
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSystematic study of SYBR green chromophore reveals major improvement with one heteroatom difference†
Ville K.
Saarnio
*,
Johanna M.
Alaranta
and
Tanja M.
Lahtinen
 *
*
Department of Chemistry, Nanoscience Center, University of Jyväskylä, Jyväskylä FI-40500, Finland. E-mail: tanja.m.lahtinen@jyu.fi
First published on 8th April 2021
Abstract
Five nucleic acid binding cyanine dyes were synthesized and their photophysical properties were evaluated. Changing a single heteroatom in the chromophore causes major differences both in brightness and photostability between the dyes. With such alteration, the brightness of the chromophore increased two-fold compared to the one found in SYBR Green I.
Detection of nucleic acids using fluorescent probes is an important tool for cell and molecular biologists in many different applications.1 These include visualising DNA using gel electrophoresis,2 and imaging RNA in microscopy,3 among others. Monomethine cyanine dyes are widely used for these purposes due to their turn-on fluorescence, selective to binding to double stranded nucleic acids.4 The fluorescence light up is induced by rigidification of the chromophore after intercalating between base pairs of a nucleic acid.5,6 As such, dyes with this type of binding offer highly selective sensing of nucleic acids in cells over other biomolecules. This class of nucleic acid stain has been studied vigorously7–9 and as a result, the SYBR family of fluorescent probes has become a popular choice for their sensing.10–12 They can be employed for general visualization of nucleic acids, from electrophoresis gels13 to microscopy applications.14 There are a few attributes that such fluorescent probes need to exhibit to be viable for sensing applications. These include high brightness combined with low background emission, high photostability under prolonged excitation, and low cytotoxicity if applied in vivo. In particular, the demand for increasingly bright nucleic acid sensors continues to increase, as the ambition for imaging singular biomolecules in a cellular environment grows.15,16
In order to meet this demand, we have synthesized five new cyanine dyes and evaluated their photophysical properties (Chart 1). The synthesis of the compounds was conducted using previously published protocols,17,18 combined with an additional nucleophilic substitution step, as shown in the ESI† Scheme S2. Our previous results indicated significant differences in the dye properties of the heteroatoms in the chromophore;18 in this study, we set out to evaluate those differences. The examined dyes were varied in structure by changing between an oxazole and thiazole moiety and varying the 2-substituent between a mercaptomethyl, dimethylamine or chloride.
 | ||
| Chart 1 Chemical structure of the studied cyanine dyes. | ||
Already the absorption and emission spectra of the synthesized dyes exhibited significant differences (Fig. 1a). The lowest energy absorption maxima of the dyes were found between 455 and 516 nm, differing slightly between solvents. The introduction of third period heteroatoms seems to decrease the absorption energy of the dyes as supported by OxN 2 showing the highest and ThzS 5 the lowest energy of absorption, with other products found half way between the two. The emission spectra behave in a similar manner with emission maxima observed at the region of 492–535 nm when bound to calf thymus DNA (ctDNA) (Fig. 1b). The excitation/emission spectra for individual dyes are presented in the ESI† Fig. S9–S13. This lead to Stoke's shifts from 16 to 28 nm with OxS 3 being responsible for the lowest value. Surprisingly, the presence of dimethylamine substitution (OxN 2 and ThzN 4) seems to increase the Stoke's shift compared to the other products OxCl 1 and ThzS 5.
