
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bi2O3 boosts brightness, biocompatibility and stability of Mn-doped Ba3(VO4)2 as NIR-II contrast agent†
Pascal M.
Gschwend
 a,
Kerda
Keevend
bc,
Marianne
Aellen
d,
Alexander
Gogos
bc,
Frank
Krumeich
e,
Inge K.
Herrmann
bc and
Sotiris E.
Pratsinis
*a
a,
Kerda
Keevend
bc,
Marianne
Aellen
d,
Alexander
Gogos
bc,
Frank
Krumeich
e,
Inge K.
Herrmann
bc and
Sotiris E.
Pratsinis
*a
aParticle Technology Laboratory, Institute of Energy and Process Engineering, Department of Mechanical and Process Engineering, ETH Zürich, Sonneggstrasse 3, CH-8092 Zurich, Switzerland. E-mail: pratsinis@ethz.ch
bParticles-Biology Interactions, Department Materials Meet Life, Swiss Federal Laboratories for Materials Science and Technology (Empa), Lerchenfeldstrasse 5, CH-9014 St. Gallen, Switzerland
cNanoparticle Systems Engineering Laboratory, Institute of Energy and Process Engineering, Department of Mechanical and Process Engineering, ETH Zürich, Sonneggstrasse 3, CH-8092 Zurich, Switzerland
dOptical Materials Engineering Laboratory, Institute of Energy and Process Engineering, Department of Mechanical and Process Engineering, ETH Zürich, Leonhardstrasse 21, CH-8092 Zurich, Switzerland
eLaboratory for Inorganic Chemistry, Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1, CH-8093 Zurich, Switzerland
First published on 11th March 2021
Abstract
Deep-tissue fluorescence imaging remains a major challenge as there is limited availability of bright biocompatible materials with high photo- and chemical stability. Contrast agents with emission wavelengths above 1000 nm are most favorable for deep tissue imaging, offering deeper penetration and less scattering than those operating at shorter wavelengths. Organic fluorophores suffer from low stability while inorganic nanomaterials (e.g. quantum dots) are based typically on heavy metals raising toxicity concerns. Here, we report scalable flame aerosol synthesis of water-dispersible Ba3(VO4)2 nanoparticles doped with Mn5+ which exhibit a narrow emission band at 1180 nm upon near-infrared excitation. Their co-synthesis with Bi2O3 results in even higher absorption and ten-fold increased emission intensity. The addition of Bi2O3 also improved both chemical stability and cytocompatibility by an order of magnitude enabling imaging deep within tissue. Taken together, these bright particles offer excellent photo-, chemical and colloidal stability in various media with cytocompatibility to HeLa cells superior to existing commercial contrast agents.
Introduction
Luminescent nanoparticles for bioimaging are most attractive for non-invasive acquisition of anatomic and functional information at high spatio-temporal resolution. Deep-tissue luminescence imaging is accomplished by probes with emission wavelengths either in the first1 (650–900 nm) or second2 (1000–1350 nm) near-infrared (NIR) windows. At these wavelengths, light absorption3 and scattering4 are reduced compared to visible light wavelengths enabling improved imaging depth. Moreover, the autofluorescence from endogenous fluorescent molecules in biological tissues is weaker.5 Tsukasaki et al.6 demonstrated how the combination of these properties favorably influences the signal to background ratio, increasing it from 1.3 (emission at 720 nm) up to 7.3 (emission at 1300 nm) in an in vivo imaging setting.An ideal fluorescent probe for deep-tissue imaging should possess high quantum yield (QY), as well as photostability, biocompatibility and chemical stability.7 Furthermore, a large Stokes shift and narrow emission lines hinder reabsorption and emission cross-talk with other emitters.7 Fluorescent contrast agents in the second NIR window include organic molecules (dyes8 and conjugated polymers9) typically suffering from limited photostability,10 and single-walled carbon nanotubes11 (SWCNT) with broad size distribution12 (200–500 nm length) and potentially cell-damaging needle-like structure.7 Furthermore, there are semiconductor quantum dots often consisting of toxic heavy metals (e.g. InAs,13 PbS14), and lanthanide-doped materials limited by the small absorption cross-section,15 which however can be improved by sensitizing via organic dyes16 or co-doping.16,17
An almost unexplored group of materials for NIR-II bioimaging are transition metal doped materials, despite several studies reporting emission above 1000 nm in relatively large crystals (micrometer to millimeter size): divalent (Ni,18 Co, V,19 and Ti20), trivalent (V21), tetravalent (Cr,22V), pentavalent (Cr,23 Mn24), and hexavalent (Mn,25 Fe26) transition metal ions exhibit attractive properties. Especially Mn5+ as an active ion doped into an otherwise NIR-inactive Ba3(VO4)2 matrix is most promising due to its broad excitation band spanning the entire first biological window and its strong and narrow emission peak around 1180 nm.27 Only recently Zhang et al.28 have reported synthesis of submicron particles (mean diameter ∼300 nm) of Ba3(VO4)2 and Ba3(PO4)2 doped with Mn5+ leading to NIR emission, however, the cytocompatibility of these materials remains unexplored, and particle sizes exceed 300 nm, making them unsuitable for intravascular applications such as passive tumor targeting.29 Specifically, particle sizes around 120 nm have been found suitable for tumor imaging,30 as well as metastatic tiny tumor detection and tumor vessel visualization.31 We recently made smaller Ba3(PO4)2:Mn5+ particles (d < 100 nm) with the potential for simultaneous deep-tissue imaging and nanothermometry.32 Despite encouraging results, these particles show partial dissolution (10% in 24 hours) in physiological media such as PBS (phosphate buffered saline), and their cellular internalization has not yet been studied.
