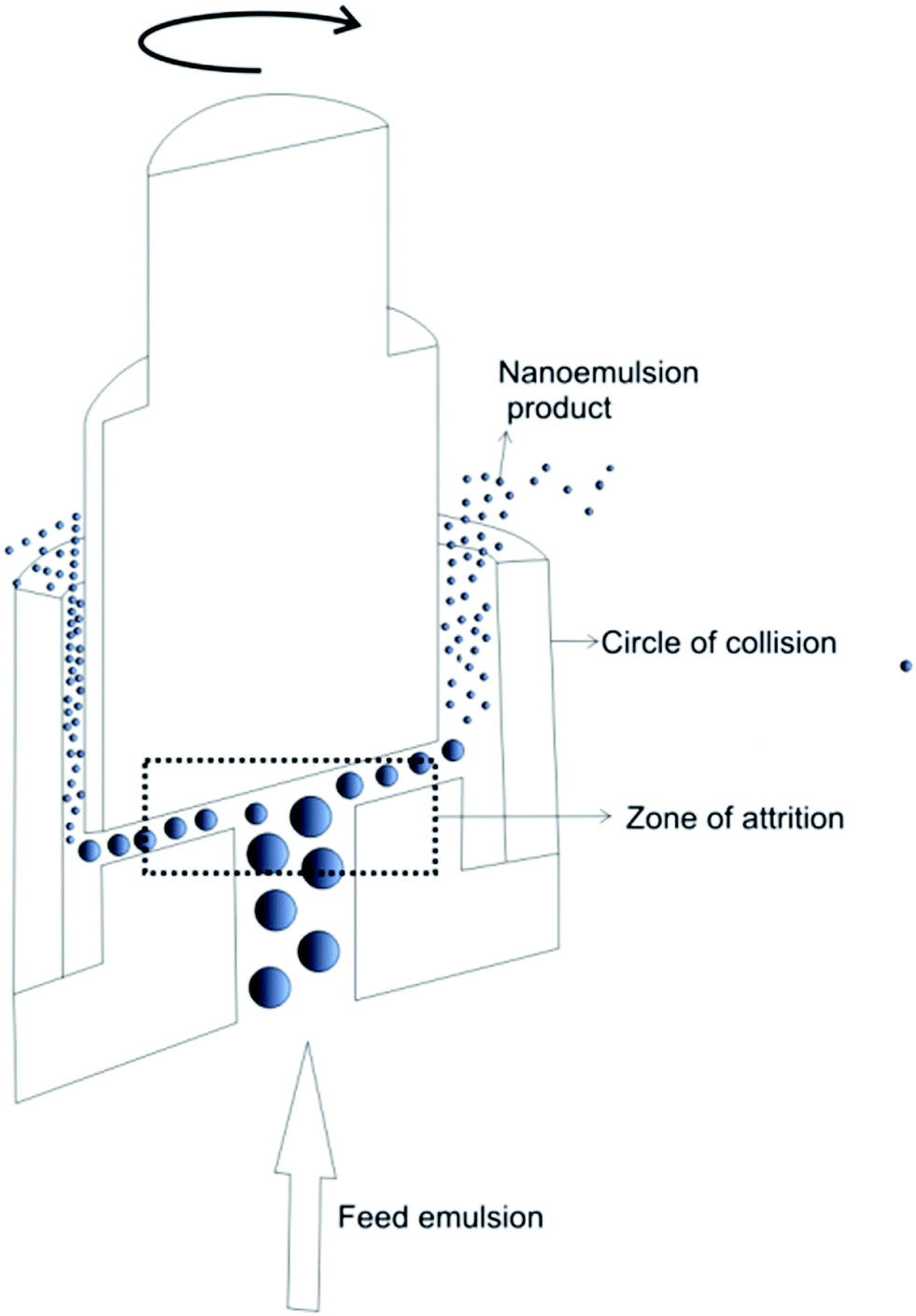 | ||
| Fig. 3 Schematic illustration of high-pressure homogenization performed in piston gap homogenizer. Copyright©2017 Elsevier B.V. Reproduced from ref. 113: https://doi.org/10.1016/j.jconrel.2017.03.008 with permission from Elsevier B.V. | ||
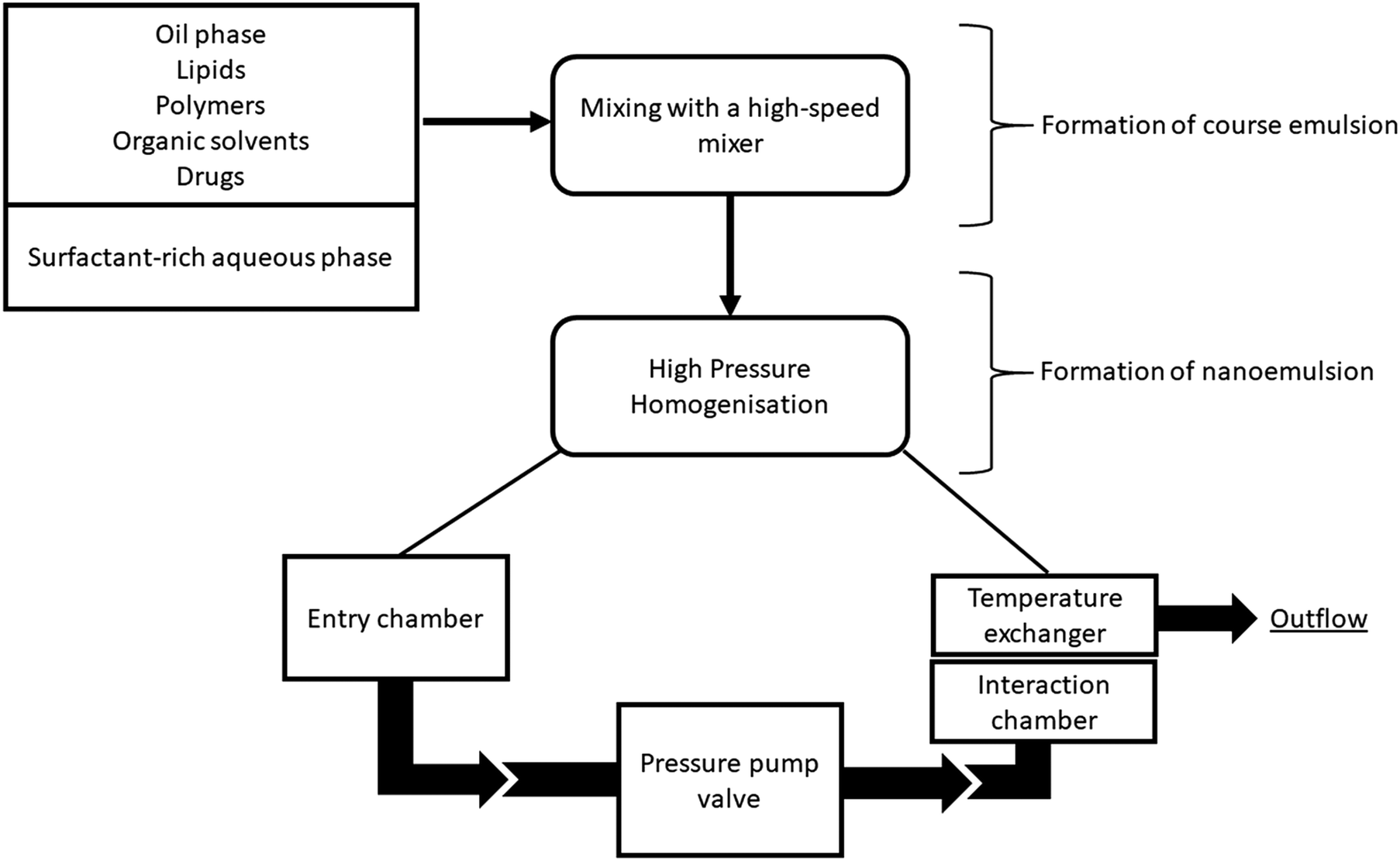 | ||
| Fig. 4 High pressure homogenization (HPH) technique. The emulsifier-rich aqueous phase is mixed with an organic phase using a high-speed mixer (e.g. Ultra Turrax disperser or Silverson mixer) to form a coarse emulsion. The resulting emulsion is introduced into a HPH device. In the homogenization chamber, hydraulic shear, intense turbulence and cavitation forces disrupt coarse emulsions into nanodroplets. In the interaction chamber, emulsifier molecules adsorb to the newly formed droplets and stabilize them. The disruption and stabilization steps are repeated several times. | ||
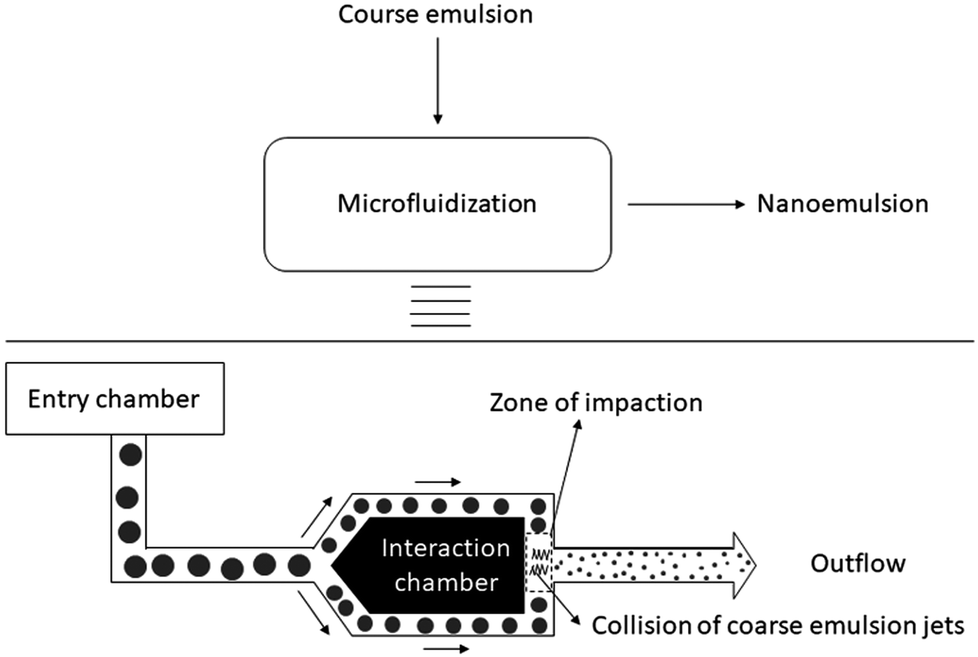 | ||
| Fig. 5 Schematic illustration of Y junction microfluidic device. Coarse emulsion is pumped under high pressure into a microchannel, which splits into two branches forming an Y junction. The two branches reconnect downstream into one channel. The shear stress generated in the interaction channel due to collision of two jets of the coarse emulsion disrupts the droplets into nanodroplets. | ||
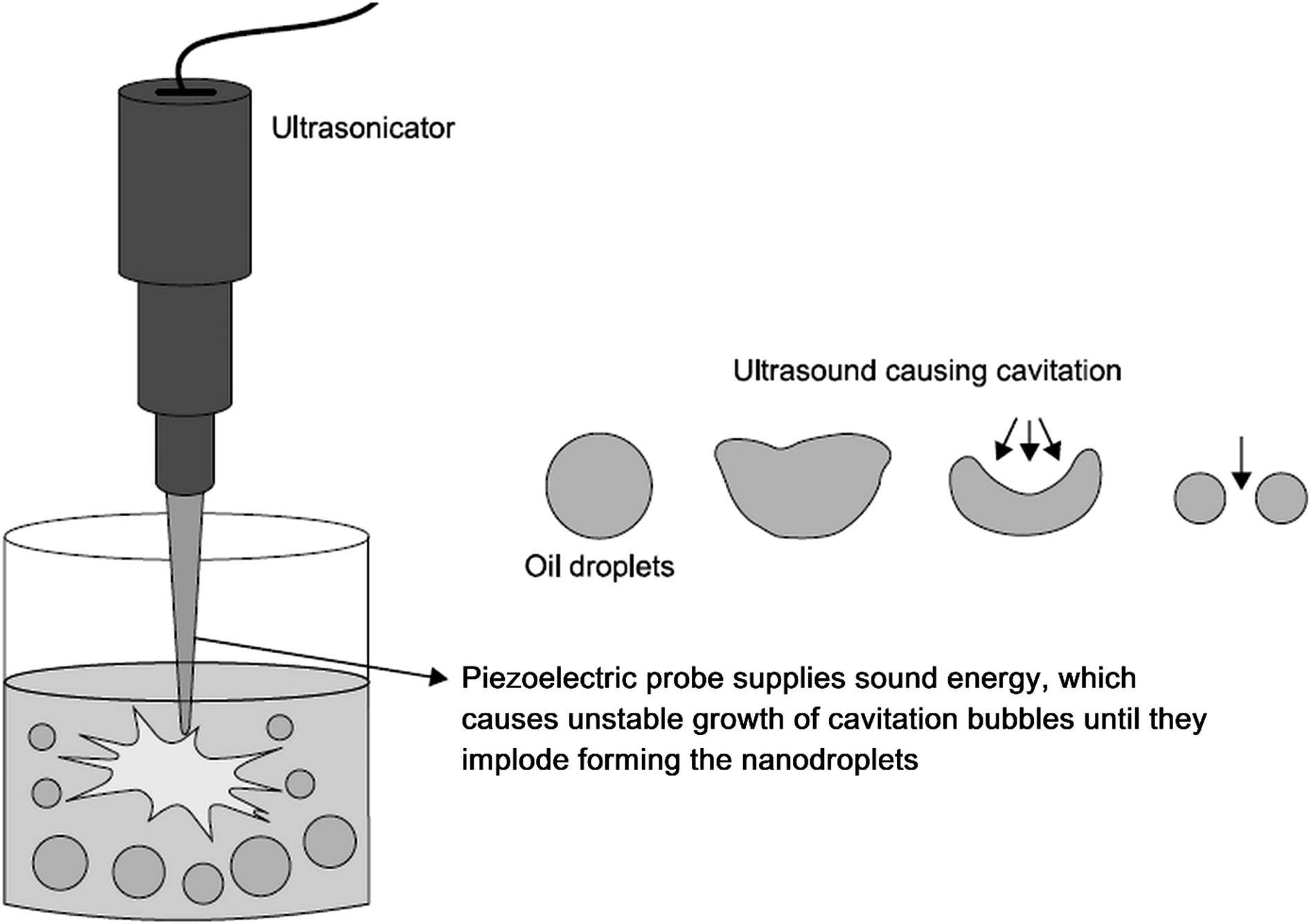 | ||
| Fig. 6 Preparation of nanoemulsion via ultrasonication. Ultrasonic waves emitted by the piezoelectric probe cause formation of cavitation bubbles, which grow in an unstable manner and ultimately implode. As a result, coarse emulsion droplets break up into nanodroplets. Copyright©2017 Elsevier B.V. Adapted from ref. 113: https://doi.org/10.1016/j.jconrel.2017.03.008 with permission from Elsevier B.V. | ||
One of the biggest advantages of high energy methods lies in their versatility. They allow effective production of nanoemulsions from oil–water mixtures of almost any composition, including mixtures without surfactants.113 The major limitation of these systems is high cost of homogenizers and large amount of heat generated by friction of the emulsions on the homogenizer surfaces, which needs to be dissipated by cooling of the device during its operation.113,124 The heat generated during homogenization rules out high energy methods from certain applications such as formulation of nanoemulsions containing thermosensitive compounds including proteins and nucleic acids.113 Another drawback of high energy nanoemulsification is degradation and fragmentation of high molecular weight compounds (e.g. polymers such as starch,125 methylcellulose,126 chitosan127) leading to reduction in their molecular weight and changes in their properties such as viscosity or degradation rate.
