
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bone-adhesive barrier membranes based on alendronate-functionalized poly(2-oxazoline)s†
María J.
Sánchez-Fernández
a,
Manon
Peerlings
a,
Rosa P.
Félix Lanao
b,
Johan C. M. E.
Bender
b,
Jan C. M.
van Hest
 c and
Sander C. G.
Leeuwenburgh
*a
c and
Sander C. G.
Leeuwenburgh
*a
aDepartment of Dentistry–Regenerative Biomaterials, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, 6525 EX Nijmegen, The Netherlands. E-mail: Sander.Leeuwenburgh@radboudumc.nl
bGATT Technologies BV, 6525 ED, Nijmegen, The Netherlands
cDepartment of Bio-Organic Chemistry, Institute for Complex Molecular Systems, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands
First published on 13th July 2021
Abstract
To create a novel generation of barrier membranes with bone-adhesive properties, three-component membranes were successfully developed using a solvent-free approach by combining an occlusive polyester backing layer with a bone-adhesive fibrous gelatin carrier impregnated with calcium-binding alendronate-functionalized poly(2-oxazoline)s (POx-Ale). The mechanical properties of these novel membranes were similar to other commercially available barrier membranes. In contrast, the adhesion of our membranes towards bone was by far superior (i.e. 62-fold) compared to conventional commercially available Bio-Gide® membranes. Moreover, alendronate-functionalized membranes retained their bone-adhesive properties under wet conditions in phosphate-buffered saline (PBS) solutions with and without collagenase. Finally, the in vitro degradation of the membranes was studied by monitoring their weight loss upon immersion in PBS solutions with and without collagenase. The membranes degraded in a sustained manner, which was accelerated by the presence of collagenase due to enzymatic degradation of the carrier. In conclusion, our results show that surface functionalization of barrier membranes with alendronate moieties renders them adhesive to bone. As such, the biomaterials design strategy presented herein opens up new avenues of research on bone-adhesive membranes for guided bone regeneration.
1. Introduction
Oral implants should be tightly fixed in alveolar bone, but this fixation is often compromised by degenerative conditions such as periodontitis, peri-implantitis, aging or trauma.1–3 To overcome this problem, guided bone regeneration (GBR) is routinely utilized in dentistry when the volume and/or dimensions of alveolar bone are insufficient.4 This procedure involves the application of barrier membranes, which support the process of bone regeneration by acting as a physical barrier against ingrowth of fast-growing soft tissues. In addition, these membranes can also stimulate the natural healing process of bone.5,6 To develop a barrier membrane for application in oral and maxillofacial surgery several requirements should be met, which include: (i) biocompatibility, (ii) mechanical stability to guarantee space maintenance required for osseous regeneration, (iii) biodegradability, (iv) occlusivity, (v) connection of the membrane to the defect site, and (vi) good clinical handling properties.7–10 So far, various types of membranes with different compositions and modifications have become commercially available. Non-degradable titanium meshes and expanded polytetrafluoroethylene (e-PTFE) membranes such as Gore-Tex® were developed first,11 followed by biodegradable membranes based on chitosan, gelatin, collagen (most notably Bio-Gide® or Collprotect®), or silk fibroin. More recently, synthetic polymers such as poly(ε-caprolactone) (PCL) and poly(lactic-co-glycolic acid) (PLGA) have also been used for GBR.12 Generally, biodegradable membranes are preferred from a clinical perspective since a second removal surgery is not required for such membranes. Nevertheless, none of the currently available biodegradable membranes meets all of the above-mentioned requirements.13 Consequently, they are still associated with drawbacks, such as poor control over their degradation rate, poor mechanical properties, and increased risk of infection. Moreover, the clinical reliability and effectiveness of biodegradable membranes are compromised due to their poor clinical manageability caused by their poor adhesion to bone. Therefore, these membranes are occasionally fixated to bone using titanium pins. However, these pins need to be removed during a second surgery due to their non-degradable nature.14–16We propose that the next generation of biodegradable barrier membranes should become adhesive to bone via a molecular mechanism to ensure fixation of the membrane. This approach will minimize gingival ingrowth, maximize the extent of bone regeneration, and improve the overall clinical handling and reliability of barrier membranes. To render barrier membranes adhesive to bone, the adhesive material should be rationally designed based on the chemical composition of bone,17 which consists of an organic collagenous matrix (≈30 wt%) reinforced by finely dispersed calcium phosphate mineral nanocrystals (CaP) (≈70 wt%). We propose that synthetic polymers such as poly(2-oxazoline)s (POx) are particularly attractive to this end since these polymers can be functionalized with pendant chemical groups of high affinity with the organic and/or inorganic components of bone tissue. In our study, we selected bisphosphonates as primary bone-bonding moiety, since bisphosphonates such as alendronate (Ale) are anti-osteoporotic molecules known for their exceptionally strong chemical affinity for calcium cations in hydroxyapatite, the mineral phase of bone.18–20
Herein, we designed novel barrier membranes consisting of three components: (i) a polyester backing layer and (ii) a fibrous gelatin carrier layer impregnated with (iii) calcium-binding alendronate-functionalized poly(2-oxazoline)s (POx-Ale). We hypothesized that this approach would allow for independent tuning of bone adhesiveness (by controlling the amount of alendronate groups) and prevention of gingival ingrowth (by controlling the degradation rate of the polyester backing layer). Ideally, this approach would ensure short-term bone adhesiveness of the membranes (in terms of days) with a long-term occlusive function (in terms of several months) of the resulting membranes. Poly(2-oxazoline)s were selected as a core polymer in regards of their high functionalization possibilities along the entire polymeric backbone, and their tunable versatility by copolymerization.21–23 POx polymers were functionalized with both alendronate (P(EtOx-Ale)) and hydroxyl moieties (P(EtOx-OH-Ale)) in view of the strong binding affinity of both moieties for calcium in hydroxyapatite bone mineral nanocrystals.24,25 A commercially available fibrous gelatin carrier (GELITA TUFT-IT®) was selected in view of the favorable biological response of gelatin-based medical devices. Furthermore, the selected fibrous gelatin carrier combines the beneficial features of gelatin-based membranes with the easy handling properties of a non-woven material.26 In addition, the fibrillar architecture of the gelatin carrier facilitated impregnation of the bone-adhesive polymers as a dry powder. Dry powder deposition was selected to improve the future shelf life of the resulting membranes. In addition, unlike impregnation with organic or inorganic solvents, dry deposition does neither affect the structural integrity nor the mechanical properties of the carrier, since this method does not create any internal mechanical stress.27 Finally, the backing layer was prepared from biodegradable polyesters to enhance the mechanical properties at early stages of bone regeneration, while enabling biodegradation of the membranes at later stages of bone regeneration. Specifically, the backing layer consisted of a blend of poly(L-lactide-co-ε-caprolactone) (P(LA-CL)), poly(D,L-lactic-co-glycolic acid) (PLGA), and poly(2-propyl-2-oxazoline) functionalized with pendant amine groups (P(PropOx-NH2)), which was processed using hot-melt extrusion. P(LA-CL) copolymer provided the desired strength and flexibility to the backing,28 whereas PLGA was added to tune the long-term degradation rate of the resulting membranes.29,30 P(PropOx-NH2) was incorporated to the polyester backing layer to facilitate its attachment to the impregnated carrier since the amine groups present in the backing could interact by hydrogen bonding with carboxylic acid groups present in the carrier as well as hydroxyl and bisphosphonate groups from the impregnated POx polymers.
The membranes were characterized in detail regarding their wettability and mechanical properties. Moreover, adhesion of these membranes to apatite-coated model surfaces as well as bone was assessed by lap-shear tests. Finally, the degradation kinetics of these membranes were evaluated in vitro in phosphate-buffered saline (PBS) with and without collagenase to study the degradation mechanism in more detail.
2. Experimental section
2.1 Materials
Anhydrous dichloromethane, anhydrous diethyl ether, and HPLC-grade methanol were purchased from Fisher Scientific. Anhydrous N,N-dimethylformamide (DMF) was acquired from Acros Organics. Dowex® 50WX4 100–200 (H) and succinic anhydride were obtained from Alfa Aesar. N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), 4-dimethylamino pyridine (DMAP), N,N′-diisopropylcarbodiimide (DIC), N-hydroxysuccinimide (NHS), and sodium alendronate trihydrate (Ale) were acquired from Fluorochem. Dimethyl sulfoxide, 2-ethanolamine, methyl-p-toluenesulfonate (MeOTs), 2-ethyl-2-oxazoline (EtOx), α-cyano-4-hydroxycinnamic acid (CHCA), and collagenase (type II) were purchased from Sigma-Aldrich. Deuterated solvents as deuterium oxide, chloroform-d, and dimethyl sulfoxide-d6 were purchased from Cambridge Isotope Laboratories and Sigma-Aldrich. Dialysis membranes, Spectra/Por® 3 (3.5 kDa cutoff) were acquired from VWR International. Sodium chloride, potassium chloride, disodium hydrogen phosphate, and potassium dihydrogen phosphate were obtained from Merck. Poly(2-ethyl-2-oxazoline) (Aquazol®, P(EtOx), 50 kDa) was purchased from Labshop. 2-Methoxycarbonylethyl-2-oxazoline (MestOx) monomer and a backing layer of 50 μm composed of poly(L-lactide-co-ε-caprolactone) (P(LA-CL), monomer ratio 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, 67 wt%) from Corbion (Purasorb 7015), poly(D,L-lactic-co-glycolic acid) (PLGA, monomer ratio 50:50, 7.8 wt%) from Corbion (Purasorb 5002A), and poly(2-propyl-2-oxazoline) functionalized with pendant amine groups (P(PropOx-NH2), monomer ratio 80:20, 25.2 wt%) were kindly provided by GATT Technologies BV. TDE™ 30 decalcifier was purchased from Tissue-Tek®. Reagents were used without purification, except for EtOx, MeOTs and 2-ethanolamine, which were distilled before use at 40 mbar and 40, 55, and 75 °C, respectively. MeCN and tetrahydrofuran (THF) were discharged under a nitrogen atmosphere using an MBraun MB SPS-800 solvent dispersing system. Ultrapure Milli-Q water set to 18.2 MΩ cm−1 was obtained from a WaterPro PS polisher. Phosphate-buffered saline (PBS) solution was prepared containing 2.7 mM KCl, 137 mM NaCl, 2 mM KH2PO4, and 8 mM Na2HPO4 (pH = 7.4).