Here, we present Ba3(VO4)2:Mn5+ nanoparticles with sizes below 100 nm for the first time made by sterile flame aerosol technology for its close control of product particle characteristics.33 This technology is most effective in overcoming the typical clinical translation barriers,34 such as scalability and reproducibility.35 Most importantly, we demonstrate how co-synthesis with Bi2O3 improves drastically both brightness and stability of Ba3(VO4)2:Mn5+. The resulting particles show high cytocompatibility (HeLa cells) due to reduced ion leaching. By assessing structure–function relationships, we provide important new insights into the mostly unexplored group of Mn5+-doped materials and their suitability for bioimaging.
Results and discussion
Optical properties of Ba3(VO4)2:Mn5+ and influence of Bi2O3 to brightness
Flame spray pyrolysis (FSP) has been employed for synthesis of Ba3(VO4)2 particles doped with Mn5+ at typical production rates of 10 g h−1, which could be scaled up to kg h−1 as shown with a variety of materials.36–38 Following extensive optimization in terms of doping concentration and particle size (Fig. S1–S11, ESI†) Ba3(VO4)2 doped with 0.5 at% Mn5+, Ba3(V0.995Mn0.005O4)2, annealed for 2 h at 600 °C (abbreviated from now on as BaVOMn) was found to be most suitable with a quantum yield (QY) of 10.6 ± 2.1% as powder and 4.6 ± 1.1% in aqueous suspension. The QY was directly measured by an absolute method using an integrating sphere (Fig. S12, ESI†) which avoids the need for reliable standard materials that are scarce in the NIR region. For example, the only available reference-dye for relative QY measurements in the NIR (IR26) has been shown to have a QY that varies by an order of magnitude between different reports.39Fig. 1a shows the excitation (dotted line) and emission spectra (solid line) for BaVOMn. The excitation spectrum features a narrow peak in the UV and a broad one spanning the entire first NIR biological window. There are three spin-allowed transitions from the 3A2 ground state that dominate the excitation and absorption spectra at 341, 654 and 888 nm, as well as a weaker spin-forbidden transition at 777 nm and a charge transfer band (CT) in the UV from the host (details in Fig. S7 and S8, ESI†). In contrast to lanthanide-based materials with narrow excitation peaks,40 the broad absorption band in NIR allows more freedom in the choice of excitation source. For example, 750 or 808 nm lasers can be employed in contrast to 980 nm (a typical excitation wavelength for Yb-based emitters41) to avoid overheating due to the strong optical absorption of water.42
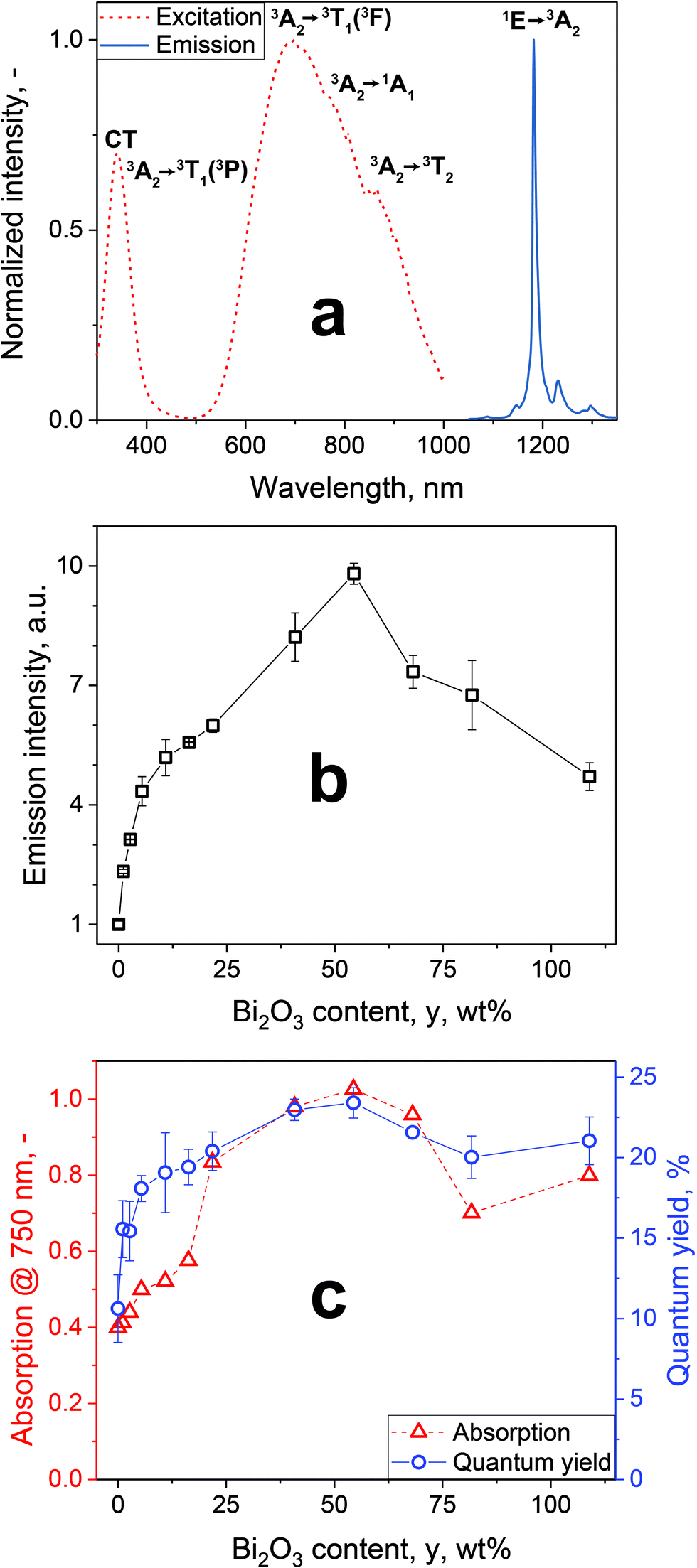 | ||
| Fig. 1 (a) Excitation (λem = 1181 nm, dotted line) and emission spectra (λex = 750 nm, solid line) of Ba3(V0.995Mn0.005O4)2 after annealing for 2 h at 600 °C (BaVOMn). (b) Emission intensity of BaVOMn – y wt% Bi2O3 (annealed 2 h at 600 °C) dispersed in water (0.5 g L−1) normalized to 0 wt% Bi2O3 (λex = 750 nm). (c) Absorption of powders in (b) at 750 nm and quantum yields (λex = 750 nm). | ||
The emission peak is centered around 1181 nm and is assigned to the 1E → 3A2 transition.24 The weak side emissions correspond to vibronic sidebands.43 These emissions lie within the second biological window, therefore allowing deeper tissue penetration compared to the first biological window. The full-width at half-maximum (FWHM) of the main emission line is only 10 nm and thus an order of magnitude lower than for QDs (around 200 nm14). This can be beneficial in avoiding unwanted cross-talk7 between several emitters in a multi-color imaging setting. It is noteworthy that the observed absorption and excitation/emission spectra unambiguously indicate the presence of Mn5+. This is further supported by the blue coloration of the powders (Fig. S6, ESI†), as well as Raman spectral fingerprints characteristic for Mn5+ (Fig. S9, ESI†).