The limitations of high energy methods such as exposure of formulation to high shear stress can be overcome by low energy nanoemulsification methods. As mentioned before, in these methods, nanoemulsion formation is induced by changes in temperature or composition of the oil–water mixture. The energy input required for nanoemulsification is generated by changes in the chemical potential of the emulsion components, which allows formation of nanoemulsions by gentle mixing of water and oil phase. Low energy methods include self-nanoemulsification method, solvent displacement method, bubble bursting method, transitional phase inversion (TPI) and catastrophic phase inversion (CPI) method. The TPI can be conducted as phase inversion temperature (PIT) or phase inversion composition (PIC), whereas the CPI is performed as emulsion inversion point (EIP).
 | ||
| Fig. 7 Schematic representation of nanoemulsion formation by phase inversion composition (PIC) method. Copyright©2012 Elsevier Ltd. Reproduced from ref. 132: https://doi.org/10.1016/j.cocis.2012.07.003 with permission from Elsevier Ltd. | ||
The PIT method offers higher emulsification efficiency and produces nanoemulsions of lower polydispersity index compared to the PIC method (described below). However, the major drawback of this technique is rapid temperature changes, which can be detrimental to the stability and bioactivity of thermosensitive compounds such as proteins. In addition, PIT method can utilize only those types of surfactants that are sensitive to changes in temperature such as polyethoxylated (POE) non-ionic surfactants. In case of POE surfactants, altering the temperature causes a change in the hydration of the poly(oxyethylene) chains, which in turn alters their molecular geometry, affinity to water and oil and the spontaneous curvature.133
The mechanism of nanodroplet formation in the PIC method is analogous to the one in the PIT technique. However, since PIC method is performed at constant temperature, it can be applied to much wider range of formulations that the PIT method, including those containing thermally sensitive compounds. PIC technique is also more suitable for large-scale production of nanoemulsions as it is easier to add water or oil to a large volume of emulsion than to produce a sudden change in its temperature. Moreover, the PIC approach is not restricted to POE surfactants. Nonetheless, this type of surfactants is broadly applied for the preparation of nanoemulsions by the PIC method.
An important advantage of spontaneous nanoemulsification is that it can be performed under mild conditions (gentle mixing and room temperature) either in the presence or absence of surfactants.144 The spontaneous nanoemulsification mechanism is often utilized in self-nanoemulsifying drug delivery systems (SNEDDS). SNEDDS are anhydrous homogenous liquid mixtures composed of an oil phase, a surfactant, a co-emulsifier and a drug, which spontaneously form an o/w nanoemulsion upon dilution with water under gentle stirring.145 When SNEDDS are introduced into the aqueous environment of the gastrointestinal tract, they form o/w nanoemulsion with a help of agitation created by stomach digestive motility.146 SNEDDS are widely used to increase adsorption and bioavailability of hydrophobic bioactive compounds.
 | ||
| Fig. 8 Schematic representation of low-energy nanoemulsification by the catastrophic phase inversion (CPI) method. The amount of water added to a W/O emulsion is progressively increased until a phase inversion occurs and an O/W emulsion is formed. Copyright©Royal Society of Chemistry 2011. Reproduced from ref. 141: https://doi.org/10.1039/C0SM00549E with permission from The Royal Society of Chemistry. | ||
 | ||
| Fig. 9 Solvent displacement method. Nanoparticles are formed by a dissolution of polymer in water-miscible organic solvent (e.g. acetone) followed by a transfer of the organic phase into an aqueous phase containing a surfactant. Rapid diffusion of organic solvent into the aqueous phase leads to spontaneous nanoemulsification. Evaporation of the solvent leads to conversion of nanoemulsion droplets into solid polymer nanoparticles. Copyright®2016 MDPI. Reproduced from ref. 150: https://doi.org/10.3390/nano6020026 available under the Creative Commons Attribution License. | ||
 | ||
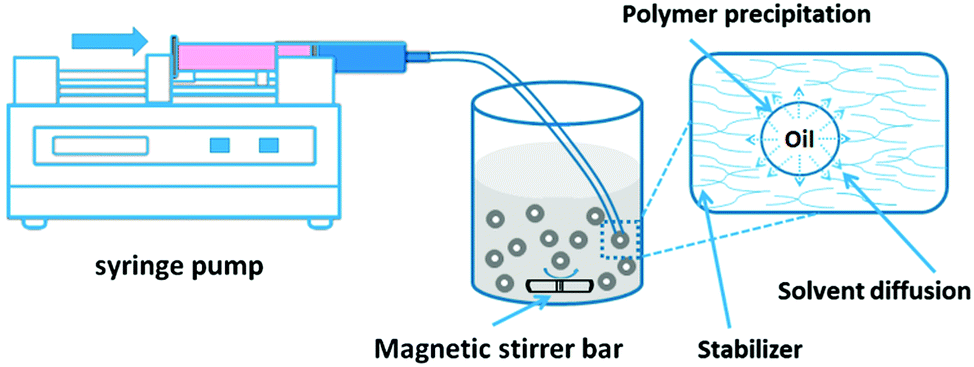
| Fig. 10 Schematic illustration of bubble bursting method. (a) Sketch of the experimental set-up. Compound interface = a thin layer of oil on the surface of water column. Inset: Close-up of the deformed compound interface. (b) After bubble bursting, we observe sub-micrometer oil droplets dispersed in the water phase. (c–f) Encapsulation of functional materials with the bubbling platform. (c) Functional materials such as amphiphilic or lipophilic compounds are dispersed in the oil layer. When a bubble reaches the interface, the oil layer is pushed up forming a bubble cap. (d) When oil layer becomes thin enough, it ruptures and generates polydisperse oil droplets that are entrained from the cavity wall into the water phase. (f) O/W nanoemulsion droplets loaded with functional compounds. Copyright©2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. Reproduced from ref. 153: https://doi.org/10.1002/adma.201505994 with permission from Wiley-VCH Verlag GmbH & Co. KGaA. | ||
The main disadvantage of low energy methods is their lack of universality. In all these approaches, formation of nanoemulsions depends fundamentally on composition of oil, water and surfactant blend. Therefore, application possibilities of these methods are strongly limited by the type of immiscible phases and emulsifier that can be used.113
Applications of nanoemulsion technology in bone regeneration
Synthesis of hydroxyapatite nanoparticles via nanoemulsion technology
Over the past three decades, much attention has been paid to applications of nanoemulsion technology in fabrication of nano-hydroxyapatite (Ca10(PO4)6(OH)2, HAp) used as inorganic component of orthopaedic and dental implants.41–44,154 Accumulating evidence shows that hydroxyapatite synthesis performed in nanoemulsion droplets, which serve as miniaturized reactors (Fig. 11), significantly improves quality of the resulting powders in terms of homogeneity,155 dispersity156 and osteoinductivity.61,62 Crystallization of HAp performed in a very limited, confined volume of nanoemulsion droplets prevents excessive growth of crystals,157–159 which allows reducing their size to the nano-level and yields more monodisperse product.160 In addition, separation of nanoreactors by the continuous oil phase prevents agglomeration and flocculation of HAp crystals,161 which is one of the main challenges of conventional wet HAp synthesis methods performed in the bulk liquid phase such as hydrothermal method,162 double decomposition/precipitation method,163–165 semi-hydrothermal method,166,167 or hydrolysis method.168,169 Hydroxyapatite powders obtained by these methods are often polydisperse, suffer from inhomogeneous phase composition58,170 and poor osteoinductivity related to large crystal size (in the micrometer range).58,59 Since hydroxyapatite nanoparticles fabricated via nanoemulsion technology display reduced polydispersity and agglomeration, they exhibit improved densification behavior during sintering, lower incidence of microstructural flaws and higher mechanical resistance of the resulting ceramics.62,171,172 Due to the nanoscale size, they also demonstrate higher resemblance to hydroxyapatite nanocrystals occurring in the natural bone58,59 and can stimulate osteogenesis48 more effectively compared to their microscale counterparts.61,62 For that reason, hydroxyapatite nanocrystals are currently the most desirable form of hydroxyapatite for bone regeneration applications. | ||
| Fig. 11 Optical microscope image of hydroxyapatite nanoparticles synthesized in double emulsion (w/o/w) droplets. Panels (a), (b) and (c) show nanoparticles obtained from precursor solutions containing different concentrations of calcium nitrate and phosphoric acid: (a) 0.1 M and 0.06 M, (b) 0.5 M and 0.3 M, and (c) 1.0 M and 0.6 M, respectively. The structure of the aggregates becomes more compact as the precursor concentration increases. Scale bars are 50 μm for (a) and (b) and 30 μm for (c). Copyright©2009 American Chemical Society. Adapted from ref. 174: https://doi.org/10.1021/cm9028935 with permission from American Chemical Society. | ||
Similarly to conventional hydroxyapatite synthesis (performed in bulk liquid reaction media), nanoemulsification-based synthesis is based on chemical reactions between calcium and phosphate sources under alkaline pH.173 In the nanoemulsion-based methods, however, the aqueous reaction medium (aqueous solution or organic solvent) is dispersed in the oil phase in the form of nanodroplets (w/o nanoemulsion). HAp synthesis in nanoemulsions is typically a two-step process. In the first step, separate reverse (w/o) nanoemulsions of reactants required for the synthesis, i.e. Ca2+ source (e.g. CaCl2,160 CaNO3175) and PO43− source (e.g. H3PO4,175 (NH4)2HPO4), are prepared. For this purpose, aqueous solution of Ca2+ or PO43− source is mixed with the oil phase composed of a non-polar solvent (e.g. cyclohexane,160n-decane175), a surfactant (e.g. CTAB,160 AOT175) and a co-surfactant (e.g. n-pentanol160). A co-surfactant is applied to increase flexibility of phase interface, which improves stability of the droplets.176 In the second step, nanoemulsions of Ca2+ and PO43− source are mixed together at an appropriate ratio (e.g. stoichiometric HAp Ca/P molar ratio 1.67) to initiate HAp synthesis reaction. Mixing of nanoemulsions leads to random collisions and coalescence of the droplets and formation of nanoreactors containing both Ca2+ and PO43− ions. The synthesis is induced by adding an alkali (e.g. NH4OH, NaOH) to the non-polar phase. Synthesis reactions can be performed at ambient177,178 or elevated temperature.160,179 Heating of nanoemulsion droplets containing HAp crystals during their growth (for example in an autoclave160 or a microwave179) provides additional process parameters allowing higher degree of control over crystal morphology.
Size and structure of HAp nanoparticles synthesized in nanoemulsion droplets is governed by Ca/P precursor molar ratio, reaction pH, temperature181 and nanoemulsion droplet size, which in turn depends on applied nanoemulsification method, composition and physicochemical properties of the water and oil phases.175 Altering these parameters allows to obtain HAp nanocrystals displaying a broad spectrum of sizes and shapes (e.g. rod-like, sphere-like, needle-like, nanofibers) as depicted in Fig. 12.
 | ||
| Fig. 12 Transmission electron microscope (TEM) images of different morphologies of hydroxyapatite nanocrystals synthesized in nanoemulsion droplets: (A) Sphere-like nanocrystals; (B) rod-like nanocrystals; (C) nanofibers, (D) needle-like crystals. Scale bars are 100 nm for (A) and (B), 1 μm for (C) and 200 nm for (D). Parts A and B of the figure were adapted from ref. 177: https://doi.org/10.1016/j.ceramint.2018.04.128 with permission from Elsevier Ltd and Techna Group S.r.l. Part (C) was adapted from ref. 160https://doi.org/10.1021/la0498197 with permission from American Chemical Society. Part (D) was adapted from ref. 180: https://doi.org/10.1016/j.jpcs.2006.11.026 with permission from Elsevier Ltd. | ||
Another disadvantage of nanoemulsion-based synthesis of hydroxyapatite is the use of surfactants, which may contaminate the obtained product and adversely affect its biocompatibility.29,30 To overcome this issue, a surfactant-free nanoemulsification methods have been devised. Formation of nanoemulsions in the absence of surfactants can be performed via direct pouring technique based on the Ouzo effect. The Ouzo effect relies on a liquid–liquid nucleation process, which occurs when a water-miscible non-polar phase (e.g. organic solvent such as acetone155,156) is poured into a surfactant-free aqueous phase.177,178 When the phases are mixed together, the water-miscible solvent becomes supersaturated, resulting in nucleation of solvent droplets and spontaneous formation of a metastable solvent-in-water nanoemulsion.187 Synthesis of HAp nanopowders based on the Ouzo effect is performed by adding the phosphorus source and alkali to the aqueous phase and calcium precursor to the non-polar phase prior to mixing of the phases as depicted in Fig. 13. This approach has the added benefit of easy functionalization of hydroxyapatite nanocrystals, typically performed by adding the functional compounds to the organic phase. The functional compound is subsequently incorporated into the structure of HAp during crystal formation. This approach has been successfully applied to obtain synthetic hydroxyapatite nanoparticles functionalized with CO32− groups (characteristic for natural carbonated hydroxyapatite),181 antibacterial agents (e.g. Ag+ ions156) or factors stimulating bone formation (e.g. Sr2+ and Mg2+ ions155,178) and angiogenesis (e.g. Co2+ ions178).
