
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceL-Dopa release from mesoporous silica nanoparticles engineered through the concept of drug-structure-directing agents for Parkinson's disease†
V.
Morales
a,
J.
McConnell
b,
M.
Pérez-Garnes
a,
N.
Almendro
a,
R.
Sanz
 a and
R. A.
García-Muñoz
*a
a and
R. A.
García-Muñoz
*a
aDepartment of Chemical and Environmental Technology, Rey Juan Carlos University, C/Tulipán s/n, 28933, Móstoles, Madrid, Spain. E-mail: rafael.garcia@urjc.es
bDepartment of Chemical & Process Engineering, University of Strathclyde, Glasgow G1 1XJ, UK
First published on 28th April 2021
Abstract
Parkinson's disease (PD) is a progressive neurodegenerative disease, the 2nd most common after Alzheimer's disease, the main effect of which is the loss of dopaminergic neurons. Levodopa or L-Dopa is an amino acid used in the treatment of PD that acts as the immediate precursor to dopamine. However, over time the efficacy of the medication gradually decreases requiring modified delivery methods. One of the major challenges for the medication to work is to achieve a gradual continuous supply of L-Dopa to the brain to minimise symptoms. Herein, mesoporous silica nanoparticles (MSNs) were engineered through the concept of drug-structure-directing agents (DSDAs) with inherent therapeutic activity. The DSDA used was L-Dopa drug modified by amidation with fatty acids to build anionic surfactants that were able to form micelles as templates for the assembly of inorganic precursors to form the silica framework. This templating route produced MSNs with tunable sizes ranging from 100 nm to 1 μm and with different shapes: spherical, with either solid structures with radial mesopores and porous shells, or hollow-shells with inside large void cavities; and elongated, characterized by long hollows covered by mesoporous shells. The concept of using DSDAs to synthesize drug nanocarriers can be used to avoid the surfactant removal and subsequent drug loading steps involved in the synthesis of conventional MSNs. We hypothesized that the L-Dopa released from MSN materials is mediated by the size and solubility of the DSDAs, and the surface chemical interactions between the DSDAs and MSN hosts. Different pHs (acidic and neutral) simulating gastrointestinal tract conditions were tested, and the results showed hardly any release for gastric conditions at pH 1.2, avoiding the premature release in the stomach typical of conventional MSNs, while for intestinal conditions of pH 7.4, the release of L-Dopa occurred in a continuous and sustained manner, which is well suited to the drug's application and delivery route, and matches well with achieving a sustained L-Dopa delivery to relief symptoms. This could open up new uses for MSNs synthesized by this approach to treat PD.
1. Introduction
Nanotechnology and nanoscience are currently amongst the most exciting and fastest moving areas of science and technology. The World's leaders in innovation focus a great deal of investment and research into nanotechnology – likely due to the wide range of applications.1 Additionally, the focus of global research and development according to the Global Innovation Index is medical innovation to create healthy lives, putting nanomedicine at the forefront of research and development.2 It is also expected that the global market for nanomedicine could reach a value of USD 350.8 billion by 2025 – providing a considerable financial incentive for research and investment into the nanomedical industry.3One of the leading areas within nanotechnology is the use of particles in the nanometric range (NPs).4–9 Specifically, in nanomedicine, particles of this size are able to mimic and alter biological processes. Since blood capillaries can be as small as 5–6 μm, the particles must be considerably smaller to prevent embolism, and are also more appropriate for intravenous administration. One of the most exciting categories of nanoparticles is Mesoporous Silica Nanoparticles (MSNs). The porous nature of the particles has several benefits, a key benefit being that it gives the particles a large surface area which can be loaded with drugs, thus making them ideal candidates for drug targeting and delivery. Also, the particles can be engineered to control the pore size for the slow and sustained release of drugs. Moreover, the easy functionalization of their surface allows for the targeting of specific sites.10–15 For certain treatments this is preferable to other conventional drug administration techniques (oral or intravenous) which result in frequent, short-term doses, which fluctuate and result in the need for large doses.16 Furthermore, as the drug is contained inside the inner porosity, it is protected – impeding its exposure to enzymes and metabolites that can modify it before targeting the objective which may cause both undesirable side effects in unwanted organs and uncontrolled effects. These characteristics make MSNs an excellent candidate for drug delivery systems (DDSs).
The typical method for MSN synthesis for drug delivery involves using surfactants such as structure-directing agents (SDA). The drug is then adsorbed onto the free space left by the surfactant following calcination or solvent extraction of the surfactant. Much research has been done following this method with the attention being directed towards controlling the pore size and ordering.17–23 It has mainly focused on release methods – implementing and utilising magnetic properties, photodynamic-enhanced chemotherapy, photothermal-enhanced chemotherapy, etc., to control and improve the treatment efficacy. Methods of release have been based on devices such as nanoimpellers, nanovalves, pH responsiveness, the use of light, gate systems and use of camouflages based on cell membranes to enhance therapeutic effects.24–39
Alternative methods to this conventional technique involve either the use of drugs that are able to co-assemble with silica40 or the modification of the drug with an appropriate chain molecule to form biocompatible and non-toxic surfactant molecules, known as the drug-structured-directing agents (DSDAs), which can in turn be dispersed and used to address the formation of MSNs resulting in achieving a fully occupied total inner porosity.41–43 This method, developed in our laboratories, not only reduces the number of steps required to make the final MSNs, but also removes the need for further functionalisation of the inner porosity of the MSNs (to promote intermolecular interactions with pharmaceutical molecules), which has previously been required to influence the adsorption and release of drugs from conventional MSNs. Additionally, this method also results in far greater loading rates of drug in the MSNs produced, enabling smaller doses of MSNs.
Just over 200 years ago the symptoms of Parkinson's disease (PD) were described by the general practitioner James Parkinson after whom the disease was eventually named. One of the key characteristics of the disease is the simultaneous loss of movement (known as hypokinesia) and gain of involuntary movement – as the disease causes a tremor. The disease is the 2nd most common progressive neurodegenerative disease after Alzheimer's and affects approximately 1–2% of people over the age of 65.44 PD results from the degeneration or death of dopaminergic neurons (neurons that synthesize dopamine) that live in the substantia nigra region of the brain, located in the midbrain at the top of the brain stem, and are projected to the striatum. These neurons release the neurotransmitter molecule dopamine, which is crucial for normal, controlled movement. However, PD cannot be treated directly with dopamine as dopamine cannot cross the blood–brain barrier (BBB). The symptoms can be controlled using a drug called L-Dopa.45L-Dopa is the most effective drug for symptomatic relief from PD; however 75% of patients experience complications after 5 years of L-Dopa therapy.46 Over time, the irregularity of L-Dopa doses take their toll on the patient, with L-Dopa treatment becoming less and less effective, resulting in the need for larger doses of L-Dopa.