To further increase the emission intensity (i.e. brightness of the particles at identical particle concentration), co-synthesis of BaVOMn with bismuth oxide has been explored, as the latter increases the emission intensity of several phosphors.44,45 A Bi-precursor was added to the precursor solution of BaVOMn prior to its FSP yielding BaVOMn – y wt% Bi2O3 with y = 0 to 108.9 wt%, relative to the BaVOMn mass, where y = 108.9 wt% corresponds to a Ba to Bi molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The fluorescence emission intensities of aqueous dispersions of these particles at constant concentration (0.5 mg ml−1) are shown in Fig. 1b, normalized to that of BaVOMn alone. The presence of Bi2O3 drastically improved the emission intensity of these already bright particles. The highest brightness was observed for 54.4 wt% Bi2O3 that exhibited an almost tenfold higher intensity than pure BaVOMn. The shape of the emission spectra was not affected by the addition of Bi2O3. Note that neither Ba3(VO4)2 (without Mn) – 1 wt% Bi2O3 nor Bi2O3:1% Mn exhibited any luminescence. The brightest sample with 54.4 wt% Bi2O3 was abbreviated with BaVOMn–BiO.
:1. The fluorescence emission intensities of aqueous dispersions of these particles at constant concentration (0.5 mg ml−1) are shown in Fig. 1b, normalized to that of BaVOMn alone. The presence of Bi2O3 drastically improved the emission intensity of these already bright particles. The highest brightness was observed for 54.4 wt% Bi2O3 that exhibited an almost tenfold higher intensity than pure BaVOMn. The shape of the emission spectra was not affected by the addition of Bi2O3. Note that neither Ba3(VO4)2 (without Mn) – 1 wt% Bi2O3 nor Bi2O3:1% Mn exhibited any luminescence. The brightest sample with 54.4 wt% Bi2O3 was abbreviated with BaVOMn–BiO.
With increasing Bi2O3 content, y, the absorption (triangles) at the excitation wavelength of 750 nm was increased (Fig. 1c). The stabilization of Mn5+ has been shown to be difficult, depending e.g. on small changes in host stoichiometry32 or annealing temperature, which was also observed here in samples without Bi2O3 (Fig. S6c, ESI†). The absorption spectra (Fig. S13, ESI†) indicate that the addition of Bi2O3 favors stabilization of Mn5+ in the host material, thereby increasing the absorption peak in the near-infrared. However, this stabilization effect appears to level off for higher Bi2O3 contents, leading to the observed peak at 54.4 wt% Bi2O3 followed by a decrease due to the addition of inactive mass.
Next, the efficiency of these particles is studied in detail with their QY, for different Bi2O3 content, as shown in Fig. 1c (circles) and compared with other reported emitters in this spectral region in Table S1 (ESI†). The QY increases with Bi2O3 content and peaks at 54.4 wt%, similar to absorption. Compared to our previously developed32 Ba3(PO4)2:Mn5+, the NIR absorption of BaVOMn–BiO is more than doubled at comparable QY.
Physicochemical characterization and colloidal stability
Motivated by the improved optical properties of BaVOMn–BiO over BaVOMn, the physicochemical properties of BaVOMn–BiO were studied in detail. The addition of Bi2O3 hardly affected the crystal size but increased the primary size (Fig. S14a, ESI†). Therefore, the annealing conditions were investigated (Fig. S14–S16, ESI†) for the most promising sample according to Fig. 1b and c, BaVOMn–BiO, to find those at which the particles show phase purity yet minor amount of aggregation. Such conditions were found at 550 °C for 30 minutes, with the crystal (dXRD = 58 nm) and primary particle (dBET = 56 nm) sizes close to each other. Due to the smaller crystal size, also the QY of this powder decreased to 8.1 ± 0.5% (2.7 ± 0.3% in water), yet represents a compromise between brightness and suitable particle size (i.e. less than 100 nm).Fig. 2a shows a high-angle annular darkfield scanning transmission electron microscopy (HAADF-STEM) image of BaVOMn–BiO nanoparticles. The Z-contrast reveals inhomogeneities in the particles, evidenced by differences in brightness. The small bright dots (<5 nm) are metallic Bi-domains that formed during imaging (Fig. S17, ESI†). In contrast, the larger inhomogeneities appeared during the thermal treatment and could not be observed in the as-prepared samples (Fig. S18, ESI†). The same particles were analyzed with energy dispersive X-ray spectroscopy (EDX) elemental mapping. Fig. 2b shows overlay of V, Ba and Bi maps (single element maps are shown in Fig. S19, ESI†). The Ba is colocated with V, while Bi is seggregated. Two areas of distinctly high (area 1) and low (area 2) Bi concentrations have been highlighted in Fig. 2b and the corresponding EDX-spectra are shown in Fig. 2c and d, respectively. They clearly confirm the strongly varying ratio of Ba to Bi, however, even in these extreme regions always both elements can be detected. The segregation of Bi-rich areas could potentially be explained by the lower melting point of Bi2O3 (825 °C)46 than Ba3(VO4)2 (1600 °C)47 and thus its higher mobility. The Mn distribution appears to be rather uniform (Fig. S19e, ESI†).