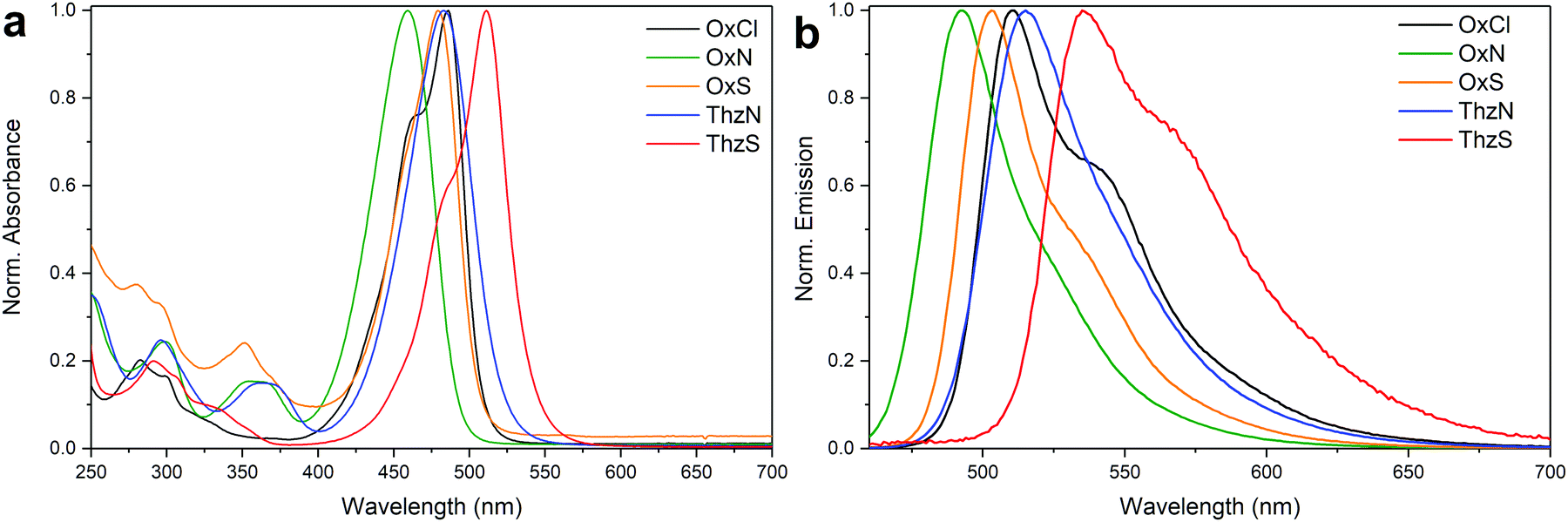 | ||
| Fig. 1 (a) Normalized absorption spectra of the dyes in 19.6 μM concentration in ethanol. (b) Normalized emission spectra of the dyes in 100 μM ctDNA in TE Buffer. | ||
The absorption coefficients (ε) of the dyes were evaluated in three different solvents; tris-EDTA (TE) buffer, ethanol and 100 μM ctDNA in TE buffer (S14–S16, ESI†). No clear relationship between the dye structure and absorptivity was observed, although the OxS 3 displayed significantly lower ε values compared to the other dyes across all media. In the ThzN 4 compound, the given absorptivity was lower by 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1, compared to molecules 1, 2, and 5. Generally, compound 2 showed the highest absorptivity of 73600 M−1 cm−1 in ethanol between the three dyes and comparable values to the two others in aqueous media. In order to evaluate which chromophore offers the best brightness, we determined the quantum yield Φ of all five dyes, when fully bound to ctDNA, using fluorescein as a standard (Fig. S17 and S18, ESI†).19 These values and the other spectral characteristics of the synthesized dyes are presented in Table 1. The benefit of maintaining a chromophore consisting of second period elements becomes apparent once again as the quantum yield of OxN 2 was determined to be 100%. Compared to this result, replacing one of the heteroatoms with sulphur in OxS 3 and ThzN 4 resulted in the quantum yields diminishing significantly, by 24% and 30%, respectively. Ohulchanskyy and co-workers found a similar trend when evaluating a series of xanthene dyes by varying a heteroatom between oxygen, sulphur and selenium in the chromophore.20 In particular, the chlorine atom in the 2-position has a dramatic effect on the quantum yield. The Φ value for OxCl 1 was found to be as low as 18% similar in magnitude to the 14% of two-sulphur-containing ThzS 5. It is possible, that the chlorine (and other halogens) atom offers additional pathways for non-radiative relaxation, perhaps due to the intersystem crossing caused by the heavy-atom effect inflicted by the substituent. Similar observations have been made by Vasilev et al.,21 who synthesized a series of halogen-containing thiazole orange (TO) derivatives, where increasing inclusion of halogens on the chromophore led exclusively to lower quantum yields. In general, the trend towards favouring second period elements to maximize the brightness seems evident among the synthesized dyes.