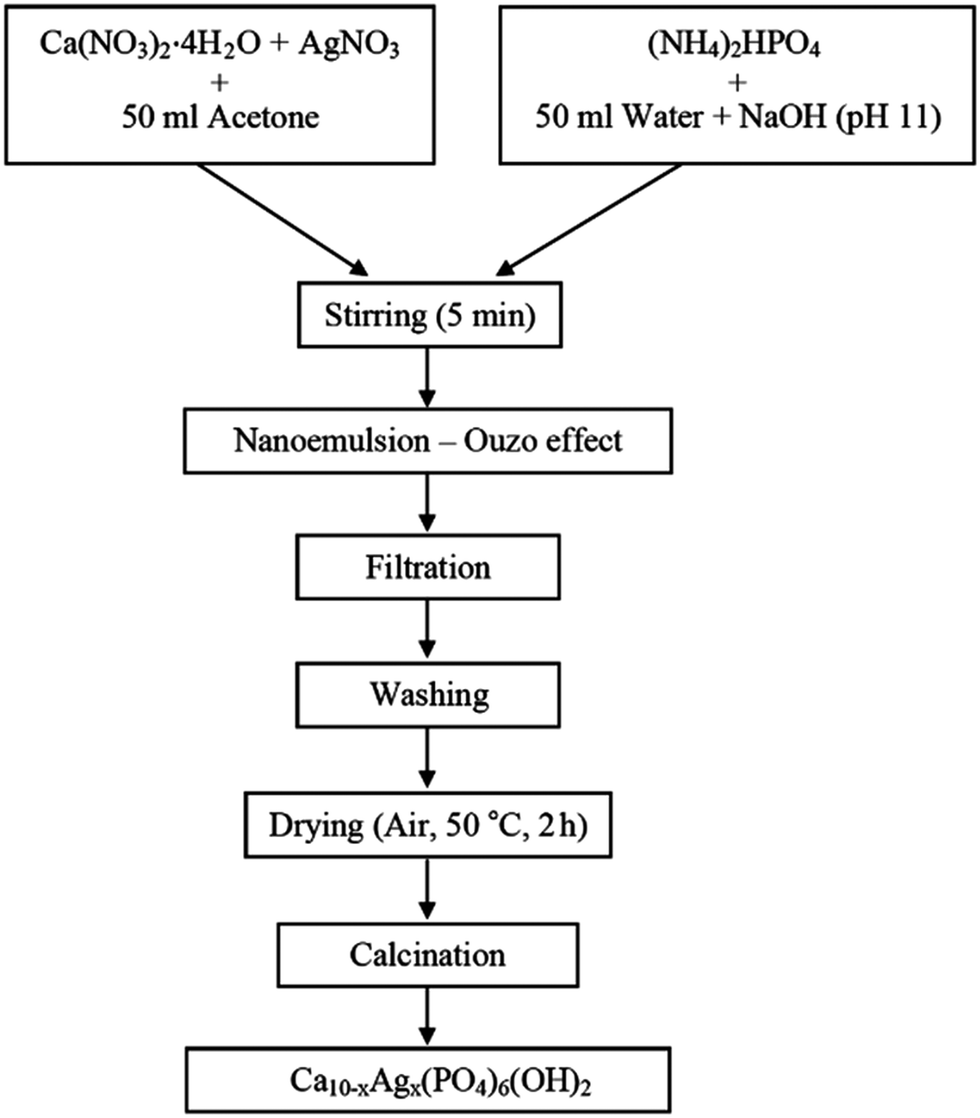 | ||
| Fig. 13 Flowchart for the synthesis of Ag+-doped nano-hydroxyapatite via direct pouring technique based on the Ouzo effect. Mixing of water-miscible organic solvent (acetone) containing calcium precursor with an alkaline aqueous phase containing phosphorus source leads to spontaneous formation of o/w nanoemulsion. Copyright©2016 Faculty of Technology, University of Novi Sad. Adapted from ref. 156: https://www.tf.uns.ac.rs/publikacije/PAC/pdf/PAC%2033%2006.pdf licensed under Creative Commons Attribution-Non Commercial-No Derivatives 4.0 International license. | ||
Methods for the synthesis of hydroxyapatite in nanoemulsions developed to date suffer also from relatively poor control over droplet size and size distribution, which may have a detrimental effect on homogeneity and batch-to-batch reproducibility of resulting nanopowders. In current approaches, the degree of control over droplet size is limited by applied nanoemulsification technique (typically vigorous mechanical agitation160,175,177–179 or ultrasonication182). More precise control over droplet size could be achieved by generating nanoemulsion droplets for HAp synthesis in nano- or microfluidic devices, which produce droplets of precisely defined size and very narrow size distribution.188 Using this type of devices could provide not only much better control over morphology and dispersity of obtained nanocrystals, but would also allow tracking and observation of individual droplets during HAp crystal growth (Fig. 14). Due to a constant flow rate of fluids in a microfluidic device, it is possible to track droplets at different stages of HAp synthesis based on the distance of the droplet from fluid injection port.174 Real-time observation of the processes occurring in the droplets could aid in tailoring synthesis conditions based on current crystal morphology and contribute to better understanding of HAp crystal formation mechanism, which is not fully understood. So far, microfluidic devices have been employed only for the synthesis of HAp in microemulsion droplets (Fig. 14).174 Combining microfluidic platforms with nanoemulsion-based synthesis of hydroxyapatite could open new perspectives for fabrication of more monodisperse nano-hydroxyapatite crystals of highly controllable size and shape.
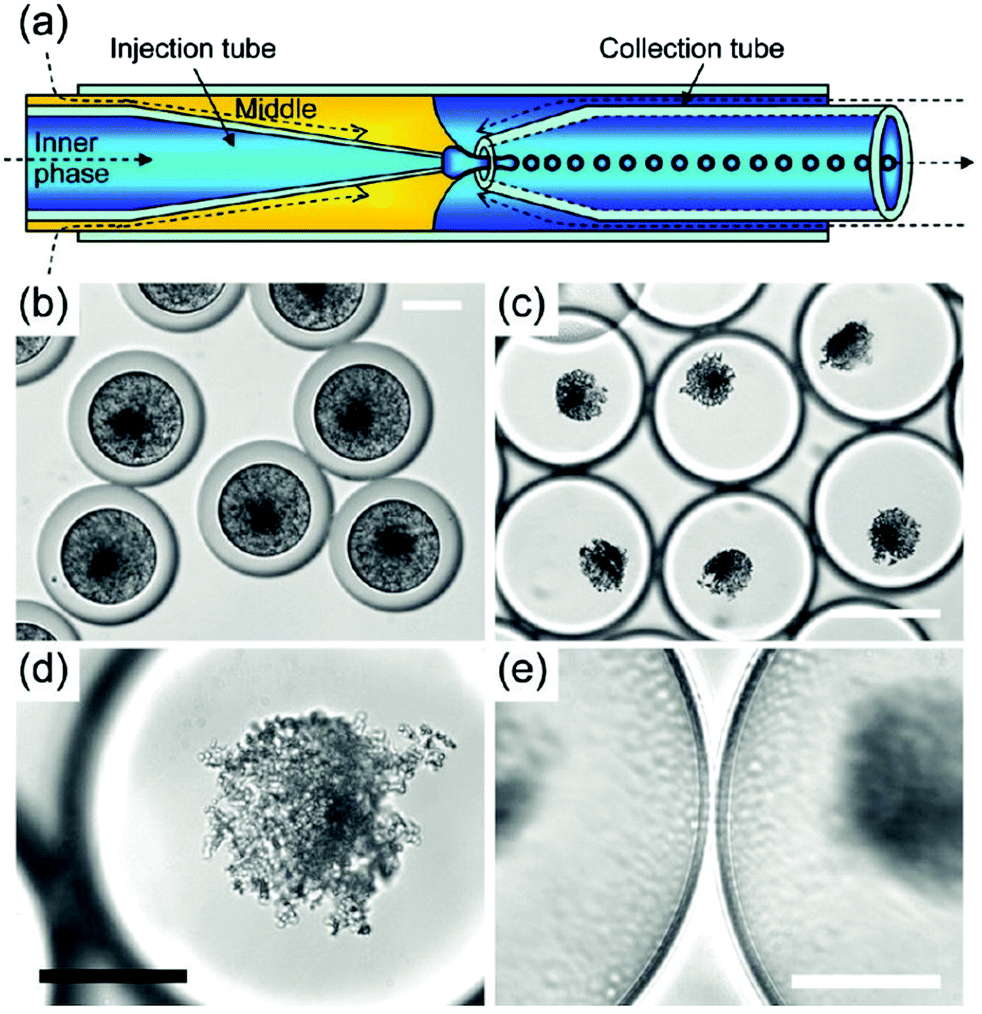 | ||
| Fig. 14 Synthesis of hydroxyapatite nanoparticles in double W/O/W microemulsion droplets produced using a microfluidic device. (a) Scheme of a microfluidic device. W/O/W double microemulsion droplets are generated by co-extrusion of an aqueous phase (aqueous solution of calcium and phosphorus precursors) and oil phase (silicon oil + surfactant) through a microcapillary into a continuous aqueous phase. The oil phase forms shells around inner aqueous phase droplets (b–e). Optical microscope images of double microemulsion droplets (b and c). Initially, HAp occupies the entire volume of microemulsion droplets (b). As water diffuses into the droplets (due to differences in osmolality), the droplets swell (c). Gradual thinning of oil shells leads to destabilization of the droplets and release of hydroxyapatite crystals. Hydroxyapatite nanoparticle aggregate (d) and oil shells at higher magnification (e). Scale bars are 50 μm for (b), (d), (e) and 150 μm for (c). Copyright©2009 American Chemical Society. Adapted from ref. 174: https://doi.org/10.1021/cm9028935 with permission from American Chemical Society. | ||
In the methods discussed above, HAp synthesis was performed in a liquid environment of water nanodroplets. Alternatively, hydrogel nanospheres can be used as nanoreactors for the crystal growth. Such approach has been employed by Ethirajan et al.,182 who conducted HAp synthesis in gelatin nanospheres fabricated via reverse nanoemulsion method. To initiate the synthesis, the cross-linked gelatin nanospheres were placed in an aqueous solution of Ca2+ source, where Ca2+ ions were bound to the carboxyl groups of gelatin molecules. Subsequent addition of phosphate ions to the suspension of gelatin nanospheres led to formation of monodisperse HAp nanoparticles inside gelatin nanospheres. This technique offers an attractive alternative to methods employing liquid nanoemulsions due to several reasons. First, it enables synthesis of HAp nanocrystals and their nanoencapsulation in one step. Second, since it can be performed under physiological conditions, it may be readily applied to co-encapsulate cells (e.g. osteogenic stem cells) with HAp crystals. Such combination could allow formation of bioartificial nanoscaffolds, in which HAp crystals are embedded in a hydrogel matrix (mimicking extracellular matrix) colonized with osteogenic cells. Also, since gelatin nanospheres can be easily assembled (e.g. via 3D-printing189) or cross-linked together190 at physiological temperature, they could be used as building blocks of modular bone tissue constructs.191 Due to their small size, HAp-loaded nanospheres could also be implanted or injected directly to the bone defect. Methods allowing synthesis of HAp directly within hydrogel nanocarriers, such as the one discussed above, used in conjunction with cell encapsulation and in vitro tissue culture hold great promise for manufacturing of nanoscaffolds and multifunctional biomimetic bone tissue constructs.
HAp nanoparticles synthesized in nanoemulsion droplets have been also used for fabrication of 3D scaffolds for cell culture.51,200,201 Zhou et al.200,201 have designed an interesting composite consisting of PLA microspheres filled with carbonated hydroxyapatite (CHAp) nanospheres. CHAp nanospheres were synthesized via o/w nanoemulsion method and used as the osteoconductive filler of PLA microspheres. CHAp-filled microspheres were then applied as the building blocks of bone tissue scaffolds.201,202 The blocks were assembled into a solid porous construct (shown in Fig. 15) by sintering using laser as a power source. The resulting construct exhibited a porous structure containing a network of interconnected macro- and micropores allowing effective transfer of nutrients/oxygen into the scaffold and providing favourable environment for the growth of cells.