 | ||
| Fig. 2 (a) HAADF-STEM image of annealed BaVOMn–BiO particles reveals inhomogeneities, where the small features (<5 nm) are caused by the e-beam. (b) Elemental mapping of V (dark blue), Ba (light blue) and Bi (magenta), with selected areas of particularly high (area 1, (c)) and low (area 2, (d)) Bi-content. Solid lines represent fitted EDX peaks. | ||
Fig. 3a shows the XRD patterns of both powders annealed at 600 °C. For BaVOMn, all peaks can be attributed to rhombohedral Ba3(VO4)2 (circles), while additional peaks corresponding to tetragonal Bi2O3 (triangles) can be observed in BaVOMn–BiO, in line with the observed presence of Bi rich areas in the elemental mapping (Fig. 2b). The XRD analysis of varying Bi2O3 concentration (Fig. S20, ESI†) reveals Bi2O3 peaks for y = 5.4 wt% that increase in intensity with increasing Bi content. For lower y either the signal is below detection limit or Bi3+ substitutes the 8-coordinated Ba2+ (ionic radii: Ba2+ = 142 pm, Bi3+ = 117 pm). Analysis of the unit cell volume (Fig. S21, ESI†) showed a slight increase from 0 to 10.9 wt% Bi2O3 leveling off above that, indicating potential incorporation of Bi. This is supported by the observed Bi-presence from electron microscopy even in barium rich areas (Fig. 2b and d). The amount of Bi in the nanoparticles was close to the nominal one, as determined by ICP-OES (Fig. S22, ESI†).
 | ||
| Fig. 3 (a) XRD patterns of BaVOMn (Ba3(V0.995Mn0.005O4)2) and BaVOMn–BiO (Ba3(V0.995Mn0.005O4)2 – 54.4 wt% Bi2O3) particles. (b) Particle size distribution of BaVOMn–BiO (annealed for 30 min at 550 °C) determined by TEM image analysis (inset: representative image) (c) hydrodynamic size distribution of HSA-coated BaVOMn–BiO in water and (d) long time stability in three media. | ||
Fig. 3b shows an exemplary TEM image and the corresponding size distribution with mean diameter and geometric standard deviation of 62 nm and 1.29, which is in good agreement with measurements by XRD (dXRD = 58 nm) and N2 adsorption (dBET = 56 nm). Their shape is slightly elongated, originating from sintering and aggregation of primary particles. In fact, elongated particles (i.e. discoidal and cylindrical) were most attractive for targeting tumors as they had accumulated more than spherical or quasi-spherical ones to organs (liver, spleen and lungs) of tumor-bearing mice.48 These particles were coated with human serum albumin (HSA) to improve their colloidal stability49 due to the extra electrostatic repulsion provided by the proteins,50 as well as the potential benefit of increased blood circulation times due to lower non-specific interaction.51 The successful functionalization was confirmed with FTIR and TGA (shown in Fig. S23 and S24, ESI†). Following differential centrifugation (Fig. S25a, ESI†) to remove the undesirable fraction (80 wt%) of largest aggregates formed during annealing (yield ∼20 wt%), hydrodynamic particle size distributions centered between 60 and 200 nm with polydispersity index around 0.1 have been obtained (Fig. S25b and c, ESI†), shown exemplarily in Fig. 3c in water. The successful separation of various size fractions by centrifugation and the removal of larger aggregates has been confirmed by electron microscopy at two magnifications (Fig. S26a–d, ESI†). The resulting aggregate size distributions of the largest Feret diameters determined by microscopy compared well with DLS measurements (Fig. S26e and f, ESI†), given the different measurement principles.52 As mentioned above, such hydrodynamic sizes are suitable for blood flow mapping (e.g. daverage = 330 nm by Bruns et al.13) as well as metastatic tumor detection, i.e. d = 30–450 nm by Gao et al.30 and d = 90–140 nm by Li et al.31 These distributions were monitored for up to 30 days in water, 0.154 M NaCl as well as RPMI cell culture medium (Fig. 3d). The hydrodynamic mode size remains constant in all media, without significant changes in size distribution (Fig. 3c: as prepared, t = 0, and after 30 days), indicating high colloidal stability (Fig. S27, ESI†). Furthermore, the evolution of their luminescence intensity in different media (water, PBS, RPMI medium) over 14 days (Fig. S28a, ESI†) showed no sign of deterioration of performance. Additionally, that intensity remained constant over pH = 5–10, for at least 14 days (Fig. S28b, ESI†).