000 M−1 cm−1, compared to molecules 1, 2, and 5. Generally, compound 2 showed the highest absorptivity of 73600 M−1 cm−1 in ethanol between the three dyes and comparable values to the two others in aqueous media. In order to evaluate which chromophore offers the best brightness, we determined the quantum yield Φ of all five dyes, when fully bound to ctDNA, using fluorescein as a standard (Fig. S17 and S18, ESI†).19 These values and the other spectral characteristics of the synthesized dyes are presented in Table 1. The benefit of maintaining a chromophore consisting of second period elements becomes apparent once again as the quantum yield of OxN 2 was determined to be 100%. Compared to this result, replacing one of the heteroatoms with sulphur in OxS 3 and ThzN 4 resulted in the quantum yields diminishing significantly, by 24% and 30%, respectively. Ohulchanskyy and co-workers found a similar trend when evaluating a series of xanthene dyes by varying a heteroatom between oxygen, sulphur and selenium in the chromophore.20 In particular, the chlorine atom in the 2-position has a dramatic effect on the quantum yield. The Φ value for OxCl 1 was found to be as low as 18% similar in magnitude to the 14% of two-sulphur-containing ThzS 5. It is possible, that the chlorine (and other halogens) atom offers additional pathways for non-radiative relaxation, perhaps due to the intersystem crossing caused by the heavy-atom effect inflicted by the substituent. Similar observations have been made by Vasilev et al.,21 who synthesized a series of halogen-containing thiazole orange (TO) derivatives, where increasing inclusion of halogens on the chromophore led exclusively to lower quantum yields. In general, the trend towards favouring second period elements to maximize the brightness seems evident among the synthesized dyes.
| Dye | OxCl 1 | OxN 2 | OxS 3 | ThzN 4 | ThzN 5 |
|---|---|---|---|---|---|
| λ exc (nm) | 491 | 464 | 487 | 488 | 514 |
| λ emi (nm) | 511 | 492 | 503 | 515 | 535 |
| Stoke's shift (nm) | 20 | 28 | 16 | 27 | 21 |
| ε max, TE (× 103 M−1 cm−1) | 59.5 ± 0.4 | 56.5 ± 0.8 | 16.0 ± 0.3 | 35.3 ± 1 | 47.5 ± 2 |
| ε max, EtOH (× 103 M−1 cm−1) | 60.7 ± 0.9 | 73.6 ± 0.6 | 25.7 ± 0.3 | 56.9 ± 0.3 | 65.4 ± 1.1 |
| ε max, ctDNA (× 103 M−1 cm−1) | 42.0 ± 0.3 | 47.3 ± 0.2 | 18.0 ± 0.3 | 36.5 ± 0.3 | 49.1 ± 0.1 |
| Quantum yield Φ (%) | 18 | 100 | 76 | 68 | 14 |
| Brightness (M−1 cm−1) | 7600 | 47300 |
13700 |
24800 |
6900 |
Next, to establish the stability of the dyes in practical use, we exposed them to a microscope light source, i.e. a 3300 K halogen lamp of 22 mW cm−2 light intensity. The use of such a light source not only provided a sufficient timeframe for observing dye decomposition but also gave a reasonable source of maximal excitation light that the dyes would experience in daily use. However, this also led to some differences in the amount of light absorbed depending on the absorption maximum of the dye, as demonstrated in Fig. S19 (ESI†). The photobleaching of the dyes was then followed over time, by measuring dye absorbance at different points of time and fitting exponential decay functions to observed absorption maxima, as shown in Fig. 2a and b. While measuring fluorescence would have granted higher sensitivity to the photobleaching, the non-fluorescent state of the dyes in TE Buffer (Fig. 2a) would have gone unmeasured in addition to a solution of ctDNA (Fig. 2b). We observed that the differences in the dye half-lives are remarkable between each other and different media (Table 2). Surprisingly, the OxCl 1 dye exhibits the highest photostability among the examined molecules in both media. The ThzN 4 on the other hand was highly unstable compared to the other dyes. In general, the dyes have significantly different half-lives free in solution and bound to ctDNA. As expected, the dyes decompose faster in the TE buffer compared to the ctDNA solution.