:30, 67 wt%) from Corbion (Purasorb 7015), poly(D,L-lactic-co-glycolic acid) (PLGA, monomer ratio 50:50, 7.8 wt%) from Corbion (Purasorb 5002A), and poly(2-propyl-2-oxazoline) functionalized with pendant amine groups (P(PropOx-NH2), monomer ratio 80:20, 25.2 wt%) were kindly provided by GATT Technologies BV. TDE™ 30 decalcifier was purchased from Tissue-Tek®. Reagents were used without purification, except for EtOx, MeOTs and 2-ethanolamine, which were distilled before use at 40 mbar and 40, 55, and 75 °C, respectively. MeCN and tetrahydrofuran (THF) were discharged under a nitrogen atmosphere using an MBraun MB SPS-800 solvent dispersing system. Ultrapure Milli-Q water set to 18.2 MΩ cm−1 was obtained from a WaterPro PS polisher. Phosphate-buffered saline (PBS) solution was prepared containing 2.7 mM KCl, 137 mM NaCl, 2 mM KH2PO4, and 8 mM Na2HPO4 (pH = 7.4).
2.2 Synthesis and characterization of alendronate-functionalized poly(2-oxazoline)s
POx polymers were synthesized and characterized following procedures described previously.24 In brief, the synthetic route consisted of five steps, as depicted in Scheme S1 (ESI†). Methyl-ester functional 2-oxazoline (MestOx) was copolymerized with ethyl-2-oxazoline (EtOx) via cationic ring opening polymerization (CROP) in the desired ratios by varying the moles of reagents, yielding methyl ester-functionalized polymer P1a. Then, methyl ester-functionalized groups were reacted with 2-aminoethanol by an amidation reaction yielding hydroxyl-functionalized polymer P1b. Next, hydroxyl-functionalized polymer was reacted with succinic anhydride either fully or partially in the desired ratios to obtain carboxylic acid-functionalized polymers P1c and P2c. Subsequently, N-hydroxysuccinimide-functionalized polymers P1d and P2d were synthesized by carbodiimide coupling with NHS. Finally, alendronate-functionalized polymers P1e and P2e were synthesized by an amidation reaction. Detailed experimental procedures and characterization of the synthesized polymers are described in S1 (ESI†).
1H NMR and 31P NMR spectroscopy were used to determine the degree of modification of the different substitutions in the polymers.24 NMR spectra were recorded on a Bruker Avance III (400 MHz) spectrometer in the indicated solvent at 25 °C. 1H NMR data are reported as chemical shifts (given in parts per million (ppm) with respect to tetramethylsilane as standard), multiplicity (br = broad), integration, and assignment. 1H NMR and 31P NMR spectra are presented in Fig. S1 (ESI†). The number average molecular weights (Mn) were recorded on a Bruker Microflex LRF matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) system as described previously.24 All mass spectra were obtained in the positive ion mode. α-Cyano-4-hydroxycinnamic acid (CHCA) was used as a matrix in THF (10 mg mL−1). Polymer samples were dissolved in THF/MeOH (1:1, 10 mg mL−1), and analyte solutions were prepared by mixing 10 μL of matrix and 1 μL of the polymer sample. Samples were applied using the dried droplet method. To determine the dispersity of the polymers (Đ), size exclusion chromatography (SEC) was performed on an automated Shimadzu HPLC system as described previously,24 with a PLgel 5 μm MIXED-D column at 50 °C, using N,N-dimethyl acetamide (DMA) containing 50 mM LiCl as the eluent at a flow rate of 0.6 mL min−1. Poly(methyl methacrylate) (PMMA) was used as standards.
The POx polymers were analyzed with regard to the amount of alendronate (Ale), hydroxyl (OH), and carboxyl (COOH) functional groups present in the polymers. Table 1 summarizes the analytical data of the POx polymers.
| Polymer | 1H NMR (mol%) | MALDI-TOF MS | Đ | ||||
|---|---|---|---|---|---|---|---|
| EtOx | OH | COOH | Ale | M n (kDa) | |||
| P(EtOx) | 100 | 50 | |||||
| P1e | P(EtOx70-Ale30) | 70 | 3 | 27 | 19.6 | 1.11 | |
| P2e | P(EtOx70-OH10-Ale20) | 70 | 11 | 1 | 18 | 14.9 | 1.11 |
The phase transition temperature of both POx-Ale P1e and P2e polymers as well as P(EtOx) was determined using differential scanning calorimetry (DSC, Mettler Toledo). The measurements were carried out at a heating rate of 10 °C min−1 under nitrogen atmosphere (sample masses ≈ 2 mg). The glass transition temperatures (Tg) were obtained as the midpoint of the intersection of the tangent before and after baseline shifting. All temperatures were determined from the second heating scan.
2.3 Preparation and characterization of adhesive membranes
Membranes were prepared by dry deposition of the POx polymers into commercially available cohesive layered gelatin fibrous carriers (GELITA TUFT-IT®) of 5 × 7.5 cm2 using an electrostatic impregnation system. First, the gelatin carrier, which consisted of approximately 8 cohesive layers, were delaminated to obtain carriers of 3 layers of approximately 300 mg and 0.3 mm thick. Then, POx polymers were processed into fine powders by ball milling at 30 Hz for 10 min, followed by sieving below 63 μm. Afterwards, polymers were dried using a rotary evaporator at 40 °C, 20 mbar for 16 h, while carriers were dried using a vacuum oven at 40 °C, 5 mbar for 16 h. To impregnate the carriers, a grid array of 5 × 7 × 0.4 cm2 consisting of 726 square wells, was filled with the polymers at a fixed polymer/carrier weight ratio of 65:35. Then, carriers were mounted on the arrays with a spacing frame of 1.5 mm thickness to allow for particle mobility. Subsequently, the construct was positioned between two dielectric disks and polymers were loaded into carriers using a high voltage electrostatic impregnation system (Fibroline SL-Preg) at 40 kV, 100 Hz for 20 seconds, obtaining a homogenous distribution of the polymers throughout the gelatin carriers with a loading efficacy of approximately 80%, which corresponded to a polymer/carrier weight ratio of 60:40. Afterwards, a polyester backing layer (P(LA-CL) 67 wt%, PLGA 7.8 wt%, and P(PropOx-NH2) 25.2%) was adhered to the carriers by two cycles of heating while compressing at 150 °C, 30 N for 3 seconds. This procedure resulted in a final weight ratio of polymer/carrier/backing of 42:28:30, as calculated by weighing the membranes before and after POx impregnation as well as after addition of the polyester backing layer. Membranes were cut using a scalpel (carbon steel, blade 20) into the required dimensions for each experiment. Further, they were placed individually in aluminum bags and dried at 40 °C, 5 mbar, with nitrogen cycles of 600 mbar for 16 h for storage. Finally, they were sealed off at 2 mbar for 4 seconds with a nitrogen flush of 600 mbar. A schematic representation of the loading method is depicted in Fig. 1. Fig. S2 (ESI†) shows representative images of a blank membrane (carrier with backing layer) and backing layer.
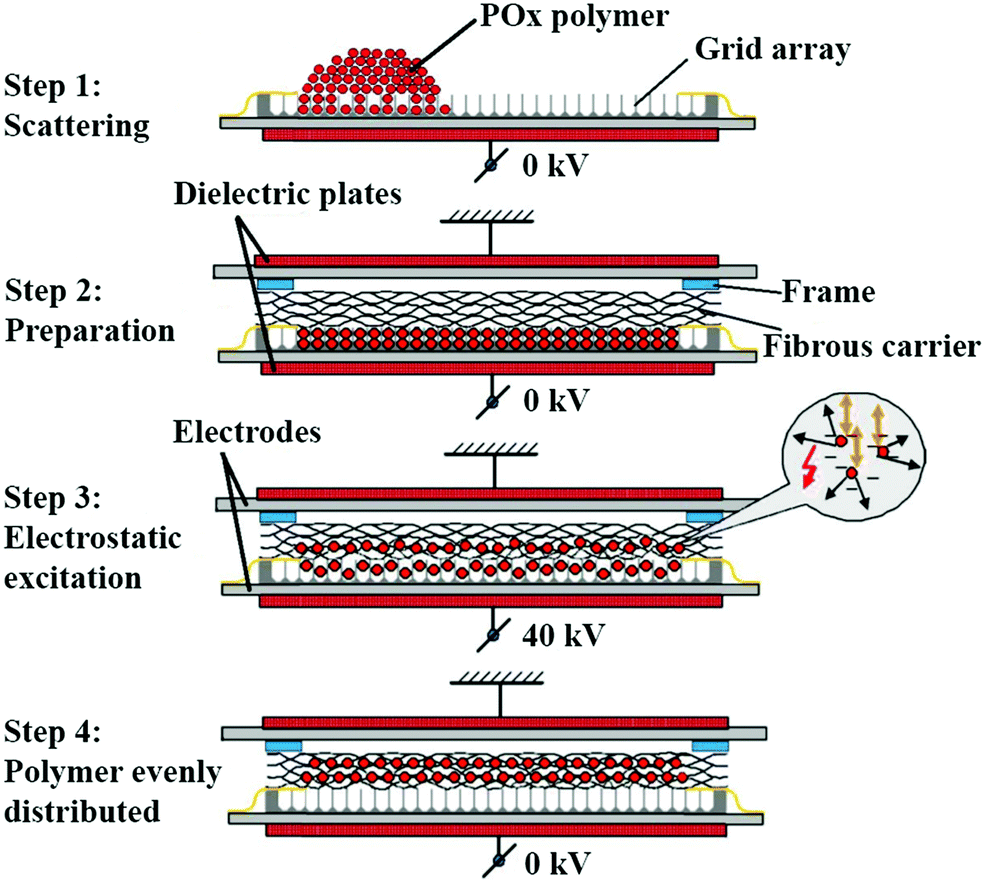 | ||
| Fig. 1 Schematic representation of electrostatic impregnation of polymers into fibrous carriers. | ||
A field emission scanning electron microscope (SEM, Zeiss Sigma 300) was used to evaluate the morphology of the membranes at an accelerating voltage of 3 kV. To this end, the membranes were placed on carbon tape and sputter-coated with a 10 nm layer of gold (Edwards Pirani 501) at 1 kV of accelerate voltage and a current of 20 mA for 120 s. The infrared spectra of the membranes were obtained by Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FTIR) spectroscopy (PerkinElmer, Spectrum Two). The density of the membranes was calculated as the average of the ratio between sample weight and volume in sextuplicate (n = 6).
The wettability of the membranes was quantified by measuring the water contact angle upon initial contact using a contact angle goniometer (Theta Lite) employing the sessile drop method. For each measurement, 2 μL of Milli-Q water was placed onto the membranes at room temperature. The spreading of the droplet was recorded using a high speed video camera and the angle was determined using Laplace Young fitting. The angle was measured after 1 second of contact as the average between the left and right angles in triplicate (n = 3).