In vitro characterization and biocompatibility
The cytocompatibility of nanoparticles (concentration: 0–1000 μg ml−1) was assessed in vitro by incubating them with HeLa cells for 24 hours. As shown in Fig. 4a, BaVOMn particles (blue bars) cause a reduction in cell viability with the LD50 around 55 μg ml−1. Similarly, also commercial PbS–CdS QDs functionalized for bio-applications (obtained from NanoOptical Materials) were tested as a benchmark, since these have been studied extensively for fluorescence microscopy including deep-tissue imaging.14 The PbS–CdS QDs (orange bars) showed a reduction in cell viability reaching below 75% at 100 μg ml−1. Higher QD concentrations could not be determined, as the concentration of supplied colloids was not high enough. Remarkably, the BaVOMn–BiO particles (red columns) exhibited superior cell compatibility. Even if corrected for the lower Ba3(VO4)2 content due to the Bi2O3 mass contribution, the nanoparticles are still better tolerated than pure BaVOMn, notably at comparable sizes (see Table S3, ESI†). The HeLa cell cytocompatibility of the BaVOMn–BiO particles is comparable to that of less bright Ba3(PO4)2:Mn5+ nanoparticlesd (up to 1000 μg ml−1).32 | ||
| Fig. 4 (a) Cytotoxicity against HeLa cells after 24 h incubation. *: not tested. (b) Leached (dissolved) fraction (wt%) of barium and vanadium ions after 24 h. +: below detection limit. (c) NIR-II fluorescence image of a stained HeLa cell incubated with NPs: green (λem = 530 nm) = cytoskeleton, blue (λem = 445 nm) = nucleus, red (λem = 1180 nm) = nanoparticles. | ||
To gain a deeper understanding, also the cytotoxicity of water soluble BaCl2 (green bars) and Na3VO4 (pink bars) was tested to investigate the toxicity of barium and orthovanadate ions. The BaCl2 hardly affected the viability of HeLa cells up to 250 μg ml−1 with significant impact only at the two highest concentrations (500 and 1000 μg ml−1). In contrast, even the lowest dose of Na3VO4 led to reduction in cell viability down to 55%, resulting in an LD50 of 30 μg ml−1. These results are in line with reported values for L929 fibroblasts, where the LD50 for vanadium salt was more than an order of magnitude lower than for BaCl2.53
To assess whether there is a difference in the amount of released ions from BaVOMn and BaVOMn–BiO nanoparticles that would explain the distinctly different cytotoxicity, leached ions were quantified in three relevant media (water, PBS andcell culture medium RPMI) by ICP-OES (Fig. 4b). Remarkably, the addition of Bi2O3 increases the stability for BaVOMn–BiO (filled columns) with leached ion concentration almost an order of magnitude lower than pure BaVOMn (open columns), in all three media. The highest fraction dissolved was in PBS with 34.6% Ba and 33.7% V in BaVOMn. In contrast, there is a distinct reduction in ion release for BaVOMn–BiO, where in PBS only 3.2% of Ba and 1.9% V were dissolved. Since the HSA-coating density determined by TGA (Fig. S24, ESI†) was similar for BaVOMn (2.4 mgHSA m−2) and BaVOMn–BiO (1.5 mgHSA m−2), this was unlikely to explain such large differences in cytotoxicity and leaching behavior.
It is also noteworthy that the leached amount of barium from the latter increased only to 4.5% after 14 days (Fig. S29, ESI†). Furthermore, the leached ion concentration from BaVOMn–BiO is about 70% lower (Fig. S30, ESI†) than our32 Ba3(PO4)2:Mn5+, attesting to the high stability of BaVOMn–BiO. Considering that typical doses for systemic injections in mice (20 g) are 0.1 to 1 mg (≈100 μl at 1 to 10 mg ml−1), for BaVOMn–BiO this would correspond to 1.9–19 μg leached barium (0.095–0.95 mg kgmouse−1) and 0.27–2.7 μg leached vanadium (0.0135–0.135 mg kgmouse−1). These values are still orders of magnitude lower than the median lethal dose (LD50) in mice of 19.2 mg kg−1 for barium54 and 18.4 mg kg−1 for vanadium55 after intravenous injection. Therefore, it can indeed be concluded that the increased cytocompabitility of BaVOMn–BiO is attributed to the reduced amount of leached ions compared to BaVOMn. The Bi2O3 appears to stabilize the nanoparticles and make them less prone to dissolution and release of Ba and V ions, thus making them very promising for in vivo applications. This could be caused by the occupation of vulnerable defect sites by Bi2O3, similar to black phosphorus/Bi2O3 heterostructures.56 Furthermore, since Bi2O3 nanoparticles functionalized with HSA have exhibited excellent biocompatibility in vivo,57 this could further explain the observed improvement in cytocompatibility.
Bright particle emission within an intracellular environment was confirmed by fluorescence microscopy, where the interaction of these particles incubated with HeLa cells was further studied. Fig. 4c shows the overlay image of three different fluorescent signals: The cytoskeleton was stained with the dye Alexa 488 Phalloidin (λem = 530 nm) shown in green, the cell nucleus was stained with the blue-emitting dye DAPI (λem = 455 nm), and the emission of the BaVOMn–BiO particles at 1180 nm is shown in red. The nanoparticles accumulate in the perinuclear region, indicating that the nanoparticles readily enter the cell. Also, some extracellular particles are observed that are typical for in vitro culture settings where particle sedimentation is a dominant mechanism for particle-cell contact.
Benchmarking with commercial fluorescent NIR-II agents
To best assess the performance of BaVOMn–BiO, it is compared to commercially available materials for fluorescence bioimaging in the NIR-II, specifically to the organic dye indocyanine green (ICG) and PbS–CdS QDs coated with diol groups on the surface. Fig. 5a shows the emission spectra of aqueous suspensions of all three samples at equal mass concentrations (0.1 g L−1) under 750 nm excitation. This wavelength is not at the optimum for any of the three emitters (excitation spectra shown in Fig. S31, ESI†) but can excite all of them while still lying in the first NIR window. The QDs (green broken line) have a strong emission between 1000 to 1350 nm. The emission spectrum of these commercial NIR-II Pbs–CdS QDs is rather broad and unattractive due to the observed shoulder, and consistent to literature14 (FWHM of ∼200 nm, their14Fig. 2a and b). The BaVOMn–BiO nanoparticles (blue solid line) show an almost equal peak intensity, while the ICG emission (red dot-broken line) peaks in the first NIR window and has only a weak emission tail in the NIR-II region. Still, it has been demonstrated that using this tail leads to better contrast than using the emission peak in the NIR-I region for bioimaging.58 | ||
| Fig. 5 Comparison of present BaVOMn–BiO to commercial fluorescent agents with emission in the NIR-II at equal mass concentration (0.1 g L−1) dispersed in water in terms of (a) emission intensity, and (b) photostability under constant illumination (750 nm, 2.9 W cm−2) at their respective peak intensity wavelength. (c) Ex vivo bioimaging: Glass capillaries filled with fluorescent contrast agents were placed under chicken skeletal muscle tissue of varying thickness, excited with a 750 nm laser and imaged. Extracted full-width at half-maximum (FWHM) (d) and signal to background ratios (e). Both BaVOMn–BiO and Pbs–CdS QDs show comparable performance. | ||
Besides brightness, also stability is crucial for fluorescent contrast agents.59 The photostability (at peak wavelength) for one hour under continuous excitation (750 nm, 2.9 W cm−2) of these three materials is shown in Fig. 5b. While the present BaVOMn–BiO shows stable emission even up to 12 hours (Fig. S32, ESI†), both ICG and the QDs drastically decrease their emission intensity within the first 20 minutes. For ICG, this is attributed to photobleaching.60 Similar behavior has been observed by Zhao et al.61 for these QDs (their61 Fig. 9a) that was attributed to quenching at unpassivated surface sites. Since no blue-shift of the emission peak was observed, changes in size of the QDs were ruled out.