This makes the treatment of PD with L-Dopa an ideal candidate for experimentation with drug delivery systems, where a longer lasting, slow and continuous release source of L-Dopa would increase the length of time that L-Dopa remains effective and hence improve the quality of life of patients in the later stages of care. To meet this goal, different drug delivery systems, such as polymeric nanoparticles and silica nanoparticles have been explored to entrap L-Dopa and improve the release of the drug, but all of them respond to conventional methodologies, in which the L-Dopa is incorporated/loaded into the corresponding matrix porosity in several steps.47–51 The novelty of this research lies in the preparation of L-Dopa@MSN nanoparticles based on the concept of DSDA containing L-Dopa, as the mainstay treatment for PD. The DSDA concept was previously introduced by our group using other different model drugs. In this work, L-Dopa was modified by amidation with fatty acids of different lengths that allowed the formation of micelles of DSDA, around which the inorganic species self-assembled to form MSNs. We demonstrated a facile one-step strategy to synthesize mesoporous solid-shell and hollow-shell MSNs that allowed the full occupation of the entire pore volume with the DSDA that after release from MSN and cleavage of the amide delivers decanoic or oleic acid and dopamine precursor drug, L-Dopa, separately, in equimolar amounts. The obtained MSNs showed different shapes, elongated or spherical, and sizes ranging from 100 nm to 1 μm. Spherical MSNs were either solid with radial mesopores and a porous shell or a hollow-shell with inside large void cavities. Otherwise, the elongated MSNs were characterized by long hollows covered by a mesoporous shell. To demonstrate the effectiveness of these DDSs, we simulated the L-Dopa in vitro release from MSNs, considering different gastrointestinal conditions after oral drug delivery. The results showed hardly any release for the gastric conditions of pH 1.2, while for the intestinal conditions of pH 7.4, the release of L-Dopa occurred in a continuous and sustained manner. This prevents the fluctuation of drug concentration in the blood caused by conventional drug administration, and makes the MSNs synthesized by this approach promising vehicles for the treatment of PD.
2. Experimental
2.1. Chemicals
L-Dopa, decanoyl chloride (98%), oleoyl chloride, sodium hydroxide (97%), sodium bicarbonate (97%), 3-(aminopropyl)trimethoxysilane (97%), tetraethyl orthosilicate (98%) and dicloromethane (99.8%) were purchased from Sigma-Aldrich. Tetrahydrofuran (99.5%), hydrochloric acid (35% w/w) and sodium chloride (99.5%) were supplied by Scharlab.The methodology for the development of hollow-shell or solid materials (named as HMSNs) can be summarized in two stages: the synthesis of DSDA surfactants and L-Dopa@MSN by a sol–gel process.
2.2. Synthesis of DSDAs
The anionic DSDAs were synthesized by the amidation reaction of L-Dopa with decanoyl chloride (C10) or oleoyl chloride (unsaturated C18). Due to the reactivity of the L-Dopa molecule, it was important to maintain an inert atmosphere during the formation of the surfactant molecules. Firstly, 50 mL of water was degassed for 5 minutes. This was performed in a water bath using sonication – the water was placed in an atmosphere of argon and sparged with helium. Once degassed, 1.91 g of Na2B4O7 (Borax) was added to the water, followed by 1 g of L-Dopa with the borax in excess. This was done to ensure that the fatty acid chlorides reacted with the amine group of the L-Dopa and not the hydroxyl groups. The pH was adjusted to 8–9 using Na2CO3 (sodium carbonate). The reactant mixture was then degassed for a further 5 minutes. The mixture was then placed in ice and allowed to cool to 0 °C. Approximately 1 mL of C10H19OCl or C18H18OCl decanoyl or oleoyl chloride, respectively, was combined with 12.5 mL of C4H8O tetrahydrofuran (THF). This was then added to the reactant mixture dropwise whilst maintaining a temperature of 0 °C. The pH was again adjusted to pH 8–9 using sodium carbonate. The reactant mixture was then degassed a third time and left in ice overnight in an argon atmosphere. Before extraction, the pH of the reaction mixture was adjusted to pH 2 using hydrochloric acid. The L-Dopa DSDA was extracted by solvent extraction using ethyl acetate. Solvent extraction was done using a separation funnel. When the ethyl acetate was added the DSDA dissolved in the organic phase and the aqueous phase could be disposed of. Ethyl acetate containing DSDA was then placed in a vacuum evaporator to remove the THF. The evaporation was done at reduced pressure and at 70 °C for approximately 2 hours and until 12.5 mL had been collected. The reaction mixture was then washed with ethyl acetate 3 times using 20–30 mL each time. Ethyl acetate containing the product was then washed once with water and then once with a saturated sodium chloride solution (brine). Ethyl acetate solution was then placed in the vacuum evaporator for 1 hour. The pressure was only slightly reduced, and the temperature was set to 60 °C. This yielded the DSDA products, N-decanoyl-L-Dopa (NDLD) and N-oleyl-L-Dopa (NOLD), respectively, which were dried overnight. The DSDA were solids with a sticky consistency and dark purple/brown colour.2.3. Preparation of L-Dopa@MSNs
The L-Dopa@MSNs were prepared in aqueous media using NDLD or NOLD as a surfactant, 3-aminopropyltrimethoxysilane (APS) as a co-surfactant and tetraethyl orthosilicate (TEOS) as a silica source. To form L-Dopa@MSN materials, DSDA was first dispersed in water at 90 °C for approximately 24 hours. After 24 hours, 3-aminopropyltrimethoxysilane (APS) was added as a co-surfactant and after 5 minutes of stirring tetraethyl orthosilicate (TEOS) was added as a silica source. The mixture was stirred for a further 10 minutes before removing the stirrer. To ensure that enough material was produced this was done using 1.4 mmol or 490 mg of DSDA – which corresponds to 0.54 mL of APS and 3.7 mL of TEOS. The mixture was left for 24 hours at 60 °C. The temperature was then adjusted to 100 °C for a further 72 hours. The ratio of substances was maintained at a molar ratio of DSDA/APS/TEOS/water equal to 1/1.65/11.8/2000. The solid was recovered by filtration, washed with abundant de-ionized water and ethanol, and dried. MSNs under different conditions, maintaining the DSDA/APS/TEOS molar ratio constant, were synthetized as shown in Table 1.| Nomenclature | DSDA | DSDA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water :water |
pH of synthesis |
|---|---|---|---|
| HMSN18-A | NOLD | 1:2000 |
7.6 |
| HMSN18-B | NOLD | 1:4000 |
7.6 |
| HMSN18-C | NOLD | 1:2000 |
8.5 |
| HMSN18-D | NOLD | 1:2000 |
9.0 |
| HMSN18-E | NOLD | 1:2000 |
9.5 |
| HMSN10-A | NDLD | 1:2000 |
7.6 |
| HMSN10-B | NDLD | 1:4000 |
7.6 |
| HMSN10-C | NDLD | 1:2000 |
8.5 |
| HMSN10-D | NDLD | 1:2000 |
9.0 |
| HMSN10-E | NDLD | 1:2000 |
9.5 |
2.4. Physicochemical characterization
The zeta potential values of the DSDAs were determined by using a NanoPlus DLS instrument. The critical micellar concentration (CMC) was determined by conductimetry using an HI2020 Edge multi-parameter meter equipped with an HI763100 electrode. The organic content (DSDA and APS) in the MSNs was quantified by thermogravimetric analysis from ambient temperature to 800 °C at 5 °C min−1 under air atmosphere. X-Ray diffraction patterns were recorded on a Philips X'PERT MPD powder diffractometer equipped with CuKα radiation. The textural properties were obtained from the N2 adsorption–desorption isotherms at −196 °C using a Micromeritics TriStar 3000 instrument. Before measurement, the materials were outgassed at 300 °C by a N2 flux. Slit pore geometry was assumed for the calculation of the mesopore size distribution using the NLDFT model. Transmission electron microscopy (TEM) images of the samples were recorded using a Philips Tecnai F20 microscope operated at 200 kV. Previously, the samples were crushed, dispersed in ethanol and deposited on a carbon-coated copper grid. The measurements of the features in nanoparticles were determined from the TEM images using ImageJ software. STEM images of the samples were recorded using a JEOL JEM3000F operated at 300 kV with a 0.14 nm resolution equipped with a HAADF detector and OXFORD INCA for EDS.2.5. L-Dopa release studies
The L-Dopa release was carried out in two different media: simulated gastric fluid, or stomach acid, at pH 1.2 (denoted as SGF) and simulated small intestinal fluid at pH 7.4 (denoted as SIF). SGF was prepared by combining 2 g of sodium chloride dissolved in 1 L of distilled water and 7 mL of 37% hydrochloric acid. SIF was prepared by combining 6.8 g of monobasic potassium phosphate dissolved in 650 mL of distilled water and 190 mL of 0.2 M sodium hydroxide, then the pH was adjusted to 7.4 using 0.2 M sodium hydroxide dropwise, and the final volume was adjusted to 1 L using deionized water.For the release study, 100 mg of dry MSNs were introduced into dialysis bags (dialyzer-weight cut-off 10000 Da). Then, the dialysis bags were immersed in 100 mL of the solution mentioned above at 37 °C. The L-Dopa release was monitored using a UV-spectrometry (JASCO V-630) at 280 nm.