2.4 Preparation and characterization of apatite-coated titanium disks
Commercially pure titanium disks (2.5 × 2.5 × 0.1 cm3, grade 2) were first grit-blasted to obtain an average roughness Ra of 1.5 μm to ensure sufficient adhesion of the apatite coating onto the disk. Subsequently, these substrates were ultrasonically cleaned with Milli-Q water, followed by isopropanol during 20 min each. Afterwards, the substrates were placed on a rotating holder and etched with argon plasma for 10 min prior to physical vapor deposition of a thin adherent coating of hydroxyapatite using a radiofrequent magnetron sputtering system (Edwards ESM 100), at a power of 400 W and an argon pressure of 5 × 10−3 mbar for 12 h. After deposition, hydroxyapatite-coated disks were crystallized by a heating treatment in a furnace at 650 °C for 2 h, with a heating and cooling rate of 1.5 °C min−1, yielding an apatitic calcium phosphate coating of 1 μm, as described and characterized previously.31,32The crystallographic structure of apatite-coated disks was determined by means of thin-film X-ray diffraction (thin-film XRD, X’Pert3 PANalytical, Philips). The measurements were performed at 45 kV acceleration and 30 mA current, scanning from 25 to 35° 2θ at a rate of 0.02° s−1. Reflection peaks were characteristic of crystalline apatite (Fig. S3A, ESI†).33,34 The infrared spectrum of the coating was obtained by ATR-FTIR spectroscopy, which also confirmed the apatitic nature of the coatings (Fig. S3B, ESI†).35 The surface morphology of the disks was examined using scanning electron microscopy, which confirmed the homogeneous coverage of the substrate (Fig. S4A, ESI†). The thickness of the coating was determined using a profilometer (Innowep, universal-surface tester).
2.5 Preparation and characterization of bone samples
Porcine ribs were obtained from a local butcher and sectioned using a circular saw machine into pieces of approximately 2.5 × 1.2 × 0.7 cm3. Each bone specimen was polished (Struers TegraPol 35) using carbon paper (P500) at 150 rpm until a flat surface of cortical bone was exposed with an average roughness Ra of 0.8 μm measured using a profilometer. Thereafter, the samples were kept frozen at −20 °C until further use.Flat bone specimens were immersed in 500 mL of Sakura reagent TDE™ 30 and decalcified using the Sakura TDETM 30 electrode system for 24 h. Thereafter, they were kept frozen at −20 °C until further use. The demineralization process was confirmed using XRD (Fig. S3A, ESI†) and ATR-FTIR spectroscopy (Fig. S3B, ESI†) and the samples were inspected visually (Fig. S4BC, ESI†).
2.6 Mechanical tests
Tensile and lap-shear tests were performed using a tensile bench (LS5, Lloyd Instruments) equipped with a 100 N load cell to determine the cohesive and substrate-adhesive strength, respectively. Experimental setups for both methods are depicted in Fig. S5 (ESI†).2.7 Bone adhesion under wet conditions
Bone substrates were moistened with a PBS soaked gauze followed by controlled attachment of membranes of 1 × 1 cm2 onto the substrates at a force of 10 N for 5 min. Then, bone specimens were immersed in 200 mL of PBS, with or without collagenase (20 μg mL−1), shaken at 150 rpm for 24 h and 72 h, after which their adhesion to bone and swelling were semi-quantitatively scored (n = 3). The degree of adhesion was graded as weak adhesion (1, corresponding to the detachment of the membranes), moderate adhesion (2, partial detachment), and strong adhesion (3, retained adhesion), whereas the swelling degree was scored as low swelling (1), medium swelling (2), and high swelling (3).2.8 In vitro degradation
Membranes were cut into 0.5 × 0.5 cm2 pieces of approximately 5 mg. They were accurately weighed and subsequently immersed in 1 mL of PBS, with or without collagenase (20 μg mL−1), in 2 mL Eppendorf tubes. Afterwards, the tubes were incubated at 37 °C at a rotation rate of 90 rpm, refreshing the medium every day. At each time point, supernatants were removed and samples were freeze dried. The degradation was calculated using the following equation:where Wi is the initial weight of the membrane and Wf is the weight after each immersion time point. All measurements were performed in triplicate (n = 3).
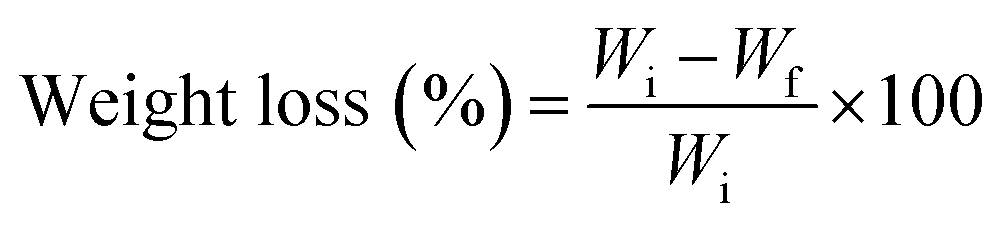
A field emission scanning electron microscope was used to evaluate the morphology of the membranes after 14 days of immersion in PBS with or without collagenase.
2.9 Statistics
The statistical analyses were performed using GraphPad InStat software. All values were presented as average ± standard deviation. Results were analyzed statistically using a one-way analysis of variance test, followed by Tukey's multiple comparison test. The significance threshold was set at P < 0.05.3. Results
3.1 Characterization of bone-adhesive membranes
The thickness of the impregnated membranes ranged from 0.25–0.31 mm and their density from 0.50–0.58 g cm−3 (Table S1, ESI†). The variability in membrane thickness (9–12%) was comparable to values reported for commercially available membranes such as Bio-Gide® (porcine dermis-derived collagen membrane; thickness variability 27%),36 Collprotect® (porcine dermis-derived collagen membrane; thickness variability 12%) and Jason® (porcine pericardium-derived collagen membrane; thickness variability 39%).37Fig. 2A–F show the surface morphology of the three-component membranes. The fibrillar architecture of the pure gelatin carriers was clearly visible in Fig. 2A, whereas the smooth appearance of the polyester backing layer was recognizable in Fig. 2B. The blank membrane (Fig. 2C), consisting of a gelatin carrier on top of a backing layer, was covered by a fibrillar gelatin layer, although the membrane structure was compacted upon adhering the backing layer to the carrier. All POx-covered membranes (Fig. 2D–F) revealed a fibrillar surface architecture. Successful impregnation of POx polymers into the fibrous gelatin carrier was evidenced by abundant coverage of gelatin fibers with POx polymers, forming a highly intertwined POx-gelatin structure. In addition, irregularly shaped POx deposits were heterogeneously dispersed throughout the superficial layer of the POx-coated membranes.
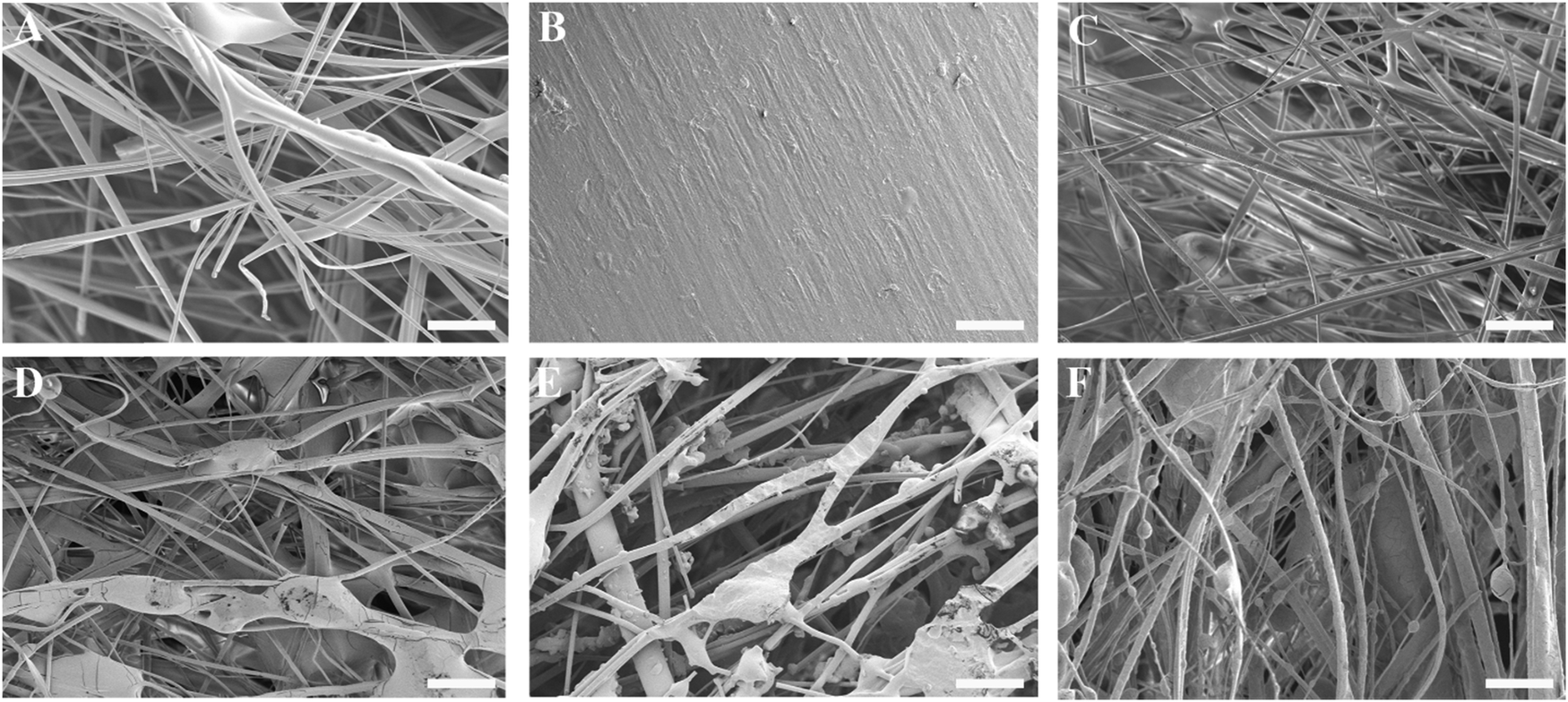 | ||
| Fig. 2 Representative scanning electron micrographs of different membranes: (A) fibrous gelatin carrier, (B) backing layer, (C) blank (carrier with backing), (D) blank impregnated with P(EtOx), (E) blank impregnated with P(EtOx-Ale), and (F) blank impregnated with P(EtOx-OH-Ale). Scale bars correspond to 50 μm. | ||
The backing layer was attached by heating at 150 °C, which was above both the glass transition temperature (60 °C) and melting point (104–114 °C) of P(LA-CL), the main component of the backing layer,38,39 and above the glass transition temperature of the POx polymers, as depicted in Table 2 and Fig. S6A (ESI†). Generally, POx polymers did not melt at temperatures below 200 °C.