Finally, the potential of these particles for deep-tissue imaging was studied ex vivo using chicken skeletal muscle tissue and a defined structure simulating a small blood vessel. Fig. 5c shows the fluorescence images of glass capillaries62 (diameter = 1.5 mm) filled with fluorescent contrast agents (BaVOMn–BiO, Pbs–CdS QDs and ICG) under layers of chicken tissue with increasing thickness (0–10 mm).
The contrast agents were excited with a 750 nm laser (37.4 mW cm−2, one order of magnitude below the exposure limit63) and images were acquired with a NIR-camera equipped with an 850 nm long-pass filter, where the exposure time was adjusted for each image close to saturation to maximize signal intensity.
The apparent width of the fluorescence image coming from the glass capillary without tissue (Fig. 5c at 0 mm tissue thickness) is clearly defined and starts broadening with increasing tissue thickness due to scattering. The latter was quantified by determining the FWHM (Fig. 5d). For both QDs and present nanoparticles, the FWHM increases from 1.3 mm up to ∼8 mm for the maximum tissue thickness of 10 mm, which is expected due to the similar spectral operating range, in agreement with phantom tissue experiments.12 In contrast, the FWHM of ICG is much more affected by tissue scattering and reaches a maximum of 19.4 mm at 10 mm depth. This demonstrates the advantage of operating within the NIR-II window, in contrast to the NIR-I, where the main emission peak of ICG lies. Furthermore, also smaller features could potentially be detected with additional optics resulting in smaller field of view and reaching sub-10 μm spatial resolutions.64
As an additional measure of image quality, the signal-to-background ratio (SBR) was quantified in Fig. 5e, using images acquired with laser excitation but without fluorescent contrast agents as background. No significant difference between the present nanoparticles and QDs can be observed, while ICG shows a much smaller ratio. It is noteworthy that even at 10 mm of tissue thickness, the signal of the BaVOMn–BiO is 40 times higher than the background. According to the Rose criterion,65 a minimum SBR of 5 is required to distinguish image features with 100% certainty. Thus, even weaker or lower lying signals down to ∼3 cm32 can still be detected reliably.
Conclusions
Sub-100 nm Mn5+-doped Ba3(VO4)2 nanoparticles with bright emission in the NIR-II and high quantum yield were produced by scalable and sterile flame aerosol technology, thereby fulfilling key requirements for bioimaging. Co-synthesis with Bi2O3 enhances their brightness up to 10-fold and drastically improves their chemical stability and cytocompatibility to HeLa cells, thus representing an attractive material for bioimaging. The aforementioned properties make BaVOMn–BiO nanoparticles a most promising NIR-II contrast agent for deep tissue imaging as it surpasses traditional fluorophores and quantum dots in photostability while exhibiting better biocompatibility than quantum dots at similar brightness and imaging capabilities. While ex vivo experiments are suited better to systematically study the penetration depth and spatial resolution of novel contrast agents due to their homogeneity, further in vivo experiments are the next step towards clinical applications to fully address and unfold the performance of these BaVOMn–BiO particles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Y. Pauli and T. Casarico (both ETH Zürich) for assistance in particle synthesis, L. Gerken for assistance in cytotoxicity experiments and M. Matter (both ETH Zürich/Empa St. Gallen) for assistance in ICP-OES analysis. We thank the Scientific Center for Optical and Electron Microscopy (ScopeM) of ETH Zurich for access to their microscopes, in particular S. Handschin for help with the liquid-nitrogen cooled holer, D. Norris for access to ICP-OES, D. Poulikakos and I. Hächler for access to UV-VIS (all ETH Zurich), and Edinburgh Instruments for fluorescence lifetime measurements. This research was mainly supported by the Particle Technology Laboratory and ETH Zürich Research Grant (ETH-43 17-1), and in part by Swiss National Science Foundation (#206021_177037 and #206021_183298).References
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS PubMed.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed.
- D. J. Naczynski, M. C. Tan, M. Zevon, B. Wall, J. Kohl, A. Kulesa, S. Chen, C. M. Roth, R. E. Riman and P. V. Moghe, Nat. Commun., 2013, 4, 2199 CrossRef CAS PubMed.
- J. A. Carr, M. Aellen, D. Franke, P. T. C. So, O. T. Bruns and M. G. Bawendi, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9080–9085 CrossRef CAS PubMed.
- S. Diao, G. S. Hong, A. L. Antaris, J. L. Blackburn, K. Cheng, Z. Cheng and H. J. Dai, Nano Res., 2015, 8, 3027–3034 CrossRef CAS.
- Y. Tsukasaki, A. Komatsuzaki, Y. Mori, Q. Ma, Y. Yoshioka and T. Jin, Chem. Commun., 2014, 50, 14356–14359 RSC.