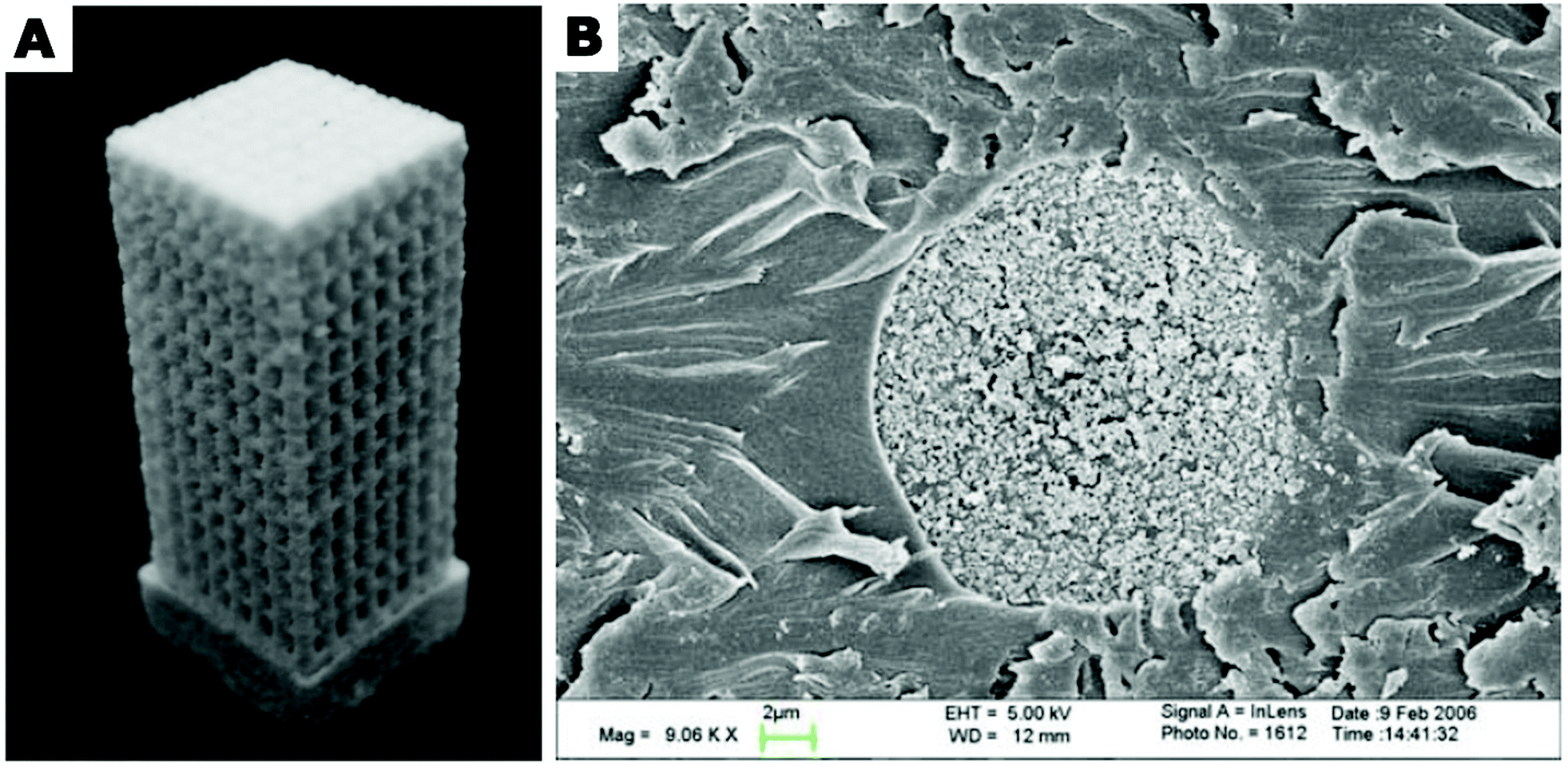 | ||
| Fig. 15 (A) A composite porous scaffold sintered from PLA composite microspheres loaded with carbonated hydroxyapatite (CHAp) nanoparticles via selective laser sintering technique. (B) Scanning electron microscopy (SEM) image of a cross-section of PLA/CHAp composite microsphere. The microsphere cross section was obtained by embedding the microsphere in epoxy resin and sectioning with a microtome. Part (A): Copyright©2007 Springer Science + Business Media, LLC. Adapted from ref. 201: https://doi.org/10.1007/s10856-007-3089-3 with permission from Springer Science + Business Media, LLC. Part (B): Copyright©2007 Springer Nature. Adapted with permission from ref. 181: https://doi.org/10.1007/s10856-007-3156-9. | ||
Nano-hydroxyapatite obtained via nanoemulsification has also been applied to improve hydrophilicity and bioactivity of hydrophobic bioinert synthetic polymers such as polycaprolactone (PCL).51 It has been found that incorporating HAp nanoparticles into such materials (e.g. electrospun PCL nonwovens51) not only significantly enhances their wettability, but also introduces ionic groups required for scaffold–cell interactions that are absent in non-modified polymer. Because of that, implants containing nano-hydroxyapatite are more readily colonized by cells (either host cells or cells seeded onto them in vitro such as stem cells).199
Fabrication of polymer nanoparticles for drug/biomolecule delivery
One of the most common applications of nanoemulsion technology in bone regeneration is fabrication of polymer nanoparticles loaded with drugs/biomolecules facilitating osteogenesis such as osteoinductive factors (e.g. bone morphogenetic proteins, BMPs,8–10 osteopontin,11 dexamethasone12), antimicrobial agents (e.g. Ag,13 CuO,14 Se,15 curcumin,16–18 chlorhexidine,19,20), anti-inflammatory factors (e.g. curcumin,16–18,21 quercitrin22,23), drugs inhibiting bone resorption (e.g. bisphosphonates such as alendronate24–26) and others. This category of nanoparticles has been extensively investigated as bioactive components of bone implant materials such as bioinert synthetic polymers (e.g. polyethylenes, PCL) or metallic substrates (titanium, aluminum oxide).203 Incorporating drug-loaded NPs into bone implants aims to enhance cell–implant interactions and cellular behaviors involved in osteogenesis and osteointegration8 or to prevent unwanted tissue response such as inflammation16–18,21 or bone resorption.24–26The key advantage of drug immobilization in NPs is controlled sustained drug release due to the presence of a polymer matrix serving as a diffusion barrier for entrapped drug molecules.16 In contrast to direct immobilization of drugs onto the surface of bone implants (e.g. via physical adsorption or covalent conjugation), functionalization of grafts with drug-eluting NPs does not require affinity between the drug and implant material and thus can be applied to much wider spectrum of bioactive agents regardless of their physicochemical properties. Such approach can also significantly increase drug loading efficiency providing more long-term therapeutic effect.16
The most widely used method for manufacturing of polymer NPs for drug/biomolecule delivery is nanoemulsion/solvent evaporation technique. In this approach, polymer (nanoparticle matrix material e.g. PLGA,17,24,25 poly(organophosphazene),204 PLA205) is dissolved in a volatile organic solvent such as chloroform, dichloromethane,206 acetone,25 DMSO,25 ethyl acetate,24etc. Polymer/organic solvent solution (the oil phase) is dispersed in an aqueous phase containing a surfactant (for example by ultrasonication207 or high-speed homogenization) forming an o/w nanoemulsion or double w/o/w nanoemulsion.206,207 Nanoemulsion droplets are subsequently converted into solid nanoparticles by evaporating the solvent from the polymer solution,112,206 which can be achieved by continuous magnetic stirring of nanoemulsion at room temperature under atmospheric or reduced pressure. During solvent evaporation, the nanodroplets can coalesce leading to uneven nanoparticle size distribution, which is one of the main limitations of this method.206 Furthermore, organic solvent present in polymer solution may lead to denaturation or loss of bioactivity of thermosensitive molecules encapsulated in the NPs.208 This method requires also the presence of surfactants, which have to be subsequently removed from the product before further use.206 Since surfactant removal procedures are time- and cost-consuming and often do not provide sufficient purity, fabrication methods performed in the absence of surfactants such as salting-out technique are particularly desirable. In salting-out method, nanoemulsion is prepared by emulsification of polymer solution in organic solvent miscible with water (e.g. acetone) in an aqueous phase containing high concentration of salt (e.g. magnesium chloride, calcium chloride) or sucrose.206,209 Dilution of nanoemulsion with excess of water causes reduction in the salt/sucrose concentration in the continuous phase and migration of the solvent from the nanoemulsion droplets, which leads to precipitation of the polymer in the form of NPs.
Among drug-eluting polymer NPs for bone regeneration applications, biodegradable NPs (e.g. PLGA,8 PLGA/PCL,212 chitosan10) loaded with bone morphogenetic proteins (BMPs)8–10 are the most intensively studied. They are being explored as an alternative to current therapeutic approaches to healing traumatic bone injuries based on supra-physiological doses of BMP-2 introduced directly into the bone defect, which often lead to adverse side effects in the surrounding tissues such as ectopic bone formation, bone resorption and inflammation.65,210 BMP-loaded NPs have been investigated as both injectable drug delivery vehicles211,212 and bioactive components of bone implant materials. Biomaterials incorporating BMP-loaded nanoparticles can be obtained via surface functionalization, which can be achieved, for example, by embedding the biomaterial in a suspension of NPs in polymer/volatile solvent solution8 or spraying such suspension onto the implant surface10 followed by solvent evaporation. The former approach has been employed by Kim et al. to obtain 3D-printed hydroxyapatite scaffolds coated with PCL layer containing PLGA nanoparticles encapsulating BMP-2.8 Coating the implant with bioactive PLGA NPs/PCL layer provided long-term sustained release of BMP-2, which improved proliferation and osteogenic differentiation of mesenchymal stem cells (MSCs) seeded onto the implant and significantly accelerated healing of bone defects in experimental animals. Due to controlled sustained biomolecule release, bone implants decorated with BMP-eluting nanoparticles could resolve both the issue of short half-life of BMPs and the problem of adverse side effects associated with high BMP doses.210 Due to very small size, drug-loaded nanoparticles can also be suspended in hydrogel precursor gelling at body temperature and injected directly into the bone defect forming in situ gelling implant.18 Such administration route allows local delivery of the drug without its systemic side effects. Drug-loaded NPs may also be incorporated into bone implants in a spatially controlled manner via 3D-printing of nanoparticle suspension in a hydrogel precursor or cell-loaded bioink.8 Combining biomolecule-loaded NPs with 3D-printing technology8,213 is considered one of the most promising strategies in bone tissue engineering. It is anticipated that this technology will allow recreating tissue-specific composition and spatial distribution of multiple biomolecules involved in osteogenesis within bone tissue constructs and enable their spatially and temporary controlled sequential release mimicking biomolecule release pattern occurring in vivo.