| Polymer | T g (°C) | |
|---|---|---|
| Onset | Midpoint | |
| P(EtOx) | 52.19 | 54.45 |
| P(EtOx-Ale) | 54.54 | 71.15 |
| P(EtOx-OH-Ale) | 54.53 | 62.94 |
In Fig. S6B (ESI†) the infrared spectra of the different membranes are combined. The small peak at 1728 cm−1 corresponds to carbonyl stretching from esters in P(EtOx-Ale) and P(EtOx-OH-Ale). The peaks at 1625, 1541, and 1238 cm−1, on the other hand, are attributed to the amides I, II, and III as present in both the gelatin carrier and the three types of POx polymers. The peaks at 1426 cm−1 and 1164 cm−1 present in all membranes correspond to bending of C–H bonds and stretching of C–N bonds, respectively. The peak at 915 cm−1 accounts for stretching of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and P–OH bonds in P(EtOx-Ale) and P(EtOx-OH-Ale).40
O and P–OH bonds in P(EtOx-Ale) and P(EtOx-OH-Ale).40
Fig. S7 (ESI†) shows the water contact angle of the different membranes after 1 s of contact. Membranes containing P(EtOx-Ale) and P(EtOx) gave a contact angle of 0 °C, absorbing the water drop within 1 s. These membranes were significantly more hydrophilic than the blank and membranes impregnated with P(EtOx-OH-Ale), which revealed contact angles of 96.3 ± 0.3 and 74.2 ± 8.2, respectively. After 6 s of contact, the water drop was absorbed by all types of membranes.
3.2 Tensile tests
Fig. 3A displays the stress–strain curves of the different membranes upon tensile testing. The initial linear-elastic region of the curves was followed by plastic deformation until the maximum tensile stress (ultimate tensile strength) was reached, which ranged from 0.5 MPa for fibrous gelatin carriers to 1–4 MPa for membranes containing a polyester backing. After reaching the maximum tensile stress, the stresses dropped gradually for all membranes, which corresponds to a ductile behavior characterized by extensive membrane stretching. This ductile behavior was more prominent in POx-free membranes.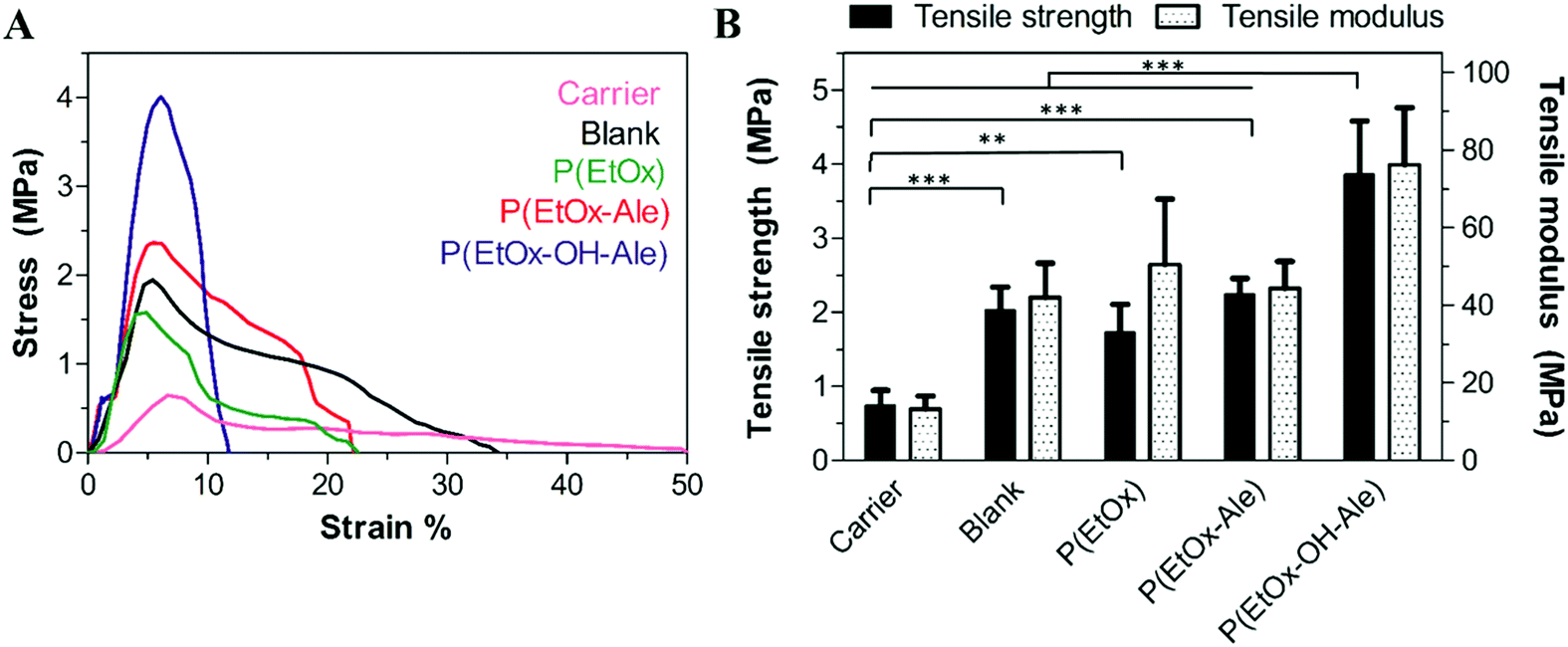 | ||
| Fig. 3 (A) Representative stress–strain curves of the different membranes. (B) Tensile strength and tensile modulus of the membranes. Values represent the mean ± standard deviation (n = 6). | ||
The tensile strength and modulus of the different membranes are depicted in Fig. 3B. Both the maximum strength and tensile modulus were 3-fold higher for membranes comprising a backing layer (strength: 2.0 ± 0.3 MPa, modulus: 42.0 ± 8.9 MPa for blank) compared to pure fibrous gelatin carriers (strength: 0.7 ± 0.2 MPa, modulus: 13.3 ± 3.4). In general, the impregnation of POx polymers into membranes comprising a polyester backing did not further reinforce these membranes. However, membranes impregnated with hydroxyl-functionalized P(EtOx-OH-Ale) polymers showed 2-fold higher tensile strength and tensile modulus (strength: 3.9 ± 0.7 MPa, modulus: 76.2 ± 14.7 MPa) compared to hydroxyl-free P(EtOx-Ale) (strength: 2.3 ± 0.2 MPa, modulus: 44.3 ± 6.9 MPa) and P(EtOx) polymers (strength: 1.7 ± 0.4 MPa and modulus: 50.5 ± 16.9 MPa). Overall, the tensile strength of hydroxyl-functionalized P(EtOx-OH-Ale) polymers was comparable to values reported for commercially available membranes such as Bio-Gide® (4.6 ± 0.9 MPa) and Ossix Plus® (5.1 ± 2.5 MPa).36
3.3 Lap-shear adhesion tests
The lap-shear adhesion strength of the different membranes to various mineralized and mineral-free substrates is depicted in Fig. 4. Membranes comprising POx polymers showed superior adhesive properties to both mineralized substrates, i.e. apatite-coated titanium substrates (Fig. 4A) and bone (Fig. 4B) (up to 64 kPa and 26 kPa, respectively). In contrast, blank membranes and commercially available Bio-Gide® membranes (which are widely used for GBR procedures) showed very poor adhesion to both mineralized and mineral-free substrates. The adhesion of POx-impregnated membranes to apatite-coated substrates was 4–8-fold higher compared to blank membranes and 10- to 18-fold higher to Bio-Gide®. Similarly, their adhesion to bone was 8- to 15-fold higher compared to blank membranes and 35- to 62-fold higher than Bio-Gide® membranes. The adhesion strength of POx-impregnated membranes to both mineral-free control substrates (i.e. Ti disks and demineralized bone specimens) was negligible relative to their corresponding mineralized control substrates. The adhesion of membranes impregnated with P(EtOx) to CaP-coated disks was similar to P(EtOx-Ale)-impregnated membranes and higher than membranes impregnated with P(EtOx-OH-Ale) polymers. All POx-impregnated membranes adhered equally strong to bone.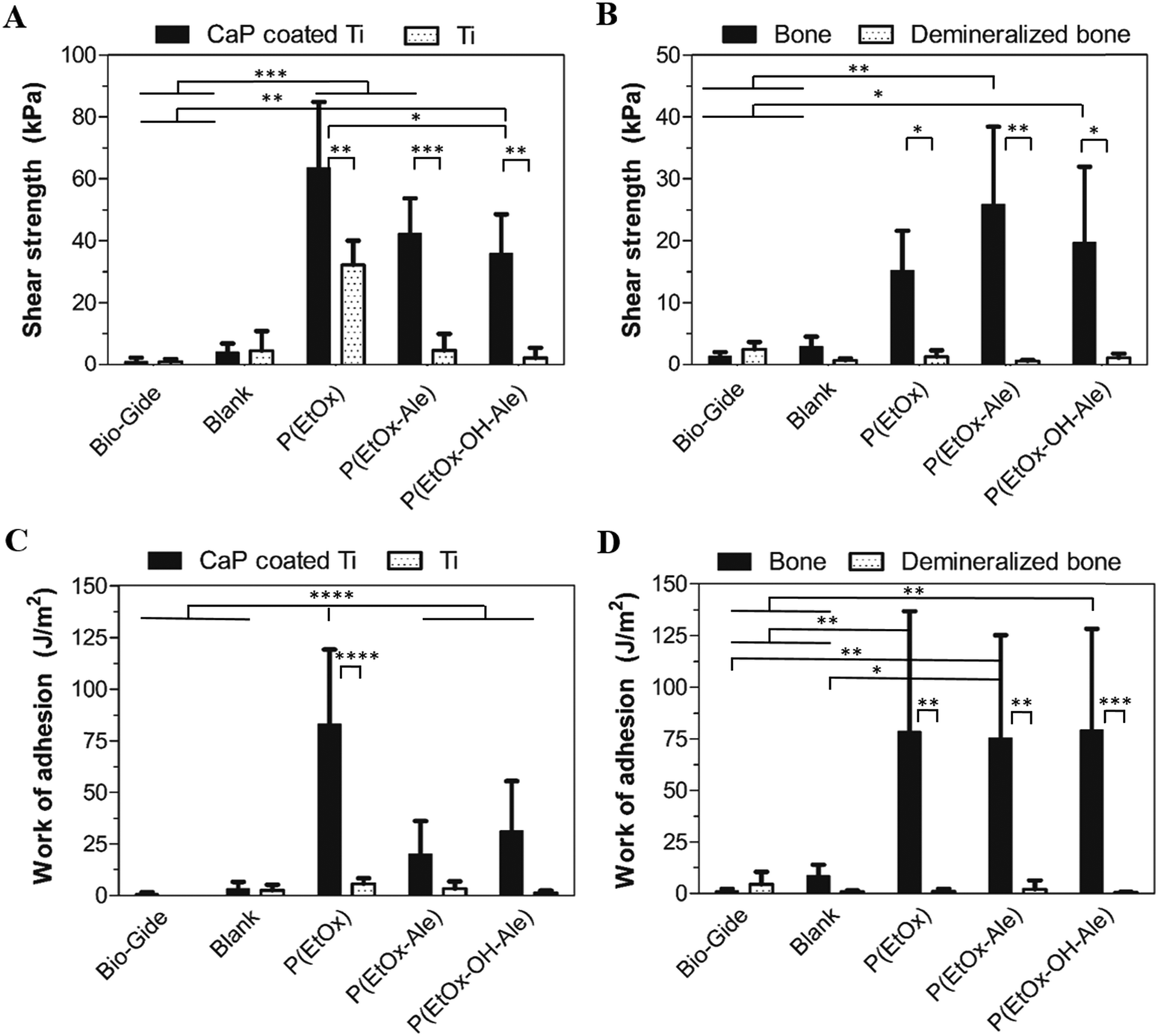 | ||
| Fig. 4 Maximum lap-shear adhesion strength of the different membranes to (A) apatite-coated and uncoated Ti disks, and (B) bone and demineralized bone specimens. Work of adhesion during lap-shear testing of the different membranes to (C) apatite-coated and uncoated Ti disks, and (D) bone and demineralized bone specimens. Values represent the mean ± standard deviation (n = 4–6). | ||
The work of adhesion, as a measure for energy dissipation during lap shearing, is shown for the various types of membranes in Fig. 4C and D. Membranes impregnated with P(EtOx) showed a significantly higher work of adhesion (up to 83 J m−2) to CaP-coated Ti disks (Fig. 4C) compared to all other experimental groups. In contrast, the work of adhesion of all POx-impregnated membranes to bone (Fig. 4D) was comparable (up to 75–79 J m−2). Generally, the work of adhesion of impregnated membranes to both types of mineralized substrates was much higher compared to mineral-free control substrates. The POx-free blank membranes and Bio-Gide® revealed extremely low amounts of energy dissipation during lap-shearing to both mineralized and mineral-free control substrates.
All POx-impregnated membranes failed due to cohesive failure upon lap-shear testing. Fig. S8 (ESI†) shows FTIR spectra of the surface of bone specimens after lap-shear testing of all experimental groups. These spectra revealed that bone samples were covered with membrane remnants for POx-impregnated membranes, which were absent on bone samples after detachment of POx-free gelatin carriers, corresponding to adhesive failure for blank controls.
The mechanical parameters of the tested membranes are summarized in Table 3.
| Prototype | Tensile strength (MPa) | Tensile modulus (MPa) | Shear strength (kPa) | Work of adhesion (J m−2) | ||
|---|---|---|---|---|---|---|
| CaP coated Ti | Bone | CaP coated Ti | Bone | |||
| Blank | 2.03 ± 0.31 | 41.95 ± 8.89 | 3.99 ± 2.87 | 2.92 ± 1.61 | 3.10 ± 3.42 | 8.62 ± 5.24 |
| P(EtOx) | 1.73 ± 0.37 | 50.50 ± 16.88 | 63.51 ± 21.36 | 15.18 ± 6.40 | 83.12 ± 36.01 | 78.44 ± 58.22 |
| P(EtOx-Ale) | 2.25 ± 0.21 | 44.29 ± 6.94 | 42.24 ± 11.36 | 25.85 ± 12.56 | 20.02 ± 16.09 | 75.44 ± 49.80 |
| P(EtOx-OH-Ale) | 3.86 ± 0.72 | 76.22 ± 14.65 | 35.88 ± 12.60 | 19.66 ± 12.27 | 31.27 ± 24.20 | 79.23 ± 49.07 |
Finally, to mimic the clinical application scenario as closely as possible, the adhesion of the various membranes was also evaluated under wet conditions in PBS for 24 and 72 h (Fig. 5A and B). Evidently, only membranes comprising alendronate groups remained adhesive to bone after 24 h immersion in PBS, which weakened with increasing immersion time. Moreover, these membranes revealed much lower degrees of swelling compared to alendronate-free groups (Fig. S9, ESI†). To investigate the biodegradation effect on the adhesion under et conditions, the adhesion of the membranes to bone was also evaluated in PBS containing collagenase for 24 and 72 h. As expected, the addition of collagenase to the immersion medium reduced the adhesion of membranes comprising both POx-Ale polymers, indicating that degradation of the fibrous gelatin layer of the membranes was accelerated by the presence of collagenase.
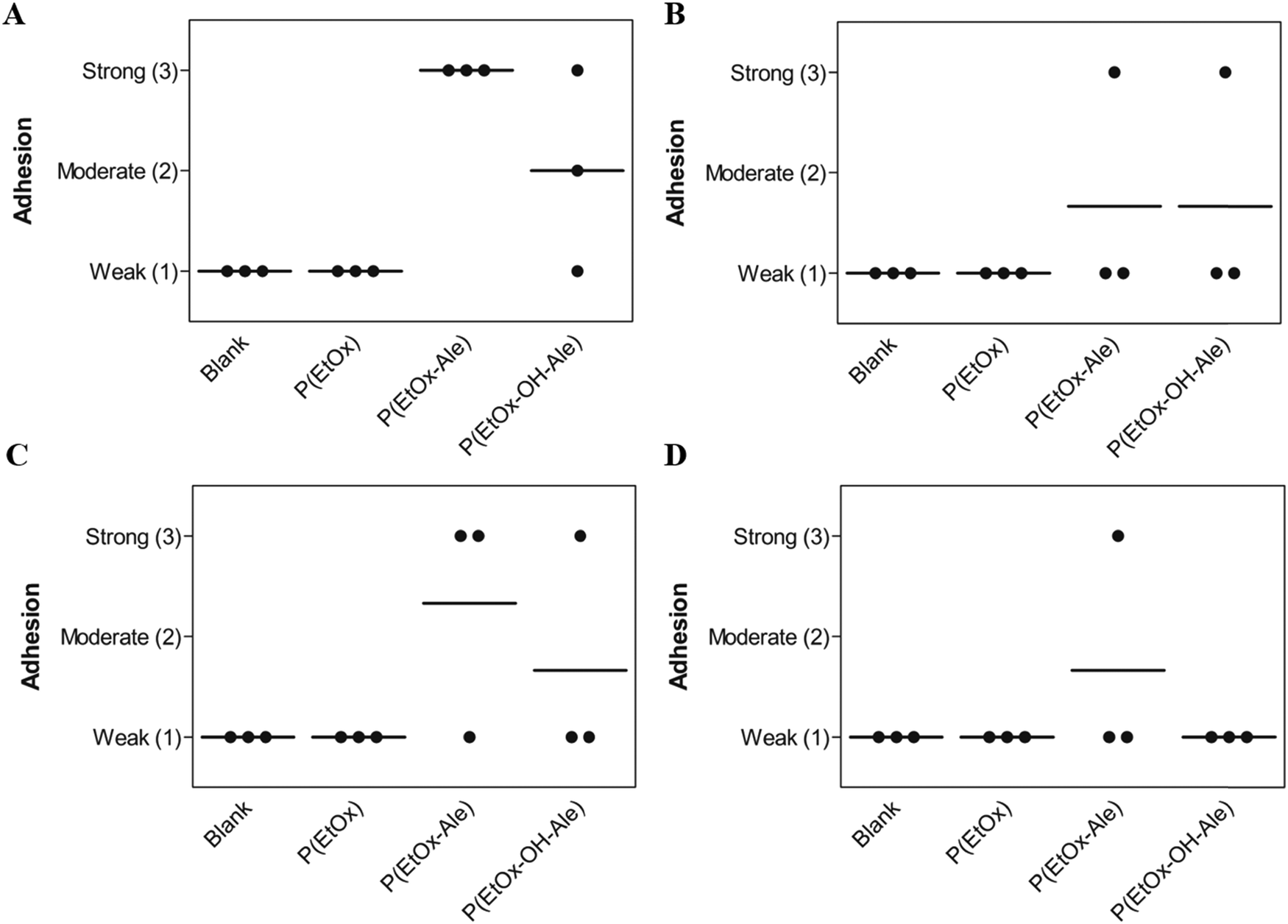 | ||
| Fig. 5 Membrane adhesion to bone under wet conditions in (A) PBS for 24 h, (B) PBS for 72 h, (C) PBS with collagenase for 24 h, and (D) PBS with collagenase for 72 h. | ||
3.4 In vitro degradation
The in vitro degradation of the various membranes is shown in Fig. 6A and B. Pure gelatin carriers completely lost their integrity by delamination upon immersion in PBS due to their high swelling ability, thereby impeding reliable quantification of their weight loss. These gelatin carriers dissolved completely after 6 h of immersion in PBS containing collagenase (weight loss of 100%), which confirmed that gelatin was degraded enzymatically. However, blank membranes consisting of a gelatin carrier and a polyester backing retained their physical integrity even after immersion in PBS containing collagenase for 14 days, losing 49% of their weight in PBS and 69% in PBS with collagenase. After a rapid initial weight loss, the polyester backings hardly degraded between day 1 and 14, and reached similar weight loss values of about 21–23% after 14 days. The POx-coated membranes lost around 35% of their weight after 6 h of immersion in PBS, which increased to around 50% when collagenase was present in the PBS. After 14 days of immersion, the weight loss of these membranes increased to considerably higher values of 58–72% in PBS and 81–88% in PBS with collagenase.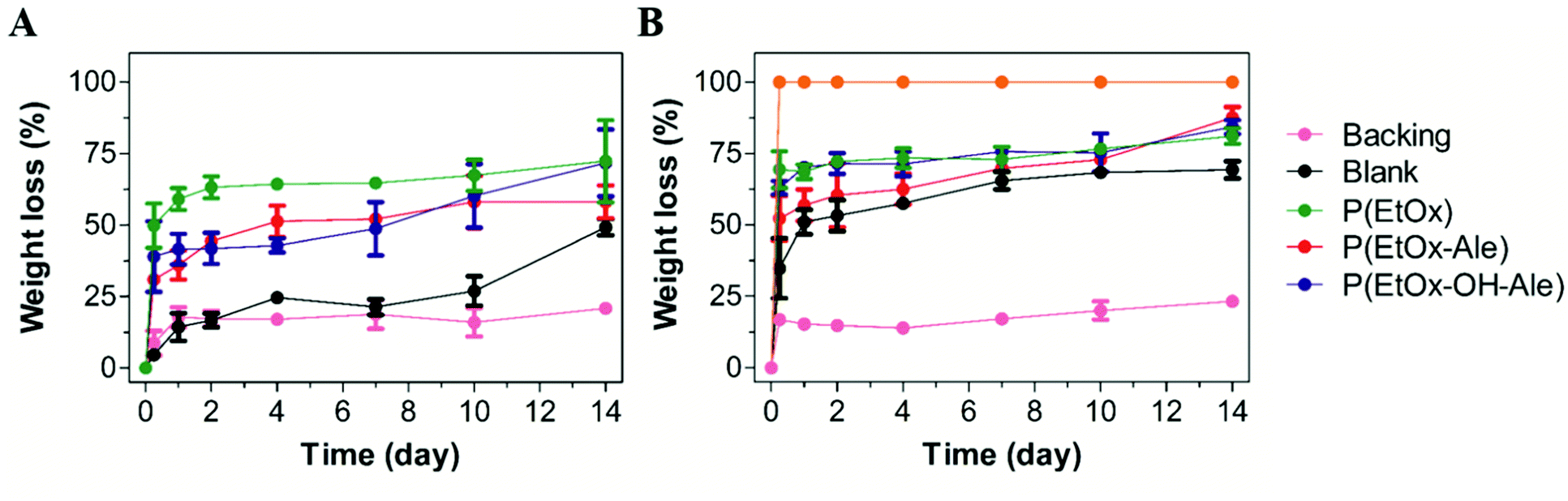 | ||
| Fig. 6 Weight loss (%) of membranes after immersion in (A) PBS and (B) PBS with collagenase. Values represent the mean ± standard deviation (n = 3). | ||
To obtain more insight into the biodegradation profile of the membranes, scanning electron microscopy was used to study the surface morphology of the membranes after 14 days of immersion in PBS with and without collagenase. As shown in Fig. 7, this SEM evaluation confirmed that blank membranes consisting of a gelatin carrier and polyester backing retained their fibrous layered structure after 14 days of soaking in PBS buffer. In PBS with collagenase, these blanks were severely degraded, as indicated by abundant formation of pores and disappearance of the fibrous surface morphology. On the contrary, the surface morphology of the polyester backing was not affected upon immersion in PBS. POx-coated membranes lost their fibrous surface morphology, revealing a smoother surface with the presence of pores after immersion in PBS with or without collagenase. With collagenase present in the solution, the pores were much larger (50–67 μm), confirming that the degradation rate of the membranes was accelerated due to enzymatic degradation of the carrier.
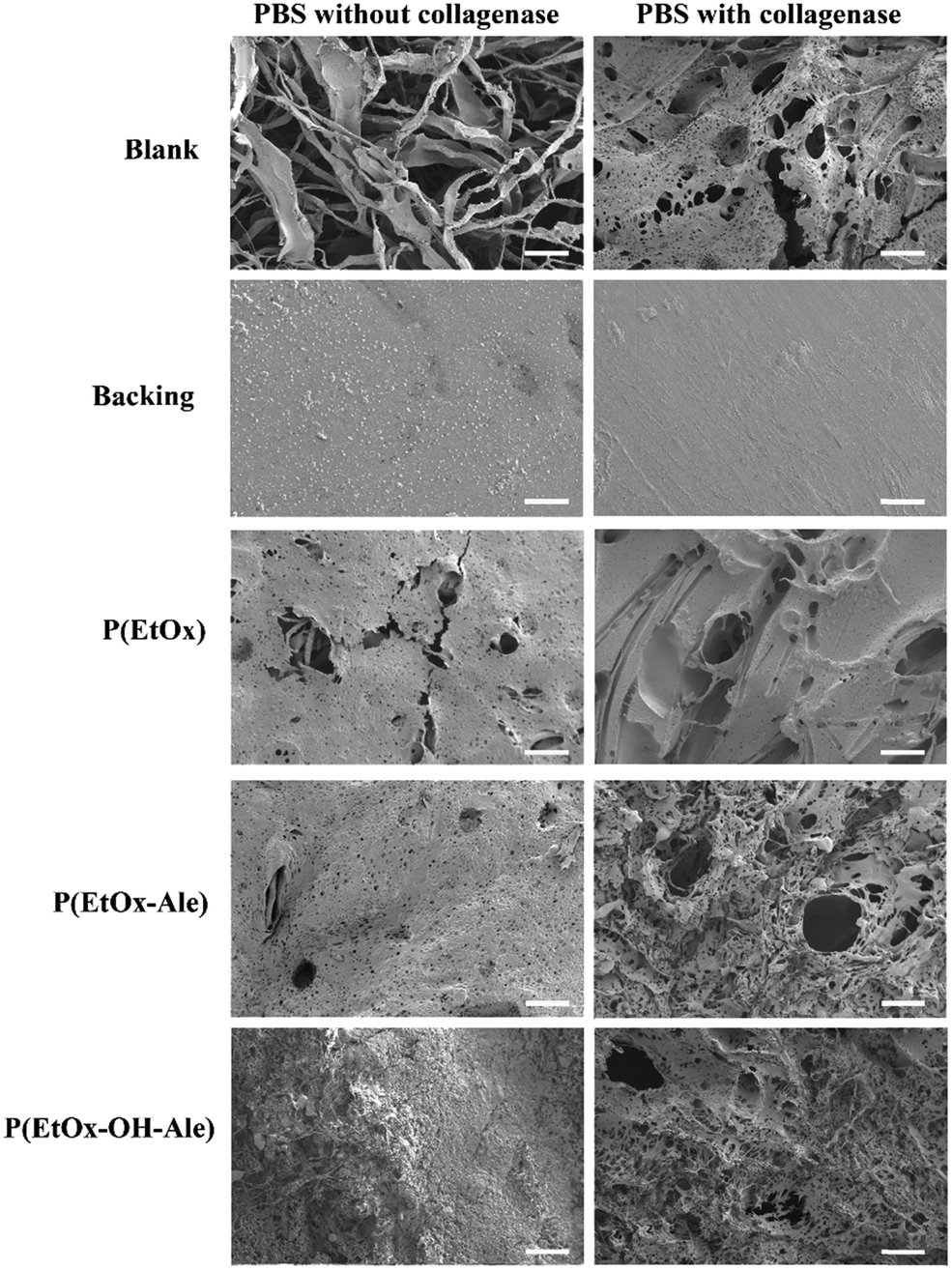 | ||
| Fig. 7 Scanning electron micrographs of the membranes after 14 days of immersion in PBS solutions with and without collagenase. Scale bars correspond to 50 μm. | ||
4. Discussion
Barrier membranes are used extensively worldwide in oral and maxillofacial surgery. However, currently available membranes are still associated with several drawbacks, including a general lack of bone adhesiveness. Although metallic pins are used to fixate barrier membranes to bone, this surgical approach comes along with disadvantages related to the non-degradable nature of these fixation devices which necessitates a second removal surgery.14–16 Hence, the goal of this study was to develop biodegradable barrier membranes with bone-adhesive properties to avoid additional surgery to remove non-degradable fixation devices and/or membranes. Recently, we found that poly(2-oxazoline)s functionalized with alendronate side groups display a strong binding affinity to calcium cations.26,27 In addition, we observed that hydroxyl side groups also contributed to the formation of robust cross-linked networks with calcium cations. Therefore, we designed bone-adhesive barrier membranes by impregnation of gelatin carriers with bone-adhesive poly(2-oxazoline)s functionalized with alendronate and hydroxyl side groups. We hypothesized that these gelatin carriers could be rendered adhesive to calcium-containing substrates such as bone by impregnation of their surface with these calcium-binding polymers.POx-polymers were homogenously dispersed throughout the fibrous gelatin carrier using dry powder deposition, yielding a loading efficacy of approximately 80%. Although polymers were homogeneously distributed throughout the carriers, the membranes showed a variability in thickness and density. However, this variability was comparable to values reported for other commercially available membranes and can be attributed to their animal-derived origin, which typically comes along with batch-to-batch variability. The polyester backing layer was attached at a temperature well above the glass transition of all polymers used, resulting in increased mobility of the polymer chains and decreased viscosity of the backing layer,41,42 thereby allowing for diffusion of P(LA-CL) and PLGA into the fibrous architecture. Consequently, abundant interconnections were formed between POx polymers, gelatin fibers and the polyester backing. Moreover, the membranes became thinner upon attachment of the polyester backing bottom layer by heating while compressing. The non-porous morphology of the backing is regarded as beneficial to minimize premature cell and tissue ingrowth into bone defects covered by these membranes, thereby providing space for osseous regeneration.
The wettability of the membranes was determined by water contact angle measurements, since we assumed that hydrophilic membranes would promote the formation of molecular interactions between membranes and substrates.43 Upon initial contact, water was completely absorbed by the most hydrophilic membranes comprising P(EtOx) and P(EtOx-Ale) polymers, since these polymers are rich in polar side groups. However, after 6 seconds of contact, all membranes absorbed the droplet due to the porous gelatin membrane.
Subsequently, we performed tensile tests in order to study the mechanical properties of the membranes. From the stress–strain curve, we observed that the carrier showed a higher strain %, which corresponded to ductile behavior. During testing, fibers were continuously pulled from the fibrous layers without any detectable failure event. Both the tensile strength and tensile modulus increased significantly when a polyester backing layer was attached to the fibrous gelatin carrier. As such, the polyester backing may act as both cell-occlusive and reinforcing layer. The incorporation of moderate amounts of LA into P(LA-CL) blends significantly increased their tensile strength and rigidity, which improved their overall mechanical performance.44,45 The impregnation of POx polymers into membranes comprising a backing layer did not lead to further reinforcement. However, membranes impregnated with hydroxyl-functionalized P(EtOx-OH-Ale) polymers showed superior tensile strength and modulus values compared to other experimental groups. This could be attributed to the interaction between the hydroxyl side groups with amines and carboxylic acid groups present in the carrier as well as amines derived from P(PropOx-NH2) present in the polyester backing layer.
The adhesion of the membranes onto mineralized substrates (i.e. CaP coated Ti disks and bone specimens) was evaluated by in vitro lap-shear tests, which are generally accepted as test methods to measure adhesion to bone. Generally, all POx-impregnated membranes showed excellent adhesion to both apatite-coated Ti substrates and bone, as evidenced by high shear strength and work of adhesion values. As expected, cohesive failure was confirmed for all POx-impregnated membranes by the presence of membrane remnants on the substrates after lap-shear testing due to the layered architecture of the membranes. The cohesion of the layers might be improved by tuning the hot melt extrusion formulation of the backing. All POx-impregnated membranes adhered more tightly to CaP-coated disks compared to bone substrates, which was attributed to the surface roughness of CaP-coated disks (Ra ≈ 1.5 μm), which was higher than the smooth bone samples (Ra ≈ 0.8 μm). The adhesion strength and work of adhesion of membranes comprising POx polymers to mineral-free control substrates (i.e. Ti and demineralized bone specimens) were significantly lower, which confirms that alendronate, hydroxyl, and amides groups present in POx polymers formed strong and specific bonds with calcium-containing substrates. In contrast, POx-free membranes as well as Bio-Gide® controls did neither adhere to mineralized (CaP-coated Ti and bone) nor mineral-free control groups (Ti and demineralized bone), since these membranes lack functional groups with bone-bonding ability. Membranes impregnated with alendronate-free P(EtOx) and P(EtOx-Ale) showed higher shear strength to CaP-coated Ti than membranes impregnated with P(EtOx-OH-Ale) polymers due to their higher hydrophilicity (lower water contact angles) causing better contact and more molecular interactions between membranes and substrate. We attributed the higher work of adhesion values of P(EtOx)-impregnated membranes than with other POx groups to the hydrophilic nature of P(EtOx).46 Importantly, all POx-impregnated membranes showed similar shear strength and work of adhesion values. Nevertheless, the standard deviations are large which compromise the statistical analysis. To test the adhesion of the membranes under clinically relevant wet conditions, we submerged the membranes adhered to bone substrate into a PBS bath with and without collagenase for 24 and 72 h. Strikingly, we observed that all P(EtOx-Ale)-impregnated membranes remained strongly adhesive to bone after 24 h, whereas two out of three P(EtOx-OH-Ale)-impregnated membranes remained adhesive to bone. We attributed this difference under wet conditions to the higher alendronate content and reduced swelling of membranes impregnated with P(EtOx-Ale). However, after 72 h, only one out of three P(EtOx-Ale) and P(EtOx-OH-Ale)-impregnated membranes remained adhesive to bone. We attribute the reduced bone adhesion after 72 h to dissolution of POx-Ale polymers caused by their high hydrophilicity. As expected, in the presence of collagenase, the adhesion of both hydroxyl and hydroxyl-free POx-Ale membranes weakened, indicating that the degradation of the membranes was accelerated in the presence of collagenase. Although it should be realized that the selected collagenase concentrations 20 μg mL−1 were relatively high, these results confirm that bone adhesiveness of our membranes decreases with enhanced biodegradation. In contrast, all blank membranes and membranes impregnated with P(EtOx) detached from bone in PBS solutions with and without collagenase, proving that long-term adhesion under wet conditions could only be achieved by formation of coordination bonds between alendronate-functionalized POx polymers and calcium in bone. In addition, the high swelling degree of P(EtOx) also weakened the interfacial adhesion, rendering this polymer unsuitable for applications requiring long-term adhesion to bone under wet conditions.
Finally, the in vitro degradation of the membranes was studied by monitoring their weight loss upon immersion in phosphate-buffered saline solutions with and without collagenase. Ideally, to ensure mechanical stability, the degradation rate of the barrier membranes should match with the rate of bone healing. Hence, the integrity and stability of membranes should be guaranteed in the range from four weeks, when bone remodeling phase starts, up to several months.17 In our study, carriers gradually disintegrated in PBS due to their high swelling ability, while they partially dissolved in PBS solutions containing collagenase, suggesting that the degradation mechanism of the membranes was mainly dependent on enzymatic degradation of the fibrous gelatin carrier. The backing layer revealed an initial weight loss of about 15%, followed by a stable phase without any substantial degradation. After 14 days, 21 and 23 wt% of the original backing layers were degraded in PBS solutions with and without collagenase, respectively, which was caused by hydrolytic cleavage of the esters bonds and dissolution of uncrosslinked P(PropOx-NH2). The backing is mainly composed of P(LA-CL), which is a slow-degrading polymer with typical in vivo degradation times of more than two years.47,48 However, PLGA was added to the backing to accelerate degradation rate of the resulting membranes by increasing the amount of hydrophilic glycolic acid monomers in the copolymer.28 This accelerating effect of glycolic acid on polyester degradation was previously reported for PLGA with different L:G ratios of 50:50 (complete degradation after 2–3 months) vs. 85:15 (6 months).49,50 Consequently, variation of parameters such as copolymer ratio and the polyester percentage in the backing will allow to tune both degradability and mechanical strength of the resulting membranes tailoring to the specific requirements of the application. Blank membranes lost ∼20% more weight after 14 days in PBS with collagenase than in collagenase-free PBS, which confirmed that the gelatin carrier degraded proteolytically. The backing layer did not only reinforce the strength of the membranes in dry state, but also it helped to keep the integrity of the membranes even after 14 days immersed in PBS solution, avoiding the delamination of the carrier. Based on the carrier/backing weight ratio and the total weight loss of the backing, we estimated that 10 wt% of gelatin remained present in the blank membranes upon 14 days of soaking in collagenase-free PBS, whereas in PBS containing collagenase, it degraded almost completely. Strikingly, only blank membranes retained their fibrous morphology after 14 days of soaking in PBS, which corresponded to the lower amounts of weight loss of blank membranes vs. POx-impregnated membranes. We attribute the accelerated degradation of gelatin in POx-impregnated membranes to a faster dissolution of POx-gelatin complexes in PBS due to their higher hydrophilicity. Overall, it can be concluded that sequential degradation of gelatin and POx polymers on short-term followed by long-term degradation of the polyester backing provides the opportunity to independently tune the duration of the initial bone-adhesive phase vs. the long-term occlusive phase where the membrane should act as a barrier against soft tissue infiltration. We speculate that sequential instead of simultaneous degradation of membrane components will allow to reduce toxicity and inflammatory responses due to sudden release of high concentrations of degradation by-products.17
5. Conclusions
A new generation of bone-adhesive barrier membranes comprising a polyester backing, a fibrous gelatin carrier impregnated with functionalized POx polymers was developed using a high voltage electrostatic impregnation system. Membranes comprising alendronate-functionalized POx polymers reacted strongly and specifically with calcium-containing substrates, which gave rise to bone adhesion under wet conditions for up to 72 hours. Moreover, our membranes adhered up to 62-fold more to bone than Bio-Gide®, a conventional membrane that is widely used in dentistry and maxillofacial surgery. After 14 days, the impregnated fibrous gelatin carriers were degraded proteolytically, while the polyester backing layers were degraded hydrolytically. This sequential degradation profile allows to tune the barrier function of the three-component membranes for several months. Overall, this study presents a novel design strategy for multi-component barrier membranes with bone-adhesive properties, thereby providing new alternatives to the use of non-degradable membrane fixation devices in oral and maxillofacial surgery.Disclosure
This work has been performed in collaboration with Johan C. M. E. Bender, CTO GATT Technologies BV. Sander C. G. Leeuwenburgh and Jan C. M. van Hest are scientific advisors for GATT Technologies BV. A patent application has been submitted by GATT Technologies BV and Radboud University Medical Center based on these results.Conflicts of interest
The authors did not receive any financial support from private partners for this research. A patent application has been submitted by GATT Technologies BV and Radboud University Medical Center based on the results of this publication, for which a license agreement has been signed between GATT Technologies BV and Radboud University Medical Center.Acknowledgements
Maarten de Wit and Bram Keereweer are acknowledged for their help on polymer synthesis and use of fibroline, respectively. We acknowledge financial support from the Netherlands Organization for Scientific Research (NWO) and GATT Technologies BV (grant number 14435).References
- V. John, J. A. Weddell, D. E. Shin and J. E. Jones, McDonald and Avery's Dentistry for the Child and Adolescent, ed. J. A. Dean, Mosby/Elsevier, Maryland Heights, MO, 10th edn, 2016, pp. 243–273 Search PubMed.
- M. K. Jeffcoat, Bone Loss in the Oral Cavity, J. Bone Miner. Res., 1993, 8, 467–473 CrossRef PubMed.
- B. L. Pihlstrom, B. S. Michalowicz and N. W. Johnson, Periodontal Diseases, Lancet, 2005, 366, 1809–1820 CrossRef.
- I. Elgali, O. Omar and C. P. Dahlin, Guided Bone Regeneration: Materials and Biological Mechanisms Revisited, Eur. J. Oral Sci., 2017, 125, 315–337 CrossRef PubMed.
- J. Caballé-Serrano, A. Munar-Frau, O. Ortiz-Puigpelat, D. Soto-Penaloza, M. Penarrocha and F. Hernández-Alfaro, On the Search of the Ideal Barrier Membrane for Guided Bone Regeneration, J. Clin. Exp. Dent., 2018, 10, 477–483 Search PubMed.
- J. Higuchi, Recent Advances in GTR/GBR Barrier Membranes Design for Periodontal Regeneration. Biomed, J. Sci. Technol. Res., 2019, 16 Search PubMed.
- Z. Sheikh, J. Qureshi, A. M. Alshahrani, H. Nassar, Y. Ikeda, M. Glogauer and B. Ganss, Collagen Based Barrier Membranes for Periodontal Guided Bone Regeneration Applications, Odontology, 2017, 105, 1–12 CrossRef PubMed.
- S. W. Lee and S. G. Kim, Membranes for the Guided Bone Regeneration, Maxillofac. Plast. Reconstr. Surg., 2014, 36, 239–246 CrossRef PubMed.
- I. A. Rodríguez, G. S. Selders, A. E. Fetz, C. J. Gehrmann, S. H. Stein, J. A. Evensky, M. S. Green and G. L. Bowling, Barrier Membranes for Dental Applications: A Review and Sweet Advancement in Membrane Developments, Mouth Teeth, 2018, 2, 2–9 Search PubMed.
- P. Bunyaratavej and H. L. Wang, Collagen Membranes: A Review, J. Periodontol., 2001, 72, 215–229 CrossRef PubMed.
- Y. D. Rakhmatia, Y. Ayukawa, A. Furuhashi and K. Koyano, Current Barrier Membranes: Titanium Mesh and other Membranes for Guided Bone Regeneration in Dental Applications, J. Prosthodont. Res., 2013, 57, 3–14 CrossRef.
- J. Wang, L. Wang, Z. Zhou, H. Lai, P. Xu, L. Liao and J. Wei, Biodegradable Polymer Membranes Applied in Guided Bone/Tissue Regeneration: A Review, Polymers, 2016, 8, 115 CrossRef PubMed.
- L. Sbricoli, R. Guazzo, M. Annunziata, L. Gobbato, E. Bressan and L. Nastri, Selection of Collagen Membranes for Bone Regeneration: A Literature Review, Materials, 2020, 13, 786–802 CrossRef PubMed.
- A. Kirsch, K. L. Ackermann, M. B. Hurzeler, W. Durr and D. Hutmacher, Development and Clinical Application of Titanium Minipins for Fixation of Nonresorbable Barrier Membranes, Quintessence Int., 1998, 29, 368–381 Search PubMed.
- C. Mehl, M. Kern, N. Meinke, Y. Açil, T. Bähr, J. Wiltfang and V. Gaßling, Can One-Wall Bone Defects Be Augmented with Xenogenic Bone Grafting Material Alone?, J. Craniomaxillofac. Surg., 2016, 44, 1137–1142 CrossRef.
- K. Sakamoto-Ozaki, Y. Matsumoto, Z. Kanno, J. Iida and K. Soma, Development of a Surgical Procedure for Biointegration of a Newly Designed Orthodontic Onplant, Orthodontics, 2012, 13, 216–225 Search PubMed.
- M. J. Sánchez-Fernández, H. Hammoudeh, R. P. Félix Lanao, M. van Erk, J. C. M. van Hest and S. C. G. Leeuwenburgh, Bone-Adhesive Materials: Clinical Requirements, Mechanisms of Action, and Future Perspective, Adv. Mater. Interfaces, 2019, 1802021 CrossRef.
- D. Wang, S. C. Miller, P. Kopeckova and J. Kopecek, Bone-Targeting Macromolecular Therapeutics, Adv. Drug Delivery Rev., 2005, 57, 1049–1076 CrossRef PubMed.
- E. Boanini, M. Gazzano, K. Rubini and A. Bigi, Composite Nanocrystals Provide New Insight on Alendronate Interaction with Hydroxyapatite Structure, Adv. Mater., 2007, 19, 2499–2502 CrossRef.
- G. A. Rodan and H. A. Fleisch, Bisphosphonates: Mechanisms of Action, J. Clin. Invest., 1996, 97, 2692–2696 CrossRef PubMed.
- M. Glassner, M. Vergaelen and R. Hoogenboom, Poly(2-oxazoline)s: A Comprehensive Overview of Polymer Structures and their Physical Properties, Polym. Int., 2018, 67, 32–45 CrossRef.
- V. R. De la Rosa, Poly(2-oxazoline)s as Materials for Biomedical Applications, J. Mater. Sci.: Mater. Med., 2014, 25, 1211–1225 CrossRef.
- J. S. Park, Y. Akiyama, F. M. Winnik and K. Kataoka, Versatile Synthesis of End-Functionalized Thermosensitive Poly(2-isopropyl-2-oxazolines), Macromolecules, 2004, 37, 6786–6792 CrossRef.
- M. J. Sánchez-Fernández, M. R. Immers, R. P. Félix Lanao, F. Yang, J. C. M. E. Bender, J. Mecinović, S. C. G. Leeuwenburgh and J. C. M. van Hest, Alendronate-Functionalized Poly(2-oxazoline)s with Tunable Affinity for Calcium Cations, Biomacromolecules, 2019, 20, 2913–2921 CrossRef.
- M. J. Sánchez-Fernández, R. P. Félix Lanao, J. Opsteen, J. C. M. van Hest, S. C. G. Leeuwenburgh and J. C. M. E. Bender, Polyoxazoline Copolymer Comprising Calcium Binding Groups, WO 2021/018922 A1, 2021 Search PubMed.
- GELITA TUFT-IT®, First Gelatin Fibrillar for Controlled Hemostasis. Retrieved on January 10, 2021 from https://www.gelitamedical.com/Products/TUFT-IT.
- V. Santos-Rosales, A. Iglesias-Mejuto and C. A. García-González, Solvent-Free Approaches for the Processing of Scaffolds in Regenerative Medicine, Polymers, 2020, 12, 533 CrossRef CAS.
- C. L. Simões, J. C. Viana and A. M. Cunha, Mechanical Properties of Poly(ε-caprolactone) and Poly(lactic acid) Blends, J. Appl. Polym. Sci., 2009, 112, 345–352 CrossRef.
- H. K. Makadia and S. J. Siegel, Poly(lactic-co-glycolic acid) (PLGA) as Biodegradable Controlled Drug Delivery Carrier, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- H. Keles, A. Naylor, F. Clegg and C. Sammon, Investigation of Factors Influencing the Hydrolytic Degradation of Single PLGA Microparticles, Polym. Degrad. Stab., 2015, 119, 228–241 CrossRef CAS.
- M. R. Nejadnik, X. Yang, M. Bongio, H. S. Alghamdi, J. J. van den Beucken, M. C. Huysmans, J. A. Jansen, J. Hilborn, D. Ossipov and S. C. G. Leeuwenburgh, Self-Healing Hybrid Nanocomposites Consisting of Bisphosphonated Hyaluronan and Calcium Phosphate Nanoparticles, Biomaterials, 2014, 35, 6918–6929 CrossRef CAS.
- M. Yoshinari, T. Hayakawa, J. G. C. Wolke, K. Nemoto and J. A. Jansen, Influence of Rapid Heating with Infrared Radiation on RF Magnetron-Sputtered Calcium Phosphate Coatings, J. Biomed. Mater. Res., 1997, 37, 60–67 CrossRef CAS.
- M. Sato, R. Tu, T. Goto, K. Ueda and T. Narushima, Hydroxyapatite Formation on Ca-P-O Coating Prepared by MOCVD, Mater. Trans., 2008, 49, 1848–1852 CrossRef CAS.
- H. Eslami, M. Solati-Hashjin and M. Tahriri, Synthesis and Characterization of Hydroxyapatite Nanocrystals Via Chemical Precipitation Technique, Iran. J. Pharm. Sci., 2008, 4, 127–134 Search PubMed.
- I. Uysal, F. Severcan and Z. Evis, Characterization by Fourier Transform Infrared Spectroscopy of Hydroxyapatite Co-doped with Zinc and Fluoride, Ceram. Int., 2013, 39, 7727–7733 CrossRef CAS.
- P. Raz, T. Brosh, G. Ronen and H. Tal, Tensile Properties of Three Selected Collagen Membranes, BioMed Res. Int., 2019, 5163603 CAS.
- L. Santo, F. Quadrini, D. Bellisario, A. Polimeni and A. Santarsiero, Variability of Mechanical Properties of Collagen Membranes Used in Dentistry, Mater. Plast., 2018, 55, 488–493 CrossRef.
- N. López-Rodríguez, A. López-Arraiza, E. Meaurio and J. R. Sarasua, Crystallization, Morphology, and Mechanical Behavior of Polylactide/Poly(ε-caprolactone) Blends, Poly. Eng. Sci., 2006, 46, 1299–1308 CrossRef.
- PURASORB PLC 7015. Retrieved on January 19, 2021 from http://www.corbion.com.
- NIST Chemistry Web Book. Available online: https://webbook.nist.gov/accessed on January 20, 2021.
- J. M. Hutchinson, Determination of the Glass Transition Temperature, J. Therm. Anal. Calorim., 2009, 98, 579–589 CrossRef CAS.
- J. S. Wang and R. S. Porter, On the Viscosity-Temperature Behavior of Polymer Melts, Rheol. Acta, 1995, 34, 496–503 CrossRef CAS.
- S. J. Marshall, S. C. Bayne, R. Baier, A. P. Tomsia and G. W. Marshall, A Review of Adhesion Science, Dent. Mater., 2010, 26, 11–16 CrossRef PubMed.
- K. Chavalitpanya and S. Phattanarudee, Poly(Lactic Acid)/Polycaprolactone Blends Compatibilized with Block Copolymer, Energy Procedia, 2013, 34, 542–548 CrossRef CAS.
- I. Fortelny, A. Ujcic, L. Fambri and M. Slouf, Phase Structure, Compatibility, and Toughness of PLA/PCL Blends: A Review, Front. Mater., 2019, 6, 206 CrossRef.
- Y. Zhang, X. Li and B. Wei, Environment-Friendly Poly(2-ethyl-2-oxazoline) as an Innovative Consolidant for Ancient Wall Paintings, Nanomaterials, 2018, 8, 649 CrossRef PubMed.
- H. Sun, L. Mei, C. Song, X. Cui and P. Wang, The in Vivo Degradation, Absorption and Excretion of PCL-Based Implant, Biomaterials, 2006, 27, 1735–1740 CrossRef CAS PubMed.
- M. Xu, C. Guo, H. Dou, Y. Zuo, Y. Sun, J. Zhang and W. Li, Tailoring the Degradation and Mechanical Properties of Poly(ε-caprolactone) Incorporating Functional ε-Caprolactone-Based Copolymers, Polym. Chem., 2019, 10, 3786–3796 RSC.
- A. S. Hussein, N. Abdullah and F. R. Ahmadun, In Vitro Degradation of Poly(D,L-lactide-co-glycolide) Nanoparticles Loaded with Linamarin, J. Nanobiotechnol., 2013, 7, 33–41 Search PubMed.
- S. H. Lee and H. Shin, Matrices and scaffolds for delivery of bioactive molecules in bone and cartilage tissue engineering, Adv. Drug Delivery Rev., 2007, 59, 339–359 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic route of alendronate-functionalized POx polymers (Scheme 1); experimental procedures and characterization of the synthesized polymers (S1); NMR spectra of synthesized polymers (Fig. S1); images of a blank membrane and backing layer (Fig. S2); thin-film X-ray diffractograms and FTIR spectra of CaP-coated titanium disks, bone, and demineralized bone specimens (Fig. S3); scanning electron micrographs of CaP-coated titanium disks and longitudinal and cross-sectional views of bone specimens before and after demineralization (Fig. S4); experimental setups for tensile and lap-shear testing (Fig. S5); thermograms of POx-polymers and FTIR spectra of various experimental groups (Fig. S6); water contact angle measurements of the membranes (Fig. S7); FTIR spectra of bone specimens after lap-shear testing of various membranes (Fig. S8); swelling of membranes under wet conditions (Fig. S9); and physical characterization of the membranes (Table S1). See DOI: 10.1039/d1tb00502b |
| This journal is © The Royal Society of Chemistry 2021 |