 | ||
| Fig. 2 Exponential decay functions fitted to the relative decrease of the initial absorption at the maximum wavelength over time in (a) TE Buffer and (b) 200 μM ctDNA. Data can be derived from Fig. S20 and S21 (ESI†). (c) Fluorescence emission maxima plotted with increasing dye concentration in 0.52 μM ctDNA solution at room temperature derived from Fig. S22 (ESI†). | ||
| Dye | OxCl 1 | OxN 2 | OxS 3 | ThzN 4 | ThzS 5 |
|---|---|---|---|---|---|
| K a (× 106 M−1) | 1.4 ± 0.04 | 8.5 ± 1.5 | 4.6 ± 1 | 5.8 ± 0.4 | 2.3 ± 0.1 |
| n (bp) | 4.3 ± 0.3 | 3.4 ± 0.4 | 1.6 ± 0.1 | 2.1 ± 0.12 | 1.7 ± 1 |
| ΔG0 (kJ mol−1) | −34.6 | −39.0 | −37.5 | −38.1 | −35.8 |
| t 1/2, TE (h) | 6.3 | 2.9 | 0.6 | 0.5 | 2.4 |
| t 1/2, 200 μM ctDNA (h) | 35.9 | 32.4 | 29.8 | 1.6 | 9.0 |
Free in solution, the observed values varied between 0.5–6.3 h, increasing to 1.6–36 h when DNA bound. Interestingly, the oxazole moiety benefits from binding to nucleic acids notably more, compared to thiazole containing products. In particular, OxS 3 that decomposes rapidly in TE Buffer, experiences a 50-fold increase in its half-life after binding to its target. The thiazole derivatives 4 and 5 only stabilized by 3.2-fold and 3.75-fold, respectively. While dyes 1 and 2 were already the most stable in TE buffer, the enhancements to photostability were further improved by 5.7-fold and 11.2-fold, respectively. The knowledge of stability in both media is highly important, as the dyes are constantly interchanging between bound and unbound states in solution.
To better understand the differences between dyes in their nucleic acid binding, we conducted two fluorescence titration experiments; one with a limited amount and the other with a high excess of ctDNA available for binding (Fig. 2c and Fig. S24b, ESI† respectively). From the relative fluorescence between the two experiments, it was possible to determine both the binding constant (Ka) and the number of nucleic acid base pairs required to accommodate a single dye molecule (n). This was made possible by employing the theoretical model of DNA–protein interactions developed by von Hippel and McGhee elucidated in more detail in our previous work.18,22 The processing of the obtained data produced a Scatchard plot (Fig. S24c, ESI†) that enabled the fitting of the McGhee and von Hippel equation for non-cooperative binding to obtain values for Ka, n and subsequent determination of the Gibbs free energy of binding involved (ΔG0, Table 2). The currently accepted model of nucleic acid binding for SYBR family (and TO related) compounds is, that the quinoline moiety intercalates between base pairs, and the indole moiety binds through electrostatic interactions to the phosphate group while the 2-substituents settle along the minor/major groove.23,24
As the synthesized dyes had a high structural similarity, we did not expect major differences among the dyes to be observed in the binding values. As expected, the binding site size between dimethylamine and methylthiol substituted dyes differs, as the extra methyl group on the amine requires additional space in binding. The effects to the binding energy of this structural change are similar between the two comparing pairs (2vs.3 and 4vs.5); this change leads to a decrease of 1.5 kJ mol−1 and 2.3 kJ mol−1, respectively. Curiously, also the changes from oxazole to thiazole had a significant effect on the binding energy. Comparing the binding energies between the two derivative pairs (2 and 4, 3 and 5) the ΔG0 is found to differ by 0.9 kJ mol−1 and 1.7 kJ mol−1, respectively. These changes can be contributed to the change of both to the electronegativity of the heteroatom, as well as to the weak binding provided by the additional methyl group in the first pair. In the case of the latter comparison pair, the differences are solely heteroatom derived. Subtracting the difference of the latter pair from the former yields 0.6 kJ mol−1 in both cases, giving a realistic estimate of the contribution of a single methyl group to the binding energy. However, another series aimed towards confirming that this estimate should be conducted. The effect on the binding energy due to the change from thiazole to