Fabrication of bone-targeting nanoparticles
Recently, many efforts have been devoted to developing bone-targeting nanoparticles able to bind specifically to bone. This type of NPs may increase therapeutic efficiency and limit adverse side effects of released drugs on other tissues/organs due to accumulation at the target site and local drug delivery.214 Bone-targeting NPs are typically obtained by modification with compounds containing functional groups with high affinity to moieties present in the bone tissue such as Ca2+ ions. An example of such compounds is bisphosphonates (BP), comprising two Ca2+-binding phosphonate groups (P-C-P) in their molecules.215 Among bisphosphonates, alendronate has emerged as a very promising agent for functionalization of NPs.24,25,204 Alendronate (ALN) is a drug approved by the FDA for clinical use in osteoporosis treatment. Its anti-osteoporotic effect is based on suppression of bone resorption through inhibition of osteoclast activity.216 ALN is also able to bind to hydroxyapatite (both natural and synthetic) via multiple Ca2+ ions present in its molecules (Ca10(PO4)6(OH)2). Furthermore, when ALN–functionalized hydroxyapatite nanoparticles are incorporated into bone implants, they act as “anchors” strengthening interaction between the implant and the host tissue.16,17 Therefore, functionalization of NPs with ALN yields three-functional delivery vehicles capable of targeting bone, inhibiting bone resorption and anchoring the implant to the host bone. For that reason, ALN has been broadly applied for functionalization of nanoparticles for bone regeneration applications including inorganic (e.g. hydroxyapatite,26 Fe3O4,217 clay218) and polymer (e.g. poly(γ-benzyl-L-glutamate),17 poly(lactic-co-glycolic acid)24,25) nanoparticles. The majority of research in this area has been focused on PLGA nanoparticles,17,24,25 which have been approved by the FDA for use in drug delivery due to sustained drug release, low toxicity and biocompatibility.53 ALN-modified PLGA nanospheres are typically obtained via previously described nanoemulsion/solvent evaporation technique.17,24,25 This type of NPs has been shown to be non-cytotoxic, haemocompatible, biocompatible and suitable for intravenous administration.25 When injected intravenously, ALN-modified PLGA nanospheres have been shown to reach the bone much faster compared to control nanospheres (non-conjugated with ALN) and accumulate in the bones of experimental animals.24 ALN-NPs were also retained in the bones for longer periods of time than control nanoparticles, which translated to significantly improved therapeutic outcome.24 Unfortunately, ALN is a rare example of a drug that can act both as a therapeutic agent and bone-targeting ligand. To provide bone-targeted delivery of drugs without innate affinity to bone, other strategies have been proposed. For example, bone-targeting drug-loaded NPs have been obtained by introducing phosphonate groups into surfactants used during nanoparticle manufacturing. Zhang et al. have proposed conjugating a bisphosphonate (pamidronate) to the terminal hydroxyl group of a non-ionic surfactant Brij 78, which was subsequently used for fabrication of nano-formulations (such as lipid NPs, hydrogel PLGA NPs and oil-in-water nanoemulsions) for bone-targeted delivery of curcumin.16 Curcumin has multiple biological effects including suppressive effect on bone resorption,219 anti-inflammatory,21 analgesic, antioxidant18 and antimicrobial activity.220 Therefore, curcumin-eluting NPs hold a lot of promise as bioactive components of multifunctional bone implants that could potentially resolve the issues of implant-related infections, pain and inflammation.18,21 Kasinathan and co-workers developed an injectable in situ gelling implant based on curcumin-loaded PCL nanoparticles suspended in PLGA sol gelling upon contact with phosphate buffer of physiological pH (pH 7.4).18 The resulting NPs-loaded gel has been demonstrated to release curcumin in a sustained manner over the period of 7 days in vitro and exert a substantially improved anti-tumor effect in mice compared to plain curcumin administered via intraperitoneal injection.18 Zambrano et al. have further confirmed therapeutic potential of injectable curcumin-loaded PLGA nanoparticles for local treatment of inflammation and inhibition of bone resorption in animal model of periodontal disease.21 Since implant-related infections and inflammation are one of the leading causes of bone implant failure, functionalization of bone substitute materials with curcumin-loaded NPs may contribute to significant improvement in their in vivo performance.21 Due to local drug release, this type of implants can also eliminate systemic side effects of drugs routinely administered in the postoperative period such as antibiotics, analgesics and anti-inflammatory drugs.221,222Challenges & outlook
Despite growing number of studies on bone implant materials functionalized with nanoparticles obtained via nanoemulsion technology, they have not yet reached the clinic and still remain at in vitro18,199 or animal testing stage.18,21 However, several bone substitute materials functionalized with nanoparticles fabricated via other techniques are currently on the market. Commercially available bone implant materials are based on ceramic nanoparticles, either hydroxyapatite nanocrystals (e.g. Ostim® injectable bone filling manufactured by Osartis, Germany or PerOssal® bone substitute by aap Implantate AG, Germany) or beta-tricalcium phosphate nanocrystals (e.g. Vitoss® Synthetic Cancellous Bone Void Filler by Orthovita/Stryker, USA).223,224 These materials have been shown to be biocompatible,224 yet some questions remain about their long-term persistence in the body and their potential chronic toxicity. Key safety concerns are associated with the release of nanoparticles from implant material over time due to its mechanical wear or matrix material degradation, which may lead to unwanted effects in the surrounding tissues such as nanoparticle-induced inflammatory response, periprosthetic osteolysis and implant loosening.225–227 Due to high surface-area-to-volume ratio nanoparticles are more biologically active compared to their micron-sized counterparts. As a result, once released from the implant, they may be able to induce much more potent adverse effects in the host. In addition, nano-dimensions open up the possibility of nanoparticle migration to distant locations in the body and crossing biological barriers such as the blood–brain barrier228,229 and endothelium,230 as well as cellular and intracellular membranes, which gives them access to various cellular compartments including the nucleus.231In vitro studies confirm that hydroxyapatite nanoparticles are easily taken up by cells,232,233 where they can exert an array of deleterious effects such as alterations in DNA structure,234 chromosome aberrations,235,236 cell membrane damage, production of reactive oxygen species/inflammatory cytokines237–241 and apoptosis.242,243 The extent of nano-hydroxyapatite cytotoxicity is strongly related to nanoparticle shape and crystallinity with low crystalline, needle-shaped crystals being the most cytotoxic.241 Needle-shaped hydroxyapatite nanoparticles show also higher tendency to penetrate through biological barriers such as mucus membranes, as demonstrated by in vitro studies on models of mucosa-like human corneal epithelial tissue.244 For these reasons, needle-shaped hydroxyapatite nanoparticles are of particular concern in relation to their systemic availability and chronic toxicity. The ability of hydroxyapatite NPs to pass through biological barriers has been also shown in a number of animal studies. Hydroxyapatite nanoparticles administered intravenously,245–247 intraperitoneally248 or orally249 have been demonstrated to migrate and accumulate in the liver, kidneys and lungs.245,247–249 It has also been found that once they reached those organs, they induced multiple histopathological changes including degeneration of nephric tubule epithelium in the kidneys,245 degeneration of hepatic adipose tissue,249 inflammatory cell infiltration,249 necrosis and apoptosis,245,248 leading to elevation of tumor necrosis factor-α (TNF-α)239 in the serum. Pathomorphological changes in the liver translated to increased serum levels of hepatic enzymes including alanine aminotransferase (ALT), alkaline phosphatase (ALP) and aspartate aminotransferase (AST).247,249 It is worth noting that the above effects have been reported for relatively high doses of nanohydroxyapatite (10.0–33.3 mg per kg body weight245,248) administered in a free (non-immobilized) form. Therefore, the informative value of these studies on in vivo toxicity of nano-HAp immobilized in composite bone implants is limited. It is likely that nano-hydroxyapatite particles released from an implant would not achieve sufficient concentration to elicit those effects. On the other hand, these findings strongly indicate that injectable therapies based on non-immobilized hydroxyapatite nanoparticles administered intravenously (e.g. bone-targeting NPs24) should be treated with particular caution in terms of nanoparticle migration/accumulation in the body. Besides the issue of NPs migration, in case of nano-hydroxyapatite administered intravenously, there is growing concern about nanoparticle-induced haemolysis250 and aggregation of the red blood cells in the blood vessels,251 which poses a serious risk of vessel occlusion and thrombotic events. It has been also shown that HAp NPs can accumulate and deposit in the blood vessels similarly to cholesterol, leading to artery calcification and atherosclerosis.252The safety of the second category of NPs fabricated via nanoemulsion technology – polymer NPs for drug/biomolecule delivery – also remains controversial. Biodegradable polymers typically used for manufacturing of this type of NPs (e.g. PLGA, PLA, PCL) are considered biocompatible and have been approved by the FDA for use in drug delivery and other biomedical applications.253 However, the distinctive physicochemical properties of nanoparticles related to their small size and high surface-area-to-volume ratio make it impossible to extrapolate the toxicity data of the raw material to polymer NPs. Although the fate of polymer NPs in the body is still not well understood, numerous in vitro studies demonstrate that they readily cross cellular membranes and are uptaken by cells.254,255 Cytotoxicity data also indicates that polymer NPs can cause cell membrane damage,256 alter DNA structure257 and induce production of inflammatory cytokines258,259 and reactive oxygen species in the cells.259–261 Further safety concerns are associated with the ability of polymer NPs to cross biological barriers (e.g. brain–blood barrier228,229 and blood–retina barrier256), migrate via the circulatory and lymphatic systems and accumulate in the distant lymph nodes262,263 or organs such as liver,228 spleen, lungs, bone marrow,264 kidney, heart and brain.228 Despite this fact, polymer NPs have not been demonstrated to exhibit significant in vivo toxicity in terms of pathomorphological changes in the target organs even at high doses administered intravenously265–267 or orally.228,260,268 In order to fully answer the question whether and to what extent nanoparticles used for bone regeneration applications are safe in the long-term, more data concerning their in vivo behavior and potential chronic toxicity is necessary.
Conclusions
Nanoemulsion technology has proven a valuable tool for fabrication of nanoparticles for bone regeneration applications. Combining nanoemulsification techniques with hydroxyapatite synthesis has enabled manufacturing of high-quality, monodisperse, agglomerate-free nanohydroxyapatite powders of significantly improved osteoinductive and mechanical properties. Polymer nanoparticles formulated via nanoemulsion technology have been extensively researched as bioactive components of orthopaedic implants providing sustained local delivery of drugs such as bone morphogenetic proteins, antimicrobial, anti-inflammatory and anti-osteoporotic agents. Further advances have been made with the development of bone-targeting nanoparticles with high affinity to bone. As a result, in the recent years bone implant materials have progressed from bioinert bone substitutes serving merely as weight-bearing prostheses to multifunctional systems able to actively interact with the host tissue and stimulate new bone formation. The emergence of advanced tissue engineering techniques such as 3D-bioprinting has allowed incorporating nanoparticles into biomimetic bone tissue constructs in a spatially controlled manner providing tissue-specific distribution of bioactive molecules. Despite these significant advances, so far nanoparticles obtained via nanoemulsion technology have not reached the clinic and in the vast majority of cases remain at in vitro or animal testing stage. The main issues impeding clinical translation of these NPs are biocompatibility and safety concerns related to their very small size and ability to pass through biological barriers such as blood–brain barrier and cell membranes. The challenge that still needs to be addressed before this category of NPs can be implemented on a larger scale is scaling up of nanoemulsion-based methods for nanoparticle fabrication. More studies exploring in vivo behavior of nanoparticles, particularly in the long term, are also necessary to allow further progress in this field.Author contributions
Barbara Kupikowska-Stobba: conceptualization, writing – original draft, visualization, writing – review & editing. Mirosław Kasprzak: conceptualization, writing – original draft, visualization, supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Funding for the present work was provided by the Foundation for Polish Science financed by the European Union under the European Regional Development Fund within the framework of the project “Multifunctional biologically active composites for applications in regenerative medicine of bone” (POIR.04.04.00-00-16D7/18) carried out within the TEAM-NET programme. The authors would like to thank Dr Vlad Dinu, Assistant Professor, School of Bioscience, University of Nottingham, UK for his constructive feedback on our manuscript.References
- H. S. Sohn and J. K. Oh, Biomater. Res., 2019, 23, 9 Search PubMed.
- T. J. Blokhuis, in Bone Substitute Biomaterials, ed. K. Mallick, Elsevier Ltd., 2014, pp. 80–92 Search PubMed.
- G. Chandra and A. Pandey, Biocybern. Biomed. Eng., 2020, 40, 596–610 Search PubMed.
- W. R. Moore, S. E. Graves and G. I. Bain, ANZ J. Surg., 2001, 71, 354–361 Search PubMed.
- A. L. Ladd and K. Wirsing, Princ. Pract. Wrist Surg., 2009, 395, 277–287 Search PubMed.
- R. R. Betz, Orthopedics, 2002, 25, S561–S570 Search PubMed.
- P. Kazimierczak and A. Przekora, Coatings, 2020, 10, 971 Search PubMed.
- B. S. Kim, S. S. Yang and C. S. Kim, Colloids Surf., B, 2018, 170, 421–429 Search PubMed.
- L. Wang, W. Xu, Y. Chen and J. Wang, Sci. Rep., 2019, 9, 18175 Search PubMed.
- N. Poth, V. Seiffart, G. Gross, H. Menzel and W. Dempwolf, Biomolecules, 2015, 5, 3–19 Search PubMed.
- T. Jensen, J. Baas, A. Dolathshahi-Pirouz, T. Jacobsen, G. Singh, J. V. Nygaard, M. Foss, J. Bechtold, C. Bünger, F. Besenbacher and K. Søballe, J. Biomed. Mater. Res., Part A, 2011, 99, 94–101 Search PubMed.
- J. M. Oliveira, R. A. Sousa, N. Kotobuki, M. Tadokoro, M. Hirose, J. F. Mano, R. L. Reis and H. Ohgushi, Biomaterials, 2009, 30, 804–813 Search PubMed.
- S. Sivolella, E. Stellini, G. Brunello, C. Gardin, L. Ferroni, E. Bressan and B. Zavan, J. Nanomater., 2012, 2012, 975842 Search PubMed.
- F. Parnia, J. Yazdani, V. Javaherzadeh and S. Maleki Dizaj, J. Pharm. Pharm. Sci., 2017, 20, 148–160 Search PubMed.
- C. Vaquette, N. Bock and P. A. Tran, ACS Appl. Mater. Interfaces, 2020, 12, 55638–55648 Search PubMed.
- J. Zhang, X. Liu, T. Deng, P. Yao, H. Song, S. Zhou and W. Yan, J. Nanomater., 2015, 2015, 393968 Search PubMed.
- L. De Miguel, M. Noiray, G. Surpateanu, B. I. Iorga and G. Ponchel, Int. J. Pharm., 2014, 460, 73–82 Search PubMed.
- N. Kasinathan, M. Amirthalingam, N. D. Reddy, H. V. Jagani, S. M. Volety and J. V. Rao, Drug Delivery, 2016, 23, 1007–1015 Search PubMed.
- W. Fan, Y. Li, Q. Sun, T. Ma and B. Fan, J. Nanobiotechnol., 2016, 14, 72 Search PubMed.
- N. J. Wood, H. F. Jenkinson, S. A. Davis, S. Mann, D. J. O’Sullivan and M. E. Barbour, J. Mater. Sci.: Mater. Med., 2015, 26, 201 Search PubMed.
- L. M. G. Zambrano, D. A. Brandao, F. R. G. Rocha, R. P. Marsiglio, I. B. Longo, F. L. Primo, A. C. Tedesco, M. R. Guimaraes-Stabili and C. Rossa, Sci. Rep., 2018, 8, 6652 Search PubMed.
- S. K. Wong, K. Y. Chin and S. Ima-Nirwana, Int. J. Mol. Sci., 2020, 21, 6448 Search PubMed.
- N. Ahmad, V. T. Banala, P. Kushwaha, A. Karvande, S. Sharma, A. K. Tripathi, A. Verma, R. Trivedi and P. R. Mishra, RSC Adv., 2016, 6, 97613–97628 Search PubMed.
- S. I. Thamake, S. L. Raut, Z. Gryczynski, A. P. Ranjan and J. K. Vishwanatha, Biomaterials, 2012, 33, 7164–7173 Search PubMed.
- E. Cenni, D. Granchi, S. Avnet, C. Fotia, M. Salerno, D. Micieli, M. G. Sarpietro, R. Pignatello, F. Castelli and N. Baldini, Biomaterials, 2008, 29, 1400–1411 Search PubMed.
- S. J. Hwang, J. S. Lee, T. K. Ryu, R. H. Kang, K. Y. Jeong, D. R. Jun, J. M. Koh, S. E. Kim and S. W. Choi, Macromol. Res., 2016, 24, 623–628 Search PubMed.
- D. J. McClements, Soft Matter, 2012, 8, 1719–1729 Search PubMed.
- T. Tadros, P. Izquierdo, J. Esquena and C. Solans, Adv. Colloid Interface Sci., 2004, 108–109, 303–318 Search PubMed.
- S. Zhang, S. Ding, J. Yu, X. Chen, Q. Lei and W. Fang, Langmuir, 2015, 31, 12161–12169 Search PubMed.