3. Results and discussion
3.1. Synthesis and characterization of L-Dopa@MSN based-DSDAs
The size of the MSN NP is extremely important for their likely application in biomedicine. Additionally, the nanoparticles morphology is a determinant in nanoparticle fluid dynamics in the plasma throughout the blood vessels. Therefore, both parameters play a key role in the pharmacokinetics, cellular uptake, biodistribution and circulation lifetime of the nanoparticles and the drugs contained within. In this work, L-Dopa@MSN nanoparticle delivery vehicles have been designed with different sizes and shapes as a suitable nanotherapy for Parkinson's disease.The MSNs, herein developed, were synthesized using the drug-structure-directing agent concept,41–43 and specifically L-Dopa-based NOLD and NDLD surfactants were used. With this alternative approach to the conventional MSN synthesis, it is possible to integrate the drug and the nanocarrier that transports it, all in one, obviating the different steps required in the synthesis of conventional MSN for therapeutic use, such as the removal of the surfactant or template and the subsequent uploading of the drug cargo on free internal porosity. Both DSDAs were prepared from the amidation reaction between L-Dopa and oleoyl or decanoyl chloride, respectively. The formation of NOLD and NDLD surfactants was qualitatively confirmed by FTIR measurements (Fig. S1, ESI†). The multiple strong peaks ranging between 2800 and 3000 cm−1 in spectrum A of Fig. S1 (ESI†) are associated with the C–H stretching vibration modes of CH2 and CH3 of hydrocarbon tails. In addition to the above peaks, infrared absorption bands assigned to NH stretching mode are clearly seen in the range of 3300–3400 cm−1. Moreover, the IR spectra A, B and C of Fig. S1 (ESI†) show infrared absorption peaks at 1680–1700 cm−1 corresponding to the strong carboxyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode of a secondary amide. To confirm these results, 1H and 13C liquid-state nuclear magnetic resonance (NMR) experiments were performed (Fig. S2 and S3, ESI†). 1H NMR spectra showed characteristic peaks of the proton of the N–H amide at 7.96 ppm as a doublet, and the proton of the α-carbon adjacent to the amide at 4.30 ppm as a triple doublet, which indicates the presence of amides. The 13C NMR spectra also exhibited clear signals at 175 ppm that can be unambiguously assigned to the presence of amides. Moreover, the synthesized L-Dopa-based surfactants contain free carboxylic acids resulting in an anionic nature, as confirmed by the Z potential measurements, in which both surfactants, NDLD and NOLD, showed values of around −40 and −55 mV at pH 7.4, respectively.
O stretching mode of a secondary amide. To confirm these results, 1H and 13C liquid-state nuclear magnetic resonance (NMR) experiments were performed (Fig. S2 and S3, ESI†). 1H NMR spectra showed characteristic peaks of the proton of the N–H amide at 7.96 ppm as a doublet, and the proton of the α-carbon adjacent to the amide at 4.30 ppm as a triple doublet, which indicates the presence of amides. The 13C NMR spectra also exhibited clear signals at 175 ppm that can be unambiguously assigned to the presence of amides. Moreover, the synthesized L-Dopa-based surfactants contain free carboxylic acids resulting in an anionic nature, as confirmed by the Z potential measurements, in which both surfactants, NDLD and NOLD, showed values of around −40 and −55 mV at pH 7.4, respectively.
To achieve the successful preparation of MSNs by using anionic NDLD and NOLD surfactants, the cationic 3-(aminopropyl)trimethoxysilane (APS) moiety was used as the co-structure directing agent (CSDA). APS moieties interact electrostatically with the head group of the NDLD and NOLD surfactants. Therefore, the development of MSNs depends on several things: firstly, on the ability of L-Dopa-based surfactants (NDLD and NOLD) to form micelles; secondly on their subsequent rearrangement after adding APS; and finally on the sol–gel process as a consequence of the TEOS addition. The conductance measurements determined the CMC of surfactants in water that resulted in values of 0.29 mM and 0.19 mM for NDLD and NOLD, respectively.
The condensation of silica was confirmed by the formation of white precipitates immediately after the addition of TEOS for all the synthesis conditions (summarized in Table 1). The pH values of the media were monitored after adding each component in the synthesis of the different MSNs. After the complete dilution of the corresponding DSDA (NDLD or NOLD), an oil phase in the aqueous media was observed, and the pH values indicate the acidic character of the media (pH values of 2.63 and 2.82 for NDLD and NOLD, respectively). This acidification was due to the acidic nature of L-Dopa, which has a pKa value of around 1.65, while the slight difference in pH between the surfactants was due to their different hydrophobic chain lengths. After adding APS (pKa between 9 and 10), the pH value underwent a sharp increase to 7.6 and 7.8, for NDLD and NOLD respectively. The further addition of TEOS and condensation of silica species caused a slight decline in the pH. Therefore, this pH was considered to be the reference for the synthesis media of L-Dopa@MSN materials.
3.2. L-Dopa@MSN synthetized using NOLD as surfactant
The general mechanism of formation of L-Dopa@MSN is the same, regardless of the DSDA employed. At high temperatures, the DSDA may form aggregates with different shapes and sizes, and has a dual function: first, it acts as surfactant, synthesized from the amidation reaction between L-Dopa and a fatty acid chain; and secondly, it acts as the oil phase dispersed in water (droplets) where the hollow internal cavities are developed. The assembly of the micellar aggregates is heavily dependent on the synthesis conditions and the nature of the surfactant. Moreover, the L-Dopa@MSN NP silica materials were synthesized through emulsification, and the effects of CSDA play a key role in the formation of both the hollow structure and the mesoporous shell framework.41–43,52 The addition of APS rearranges and stabilizes these aggregates through electrostatic interactions. The alkoxysilane groups of TEOS and APS hydrolyze and further condense around the surface and interstices of the micellar aggregates. Finally, the external silica shell around the systems is developed by the condensation of silica. As the reference pH of the synthesis is modified, increased by adding NaOH, the condensation rate of TEOS increases, promoting the formation of hollow-shell materials or solid mesostructures, termed as HMSNs.First of all, two different L-Dopa@MSN materials (HMSN18-A and HMSN18-B) were synthesized using NOLD as the surfactant and APS as CSDA, and modifying the DSDA(NOLD):water ratio, while the other conditions remained unchanged, including the pH. Fig. 1A–H shows the SEM and TEM images of the L-Dopa@MSN NP silica materials. SEM and TEM micrographs indicate that the L-Dopa@MSN materials are characterized by a hollow-shell elongated morphology with diameters between 112.6 and 96.7 nm, and lengths of 465.2 and 286.9 nm, corresponding to HMSN18-A and HMSN18-B, respectively. In the TEM images, the contrast between the dark edges and the clear centre confirms the hollow shelled structure. The morphology and structure of both samples remained unaffected, but interestingly, the size and length of the materials varied as the molar ratio of DSDA:water was modified, which can be clearly observed in SEM images (Fig. 1A–D). Thus, HMSN18-B, synthesized at a lower DSDA concentration, yielded elongated hollow-shell silica nanoparticles with smaller size and length than HMSN18-A obtained at higher DSDA concentration. These peculiar, elongated, peapod-like structures are quite similar to those found in previous studies with other drugs,41,42 but were prepared using oleoyl species as the surfactant tails. It is likely that oleoyl moieties can reduce the surface tension resulting in the formation of elongated microaggregates. The materials display a hollow-shell structure characterized by a mesoporous shell and inner cavities, which are separated by mesoporous lamellar wall-like structures, providing the materials with void compartments separated by lamellar walls of condensed silica. The mesoporous shell thicknesses determined from TEM images were approximately 15.9 and 18.4 nm (Table 2), respectively, and it was observed that the thickness of the silica shell was uniform.
 | ||
| Fig. 1 Structural characterization of L-Dopa@MSNs. SEM and TEM images of L-Dopa@MSN synthetized using different NOLD:water molar ratios: HMSN18-A (A, B and E, F) and HMSN18-B (C, D and G, H). The scale bar is 1 μm in (A and C) and 100 nm in (B and D). | ||
| Nomenclature | V p (cm3 g−1) | S BET (m2 g−1) | D PORE (nm) | D N (nm) | Shell thickness (nm) |
|---|---|---|---|---|---|
| a Range of the pore diameter and more frequent pore width (in brackets). b Mean length of the transversal section. c Samples with no defined shape. | |||||
| HMSN18-A | 0.234 | 230 | 2.3–12.1 (3.1) | 112.6 ± 56.1a | 15.9 ± 1.8b |
| HMSN18-B | 0.287 | 168 | 2.5–14.3 (3.0) | 96.7 ± 29.5a | 18.4 ± 1.8b |
| HMSN18-C | 0.410 | 182 | 2.5–17.5 (3.0) | — | |
| HMSN18-D | 0.247 | 124 | 2.5–11.3 (2.9) | — | |
| HMSN18-E | 0.401 | 156 | 2.6–19.5 (3.0) | — | |
| HMSN10-A | 0.110 | 140 | 2.2–3.1 (2.4) | 986.8 ± 158.6 | 141.8 ± 16.0 |
| HMSN10-B | 0.154 | 204 | 2.0–3.3 (2.7) | 923.4 ± 81.9 | 130.9 ± 15.5 |
| HMSN10-C | 0.161 | 303 | 2.2–3.1 (2.4) | 116.7 ± 16.5 | 17.6 ± 2.1 |
| HMSN10-D | 0.115 | 194 | 2.0–2.6 (2.5) | 234.7 ± 82.2 | — |
| HMSN10-E | 0.085 | 157 | 2.0–3.0 (2.5) | 244.4 ± 80.7 | — |
Fig. 2A shows the thermogravimetric analysis of the L-Dopa@MSN synthesized. The organic content in these materials (DSDA and APS) was determined to be between 20 and 25%, deduced from the remaining weight after a temperature of 700 °C was reached. HMSN18-A and HMSN18-B showed three thermal degradation stages between 100 and 700 °C, centred at 200, 300 and 500 °C. The thermal decomposition, albeit indistinctly, corresponds approximately to the DSDA and APS. The XRD patterns of the as-made materials are shown in Fig. 2B. No significant XRD peaks are observed, most likely because of the presence of the disordered pores in the shell.
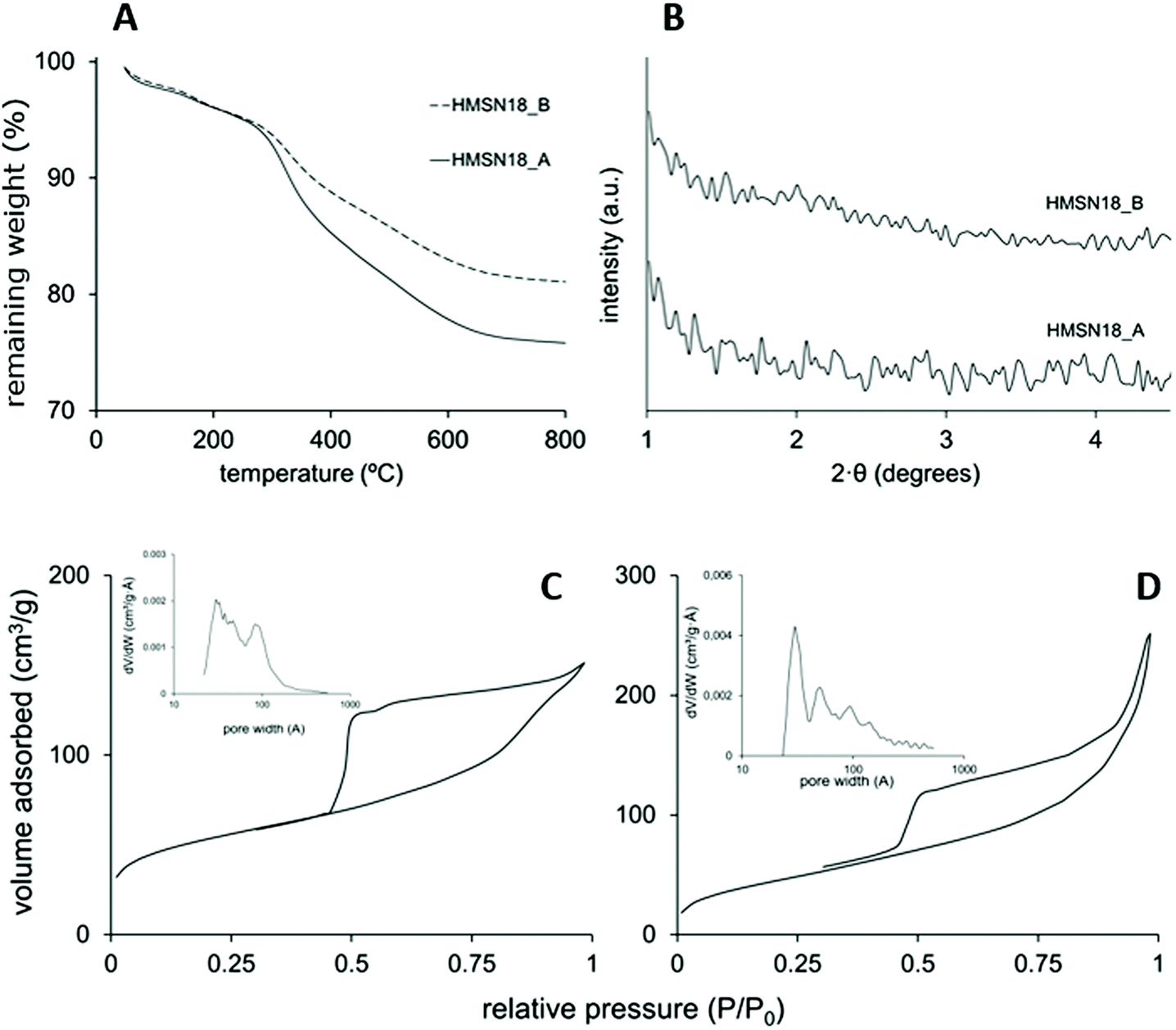 | ||
| Fig. 2 (A) Thermogravimetric analysis and (B) low-angle X-ray diffraction patterns of L-Dopa@MSN synthetized using different concentrations of NOLD. N2 adsorption–desorption isotherms of the (C) HMSN18-A and (D) HMSN1-B materials. | ||
Fig. 2C and D shows the N2 adsorption–desorption isotherms of HMSN18-A and HSMN18-B materials. The isotherms are characterized by curves that fit the type IV isotherm according to the IUPAC classification. The capillary condensation step at a relative pressure of 0.5–0.7 confirms a significant filling of mesopores. At a relative pressure near 0.9 another step that may be attributed to filling of the hollow cavity or interparticle space is observed. Furthermore, both materials display a type H3 hysteresis cycle which suggests the formation of hollows, as previously reported.41,42 The SBET of these nanoparticles achieved values of 230 and 168 m2 g−1 for HMSN18-A and HSMN18-B materials, respectively (Table 2). On the other hand, pore volumes with values between 0.234 and 0.287 cm3 g−1 were observed for HMSN18-A and HSMN18-B samples respectively. In addition, both materials showed a relatively broad pore size distribution (PSD) with porous diameters in the range of 2–14.5 nm, with a maximum peak around 3 nm which corresponds to the shell mesoporosity. The broad PSD of these nanoparticles is concordant with the hollow structure shown in Fig. 1.
Furthermore, the synthesis pH was modified to study its influence on the morphology and structure of the materials, samples HMSN18-C, D and E vs. HMSN18-A and B. However, the variation of the pH of the medium of synthesis (pH values between 8.5 and 9.5 for samples HMSN18-C, D and E) did not lead to well-defined structures using NOLD as the surfactant (see Fig. S4 (ESI†) and Table 2). This phenomenon might be associated with the intrinsic arrangement of the NOLD micellar aggregates and the loss of morphology when the pH is altered.
3.3. L-Dopa@MSN synthetized using NDLD as surfactant
Different L-Dopa@MSN materials were synthesized by using NDLD as the surfactant and APS as CSDA, and modifying the DSDA (NDLD):water ratio, and the reference synthesis pH.
The TEM and SEM images of the resulting materials prepared with various NDLD:water molar ratios and pH modifications are shown in Fig. 3 and 4, respectively. It can be seen that all of the materials possess a spherical morphology with different sizes depending on the synthesis conditions. Fig. 3A–C shows the micrographies of the as-synthesized HMSN10-A material (NDLD:water ratio 1:2000 and at the reference pH of 7.6). The HMSN10-A material shows a well-defined spherical shape with average particle sizes of around 980 nm (Table 2) and display a hollow-shell structure. The material shows a particular hollow-shell framework, in which multiple small inner hollows are separated by radial-like lamellar mesoporous walls. As the concentration of surfactant decreases (NDLD:water ratio of 1:4000) the resulting material, sample HMSN10-B, did not experience meaningful changes in its morphology and structure (Fig. 3D–F). Fig. 4A, B and D, E show the SEM images of HMSN-A and -B. Both materials have spherical morphology. However, the nanoparticle size distribution measured by nanoparticle sizing instrument (Fig. 4C and F) shows that average particle sizes undergo a slight decline yielding particles with diameters of around 900 nm, leading to the inference that the particle size of hollow mesoporous silica nanoparticle can be tuned by controlling the NDLD:water ratio. Although the average diameter reduces, the thickness of the L-Dopa@MSN shells maintains similar values between 130.9 and 141.8 nm (Table 2). Fig. 5C shows the XRD patterns of both materials. Although no significant, well-resolved XRD peaks are visible, an incipient reflection at 2θ = 2.1° confirms some degree of ordering.
 | ||
| Fig. 3 TEM images of HMSN10-A (A–C), HMSN10-B (D–F), HMSN10-C (G–I), HMSN10-D (J–L) and HMSN10-E (M–O) samples. | ||
 | ||
| Fig. 4 SEM images and corresponding nanoparticle size distribution of L-Dopa@MSN synthetized using NDLD: HMSN10-A (A–C), HMSN10-B (D–F), HMSN10-C (G–I), HMSN10-D (J–L) and HMSN10-E (M–O) samples. The scale bar is 1 μm in SEM images. | ||
 | ||
| Fig. 5 Thermogravimetric analysis of HMSN10 samples (A and B), and low-angle X-ray diffraction patterns of HMSN10 samples (C and D). N2 adsorption–desorption isotherms of (E) HMSN-A, (F) HMSN-B, (G) HMSN-C, (H) HMSN-D and (I) HMSN-E samples. | ||
The modification of the synthesis pH between a value of 8.5 and 9.5 by adding sodium hydroxide in a controlled way did not only lead to a drastic drop in the average nanoparticle size (Fig. 4I, L and O), but also to a clear transformation of the nanoparticle architecture from hollow-shell spheres to solid spheres with different degrees of mesoporosity and compactness. TEM images of HMSN10-D and HMSN10-E materials (Fig. 3J–O), synthesized at pH values of 9 and 9.5 respectively, show nonporous contrast and the complete absence of a mesoporous shell. No significant XRD peaks are visible (Fig. 4D), in agreement with the TEM images. TEM (Fig. 3J–O) and SEM (Fig. 4J, K and M, N) images show that both materials are characterized by a spherical morphology with a compact structure. As the pH is close to the pKa of APS (pKa 10.2), APS may mostly exist as neutral 3-aminopropyl trimethoxysilane molecules that mismatch with the NDLT negative charge, hindering their interactions. Under these conditions, the quick hydrolysis and condensation of TEOS is favoured and the formation of small, solid nanospheres is promoted.43
In contrast, at a pH of 8.5, the obtained HMSN10-C material is composed of solid, spherical nanoparticles with a high degree of mesoporosity, and is encompassed by a mesoporous shell, identified from the contrast of the inner and outer surface of the material, shown in the TEM images (Fig. 3G–I). These nanoparticles have a narrow distribution of sizes with sphere diameters of around 100 nm (Table 2 and Fig. 4I). XRD patterns, shown in Fig. 5D, exhibited a well-defined reflection peak at 2θ = 2.1°, which indicates that the silica spheres are mesoporous. The formation of this ordered mesostructured material might be associated with the optimal interaction between NDLD and APS at pH 8.5. Moreover, at this pH, further TEOS hydrolysis and condensation mediates the formation of mesoporous solid nanospheres due to the diffusion of silica species into the oil micellar aggregates of NDLD/APS.41–43 As shown in Fig. 4G–I, the HMSN10-C material is characterized by spherical morphology with a compact structure, exhibiting excellent uniformity with very similar sizes, as deduced from the SEM images and narrow nanoparticle size distribution.
The thermal degradation and the organic content in the L-Dopa@MSN (amount of DSDA and APS) were determined by thermogravimetric analysis. As previously observed in the nanoparticles prepared using NOLD, no matter what the length of hydrophobic tail of surfactant, the nanoparticles arranged using NDLD as the surfactant showed similar thermograms (Fig. 5A and B), with three overlapped degradation stages from 100 to 700 °C, and maximum degradation rates at 200, 300 and 500 °C. These stages correspond to the thermal decomposition of both DSDA and APS. Beyond 700 °C, the weight stabilized at values between 70 and 85% of its initial, which corresponds to the amount of condensed silica. The HMSN10-C sample showed the highest weight loss, meaning it had the largest loading of organic moieties inside the nanostructure, as a result of the optimal interactions mentioned above, between NDLD-APS and TEOS at pH of 8.5.
Fig. 5E–I shows the N2 adsorption–desorption isotherms of the nanoparticles, and pore size distributions of the calcined samples, while their porous properties are summarized and shown in Table 2. All of the materials fitted well with type IV isotherms according to the IUPAC classifications, which is associated with the formation of mesopores. Specifically, samples HMSN10-A and -B are characterized by a type H3 hysteresis cycle that may be attributed to the filling of the hollow cavity, while samples HMS10-C, -D and -E exhibit a reduced (or no) hysteresis cycle in the mesoporous range, which indicates the absence of inner hollows. The BET surface area (SBET) in the reference material, HMSN10-A, has a low value of 140 m2 g−1. However, this value is increased both when the synthesis was carried out in a more diluted surfactant solution (HMSN10-B sample), and also when pH increases from 8.5 up to 9.5 (HMSN10-C, HMSN10-D and HMSN10-E samples). It is worth noting that the sample with the highest SBET, HMSN10-C material, reaches a value of 300 m2 g−1. This result is likely due to the improved interaction between DSDA and APS when the pH of the medium synthesis is adjusted to 8.5, as explained above. On the other hand, insets in Fig. 5 show that the nanoparticles contain a narrow pore size distribution. In all samples using NDLD as the surfactant, the mesopores have pore diameters ranging between 2 and 3.3 nm.
The C and N atomic distribution in the L-Dopa@MSN was confirmed by advanced STEM-XEDS analysis. Fig. 6B and D show that regardless of the approach chosen for synthesizing L-Dopa@MSN (NOLD or NDLD surfactants), the C and N atoms are successfully incorporated inside the silica nanoparticles for both HMSN18-A and HMSN10-C materials (selected to represent each NP material). Indistinctly, for both systems, C and N seems to be uniformly dispersed inside the porous network of the materials. Additionally, the high angle annular dark field images, in Fig. 6A and C, confirmed the elongated and spherical morphology of the HMSN18-A and HMSN10-C nanoparticles respectively, as well as the remarkable porosity envisioned from the contrast between the dark and clear forms. Radial mesopores are clearly observed in the sample HMSN10-C, and a denser clear core in the centre of all the nanoparticles is also detected. Otherwise, a completely porous shell evidenced by dark dots, indicative of mesopores throughout the whole shell covering the elongated nanoparticles, is clearly seen in sample HMSN18-A (red arrows in Fig. 6A).
 | ||
| Fig. 6 STEM images and elemental analysis of Si, C and N in the samples HMSN10-C (A and B) and HMSN18-B (C and D). The scale bar is 50 nm in (A and C) and 100 nm in (B and D). | ||
3.4. L-Dopa release from MSNs
The release studies were carried out at 37 °C in both gastric and small intestinal simulating fluids, since orally ingested MSN would stay in the stomach for 2–4 hours before entering in the duodenum (∼1 h) and reaching the intestinal environment (>8 h). Although the HMSN could remain in the intestine for a longer digestion time – which can be anywhere from a few hours to several days – a time of two days was set as a regular digestion time. After release from the DSDA and cleavage of the amide,41 the DSDA renders decanoic or oleic acid and dopamine precursor drug, L-Dopa, separately, in equimolar amounts. The L-Dopa release concentration, with respect to the total amount of HMSN material from the nanoparticles, was plotted versus the release time, and thus the L-Dopa release profile results are shown in Fig. 7. It can be observed that at both conditions, both the L-Dopa release and the release rate from HMSN-10-C material are higher than those of the HMSN-18-B. In addition, all the experiments show that the total release and release rate are significantly lower in gastric fluid (acid conditions) than in intestinal fluid (neutral media).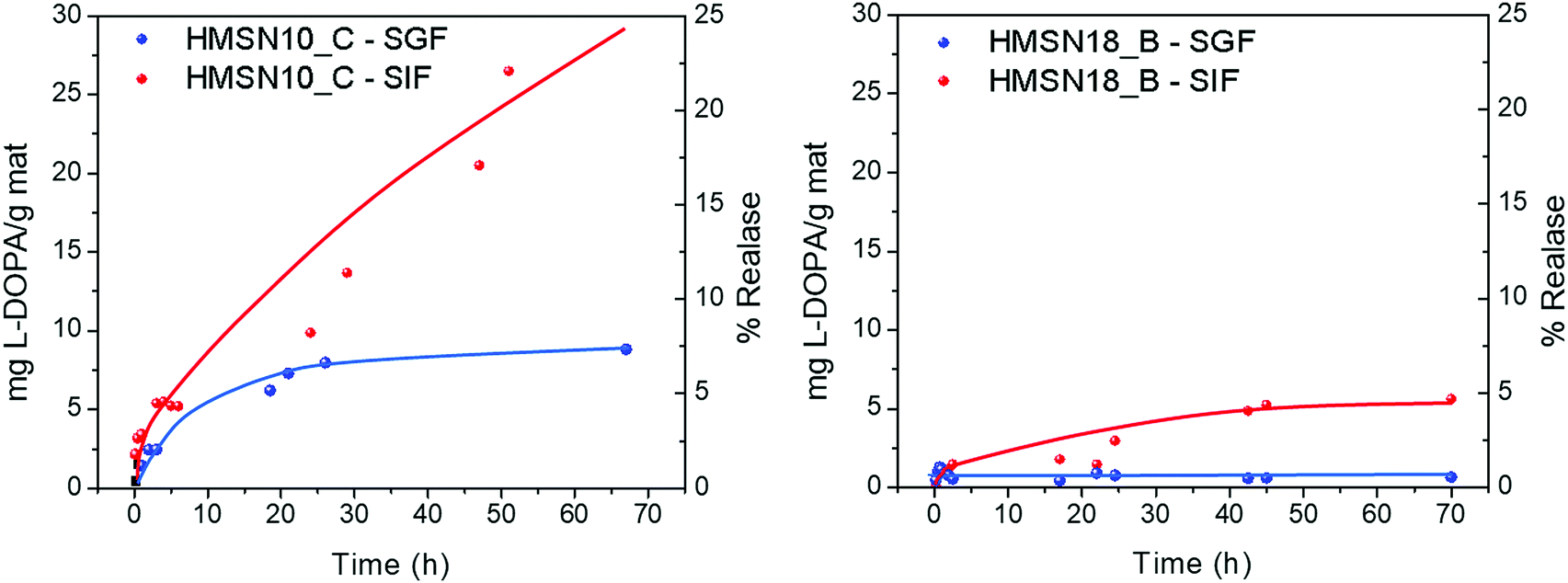 | ||
| Fig. 7 L-Dopa release as a function of time from the HMSNs synthetized using (left) NDLD (HMSN-10-C sample) and (right) NOLD (HMSN-18-B sample) as surfactants. | ||
At pH 1.2, the profiles of the L-Dopa release from HMSN using NDLD and NOLD as the surfactant, HMSN10-C and HMSN-18-B respectively, exhibited an almost negligible L-Dopa release. In this gastric or stomach media, a mass of 2.5 mg g−1 and 0.5–0.75 mg g−1 is released after the first 3 hours from the material HMSN-10-C and HMSN18-B, respectively. This is probably because the larger size of the DSDA NOLD compared to the DSDA NDLD with very similar pore sizes (average pore size of 3.5 vs. 2.4 nm) presents greater steric restrictions for intraparticular diffusion to occur from inside the pores to outside. Moreover, the nonpolar nature of both DSDA type L-Dopa surfactants, and particularly the NOLD composed of long oleyl fatty acid chains, hinders the release capacity from HMSN hosts to the aqueous simulated body fluid system. Additionally, zeta-potential measurements at pH 1.2 show that the surface charges of both samples, HSMN10-C and HMSN18-B (after calcination), exhibit an electropositive nature (10 and 17 mV, respectively). However, both DSDA, NDLT and NOLD, present different surface charges (2 and −6 mV, respectively). The similar electropositive nature of HMSN10-C and NDLD results in repulsion of the NDLD from HMSN10-C, and as a result the drug release capacity increases. Conversely, HMSN18-B and NOLD exhibited opposite charges, and therefore higher surface electrostatic interactions between them, which results in lesser NOLD DSDA release capacities.
As the pH of the medium increases (pH of 7.4), simulating the intestinal conditions, the global interaction between the DSDA and HMSN hosts becomes weak and the release of L-Dopa commences. The experimental results show that better L-Dopa release capacities are exhibited at higher pH values, which can be observed in a remarkable linear delivery of L-Dopa within the first 48 hours compared to a pH of 1.2. The mass of L-Dopa released is 5.4 and 1.5 mg g−1 after the first 3 hours, 9.9 and 3.0 mg mg−1 after 24 hours, and 26.5 and 5.3 mg g−1 after 48 hours, for HMSN10-C and HMSN18-B, respectively. Again, the L-Dopa released from HMSN materials is mediated by the influence of surface chemical properties. The release performances at pH 7.4 are increased. A likely interpretation is that electrostatic repulsions exist between DSDAs and HMSN adsorbents. When the pH increases, the electronegative nature of both HMSN10-C and NDLD DSDA (−14 and −40 mV, respectively) results in the repulsion of the L-Dopa from the HMSN10-C and NOLD DSDA (−20 and −55 mV, respectively) hosts, and therefore, the release capacity increases compared to a pH of 1.2. However, the L-Dopa released is mainly dictated by the solubility of the different DSDAs in the simulated body fluid system, clearly lower for the NOLD DSDA surfactant (0.0108 and 30 mg mL−1 at pH = 7.4 for NOLD and NDLD, respectively) that makes the L-Dopa release slower than that of the HSMN10-C.
The results obtained confirm that, ideally, almost no release occurs at a gastric pH of 1.2, while at intestinal pH, the release of L-Dopa from HMSNs occurs in a continuous and sustained manner, without reaching any plateau and releasing only 25% of its capacity after 48 hours, which makes these materials promising for their application in Parkinson's disease.
4. Conclusions
We have found that it is possible to synthesize MSNs with inherent therapeutic activity through the concept of drug-structure-directing agents (DSDAs) containing L-Dopa, central for the treatment of PD. L-Dopa was modified by amidation with fatty acids to build anionic surfactants that were able to form micelles as templates for the assembly of inorganic precursors to form the silica framework, thus addressing the structure of the MSN nanocarriers. The obtained MSNs showed different shapes, elongated or spherical, and achieving sizes ranging from 100 nm to 1 μm and with the adequate internal porosity provided by the DSDA containing L-Dopa. L-Dopa modified with decanoyl chloride yielded N-decanoyl-L-Dopa (NDLD) as DSDA, that allowed the formation of spherical MSNs with either solid with radial mesopores and a porous shell or hollow-shell with inside large void cavities. Otherwise, L-Dopa modified with oleyl chloride yielded N-oleyl-L-Dopa (NOLD) as DSDA that facilitated the formation of elongated MSNs characterized by long hollows covered by a mesoporous shell. The materials thus synthesized were proved to be pH responsive drug delivery systems. We simulated the DSDA containing L-Dopa in vitro release from MSNs, considering different gastrointestinal conditions after oral drug delivery. The results showed hardly any release for gastric conditions of pH 1.2, avoiding premature delivery in the stomach typical of conventional MSNs, while for intestinal conditions of pH 7.4, the release of L-Dopa occurred in a continuous and sustained manner, demonstrating that these MSNs could be useful as programmed oral L-Dopa release nanosystems for the treatment of Parkinson's disease.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The financial support of the Spanish government (Grant No. CTQ2014-57858-R and CTQ2017-88642-R) is gratefully acknowledged.References
- UNESCO, “Media Services,” UNESCO, 12 07 2016. [Online]. Available: http://www.unesco.org/new/en/media-services/single-view/news/nanotechnology_is_a_growing_research_priority/ [accessed 12 02 2020].
- Global Innovation Index, “Global Innovation Index 2019: Creating Healthy Lives – The Future of Medical Innovation,” Global Innovation Index, 2019.
- Grand View Research, “Nanomedicine Market Analysis By Products, (Therapeutics, Regenerative Medicine, Diagnostics), By Application, (Clinical Oncology, Infectious diseases), By Nanomolecule (Gold, Silver, Iron Oxide, Alumina), & Segment Forecasts, 2018–2025,” Grand View Research, 2017.
- S. M. P. Anu Mary Ealias, A review on the classification, characterisation, synthesis of nanoparticles and their application, in IOP Conference Series: Materials Science and Engineering, 2017 Search PubMed.
- S. Hasan, Res. J. Recent Sci., 2015, 4, 9 Search PubMed.
- R. K. Kankala, Y.-H. Han, J. Na, C.-H. Lee, Z. Sun, S.-B. Wang, T. Kimura, Y. S. Ok, Y. Yamauchi, A.-Z. Chen and K. C.-W. Wu, Adv. Mater., 2020, 32, 1907035 CrossRef CAS PubMed.
- E. Doustkhah, J. Lin, S. Rostamnia, C. Len, R. Luque, X. Luo, Y. Bando, K. C.-W. Wu, J. Kim, Y. Yamauchi and Y. Ide, Chem. – Eur. J., 2019, 25, 1614 CrossRef CAS PubMed.
- J. Wang, Y. Xu, B. Ding, Z. Chang, X. Zhang, Y. Yamauchi and K. C.-W. Wu, Angew. Chem., Int. Ed., 2018, 57, 2894 CrossRef CAS PubMed.
- R. A. García-Muñoz, V. Morales, M. Linares, P. E. González, R. Sanz and D. P. Serrano, J. Mater. Chem. B, 2014, 2, 7996 RSC.
- I. I. Slowing, J. L. Vivero-Escoto, B. G. Trewyn and V. S.-Y. Lin, J. Mater. Chem., 2010, 20, 7924 RSC.
- J. Andersson, J. Rosenholm, S. Areva and M. Linden, Chem. Mater., 2004, 16, 4160 CrossRef CAS.
- S.-W. Song, K. Hidajat and S. Kawi, Langmuir, 2005, 21, 9568 CrossRef CAS PubMed.
- F. Chen, H. Hong, S. Shi, S. Goel, H. F. Valdovinos, R. Hernandez, C. P. Theuer, T. E. Barnhart and W. Cai, Sci. Rep., 2014, 4, 5080 CrossRef CAS PubMed.
- V. Morales, M. N. Idso, M. Balabasquer, B. F. Chmelka and R. A. García-Muñoz, J. Phys. Chem. C, 2016, 120(30), 16887 CrossRef CAS.
- R. A. García-Muñoz, V. Morales, M. Linares, P. E. González, R. Sanz and D. P. Serrano, J. Mater. Chem. B, 2014, 2(45), 7996 RSC.
- D. Arcos and M. Vallet-Regí, Acta Mater., 2013, 61, 890 CrossRef CAS.
- P. P. Pednekar, S. C. Godiyal, K. R. Jadhav and V. J. Kadam, Mesoporous silica nanoparticles: a promising multifunctional drug delivery system, in Nanostructures for Cancer Therapy, Elsevier, 2017, vol. 3, p. 593 Search PubMed.
- Y. Wang, B. Li, L. Zhang and H. Song, Langmuir, 2013, 29, 273 Search PubMed.
- V. Morales, A. Martín, J. Ortiz-Bustos, R. Sanz and R. A. García-Muñoz, J. Mater. Sci., 2019, 54, 11635 CrossRef CAS.
- J. Ortiz-Bustos, A. Martín, V. Morales, R. Sanz and R. A. García-Muñoz, Microporous Mesoporous Mater., 2017, 240, 236 CrossRef CAS.
- A. Martín, V. Morales, J. Ortiz-Bustos, M. Pérez-Garnes and L. F. Bautista, Microporous Mesoporous Mater., 2018, 262, 23 CrossRef.
- J. Zhang, Z.-F. Yuan, Y. Wang, W.-H. Chen, G.-F. Luo, S.-X. Cheng, R.-X. Zhuo and X.-Z. Zhang, J. Am. Chem. Soc., 2013, 135, 5068 CrossRef CAS PubMed.
- A. Martín, R. A. García, D. S. Karaman and J. M. Rosenholm, J. Mater. Sci., 2014, 49, 1437 CrossRef.
- J. Zhang, D. Desai and J. M. Rosenholm, Adv. Funct. Mater., 2013, 24(16), 2352 CrossRef.
- J. Lu, E. Choi, F. Tamanoi and J. I. Zink, Small, 2008, 4, 421 CrossRef CAS PubMed.
- T. D. NguyenKen, C.-F. Leung, M. Liong, C. D. Pentecost, J. F. Stoddart and J. I. Zink, Org. Lett., 2006, 8(15), 3363 CrossRef PubMed.
- C. Park, K. Oh, S. L. Dr and C. K. Prof, Angew. Chem., Int. Ed., 2007, 46(9), 1455 CrossRef CAS PubMed.
- M. Gisbert-Garzarán, M. Manzano and M. Vallet-Regí, Bioengineering, 2017, 4(3), 2017 Search PubMed.
- R. Casasús, M. D. Marcos, R. Martínez-Máñez, J. V. Ros-Lis, J. Soto, L. A. Villaescusa, P. Amorós, D. Beltrán, C. Guillem and J. Latorre, J. Am. Chem. Soc., 2004, 126(28), 8612 CrossRef PubMed.
- E. Ruiz-Hernández, A. Baeza and M. Vallet-Regí, ACS Nano, 2011, 5(2), 1259 CrossRef PubMed.
- Z. Li, D. L. Clemens, B.-Y. Lee, B. J. Dillon, M. A. Horwitz and A. J. I. Zink, ACS Nano, 2015, 9(11), 10778 CrossRef CAS PubMed.
- M. Frasconi, Z. Liu, J. Lei, Y. Wu, E. Strekalova, D. Malin, M. W. Ambrogio, X. Chen, Y. Y. Botros, V. L. Cryns, J.-P. Sauvage and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135(31), 11603 CrossRef CAS PubMed.
- M. Chen, X. He, K. Wang, D. He, S. Yang, P. Qiu and S. Chen, J. Mater. Chem. B, 2014, 2, 428 RSC.
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2(5), 889 CrossRef CAS PubMed.
- L. Sun, X. Zhang, C. Zheng, Z. Wu and C. Li, J. Phys. Chem. B, 2013, 117, 3852 CrossRef CAS PubMed.
- W. Wang, P. Wang, X. Tang, A. A. Elzatahry, S. Wang, D. Al-Dahyan, M. Zhao, C. Yao, C.-T. Hung, X. Zhu, T. Zhao, X. Li, F. Zhang and D. Zhao, ACS Cent. Sci., 2017, 3(8), 839 CrossRef CAS PubMed.
- W. Xie, W.-W. Deng, M. Zan, L. Rao, G.-T. Yu, D.-M. Zhu, W.-T. Wu, B. Chen, L.-W. Ji, L. Chen, K. Liu, S.-S. Guo, H.-M. Huang, W.-F. Zhang, X. Zhao, Y. Yuan, W. Dong, Z.-J. Sun and W. Liu, ACS Nano, 2019, 13, 2849 CrossRef CAS PubMed.
- D. Nie, Z. Dai, J. Li, Y. Yang, Z. Xi, J. Wang, W. Zhang, K. Qian, S. Guo, C. Zhu, R. Wang, Y. Li, M. Yu, X. Zhang, X. Shi and Y. Gan, Nano Lett., 2020, 20, 936 CrossRef CAS PubMed.
- W.-H. Chen, G.-F. Luo, W.-X. Qiu, Q. Lei, L.-H. Liu, S.-B. Wang and X.-Z. Zhang, Biomaterials, 2017, 117, 54 CrossRef CAS PubMed.
- C. A. Stewart, Y. Finer and B. D. Hatton, Sci. Rep., 2018, 8, 895 CrossRef PubMed.
- V. Morales, M. Gutiérrez-Salmerón, M. Balabasquer, J. Ortiz-Bustos, A. Chocarro-Calvo, C. García-Jiménez and R. A. García-Muñoz, Adv. Funct. Mater., 2016, 26(40), 7291 CrossRef CAS.
- V. Morales, M. Pérez-Garnes, M. Balabasquer, J. González-Casablanca and R. A. García-Muñoz, Microporous Mesoporous Mater., 2018, 264, 43 CrossRef CAS.
- M. Pérez-Garnes, M. Gutiérrez-Salmerón, V. Morales, A. Chocarro-Calvo, R. Sanz, C. García-Jiménez and R. A. García-Muñoz, Mater. Sci. Eng., C, 2020, 112, 110935 CrossRef PubMed.
- P. Johns, Parkinson's disease in Clinical Neuroscience, Churchill Livingstone, 2014, vol. 13, p. 163 Search PubMed.
- D. G. Waller and A. P. Sampson, Extrapyramidal movement disorders and spasticity, in Medical Pharmacology & Therapeutics (Fifth Edition), Elsevier, 2018, vol. 24, p. 325 Search PubMed.
- S. Fahn, J. Jankovic, M. Hallett and P. Jenner, Medical Treatment of Parkinson Disease in Principles and Practice of Movement Disorders, Churchill Livingstone, 2007, vol. 6, p. 129 Search PubMed.
- S. Swar, V. Máková and I. StiborSwar, Materials, 2019, 12, 3202 CrossRef CAS PubMed.
- T. López, D. Esquivel, G. Mendoza-Díaz, E. Ortiz-Islas, R. D. González and O. Novaro, Mater. Lett., 2015, 161, 160 CrossRef.
- G. R. Bardajee, N. Khamoushi, S. Nasri and C. Vancaeyzeele, Int. J. Biol. Macromol., 2020, 153, 180 CrossRef CAS PubMed.
- F. Trotta, F. Caldera, R. Cavalli, M. Soster, C. Riedo, M. Biasizzo, G. U. Barretta, F. Balzano and V. Brunella, Expert Opin. Drug Delivery, 2016, 13, 1671 CrossRef CAS PubMed.
- B. A. Sabel, P. Dominiak, W. Hauser, M. J. During and A. Freese, Ann. Neurol., 1990, 28, 714 CrossRef CAS PubMed.
- L. Han, C. Gao, X. Wu, Q. Chen, P. Shu, Z. Ding and S. Che, Solid State Sci., 2011, 13, 721 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb00481f |
| This journal is © The Royal Society of Chemistry 2021 |