- C. Y. Li and Q. B. Wang, ACS Nano, 2018, 12, 9654–9659 CrossRef CAS PubMed.
- K. Q. Shou, C. R. Qu, Y. Sun, H. Chen, S. Chen, L. Zhang, H. B. Xu, X. C. Hong, A. X. Yu and Z. Cheng, Adv. Funct. Mater., 2017, 27, 1700995 CrossRef PubMed.
- S. J. Zhu, S. Herraiz, J. Y. Yue, M. X. Zhang, H. Wan, Q. L. Yang, Z. R. Ma, Y. Wang, J. H. He, A. L. Antaris, Y. T. Zhong, S. Diao, Y. Feng, Y. Zhou, K. A. Yu, G. S. Hong, Y. Y. Liang, A. J. Hsueh and H. J. Dai, Adv. Mater., 2018, 30, 1705799 CrossRef PubMed.
- A. L. Antaris, H. Chen, K. Cheng, Y. Sun, G. S. Hong, C. R. Qu, S. Diao, Z. X. Deng, X. M. Hu, B. Zhang, X. D. Zhang, O. K. Yaghi, Z. R. Alamparambil, X. C. Hong, Z. Cheng and H. J. Dai, Nat. Mater., 2016, 15, 235–242 CrossRef CAS PubMed.
- K. Welsher, Z. Liu, S. P. Sherlock, J. T. Robinson, Z. Chen, D. Daranciang and H. Dai, Nat. Nanotechnol., 2009, 4, 773–780 CrossRef CAS PubMed.
- K. Welsher, S. P. Sherlock and H. J. Dai, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8943–8948 CrossRef CAS PubMed.
- O. T. Bruns, T. S. Bischof, D. K. Harris, D. Franke, Y. X. Shi, L. Riedemann, A. Bartelt, F. B. Jaworski, J. A. Carr, C. J. Rowlands, M. W. B. Wilson, O. Chen, H. Wei, G. W. Hwang, D. M. Montana, I. Coropceanu, O. B. Achorn, J. Kloepper, J. Heeren, P. T. C. So, D. Fukumura, K. F. Jensen, R. K. Jain and M. G. Bawendi, Nat. Biomed., 2017, 1, 0056 CrossRef CAS PubMed.
- Y. Tsukasaki, M. Morimatsu, G. Nishimura, T. Sakata, H. Yasuda, A. Komatsuzaki, T. M. Watanabe and T. Jin, RSC Adv., 2014, 4, 41164–41171 RSC.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC.
- W. Shao, G. Y. Chen, A. Kuzmin, H. L. Kutscher, A. Pliss, T. Y. Ohulchanskyy and P. N. Prasad, J. Am. Chem. Soc., 2016, 138, 16192–16195 CrossRef CAS PubMed.
- C. Cao, M. Xue, X. J. Zhu, P. Y. Yang, W. Feng and F. Y. Li, ACS Appl. Mater. Interfaces, 2017, 9, 18540–18548 CrossRef CAS PubMed.
- C. Matuszewska, K. Elzbieciak-Piecka and L. Marciniak, J. Phys. Chem. C, 2019, 123, 18646–18653 CrossRef CAS.
- L. F. Johnson, H. J. Guggenheim and R. A. Thomas, Phys. Rev., 1966, 149, 179–186 CrossRef CAS.
- S. M. Jacobsen, W. E. Smith, C. Reber and H. U. Gudel, J. Chem. Phys., 1986, 84, 5205–5206 CrossRef CAS.
- C. Reber and H. U. Gudel, J. Lumin., 1988, 42, 1–13 CrossRef CAS.
- C. Deka, B. H. T. Chai, Y. Shimony, X. X. Zhang, E. Munin and M. Bass, Appl. Phys. Lett., 1992, 61, 2141–2143 CrossRef CAS.
- T. C. Brunold, M. F. Hazenkamp and H. U. Gudel, J. Lumin., 1997, 72-4, 164–165 CrossRef.
- L. D. Merkle, A. Pinto, H. R. Verdun and B. Mcintosh, Appl. Phys. Lett., 1992, 61, 2386–2388 CrossRef CAS.
- T. C. Brunold and H. U. Gudel, Inorg. Chem., 1997, 36, 1946–1954 CrossRef CAS PubMed.
- M. Herren and H. U. Gudel, Inorg. Chem., 1992, 31, 3683–3684 CrossRef CAS.
- R. Moncorgé and H. Manaa, Ann. Chim.-Sci. Mat., 1995, 20, 241–248 Search PubMed.
- X. W. Zhang, Y. Li, Z. L. Hu, Z. Chen and J. R. Qiu, RSC Adv., 2017, 7, 10564–10569 RSC.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- S. Gao, G. Wei, S. Zhang, B. Zheng, J. Xu, G. Chen, M. Li, S. Song, W. Fu, Z. Xiao and W. Lu, Nat. Commun., 2019, 10, 2206 CrossRef PubMed.
- Y. Li, S. Zeng and J. Hao, ACS Nano, 2019, 13, 248–259 CrossRef CAS PubMed.
- P. M. Gschwend, D. Niedbalka, L. R. H. Gerken, I. K. Herrmann and S. E. Pratsinis, Adv. Sci., 2020, 2000370 CrossRef CAS PubMed.
- L. Madler, W. J. Stark and S. E. Pratsinis, J. Mater. Res., 2002, 17, 1356–1362 CrossRef CAS.
- P. M. Valencia, O. C. Farokhzad, R. Karnik and R. Langer, Nat. Nanotechnol., 2012, 7, 623–629 CrossRef CAS PubMed.
- F. H. L. Starsich, I. K. Herrmann and S. E. Pratsinis, Annu. Rev. Chem. Biomol., 2019, 10, 155–174 CrossRef PubMed.
- R. Mueller, L. Madler and S. E. Pratsinis, Chem. Eng. Sci., 2003, 58, 1969–1976 CrossRef CAS.
- K. Wegner and S. E. Pratsinis, Powder Technol., 2005, 150, 117–122 CrossRef CAS.
- A. J. Grohn, S. E. Pratsinis, A. Sanchez-Ferrer, R. Mezzenga and K. Wegner, Ind. Eng. Chem. Res., 2014, 53, 10734–10742 CrossRef CAS.
- O. E. Semonin, J. C. Johnson, J. M. Luther, A. G. Midgett, A. J. Nozik and M. C. Beard, J. Phys. Chem. Lett., 2010, 1, 2445–2450 CrossRef CAS.
- P. M. Gschwend, F. H. L. Starsich, R. C. Keitel and S. E. Pratsinis, Chem. Commun., 2019, 55, 7147–7150 RSC.
- N. Venkatachalam, T. Yamano, E. Hemmer, H. Hyodo, H. Kishimoto and K. Soga, J. Am. Ceram. Soc., 2013, 96, 2759–2765 CrossRef CAS.
- Y. F. Wang, G. Y. Liu, L. D. Sun, J. W. Xiao, J. C. Zhou and C. H. Yan, ACS Nano, 2013, 7, 7200–7206 CrossRef CAS PubMed.
- L. D. Merkle, OSA Proc., 1993, 15, 310–314 Search PubMed.
- A. Spyrogianni, P. Tiefenboeck, F. H. L. Starsich, K. Keevend, F. Krumeich, I. K. Herrmann, J. C. Leroux and G. A. Sotiriou, AlChE J., 2018, 64, 2947–2957 CrossRef CAS.
- X. C. Zhou, L. P. Zhong, Q. P. Liu, R. Y. Kuang and H. M. Chen, Inorg. Mater., 2009, 45, 1295–1298 CrossRef CAS.
- E. M. Levin and R. S. Roth, J. Res. Natl. Bur. Stand. A Phys. Chem., 1964, 68, 189–195 CrossRef PubMed.
- A. A. Fotiev, V. V. Makarov, V. L. Volkov and L. L. Surat, Russ. J. Inorg. Chem., 1969, 14, 144–146 Search PubMed.
- P. Decuzzi, B. Godin, T. Tanaka, S. Y. Lee, C. Chiappini, X. Liu and M. Ferrari, J. Controlled Release, 2010, 141, 320–327 CrossRef CAS PubMed.
- P. M. Gschwend, S. Conti, A. Kaech, C. Maake and S. E. Pratsinis, ACS Appl. Mater. Interfaces, 2019, 11, 22550–22560 CrossRef CAS PubMed.
- T. L. Moore, L. Rodriguez-Lorenzo, V. Hirsch, S. Balog, D. Urban, C. Jud, B. Rothen-Rutishauser, M. Lattuada and A. Petri-Fink, Chem. Soc. Rev., 2015, 44, 6287–6305 RSC.
- J. Mariam, S. Sivakami and P. M. Dongre, Drug Delivery, 2015, 23, 2668–2676 CrossRef.
- B. H. Kaye, Characterization of Powders and Aerosols, Wiley-VCH, Weinheim, 1998 Search PubMed.
- A. Yamamoto, R. Honma and M. Sumita, J. Biomed. Mater. Res., 1998, 39, 331–340 CrossRef CAS.
- I. B. Syed and F. Hosain, Toxicol. Appl. Pharmacol., 1972, 22, 150–152 CrossRef CAS PubMed.
- J. M. Llobet and J. L. Domingo, Toxicol. Lett., 1984, 23, 227–231 CrossRef CAS.
- H. Huang, L. He, W. Zhou, G. Qu, J. Wang, N. Yang, J. Gao, T. Chen, P. K. Chu and X.-F. Yu, Biomaterials, 2018, 171, 12–22 CrossRef CAS PubMed.
- H. Aviv, S. Bartling, I. Grinberg and S. Margel, J. Biomed. Mater. Res., Part B, 2013, 101, 131–138 CrossRef.
- J. A. Carr, D. Franke, J. R. Caram, C. F. Perkinson, M. Saif, V. Askoxylakis, M. Datta, D. Fukumura, R. K. Jain, M. G. Bawendi and O. T. Bruns, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4465–4470 CrossRef CAS PubMed.
- E. Thimsen, B. Sadtler and M. Y. Berezin, Nanophotonics, 2017, 6, 1043–1054 CAS.
- J. T. Alander, I. Kaartinen, A. Laakso, T. Patila, T. Spillmann, V. V. Tuchin, M. Venermo and P. Valentina Caudalisuo, Int. J. Biomed. Imaging, 2012, 2012, 1–26 CrossRef PubMed.
- H. G. Zhao, M. Chaker and D. L. Ma, J. Mater. Chem., 2011, 21, 17483–17491 RSC.
- K. Welsher, S. P. Sherlock and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8943–8948 CrossRef CAS PubMed.
- International Commission on Non-Ionizing Radiation Protection, 2013, vol. 105, pp. 271–295.
- H. Wan, J. Y. Yue, S. J. Zhu, T. Uno, X. D. Zhang, Q. L. Yang, K. Yu, G. S. Hong, J. Y. Wang, L. L. Li, Z. R. Ma, H. P. Gao, Y. T. Zhong, J. Su, A. L. Antaris, Y. Xia, J. Luo, Y. Y. Liang and H. J. Dai, Nat. Commun., 2018, 9, 1171 CrossRef PubMed.
- J. T. Bushberg, The Essential Physics of Medical Imaging, Lippincott Williams & Wilkins, Philadelphia, 3rd edn, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimization of BaVOMn, N2 adsorption measurements, TEM images and size distributions, TGA analysis, XRD patterns, ICP-OES measurements, HAADF-STEM images, FTIR spectra, hydrodynamic sizes, photostability, dissolution data, Table of quantum yields reported in the NIR-II, and experimental section. See DOI: 10.1039/d0tb02792h |
| This journal is © The Royal Society of Chemistry 2021 |