- N. Vlachy, D. Touraud, J. Heilmann and W. Kunz, Colloids Surf., B, 2009, 70, 278–280 Search PubMed.
- A. W. Khan, S. Kotta, S. H. Ansari, R. K. Sharma and J. Ali, Expert Opin. Drug Delivery, 2012, 9, 1305–1317 Search PubMed.
- S. Vieira, S. Vial and J. M. Oliveira, Biotechnol. Prog., 2017, 33, 590–611 Search PubMed.
- J. G. Lyons, M. A. Plantz, W. K. Hsu, E. L. Hsu and S. Minardi, Front. Bioeng. Biotechnol., 2020, 8, 922 Search PubMed.
- G. G. Walmsley, A. McArdle, R. Tevlin, A. Momeni, D. Atashroo, M. S. Hu, A. H. Feroze, V. W. Wong, P. H. Lorenz, M. T. Longaker and D. C. Wan, Nanomedicine, 2015, 11, 1253–1263 Search PubMed.
- A. Tautzenberger, A. Kovtun and A. Ignatius, Int. J. Nanomed., 2012, 7, 4545–4557 Search PubMed.
- N. Monteiro, A. Martins, R. L. Reis and N. M. Neves, Regener. Ther., 2015, 1, 109–118 Search PubMed.
- M. E. Marti, A. D. Sharma, D. S. Sakaguchi and S. K. Mallapragada, in Nanomaterials in Tissue Engineering: Fabrication and Applications, ed. A. K. Gaharwar, S. Sant, M. J. Hancock and S. A. Hacking, Woodhead Publishing Limited, 2013, pp. 275–301 Search PubMed.
- D. R. Amin, E. Sink, S. P. Narayan, M. Abdel-Hafiz, L. Mestroni and B. Peña, Molecules, 2020, 25, 5189 Search PubMed.
- C. Chu, J. Deng, L. Liu, Y. Cao, X. Wei, J. Li and Y. Man, RSC Adv., 2016, 6, 90856–90872 Search PubMed.
- M. Fathi-Achachelouei, H. Knopf-Marques, C. E. Ribeiro da Silva, J. Barthès, E. Bat, A. Tezcaner and N. E. Vrana, Front. Bioeng. Biotechnol., 2019, 7, 113 Search PubMed.
- A. Szcześ, L. Hołysz and E. Chibowski, Adv. Colloid Interface Sci., 2017, 249, 321–330 Search PubMed.
- H. L. Kim, G. Y. Jung, J. H. Yoon, J. S. Han, Y. J. Park, D. G. Kim, M. Zhang and D. J. Kim, Mater. Sci. Eng., C, 2015, 54, 20–25 Search PubMed.
- D. Arcos and M. Vallet-Regí, J. Mater. Chem. B, 2020, 8, 1781–1800 Search PubMed.
- M. Sharafipour, H. Oveisi and A. Meshkini, J. Biomed. Mater. Res., Part A, 2020, 108, 882–894 Search PubMed.
- M. D. Calasans-Maia, B. R. Melo, A. T. N. N. Alves, R. F. B. Resende, R. S. Louro, S. C. Sartoretto, J. M. Granjeiro and G. G. Alves, J. Appl. Oral Sci., 2015, 23, 599–608 Search PubMed.
- S. L. Aktuğ, S. Durdu, E. Yalçın, K. Çavuşoğlu and M. Usta, Mater. Sci. Eng., C, 2017, 71, 1020–1027 Search PubMed.
- H.-J. Lo, G. Rau, Y.-J. Kuo, C. J. Liao, C.-H. Yang and Y.-H. Tsuang, J. Orthop. Surg., 2009, 26, 111–119 Search PubMed.
- X. Yang, Y. Li, X. Liu, R. Zhang and Q. Feng, Stem Cells Int., 2018, 2018, 2036176 Search PubMed.
- L. Lin, K. L. Chow and Y. Leng, J. Biomed. Mater. Res., Part A, 2009, 89, 326–335 Search PubMed.
- L. Cheng, F. Ye, R. Yang, X. Lu, Y. Shi, L. Li, H. Fan and H. Bu, Acta Biomater., 2010, 6, 1569–1574 Search PubMed.
- M. I. Hassan, N. Sultana and S. Hamdan, J. Nanomater., 2014, 2014, 573238 Search PubMed.
- Y. M. Baba Ismail, I. Wimpenny, O. Bretcanu, K. Dalgarno and A. J. El Haj, J. Biomed. Mater. Res., Part A, 2017, 105A, 1775–1785 Search PubMed.
- I. Bala, S. Hariharan and M. N. V. Ravi Kumar, Crit. Rev. Ther. Drug Carrier Syst., 2004, 21, 387–422 Search PubMed.
- Y. M. Khan, D. S. Katti and C. T. Laurencin, J. Biomed. Mater. Res., Part A, 2004, 69, 728–737 Search PubMed.
- M. Kikuchi, Y. Koyama, K. Takakuda, H. Miyairi, N. Shirahama and J. Tanaka, J. Biomed. Mater. Res., 2002, 62, 265–272 Search PubMed.
- T. Chae, H. Yang, V. Leung, F. Ko and T. Troczynski, J. Mater. Sci.: Mater. Med., 2013, 24, 1885–1894 Search PubMed.
- J. Qian, W. Xu, X. Yong, X. Jin and W. Zhang, Mater. Sci. Eng., C, 2014, 36, 95–101 Search PubMed.
- J. Norton, K. R. Malik, J. A. Darr and I. U. Rehman, Adv. Appl. Ceram., 2006, 105, 113–139 Search PubMed.
- T. Janicki, A. Sobczak-Kupiec, P. Skomro and Z. Wzorek, Acta Bioeng. Biomech., 2012, 14, 31–38 Search PubMed.
- P. Malmberg and H. Nygren, Proteomics, 2008, 8, 3755–3762 Search PubMed.
- X. Gao, C. Dai, W. Liu, Y. Liu, R. Shen, X. Zheng, K. Duan, J. Weng and S. Qu, J. Mater. Sci.: Mater. Med., 2017, 28, 83 Search PubMed.
- S. V. Dorozhkin, Acta Biomater., 2010, 6, 715–734 Search PubMed.
- V. Ogay, E. A. Mun, G. Kudaibergen, M. Baidarbekov, K. Kassymbek, Z. Zharkinbekov and A. Saparov, Polymers, 2020, 12, 2881 Search PubMed.
- A. Peeters, G. Putzeys and L. Thorrez, J. Bone Jt. Infect., 2019, 4, 245–253 Search PubMed.
- K. S. Cahill, J. H. Chi, A. Day and E. B. Claus, JAMA, 2009, 302, 58–66 Search PubMed.
- K. Gurpret and S. K. Singh, Indian J. Pharm. Sci., 2018, 80, 781–789 Search PubMed.
- T. F. Tadros, in An Introduction to Surfactants, ed. T. F. Tadros, De Gruyter, 2014, pp. 135–152 Search PubMed.
- F. Goodarzi and S. Zendehboudi, Can. J. Chem. Eng., 2019, 97, 281–309 Search PubMed.
- R. L. McMullen, M. Gorcea and S. Chen, in Handbook of Formulating Dermal Applications: A Definitive Practical Guide, ed. N. Dayan, Scrivener Publishing LLC., 2016, pp. 129–153 Search PubMed.
- T. F. Tadros, in Emulsion Formation and Stability, ed. T. F. Tadros, Wiley-VCH Verlag GmbH & Co. KGaA, 1st edn, 2013, pp. 1–75 Search PubMed.
- S. L. Ee, X. Duan, J. Liew and Q. D. Nguyen, Chem. Eng. J., 2008, 140, 626–631 Search PubMed.
- A. Gupta, H. B. Eral, T. A. Hatton and P. S. Doyle, Soft Matter, 2016, 12, 2826–2841 Search PubMed.
- D. J. McClements, Crit. Rev. Food Sci. Nutr., 2007, 47, 611–649 Search PubMed.
- R. Botet, J. Phys.: Conf. Ser., 2012, 352, 012047 Search PubMed.
- Q. Huang, H. Yu and Q. Ru, J. Food Sci., 2010, 75, R50–R57 Search PubMed.
- M. Koroleva, T. Nagovitsina and E. Yurtov, Phys. Chem. Chem. Phys., 2018, 20, 10369–10377 Search PubMed.
- R. Parveen, S. Baboota, J. Ali, A. Ahuja and S. Ahmad, J. Pharm. BioAllied Sci., 2015, 7, 321–324 Search PubMed.
- T. Sheth, S. Seshadri, T. Prileszky and M. E. Helgeson, Nat. Rev. Mater., 2020, 5, 214–228 Search PubMed.
- C. Liu, M. Li, R. Han, J. Li and C. Liu, J. Dispersion Sci. Technol., 2016, 37, 333–344 Search PubMed.
- C. Qian and D. J. McClements, Food Hydrocolloids, 2011, 25, 1000–1008 Search PubMed.
- L. Mao, J. Yang, D. Xu, F. Yuan and Y. Gao, J. Dispersion Sci. Technol., 2010, 31, 986–993 Search PubMed.
- C. P. Tan and M. Nakajima, Food Chem., 2005, 92, 661–671 Search PubMed.
- D. J. McClements, Food Emulsions. Principles, Practices, and Techniques, CRC Press, 2nd edn, 2004 Search PubMed.
- D. H. Oh, P. Balakrishnan, Y. K. Oh, D. D. Kim, C. S. Yong and H. G. Choi, Int. J. Pharm., 2011, 404, 191–197 Search PubMed.
- J. Rao and D. J. McClements, J. Agric. Food Chem., 2011, 59, 5026–5035 Search PubMed.
- H. Schubert and R. Engel, Chem. Eng. Res. Des., 2004, 82, 1137–1143 Search PubMed.
- L. Mao, D. Xu, J. Yang, F. Yuan, Y. Gao and J. Zhao, Food Technol. Biotechnol., 2009, 47, 336–342 Search PubMed.
- S. M. Jafari, E. Assadpoor, Y. He and B. Bhandari, Food Hydrocolloids, 2008, 22, 1191–1202 Search PubMed.
- A. Håkansson, C. Trägårdh and B. Bergenståhl, Chem. Eng. Sci., 2009, 64, 2915–2925 Search PubMed.
- Y. T. Hu, Y. Ting, J. Y. Hu and S. C. Hsieh, J. Food Drug Anal., 2017, 25, 16–26 Search PubMed.
- K. Rahn-Chique, A. M. Puertas, M. S. Romero-Cano, C. Rojas and G. Urbina-Villalba, Adv. Colloid Interface Sci., 2012, 178, 1–20 Search PubMed.
- H. H. Tayeb and F. Sainsbury, Nanomedicine, 2018, 13, 2507–2525 Search PubMed.
- Y. Zheng, M. Zheng, Z. Ma, B. Xin, R. Guo and X. Xu, in Polar Lipids, ed. M. U. Ahmad and X. Xu, Biology, Chemistry, and Technology, Elsevier Inc., 2015, pp. 215–243 Search PubMed.
- I. Kralova and J. Sjöblom, J. Dispersion Sci. Technol., 2009, 30, 1363–1383 Search PubMed.
- N. Ng and M. A. Rogers, in Encyclopedia of Food Chemistry, ed. L. Melton, F. Shahidi and P. Varelis, Elsevier Inc., 2019, pp. 276–282 Search PubMed.
- A. Witthayapanyanon, J. H. Harwell and D. A. Sabatini, J. Colloid Interface Sci., 2008, 325, 259–266 Search PubMed.
- M. J. Rosen and J. T. Kunjappu, Surfactants and Interfacial Phenomena, John Wiley & Sons, Inc., 4th edn, 2012 Search PubMed.
- P. Taylor, J. Salager, J. Bullón, A. Pizzino, M. Rondón-González and L. Tolosa, in Encyclopedia of Surface and Colloid Science, ed. P. Somasundaran, Taylor & Francis, 2nd edn, 2010, pp. 1–16 Search PubMed.
- Handbook of Detergents, Part A: Properties, ed. G. Broze, CRC Press, 1st edn, 1999 Search PubMed.
- E. J. Acosta, J. S. Yuan and A. S. Bhakta, J. Surfactants Deterg., 2008, 11, 145–158 Search PubMed.
- P. Izquierdo, J. Esquena, T. F. Tadros, C. Dederen, M. J. Garcia, N. Azemar and C. Solans, Langmuir, 2002, 18, 26–30 Search PubMed.
- L. Salvia-Trujillo, R. Soliva-Fortuny, M. A. Rojas-Graü, D. J. McClements and O. Martín-Belloso, Annu. Rev. Food Sci. Technol., 2017, 8, 439–466 Search PubMed.
- Colloidal Suspension Rheology, ed. J. Mewis and N. J. Wagner, Cambridge University Press, 2011 Search PubMed.
- G. Chen and D. Tao, Fuel Process. Technol., 2005, 86, 499–508 Search PubMed.
- A. Forgiarini, J. Esquena, C. González and C. Solans, in Trends in Colloid and Interface Science XIV, ed. Buckin, Progress in Colloid and Polymer Science, Springer, Berlin, Heidelberg, 2000, vol. 115, pp. 36–39 Search PubMed.
- P. Izquierdo, J. Esquena, T. F. Tadros, J. C. Dederen, J. Feng, M. J. Garcia-Celma, N. Azemar and C. Solans, Langmuir, 2004, 20, 6594–6598 Search PubMed.
- Foundations of Colloid Science, ed. R. J. Hunter, Oxford University Press, 2nd edn, 2001 Search PubMed.
- Y. Takahashi and Y. Kondo, in Stimuli-Responsive Interfaces: Fabrication and Application, ed. T. Kawai and M. Hashizume, Springer, Singapore, 1st edn, 2017, pp. 5–17 Search PubMed.