oxazole can be explained by the electronegativities of the other heteroatom present in the 2-position. Oxygen, nitrogen and sulphur have electronegativities of 3.44, 3.04, and 2.58 in the Pauling scale, respectively. An oxygen atom in the oxazole is able to draw a greater amount of electron density from a sulphur atom vs. a nitrogen that is more negatively charged. As the DNA bases are electronegative in nature, it is logical that depleting the intercalating molecule of electron density would increase the binding affinity, as was observed. Furthermore, by examining the electronegativity difference between the heteroatoms in the comparison of 2 and 4, as well as 3 and 5, we found a relation to the binding energy differences between the two pairs. Multiplying the 1.7 kJ mol−1 with the electronegativity difference gives 0.8 kJ mol−1, very close to the 0.9 kJ mol−1 value of the 2 and 4 pair. The low binding energy determined for OxCl 1 also supports this notion, although part of this might be due to chlorine hindering the intercalation itself, as observed with the high n value for the compound. The idea of manipulating electron density in the intercalating part of the molecule to increase target binding warrants high interest when designing new molecules in the future.
To summarize, in this work, we set out to synthesize five nucleic acid binding cyanine dyes with different heteroatomic constitutions in order to evaluate their photophysical properties. The molecules were designed so that the effects of single atom differences in the chromophore could be evaluated. In addition to the determination of the brightness related parameters, also the photostability, binding energy, and binding site size with ctDNA were established for all dyes. Among the evaluated products one dye, OxN 2, showed outstanding performance compared to the others. These properties include brightness of 47300 M−1 cm−1, −39 kJ mol−1 binding energy, and over 32 h photobleaching half-life in complex with ctDNA, coupled with a 28 nm Stoke's shift. Generally, the introduction of second period heteroatoms (S and Cl) seemed to decrease the performance in all aspects compared to OxN 2. In particular, the introduction of the chlorine (1) or two sulphur atoms (5) had a dramatic effect on the quantum yield of the dyes, albeit the photostability of 1 was the best among the compounds herein. The binding studies on the synthesized molecules indicated a benefit from introducing more electronegative elements to the intercalating moiety in the dyes. As researchers aspire towards the detection of diminishing amounts of nucleic acids in many biological contexts, the demand for increasingly sensitive probes rises. While another approach to this problem has been preparing artificial nucleosides with fluorescent properties, this method requires always synthetically altering the nucleic acid strand in question.25 Intercalating turn-on fluorescent probes offers a universal solution for all solution based nucleic acid detection. The molecules presented in this work should be applicable in a similar manner to the SYBR family dyes in imaging and otherwise. This is why such probes with continuously improving photophysical properties are highly enticing and hence, understanding the underlying patterns leading to the best possible results should be of interest to every chemist, biochemist and biologist involved in their use.
This work was supported by Jane and Aatos Erkko Foundation project Novel Probes for Discovering Anti-Virals and Vilho, Yrjö and Kalle Väisälä Fund. We are thankful for the assistance provided by Dr Elina Kalenius with mass spectroscopy. The authors thank Prof. Kari Rissanen for his contribution to the fruitful discussion.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. J. Kricka, Ann. Clin. Biochem., 2002, 39, 114–129 CrossRef CAS PubMed.
- A. M. Haines, S. S. Tobe, H. J. Kobus and A. Linacre, Electrophoresis, 2015, 36, 941–944 CrossRef CAS PubMed.
- S. Ray, J. R. Widom and N. G. Walter, Chem. Rev., 2018, 118, 4120–4155 CrossRef CAS PubMed.
- A. Bruce, in Topics in Heterocyclic Chemistry, ed. L. Strekowski, Springer, Pittsburgh, 14th edn, 2008, pp. 11–29 Search PubMed.
- H. Ihmels and D. Otto, Top. Curr. Chem., 2005, 258, 161–204 CrossRef CAS.
- V. Karunakaran, J. L. Pérez Lustres, L. Zhao, N. P. Ernsting and O. Seitz, J. Am. Chem. Soc., 2006, 128, 2954–2962 CrossRef CAS PubMed.
- N. I. Shank, H. H. Pham, A. S. Waggoner and B. A. Armitage, J. Am. Chem. Soc., 2013, 135, 242–251 CrossRef CAS PubMed.
- H. J. Karlsson, M. H. Bergqvist, P. Lincoln and G. Westman, Bioorg. Med. Chem., 2004, 12, 2369–2384 CrossRef CAS PubMed.
- T. G. Deligeorgiev, N. I. Gadjev, A. A. Vasilev, V. A. Maximova, I. I. Timcheva, H. E. Katerinopoulos and G. K. Tsikalas, Dyes Pigm., 2007, 75, 466–473 CrossRef CAS.
- X. Jin, S. Yue, V. L. Singer, L. J. Jones, M. P. Beaudet, C.-Y. Cheung and R. S. Tuma, Anal. Biochem., 2003, 288, 278–288 Search PubMed.
- H. Zipper, H. Brunner, J. Bernhagen and F. Vitzthum, Nucleic Acids Res., 2004, 32, e103 CrossRef.
- W. E. Evenson, L. M. Boden, K. A. Muzikar and D. J. Oleary, J. Org. Chem., 2012, 77, 10967–10971 CrossRef CAS.
- L. J. Jones, S. T. Yue, C. Y. Cheung and V. L. Singer, Anal. Biochem., 1998, 265, 368–374 CrossRef CAS PubMed.
- T. Suzuki, K. Fujikura, T. Higashiyama and K. Takata, J. Histochem. Cytochem., 1997, 45, 49–53 CrossRef CAS PubMed.
- A. S. Boutorine, D. S. Novopashina, O. A. Krasheninina, K. Nozeret and A. G. Venyaminova, Molecules, 2013, 18, 15357–15397 CrossRef CAS PubMed.
- A. D. Cawte, P. J. Unrau and D. S. Rueda, Nat. Commun., 2020, 11, 1283 CrossRef CAS PubMed.
- L. Ying, US Pat., US20130137875A1, 2013 Search PubMed.
- V. K. Saarnio, K. Salorinne, V. P. Ruokolainen, J. R. Nilsson, T.-R. Tero, S. Oikarinen, L. M. Wilhelmsson, T. M. Lahtinen and V. S. Marjomäki, Dyes Pigm., 2020, 177, 108282 CrossRef CAS.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535–1550 CrossRef PubMed.
- T. Y. Ohulchanskyy, D. J. Donnelly, M. R. Detty and P. N. Prasad, J. Phys. Chem. B, 2004, 108, 8668–8672 CrossRef CAS.
- A. A. Vasilev, M. I. Kandinska, S. S. Stoyanov, S. B. Yordanova, D. Sucunza, J. J. Vaquero, O. D. Castaño, S. Baluschev and S. E. Angelova, Beilstein, J. Org. Chem., 2017, 13, 2902–2914 CAS.
- J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 469–489 CrossRef CAS.
- C. D. Geddes, A. I. Dragan, J. R. Casas-Finet, M. A. Schenerman, J. B. McGivney, R. J. Strouse, R. Pavlovic and E. S. Bishop, J. Fluoresc., 2012, 22, 1189–1199 CrossRef PubMed.
- A. I. Dragan, J. R. Casas-Finet, E. S. Bishop, R. J. Strouse, M. A. Schenerman and C. D. Geddes, Biophys. J., 2010, 99, 3010–3019 CrossRef CAS PubMed.
- R. T. Ranasinghe and T. Brown, Chem. Commun., 2011, 47, 3717–3735 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis and characterization of the products, raw absorption/fluorescence spectra and insight into the binding characterization. See DOI: 10.1039/d1tb00312g |
| This journal is © The Royal Society of Chemistry 2021 |