- S. Li, F. Wang, Z. Yang, J. Xu, H. Liu, L. Zhang and W. Xu, Powder Technol., 2020, 359, 17–26 Search PubMed.
- F. Wang, X. Yu, Z. Yang, H. Duan, Z. Zhang and H. Liu, J. Phys. Chem. C, 2018, 122, 18995–19003 Search PubMed.
- A. Maali and M. T. H. Mosavian, J. Dispersion Sci. Technol., 2013, 34, 92–105 Search PubMed.
- N. Anton, J. P. Benoit and P. Saulnier, J. Controlled Release, 2008, 128, 185–199 Search PubMed.
- Y. Singh, J. G. Meher, K. Raval, F. A. Khan, M. Chaurasia, N. K. Jain and M. K. Chourasia, J. Controlled Release, 2017, 252, 28–49 Search PubMed.
- M. Stang, H. Schuchmann and H. Schubert, Eng. Life Sci., 2001, 1, 151–157 Search PubMed.
- J. B. Aswathanarayan and R. R. Vittal, Front. Sustain. Food Syst., 2019, 3, 95 Search PubMed.
- S. Schultz, G. Wagner, K. Urban and J. Ulrich, Chem. Eng. Technol., 2004, 27, 361–368 Search PubMed.
- M. Kumar, R. S. Bishnoi, A. K. Shukla and C. P. Jain, Prev. Nutr. Food Sci., 2019, 24, 225–234 Search PubMed.
- E. B. Souto, C. Anselmi, M. Centini and R. H. Müller, Int. J. Pharm., 2005, 295, 261–268 Search PubMed.
- O. Behrend, K. Ax and H. Schubert, Ultrason. Sonochem., 2000, 7, 77–85 Search PubMed.
- T. S. H. Leong, T. J. Wooster, S. E. Kentish and M. Ashokkumar, Ultrason. Sonochem., 2009, 16, 721–727 Search PubMed.
- S. Kentish, T. J. Wooster, M. Ashokkumar, S. Balachandran, R. Mawson and L. Simons, Innovative Food Sci. Emerging Technol., 2008, 9, 170–175 Search PubMed.
- K. Landfester, J. Eisenblätter and R. Rothe, J. Coat. Technol. Res., 2004, 1, 65–68 Search PubMed.
- S. G. Gaikwad and A. B. Pandit, Ultrason. Sonochem., 2008, 15, 554–563 Search PubMed.
- I. O. Ocampo-Salinas, D. I. Ellez-Medina, C. Jimenez-Martinez and G. Davila-Ortiz, Int. J. Environ. Agric. Biotechnol., 2016, 1, 646–662 Search PubMed.
- G. Modig, L. Nilsson, B. Bergenståhl and K. G. Wahlund, Food Hydrocolloids, 2006, 20, 1087–1095 Search PubMed.
- J. Floury, A. Desrumaux, M. A. V. Axelos and J. Legrand, Food Hydrocolloids, 2002, 16, 47–53 Search PubMed.
- M. R. Kasaai, G. Charlet, P. Paquin and J. Arul, Innovative Food Sci. Emerging Technol., 2003, 4, 403–413 Search PubMed.
- I. Solè, C. M. Pey, A. Maestro, C. González, M. Porras, C. Solans and J. M. Gutiérrez, J. Colloid Interface Sci., 2010, 344, 417–423 Search PubMed.
- N. Anton and T. F. Vandamme, Int. J. Pharm., 2009, 377, 142–147 Search PubMed.
- M. Pathak, in Nanotechnology Applications in Food: Flavor, Stability, Nutrition and Safety, ed. A. E. Oprea and A. M. Grumezescu, Elsevier Inc., 2017, pp. 87–106 Search PubMed.
- C. Jadhav, V. Kate and S. A. Payghan, J. Nanostruct. Chem., 2015, 5, 107–113 Search PubMed.
- C. Solans and I. Solé, Curr. Opin. Colloid Interface Sci., 2012, 17, 246–254 Search PubMed.
- P. Izquierdo, J. Feng, J. Esquena, T. F. Tadros, J. C. Dederen, M. J. Garcia, N. Azemar and C. Solans, J. Colloid Interface Sci., 2005, 285, 388–394 Search PubMed.
- D. J. McClements and J. Rao, Crit. Rev. Food Sci. Nutr., 2011, 51, 285–330 Search PubMed.
- A. Maestro, I. Solè, C. González, C. Solans and J. M. Gutiérrez, J. Colloid Interface Sci., 2008, 327, 433–439 Search PubMed.
- J. C. L. Liew, Q. D. Nguyen and Y. Ngothai, Asia-Pac. J. Chem. Eng., 2010, 5, 570–576 Search PubMed.
- A. Perazzo and V. Preziosi, in Nanoemulsions: Formulation, Applications, and Characterization, ed. S. M. Jafari and D. J. McClements, Academic Press, 1st edn, 2018, pp. 53–76 Search PubMed.
- T. S. Dunstan, P. D. I. Fletcher and S. Mashinchi, Langmuir, 2012, 28, 339–349 Search PubMed.
- R. K. Thakur, C. Villette, J. M. Aubry and G. Delaplace, Colloids Surf., A, 2008, 315, 285–293 Search PubMed.
- S. M. Jafari, P. Paximada, I. Mandala, E. Assadpour and M. A. Mehrnia, in Nanoencapsulation Technologies for the Food and Nutraceutical Industries, ed. S. M. Jafari, Academic Press, 1st edn, 2017, pp. 36–73 Search PubMed.
- D. J. McClements, Soft Matter, 2011, 7, 2297–2316 Search PubMed.
- K. Bouchemal, S. Briançon, E. Perrier and H. Fessi, Int. J. Pharm., 2004, 280, 241–251 Search PubMed.
- N. A. N. Azmi, A. A. M. Elgharbawy, S. R. Motlagh, N. Samsudin and H. M. Salleh, Processes, 2019, 7, 617 Search PubMed.
- M. Safaya and Y. C. Rotliwala, Mater. Today: Proc., 2020, 27, 454–459 Search PubMed.
- A. A. Date, N. Desai, R. Dixit and M. Nagarsenker, Nanomedicine, 2010, 5, 1595–1616 Search PubMed.
- A. W. Khan, S. Kotta, S. H. Ansari, R. K. Sharma and J. Ali, Drug Delivery, 2015, 22, 552–561 Search PubMed.
- M. Dahiya and P. Pandey, Int. J. Pharm. Integr. Life Sci., 2016, 4, 43–61 Search PubMed.
- S. Parveen, R. Misra and S. K. Sahoo, Nanomedicine, 2012, 8, 147–166 Search PubMed.
- Z. Urbán-Morlán, S. E. Mendoza-Elvira, R. S. Hernández-Cerón, S. Alcalá-Alcalá, H. Ramírez-Mendoza, A. Ciprián-Carrasco, E. Piñón-Segundo and D. Quintanar-Guerrero, J. Mex. Chem. Soc., 2015, 59, 173–180 Search PubMed.
- Y. Wang, P. Li, T. T.-D. Tran, J. Zhang and L. Kong, Nanomaterials, 2016, 6, 26 Search PubMed.
- S. Grandhi, A. Prameela Rani and R. Pathuri, Res. J. Pharm. Technol., 2018, 11, 2829–2835 Search PubMed.
- J. Feng, M. Roché, D. Vigolo, L. N. Arnaudov, S. D. Stoyanov, T. D. Gurkov, G. G. Tsutsumanova and H. A. Stone, Nat. Phys., 2014, 10, 606–612 Search PubMed.
- J. Feng, J. K. Nunes, S. Shin, J. Yan, Y. L. Kong, R. K. Prud’homme, L. N. Arnaudov, S. D. Stoyanov and H. A. Stone, Adv. Mater., 2016, 28, 4047–4052 Search PubMed.
- M. M. Villa, L. Wang, J. Huang, D. W. Rowe and M. Wei, J. Biomed. Mater. Res., Part B, 2015, 103, 243–253 Search PubMed.
- M. Prekajski, M. Mirković, B. Todorović, A. Matković, M. Marinović-Cincović, J. Luković and B. Matović, J. Eur. Ceram. Soc., 2016, 36, 1293–1298 Search PubMed.
- M. Prekajski, B. Jokić, A. Kalijadis, J. Maletaškić, N. Stanković, J. Luković and B. Matović, Process. Appl. Ceram., 2016, 10, 169–174 Search PubMed.
- M. P. Pileni, J. Phys. Chem., 1993, 97, 6961–6973 Search PubMed.
- G. K. Lim, J. Wang, S. C. Ng, C. H. Chew and L. M. Gan, Biomaterials, 1997, 18, 1433–1439 Search PubMed.
- G. K. Lim, J. Wang, S. C. Ng and L. M. Gan, Mater. Lett., 1996, 28, 431–436 Search PubMed.
- M. Cao, Y. Wang, C. Guo, Y. Qi and C. Hu, Langmuir, 2004, 20, 4784–4786 Search PubMed.
- A. Banerjee, A. Bandyopadhyay and S. Bose, Mater. Sci. Eng., C, 2007, 27, 729–735 Search PubMed.
- K. Suchanek, A. Bartkowiak, A. Gdowik, M. Perzanowski, S. Kąc, B. Szaraniec, M. Suchanek and M. Marszałek, Mater. Sci. Eng., C, 2015, 51, 57–63 Search PubMed.
- L. T. Bang, S. Ramesh, J. Purbolaksono, Y. C. Ching, B. D. Long, H. Chandran and R. Othman, Mater. Des., 2015, 87, 788–796 Search PubMed.
- N. Jing, A. N. Zhou and Q. H. Xu, J. Hazard. Mater., 2018, 353, 89–98 Search PubMed.
- S. E. Cahyaningrum, N. Herdyastuty, B. Devina and D. Supangat, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 299, 012039 Search PubMed.
- M. Biernat, Z. Jaegermann, P. Tymowicz-Grzyb and G. Konopka, Process. Appl. Ceram., 2019, 13, 57–64 Search PubMed.
- A. C. Tas, J. Am. Ceram. Soc., 2007, 90, 2358–2362 Search PubMed.
- O. V. Sinitsyna, A. G. Veresov, E. S. Kovaleva, Y. V. Kolen’ko, V. I. Putlyaev and Y. D. Tretyakov, Russ. Chem. Bull., 2005, 54, 79–86 Search PubMed.
- W. J. Shih, Y. F. Chen, M. C. Wang and M. H. Hon, J. Cryst. Growth, 2004, 270, 211–218 Search PubMed.
- M. G. S. Murray, J. Wang, C. B. Ponton and P. M. Marquis, J. Mater. Sci., 1995, 30, 3061–3074 Search PubMed.
- N. Y. Mostafa, Mater. Chem. Phys., 2005, 94, 333–341 Search PubMed.
- M. Eriksson, Y. Liu, J. Hu, L. Gao, M. Nygren and Z. Shen, J. Eur. Ceram. Soc., 2011, 31, 1533–1540 Search PubMed.
- N. A. S. Mohd Pu’ad, R. H. Abdul Haq, H. Mohd Noh, H. Z. Abdullah, M. I. Idris and T. C. Lee, Mater. Today: Proc., 2019, 29, 233–239 Search PubMed.
- H. C. Shum, A. Bandyopadhyay, S. Bose and D. A. Weitz, Chem. Mater., 2009, 21, 5548–5555 Search PubMed.
- T. Y. Podlipskaya and A. I. Bulavchenko, J. Struct. Chem., 2019, 60, 1133–1142 Search PubMed.
- A. Amani and M. Amani, J. Contemp. Med. Sci., 2015, 1, 1–6 Search PubMed.
- I. Ezekiel, S. R. Kasim, Y. M. B. Ismail and A. F. M. Noor, Ceram. Int., 2018, 44, 13082–13089 Search PubMed.
- M. N. Muhammad Syazwan and B. I. Yanny Marliana, Ceram. Int., 2019, 45, 14783–14788 Search PubMed.
- W. Amer, K. Abdelouahdi, H. R. Ramananarivo, M. Zahouily, A. Fihri, Y. Coppel, R. S. Varma and A. Solhy, Mater. Lett., 2013, 107, 189–193 Search PubMed.
- Y. Sun, G. Guo, D. Tao and Z. Wang, J. Phys. Chem. Solids, 2007, 68, 373–377 Search PubMed.
- W. Y. Zhou, M. Wang, W. L. Cheung, B. C. Guo and D. M. Jia, J. Mater. Sci.: Mater. Med., 2008, 19, 103–110 Search PubMed.
- A. Ethirajan, U. Ziener, A. Chuvilin, U. Kaiser, H. Cölfen and K. Landfester, Adv. Funct. Mater., 2008, 18, 2221–2227 Search PubMed.
- K. Mitri, C. Vauthier, N. Huang, A. Menas, C. Ringard-Lefebvre, C. Anselmi, M. Stambouli, V. Rosilio, J. J. Vachon and K. Bouchemal, J. Pharm. Sci., 2012, 101, 4240–4247 Search PubMed.
- S. A. Galindo-Rodríguez, F. Puel, S. Briançon, E. Allémann, E. Doelker and H. Fessi, Eur. J. Pharm. Sci., 2005, 25, 357–367 Search PubMed.
- Y. Liu, C. Cheng, Y. Liu, R. K. Prud’homme and R. O. Fox, Chem. Eng. Sci., 2008, 63, 2829–2842 Search PubMed.
- D. L. Marchisio, L. Rivautella and A. A. Barresi, AIChE J., 2006, 52, 1877–1887 Search PubMed.
- F. Ganachaud and J. L. Katz, ChemPhysChem, 2005, 6, 209–216 Search PubMed.
- B. Kupikowska-Stobba and D. Lewińska, Biomater. Sci., 2020, 8, 1536–1574 Search PubMed.
- J. Chen, D. Huang, L. Wang, J. Hou, H. Zhang, Y. Li, S. Zhong, Y. Wang, Y. Wu and W. Huang, J. Colloid Interface Sci., 2020, 574, 162–173 Search PubMed.
- M. A. Vandelli, F. Rivasi, P. Guerra, F. Forni and R. Arletti, Int. J. Pharm., 2001, 215, 175–184 Search PubMed.
- A. Totaro, A. Salerno, G. Imparato, C. Domingo, F. Urciuolo and P. A. Netti, J. Tissue Eng. Regener. Med., 2017, 11, 1865–1875 Search PubMed.
- S. Itoh, M. Kikuchi, Y. Koyama, K. Takakuda, K. Shinomiya and J. Tanaka, Biomaterials, 2002, 23, 3919–3926 Search PubMed.
- L. Gritsch, M. Maqbool, V. Mouriño, F. E. Ciraldo, M. Cresswell, P. R. Jackson, C. Lovell and A. R. Boccaccini, J. Mater. Chem. B, 2019, 7, 6109–6124 Search PubMed.
- Y. Chen, A. F. T. Mak, M. Wang, J. S. Li and M. S. Wong, J. Mater. Sci.: Mater. Med., 2008, 19, 2261–2268 Search PubMed.
- G. Wei and P. X. Ma, Biomaterials, 2004, 25, 4749–4757 Search PubMed.
- L. Jiang, Y. Li, C. Xiong, S. Su and H. Ding, ACS Appl. Mater. Interfaces, 2017, 9, 4890–4897 Search PubMed.
- P. Habibovic, Biomatter, 2014, 4, e27664 Search PubMed.
- M. Wang, Biomaterials, 2003, 24, 2133–2151 Search PubMed.
- Y. M. Baba Ismail, A. M. Ferreira, O. Bretcanu, K. Dalgarno and A. J. El Haj, Colloids Surf., B, 2017, 159, 445–453 Search PubMed.
- W. Y. Zhou, M. Wang and W. L. Cheung, in Advances in Diverse Industrial Applications of Nanocomposites, ed. B. Reddy, IntechOpen, 2011, pp. 203–230 Search PubMed.
- W. Y. Zhou, S. H. Lee, M. Wang, W. L. Cheung and W. Y. Ip, J. Mater. Sci.: Mater. Med., 2008, 19, 2535–2540 Search PubMed.
- W. Y. Zhou, M. Wang and W. L. Cheung, Key Eng. Mater., 2007, 334–335, 1221–1224 Search PubMed.
- J. F. Mano, R. A. Sousa, L. F. Boesel, N. M. Neves and R. L. Reis, Compos. Sci. Technol., 2004, 64, 789–817 Search PubMed.
- J. Vandorpe, E. Schacht, S. Stolnik, M. C. Garnett, M. C. Davies, L. Illum and S. S. Davis, Biotechnol. Bioeng., 1996, 52, 89–95 Search PubMed.
- Q. Li, K. Li, Q. Han, M. Wu and N. Zhang, Int. J. Clin. Exp. Med., 2020, 13, 4035–4041 Search PubMed.
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887–913 Search PubMed.
- U. Bilati, E. Allémann and E. Doelker, Pharm. Dev. Technol., 2003, 8, 1–9 Search PubMed.
- U. Bilati, E. Allémann and E. Doelker, AAPS PharmSciTech, 2005, 6, E594–E604 Search PubMed.
- Z. Zhang, D. W. Grijpma and J. Feijen, J. Controlled Release, 2006, 111, 263–270 Search PubMed.
- A. W. James, G. LaChaud, J. Shen, G. Asatrian, V. Nguyen, X. Zhang, K. Ting and C. Soo, Tissue Eng., Part B, 2016, 22, 284–297 Search PubMed.
- B. B. Seo, H. Choi, J. T. Koh and S. C. Song, J. Controlled Release, 2015, 209, 67–76 Search PubMed.
- A. H. Hassan, K. M. Hosny, Z. A. Murshid, A. Alhadlaq, A. Alyamani and G. Naguib, Drug Des., Dev. Ther., 2015, 9, 3599–3606 Search PubMed.
- K. H. Huang, Y. H. Lin, M. Y. Shie and C. P. Lin, J. Formosan Med. Assoc., 2018, 117, 879–887 Search PubMed.
- H. Cheng, A. Chawla, Y. Yang, Y. Li, J. Zhang, H. L. Jang and A. Khademhosseini, Drug Discovery Today, 2017, 22, 1336–1350 Search PubMed.
- V. Hengst, C. Oussoren, T. Kissel and G. Storm, Int. J. Pharm., 2007, 331, 224–227 Search PubMed.
- C. T. Leu, E. Luegmayr, L. P. Freedman, G. A. Rodan and A. A. Reszka, Bone, 2006, 38, 628–636 Search PubMed.
- M. S. Lee, C. M. Su, J. C. Yeh, P. R. Wu, T. Y. Tsai and S. L. Lou, Int. J. Nanomed., 2016, 11, 4583–4594 Search PubMed.
- H. Piao, M. H. Kim, M. Cui, G. Choi and J. H. Choy, J. Korean Med. Sci., 2019, 34, e37 Search PubMed.
- M. Hie, M. Yamazaki and I. Tsukamoto, Eur. J. Pharmacol., 2009, 621, 1–9 Search PubMed.
- B. B. Aggarwal and K. B. Harikumar, Int. J. Biochem. Cell Biol., 2009, 41, 40–59 Search PubMed.
- B. A. Cunha, Med. Clin. North Am., 2001, 85, 149–185 Search PubMed.
- S. Heta and I. Robo, Med. Sci., 2018, 6, 6 Search PubMed.
- K. Brannigan and M. Griffin, Open Orthop. J., 2017, 10, 808–823 Search PubMed.
- M. L. Etheridge, S. A. Campbell, A. G. Erdman, C. L. Haynes, S. M. Wolf and J. McCullough, Nanomedicine, 2013, 9, 1–14 Search PubMed.
- L. Zhang, E. M. Haddouti, K. Welle, C. Burger, K. Kabir and F. A. Schildberg, Int. J. Nanomed., 2020, 15, 6705–6720 Search PubMed.
- S. B. Goodman and J. Gallo, J. Clin. Med., 2019, 8, 2091 Search PubMed.
- K. Fox, P. A. Tran and N. Tran, ChemPhysChem, 2012, 13, 2495–2506 Search PubMed.
- B. Semete, L. Booysen, Y. Lemmer, L. Kalombo, L. Katata, J. Verschoor and H. S. Swai, Nanomedicine, 2010, 6, 662–671 Search PubMed.
- P. R. Lockman, R. J. Mumper, M. A. Khan and D. D. Allen, Drug Dev. Ind. Pharm., 2002, 28, 1–13 Search PubMed.
- S. Barua and S. Mitragotri, Nano Today, 2014, 9, 223–243 Search PubMed.
- W. H. De Jong and P. J. A. Borm, Int. J. Nanomed., 2008, 3, 133–149 Search PubMed.
- I. W. Bauer, S. P. Li, Y. C. Han, L. Yuan and M. Z. Yin, J. Mater. Sci.: Mater. Med., 2008, 19, 1091–1095 Search PubMed.
- L. Chen, J. M. Mccrate, J. C. M. Lee and H. Li, Nanotechnology, 2011, 22, 105708 Search PubMed.
- M. Banik and T. Basu, Dalton Trans., 2014, 43, 3244–3259 Search PubMed.
- H. Turkez, M. I. Yousef, E. Sönmez, B. Togar, F. Bakan, P. Sozio and A. D. Stefano, J. Appl. Toxicol., 2014, 34, 373–379 Search PubMed.
- V. Opačić-Galić, V. Petrović, S. Živković, V. Jokanović, B. Nikolić, J. Knežević-Vukčević and D. Mitić-Ćulafić, Int. Endod. J., 2013, 46, 506–516 Search PubMed.
- J. Xu, P. Xu, Z. Li, J. Huang and Z. Yang, J. Biomed. Mater. Res., Part A, 2012, 100, 738–745 Search PubMed.
- C. S. Geetha, N. S. Remya, K. B. Leji, S. Syama, S. C. Reshma, P. J. Sreekanth, H. K. Varma and P. V. Mohanan, Colloids Surf., B, 2013, 112, 204–212 Search PubMed.
- X. Liu and J. Sun, Int. J. Nanomed., 2014, 9, 1261–1273 Search PubMed.
- P. Laquerriere, A. Grandjean-Laquerriere, E. Jallot, G. Balossier, P. Frayssinet and M. Guenounou, Biomaterials, 2003, 24, 2739–2747 Search PubMed.
- S. Dey, M. Das and V. K. Balla, Mater. Sci. Eng., C, 2014, 39, 336–339 Search PubMed.
- X. Chen, C. Deng, S. Tang and M. Zhang, Biol. Pharm. Bull., 2007, 30, 128–132 Search PubMed.
- S.-H. Chu, D.-F. Feng, Y.-B. Ma and Z.-Q. Li, Int. J. Nanomed., 2012, 7, 3659–3666 Search PubMed.
- J. Scheel and M. Hermann, Regul. Toxicol. Pharmacol., 2011, 59, 310–323 Search PubMed.
- T. Ding, Y. Xue, H. Lu, Z. Huang and J. Sun, IEEE Trans Nanobioscience, 2012, 11, 336–340 Search PubMed.
- L. Liu, Y. Xiao, Z. Xiao, Z. Wang, C. Li and X. Gong, J. Hyg. Res., 2005, 34, 474–476 Search PubMed.
- Q. Chen, Y. Xue and J. Sun, J. Appl. Toxicol., 2014, 34, 1256–1264 Search PubMed.
- L. Wang, G. Zhou, H. Liu, X. Niu, J. Han, L. Zheng and Y. Fan, Nanoscale, 2012, 4, 2894–2899 Search PubMed.
- B. Lin, Z. Xi, Y. Zhang and H. Zhang, Weisheng Yanjiu, 2008, 37, 651–653 Search PubMed.
- K. M. de la Harpe, P. P. D. Kondiah, Y. E. Choonara, T. Marimuthu, L. C. du Toit and V. Pillay, Cells, 2019, 8, 1209 Search PubMed.
- Y. Han, X. Wang, H. Dai and S. Li, ACS Appl. Mater. Interfaces, 2012, 4, 4616–4622 Search PubMed.
- I. Nadra, A. R. Boccaccini, P. Philippidis, L. C. Whelan, G. M. McCarthy, D. O. Haskard and R. C. Landis, Atherosclerosis, 2008, 196, 98–105 Search PubMed.
- E. M. Elmowafy, M. Tiboni and M. E. Soliman, J. Pharm. Invest., 2019, 49, 347–380 Search PubMed.
- P. Foroozandeh and A. A. Aziz, Nanoscale Res. Lett., 2018, 13, 339 Search PubMed.
- C. Fornaguera, C. Castells-Sala and S. Borrós, in Cell Biology and Translational Medicine IX, ed. K. Turksen, Advances in Experimental Medicine and Biology, Springer, 2019, vol. 1288, pp. 117–138 Search PubMed.
- N. Voigt, P. Henrich-Noack, S. Kockentiedt, W. Hintz, J. Tomas and B. A. Sabel, J. Nanopart. Res., 2014, 16, 2379 Search PubMed.
- S. Jesus, M. Schmutz, C. Som, G. Borchard, P. Wick and O. Borges, Front. Bioeng. Biotechnol., 2019, 7, 261 Search PubMed.
- N. Grabowski, H. Hillaireau, J. Vergnaud-Gauduchon, V. Nicolas, N. Tsapis, S. Kerdine-Römer and E. Fattal, J. Biomed. Nanotechnol., 2016, 12, 135–146 Search PubMed.
- N. Grabowski, H. Hillaireau, J. Vergnaud, N. Tsapis, M. Pallardy, S. Kerdine-Römer and E. Fattal, Int. J. Pharm., 2015, 482, 75–83 Search PubMed.
- M. Sharma, S. Sharma, V. Sharma, K. Sharma, S. K. Yadav, P. Dwivedi, S. Agrawal, S. K. Paliwal, A. K. Dwivedi, J. P. Maikhuri, G. Gupta, P. R. Mishra and A. K. S. Rawat, Int. J. Biol. Macromol., 2017, 104, 1345–1358 Search PubMed.
- R. P. Singh and P. Ramarao, Toxicol. Sci., 2013, 136, 131–143 Search PubMed.
- K. A. V. Zubris, O. V. Khullar, A. P. Griset, S. Gibbs-Strauss, J. V. Frangioni, Y. L. Colson and M. W. Grinstaff, ChemMedChem, 2010, 5, 1435–1438 Search PubMed.
- O. V. Khullar, A. P. Griset, S. L. Gibbs-Strauss, L. R. Chirieac, K. A. V. Zubris, J. V. Frangioni, M. W. Grinstaff and Y. L. Colson, J. Am. Coll. Surg., 2012, 214, 328–337 Search PubMed.
- R. Löbenberg and J. Kreuter, AIDS Res. Hum. Retroviruses, 1996, 12, 1709–1715 Search PubMed.
- M. A. Radwan, B. T. AlQuadeib, L. Šiller, M. C. Wright and B. Horrocks, Drug Delivery, 2017, 24, 40–50 Search PubMed.
- H. Fasehee, R. Dinarvand, A. Ghavamzadeh, M. Esfandyari-Manesh, H. Moradian, S. Faghihi and S. H. Ghaffari, J. Nanobiotechnol., 2016, 14, 32 Search PubMed.
- S. Vasanthakumar, H. N. Ahamed and R. N. Saha, Eur. J. Drug Metab. Pharmacokinet., 2015, 40, 137–161 Search PubMed.
- T. C. Moraes Moreira Carraro, C. Altmeyer, N. Maissar Khalil and R. Mara Mainardes, J. Mycol. Med., 2017, 27, 519–529 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |