
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence“Sweet tooth”-oriented SN38 prodrug delivery nanoplatform for targeted gastric cancer therapy†
Ning
Ding‡
 abc,
Shengjun
Xu‡
de,
Sheng
Zheng
abc,
Qianwei
Ye
de,
Li
Xu
e,
Sunbin
Ling
de,
Shanshan
Xie
f,
Wenwen
Chen
abc,
Zizhen
Zhang
abc,
Meng
Xue
abc,
Zhenghua
Lin
abc,
Xiao
Xu
*de and
Liangjing
Wang
*abc
abc,
Shengjun
Xu‡
de,
Sheng
Zheng
abc,
Qianwei
Ye
de,
Li
Xu
e,
Sunbin
Ling
de,
Shanshan
Xie
f,
Wenwen
Chen
abc,
Zizhen
Zhang
abc,
Meng
Xue
abc,
Zhenghua
Lin
abc,
Xiao
Xu
*de and
Liangjing
Wang
*abc
aDepartment of Gastroenterology, the Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou 310020, China. E-mail: wangljzju@zju.edu.cn
bInstitution of Gastroenterology, Zhejiang University, Hangzhou 310000, China
cZhejiang University Cancer Center, Hangzhou 310000, China
dDepartment of Hepatobiliary and Pancreatic Surgery, Affiliated Hangzhou First People's Hospital, Zhejiang University School of Medicine, Hangzhou 310006, China. E-mail: zjxu@zju.edu.cn
eNHC Key Laboratory of Combined Multi-organ Transplantation, Hangzhou 310003, China
fThe Children's Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou 310052, China
First published on 4th March 2021
Abstract
Most cancer cells employ overexpression of glucose transports (GLUTs) to satisfy glucose demand (“Sweet Tooth”) for increased aerobic glycolysis rates. GLUT1, one of the most widely expressed GLUTs in numerous cancers, was identified as a prognosis-related biomarker of gastric cancer via tissue array analysis. Herein, a “Sweet Tooth”-oriented SN38 prodrug delivery nanoplatform (Glu-SNP) was developed for targeted gastric cancer therapy. For this purpose, a SN38-derived prodrug (PLA-SN38) was synthesized by tethering 7-ethyl-10-hydroxycamptothecin (SN38) to biocompatible polylactic acid (PLA) with the appropriate degree of polymerization (n = 44). The PLA-SN38 conjugate was further assembled with glycosylated amphiphilic lipid to obtain glucosamine-decorated nanoparticles (Glu-SNP). Glu-SNP exhibited potent antitumor efficiency both in vitro and in vivo through enhanced cancer cell-specific targeting associated with the overexpression of GLUT1, which provides a promising approach for gastric cancer therapy.
1. Introduction
The Warburg effect is a phenomenon that cancer cells mainly rely on aerobic glycolysis rather than mitochondrial oxidative phosphorylation to generate energy.1 To adapt to inefficient ATP generation via aerobic glycolysis and to further satisfy the energy demand of cancer cells for cellular processes and infinite proliferation, cancer cells employ various mechanisms, such as upregulated glycolytic rate, vascularization and overexpression of nutrient transporters.2–5 As glucose is the predominant cellular energy resource, most cancer cells overexpress glucose transporters (GLUTs) for their increased glucose demand (“Sweet Tooth”).4 The recognition of “Sweet Tooth” in cancer cells has attracted researchers to focus on targeting glucose uptake as a potent strategy for cancer diagnosis and therapy. Accordingly, 2-[fluorine-18]-fluoro-2-deoxy-D-glucose (18F-FDG), the radiolabeled glucose analogue, has already been used in clinical practice for imaging primary tumor and tumor metastases.6,7GLUT1, encoded by SLC2A1, is one of the most widely expressed GLUTs in numerous cancers (e.g. hepatocellular carcinoma, renal cell carcinoma and gastric cancer).8–11 Previous studies have revealed that the rate of glucose metabolism in cancer cells is associated with the expression level of GLUT1.12 Recently, Ning Yan et al. reported the crystal structure of human GLUT1, which had led to the development of small molecule GLUT1 inhibitors for cancer therapeutics.13–15 Moreover, glucose-conjugated paclitaxel and other glycoconjugated taxoids have been synthesized.16–18 For example, 2′-paclitaxel methyl 2-glucopyranosyl succinate, fabricated by Shui-Tein Chen and coworkers, exhibited excellent selective cytotoxicity against MCF-7 breast cancer cells without inducing toxicity to normal cells.18
Gastric cancer is the fifth most prevalent cancer diagnosed and the third leading cause of cancer deaths worldwide.19 Although recent years have witnessed great progress in the early diagnosis and treatment for gastric cancer with the development of endoscopic techniques, a large proportion of patients with gastric cancer were diagnosed at advanced stages owing to the lack of specific clinical symptoms for early disease. Currently, for patients with unresectable gastric cancer, the standard choice of the chemotherapy regimen shows limited effectiveness and the median overall survival is merely ∼11 months.20–22 Several studies based on Germany and Japanese population indicated that an elevated expression of GLUT1 was associated with a poor prognosis in gastric cancer via immunohistochemistry (IHC) staining of surgical resection specimens.10,23 Therefore, GLUT1-targeting therapeutics could be a promising approach against gastric cancer.
Nanomedicines targeting specifically cancer cells, with low systemic toxicity and enhanced intratumoral accumulation of therapeutics, have been a hot spot recently.24–26 Glucose and its derivatives (glucosamine and 2-deoxyglucose) are adopted as specific tumor-homing ligands for cancer therapeutics.26–29 Alejandro Sosnik et al. developed glucosylated polymeric nanomicelles to actively deliver dasatinib for therapy against glucose-avid pediatric sarcomas.28 Motivated by “Sweet Tooth” of gastric cancer and translational potential of glucose-decorated nanoformulations, we here applied glucosamine, a glucose derivative, as a specific gastric cancer-homing ligand. 7-Ethyl-10-hydroxycamptothecin (SN38), an active metabolite of irinotecan (CPT-11, the preferred treatment option for unresectable gastric cancer), was tethered to biocompatible polylactic acid (PLA) with the appropriate degree of polymerization (n = 44).30 PLA-SN38 was further formulated in glycosylated amphiphilic lipids to obtain glucosamine-decorated nanoparticles (Glu-SNP). Glu-SNP exhibited cancer cell-specific targetable capability and potent antitumor efficiency in the in vitro experiments. In vivo antitumor efficiency and biosafety of Glu-SNP were further evaluated in an MKN45 cell-derived xenograft model. Therefore, we developed a “Sweet Tooth”-oriented SN38 prodrug delivery nanoplatform, which is promising for gastric cancer treatment (Scheme 1).
 | ||
| Scheme 1 Schematic illustration of chemical structure and assembly process of SN38-loaded nanoparticle (SNP and Glu-SNP) for efficient in vivo drug delivery. The SN38 prodrug (PLA-SN38) was enfolded into amphiphilic copolymers (DSPE-PEG2000) with or without glucose decoration (DSPE-PEG2000-Glu). The glucose-decorated SN38-loaded nanoparticles (Glu-SNP) could accumulate in tumor sites through both the passive (enhanced permeability and retention [EPR] effect) and active (glucose transporters- [GLUTs-] specific binding) targeting effect. | ||
2. Materials and methods
2.1 Materials
7-Ethyl-10-hydroxycamptothecin (SN38) was purchased from Tokyo Chemical Industry (Shanghai, China). Irinotecan hydrochloride (CPT-11) was purchased from J&K Scientific (Shanghai, China). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol) 2000] (DSPE-PEG2000) was purchased from A.V.T. Pharmaceutical (Shanghai, China). The glucosamine-terminal DSPE-PEG2000 (DSPE-PEG2000-Glu) was customized by Ruixi Biological Technology (Xi’an, China). Carboxy-terminated polylactic acid (PLA-COOH) was purchased from Daigang Biological Technology (Jinan, China). All other compounds and solvent were purchased from J&K Science (Shanghai, China).2.2 Cell lines and cell culture
Gastric cancer cell lines MKN45 (obtained from the Chinese Academy of Medical Sciences) and SGC-7901 (obtained from Beijing Cancer Hospital) were cultured in Roswell Park Memorial Institute 1640 (RPMI-1640) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics. Normal gastric epithelial cell line GES-1 was obtained from the Chinese Academy of Medical Sciences and cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS and 1% antibiotics. All cell lines were maintained at 37 °C in a humidified atmosphere with 5% CO2.2.3 Preparation of SN38 prodrug loaded nanoparticles
SN38 prodrug (PLA-SN38) loaded nanoparticles were fabricated via nanoprecipitation. For the preparation of nontargeted PLA-SN38-loaded nanoparticles (SNP), PLA-SN38 (at an SN38 equivalence) to DSPE-PEG2000 was premixed at a mole ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. While in the preparation of glucosamine-decorated SN38 prodrug-loaded nanoparticles (Glu-SNP), the mole ratio of PLA-SN38 (at an SN38 equivalence) to the two matrices (DSPE-PEG2000 and DSPE-PEG2000-Glu) was fixed at 3:2:2. Predetermined amounts of DSPE-PEG2000-Glu and/or DSPE-PEG2000 and PLA-SN38 (1 mg, at an SN38 equivalence) were dissolved in 1 mL acetone and then added dropwise into 5 mL deionized water while stirring. After stirring at room temperature for 30 min, the remaining acetone was removed by rotary evaporation at reduced pressure. The nanoparticle solution was further concentrated with an Amicon Ultra-4 centrifugal filter device (10k MWCO, Millipore Corp.), and washed with deionized water for further experiments. The hydrodynamic diameters and zeta potentials of SNP and Glu-SNP were measured using Dynamic Light Scattering (DLS, Malvern, U.K.). And the morphology of SNP and Glu-SNP was observed and imaged by Transmission Electron Microscopy (TEM, TECNAL 10, Philips). The long-term stabilities of nanoparticles were assessed over 2 weeks by incubating the nanoparticle solutions in the deionized water supplemented with 10% (v/v) fetal bovine serum (FBS) at 37 °C. The sizes were recorded every day through DLS analysis.
:4. While in the preparation of glucosamine-decorated SN38 prodrug-loaded nanoparticles (Glu-SNP), the mole ratio of PLA-SN38 (at an SN38 equivalence) to the two matrices (DSPE-PEG2000 and DSPE-PEG2000-Glu) was fixed at 3:2:2. Predetermined amounts of DSPE-PEG2000-Glu and/or DSPE-PEG2000 and PLA-SN38 (1 mg, at an SN38 equivalence) were dissolved in 1 mL acetone and then added dropwise into 5 mL deionized water while stirring. After stirring at room temperature for 30 min, the remaining acetone was removed by rotary evaporation at reduced pressure. The nanoparticle solution was further concentrated with an Amicon Ultra-4 centrifugal filter device (10k MWCO, Millipore Corp.), and washed with deionized water for further experiments. The hydrodynamic diameters and zeta potentials of SNP and Glu-SNP were measured using Dynamic Light Scattering (DLS, Malvern, U.K.). And the morphology of SNP and Glu-SNP was observed and imaged by Transmission Electron Microscopy (TEM, TECNAL 10, Philips). The long-term stabilities of nanoparticles were assessed over 2 weeks by incubating the nanoparticle solutions in the deionized water supplemented with 10% (v/v) fetal bovine serum (FBS) at 37 °C. The sizes were recorded every day through DLS analysis.
2.4 In vitro SN38 release kinetics study
SNP and Glu-SNP solution with a 0.1 mg mL−1 SN38 equivalent concentration with/without 50 U mL−1 esterase were dialyzed against phosphate buffer saline (PBS, pH = 7.4) containing 0.2% Tween 80. The dialysis bags (spectrum, Mw cutoff = 2 kDa) were continuously and gently stirred in an orbital shaking water bath at 37 °C. At predetermined timepoints, 1 mL release media were collected and the same volume of fresh media was supplemented. The amounts of released SN38 were determined by UV-vis spectrometer (UH5300, Hitachi) at 378 nm.2.5 In vitro cytotoxicity study
MKN45 and SGC-7901 cells were seed into 96-well plates at a density of 3000 cell per well and incubated overnight. Cells were treated with SN38, SNP and Glu-SNP with serial dilutions and incubated for 72 h. After exposure, the medium in each well was replaced with 110 μL fresh medium containing 10 μL CCK-8 solution (MCE). After 2 h incubation at 37 °C, the absorbance at 450 nm was measured with a microplate reader (Multiskan FC, Thermo Scientific). The IC50 of SN38, SNP and Glu-SNP against MKN45 and SGC-7901 cells were calculated via Prism software.2.6 Cell proliferation study
Cell proliferation was studied by the 5′-ethynyl-2′-deoxyuridine (EdU) incorporation assay. MKN45 and SGC-7901 cells were respectively seeded into 48-well plates with 10000 cell per well and incubated overnight. SN38, SNP and Glu-SNP were added into well at SN38 concentration of 20 nM. After incubated for 24 h, DNA synthesis was quantified by a Click-iT® EdU Alexa Fluor® 488 Assay Kit (Invitrogen) according to the manufacturers’ protocol. Briefly, EdU was added to each well and incubated for 2 h at 37 °C, then the cells were washed with PBS and fixed by 4% formaldehyde at room temperature for 15 min. After that, 0.5% Triton X-100 was added and incubated for 10 min. Alexa Fluor® 488 azide was added into the cells and incubated for 30 min in dark. Finally, cells were observed by fluorescence microscopy (Olympus, IX71) after staining the nuclear with Hoechst 33342 (Invitrogen) for 15 min. More than 5 regions with 1500–2000 total cells were counted to assess the presence of the cell proliferation.
2.7 Cell apoptosis study
Cell apoptosis was examined by flow cytometry and western blot assay. For flow cytometry analysis, MKN45 cells were seeded into 6-well plates with 100000 cells per well and incubated overnight. Then these cells were exposed to SN38, SNP, Glu-SNP at SN38 concentration of 200 nM for 36 h. After exposure, cells were harvested and stained with Annexin V-FITC and propidium iodide (PI) (Keygen, China). The apoptosis rates were detected by a flow cytometer (BD FACSCanto II).
For the detection of expressions of apoptotic protein (cleaved PARP and cleaved caspase 3), MKN45 cells were seeded and treated with SN38, SNP, Glu-SNP at SN38 concentration of 100 nM for 48 h. At the end of exposure, cells were harvested and whole-cell extracts were generated in RIPA buffer (Fude Biological Tech.) supplemented with protease inhibitors. Western blot analysis was performed as reported previously.31 Bands were incubated with primary antibody dilution overnight, followed by incubation with corresponding secondary antibody dilution for 2 h. Finally, bands were visualized with ECL detection reagents (Fude Biological Tech.). All antibodies were diluted according to manufacturers’ recommendations and supplied in Table S1 (ESI†).
2.8 Cell cycle analysis
For the detection of changes in the cell cycle after different drug exposure, MKN45 cells were seeded in 6-well plates with 100000 cells per well. After treatment with free SN38, SNP, Glu-SNP at SN38 concentration of 20 nM for 24 h, cells were harvested and washed with PBS, then fixed with 70% cold ethanol for 2 h. Then the cells were washed again with PBS and resuspended with 500 μL PI binding buffer (Sigma Aldrich), incubated in dark for 30 min. Finally, changes in the cell cycle were analyzed by flow cytometer (BD FACSCanto II).
2.9 Intracellular uptake of nanoparticles
Cy5.5, a near-infrared (NIR) fluorescence probe, was covalently tethered to PLA as reported previously and further co-assembled with PLA-SN38, DSPE-PEG2000 and DSPE-PEG2000-Glu to afford Cy5.5 labeled SNP (Cy5.5@SNP) and Glu-SNP (Cy5.5@Glu-SNP) for visualization of intracellular uptake of nanoparticles.32 MKN45, SGC-7901 and GES-1 cells were incubated in glass bottom Petri dishes at a suitable cell density and then treated with Cy5.5@SNP and Cy5.5@Glu-SNP at SN38 concentration of 10 μM. At determined timepoints (1 h, 3 h and 6 h), cells were fixed and then stained with Hoechst 33342, finally imaged by CLSM (FV3000, Olympus). The fluorescence intensity was quantified by Image J software at the same parameters. At the same time, cells were harvested and subjected for flow cytometry analysis to quantify the intracellular uptake of nanoparticles.For further analyzing the role of GLUT1 in the intracellular uptake of SNP and Glu-SNP, 10000 MKN45 cells were seeded in glass bottom Petri dishes and cultured overnight. And then the medium was replaced with fresh RPMI-1640 medium containing 10 μM WZB-117, RPMI-1640 medium without glucose and RPMI-1640 medium with high glucose (25 mM), respectively. After incubated for 3 h, Cy5.5@SNP and Cy5.5@Glu-SNP at SN38 concentration of 10 μM were added. At predetermined timepoints, CLSM observation and flow cytometry analysis were carried out as mentioned above.
2.10 In vivo and ex vivo imaging study
5 week-old male BALB/c nude mice, obtained from the Gempharmatech Biotechnology Company, were maintained in specific pathogen-free facilities. All animal studies were conducted following the National Institute Guide for the Care and Use of Laboratory Animals. The experimental protocols were approved by the Ethics Committee of the Second Affiliated Hospital, Zhejiang University School of Medicine.5000000 MKN45 cells were subcutaneously injected into the right flank of nude mice to establish an MKN45 xenograft nude mouse model. Cy5.5@SNP and Cy5.5@Glu-SNP, prepared above, were used to track biodistribution of SNP and Glu-SNP, respectively. When the tumor volume reached ∼100 mm3, the mice were randomly divided into two groups (n = 3 in each group). The mice were injected with Cy5.5@SNP and Cy5.5@Glu-SNP at a Cy5.5 dose of 20 μg per mouse via the tail vein, respectively. In vivo imaging was performed at 1, 4, 8, 24, 48 h post-injection. At 48 h post-injection, mice were sacrificed in a humanitarian way. Tumors and major organs (heart, liver, spleen, lung and kidney) were collected for ex vivo imaging. In vivo and ex vivo imaging was accomplished using IVIS spectrum in vivo imaging system (PerkinElmer, Waltham), and the imaging parameters were set to 1 s exposure, 1 f/stop, 1 binning.
2.11 In vivo antitumor study in MKN45 xenograft nude mouse model
5000000 MKN45 cells were subcutaneously injected in the right flank of nude mice to establish an MKN45 xenograft nude mouse model on Day 0. When the tumor volume reached 100–150 mm3, the mice were randomized into four groups (n = 6 in each group). The mice were injected with saline, CPT-11 (15 mg kg−1), the solution containing SNP and Glu-SNP (10 mg kg−1, at SN38 equivalent dose) via tail vein on Day 9, 11, 13. The tumor volume and weight of mice were monitored and recorded every day. The tumor volume was evaluated by measuring the length (L) and width (W) with a caliper and calculated using the following formula: V = (L × W2)/2, with W smaller than L.
On Day 18, mice were sacrificed in a humanitarian way and tumors and major organs were excised and collected. After being fixed with 4% formaldehyde, tumor and other tissues were embedded in paraffin, cut into 5 μm slices and stained with hematoxylin and eosin (H&E, Sigma) for histological analysis. Immunohistochemistry staining of Ki67 and TUNEL assay were carried out to detect the proliferation and apoptosis of cancer cells. Meanwhile, blood samples were collected and subjected for whole blood count, renal and liver function tests.
2.12 Statistical analysis
All quantitative data are represented as the mean ± SD. IBM SPSS Statistics 20.0 was used for the statistical analysis. Without specific instrument, unpaired two-tailed Student's t-test was applied to assess the statistical significance between two groups. Multivariate Cox regression analysis was applied for prognosis analysis. Statistical significance was defined as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 and ‘ns’ represented no significant difference (p > 0.05).3. Results
3.1 Role of glucose transporter-1 (GLUT1) in gastric cancer
GLUT1 belongs to glucose transporters (GLUTs) family and mediates cellular uptake of glucose. It has been widely reported that GLUT1 was involved in the tumorigenesis and aggressiveness of numerous cancers.9,11,33,34 Toshimitsu Suzuki et al. found that GLUT1, overexpressed in gastric cancer, connected tightly with invasive depth, metastasis and clinical stages of gastric cancer, thus influencing the survival of patients.10 Herein, we conducted immunohistochemistry (IHC) analysis of GLUT1 in a tissue array (TA) containing a total of 87 pairs of gastric cancer and corresponding paracancerous tissue (normal stomach tissue), to analyze the protein level of GLUT1 expression in gastric cancer. IHC staining revealed that GLUT1 localized in the cell membrane of gastric cancer and normal stomach epithelium cells (Fig. 1A). Notably, we found that 72.4% (63/87) of gastric cancer tissues showed predominant positive staining, whereas the GLUT1-positive percentage of normal tissues was only 23.0% (20/87) (Fig. 1B), which was consistent with previous reports10,23 and the mRNA expression level analysis of GLUT1 using TCGA data (Fig. 1D and Fig. S1, ESI†). Based on differences of GLUT1 H-score between cancer and corresponding paracancerous tissue (whether above 0.5), patients with gastric cancer were divided into the non-elevated expression of GLUT1 and elevated expression of GLUT1 subgroups. Kaplan–Meier overall survival analysis indicated that patients with elevated expression of GLUT1 had a poorer prognosis (Fig. 1C). All these data provided compelling evidence that gastric cancer was apt to overexpress GLUT1 and the elevation of GLUT1 negatively correlated with the prognosis of gastric cancer patients, which further prompted that GLUT1 was a potential treatment target for gastric cancer. | ||
| Fig. 1 Role of GLUT1 in gastric cancer. (A) Representative anti-GLUT1 immunohistochemistry (IHC) images of the tissue array containing 87 pairs of gastric cancer samples and corresponding paracancerous tissue (normal stomach epithelium) samples. Para represented the paracancerous tissue. Scale bars = 500 μm and 50 μm. (B) H-score of GLUT1 protein level expression quantified by IHC staining in the tissue array. (C) Kaplan–Meier overall survival analysis of gastric cancer patients in the non-elevated expression of GLUT1 and elevated expression of GLUT1 subgroups. The Multivariate Cox regression analysis was applied for significant difference and Hazard Ratio, in which gender and age differences between groups were adjusted (p = 0.015, adjusted HR = 2.916, 95%CI = 1.235–6.887, n = 80). (D) The RNA level of GLUT1 expression analysed in paired gastric cancer samples and normal gastric mucosal samples (n = 32) using TCGA cohort data. Paired two-tailed Student's t-test was applied to assess the statistical significance. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001 and ‘ns’ represents p > 0.05. | ||
3.2 Construction and characterization of PLA-SN38 prodrug and glucose-decorated nanoparticles
SN38, a semi-synthetic derivative of camptothecin, exhibited 100- to 1000-fold higher in vitro cytotoxicity than CPT-11 as reported previously.30,35 However, SN38 cannot be administrated orally or intravenously owing to its poor solubility and instability in the physiological environment, which obstructs its clinical application. Prodrug design and nanoformulation strategy provided us with an ideal way to overcome the hydrophobicity and instability of SN38 to deliver efficiently to tumor sites. In this study, biocompatible polylactic acid (PLA) was applied to conjugate with SN38 via an ester bond. 1H NMR spectrum and RP-HPLC chromatogram unambiguously confirmed the successful synthesis of PLA-SN38 prodrug (Fig. 2A and Fig. S2, ESI†), with grafting rate ∼90%. To improve water solubility and pharmacokinetic properties of PLA-SN38, DSPE-PEG2000 and its targeting derivation (DSPE-PEG2000-Glu) were utilized to encapsulate PLA-SN38 via a one-step nanoprecipitation method. Contributed by the PLA-tethered prodrug design and encapsulation by DSPE-PEG2000-derived matrixes, hydrophobic SN38 can be formulated into nanoparticles with ∼50‰ DL and ∼95% EE; whereas, free SN38, without conjugation to PLA segment, was not able to assembly with DSPE-PEG2000 to form uniform nanoparticles (Table S1 and Fig. S3, ESI†). Results of DLS analysis and TEM images exhibited SNP and Glu-SNP uniform nanostructures, with average diameters of ∼140 nm and zeta potentials of ∼−35 mV (Fig. 2B, C and Table S1, ESI†). Both SNP and Glu-SNP exhibited long-term stability against incubation in 10% FBS solution for 2 weeks through monitoring size changes of the nanoparticles (Fig. 2D). | ||
| Fig. 2 Characterization of SN38-derived prodrug (PLA-SN38) and SN38-loaded nanoparticles (SNP and Glu-SNP). (A) 1H NMR spectrum of PLA-SN38. (B and C) Representative transmission electron microscopy (TEM) images, hydrodynamic diameter (DH) distribution and polydispersity index (PDI) of SNP and Glu-SNP. (D) Diameter changes of SNP and Glu-SNP over 2 weeks, indicating the stability of nanoparticles. (E) In vitro drug release profiles of total SN38 from SNP and Glu-SNP under dialyzing against PBS (pH 7.4) containing 0.2% Tween 80 with/without 50 U mL−1 esterase at 37 °C for 2 weeks. The data are presented as the mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001 and ‘ns’ represents p > 0.05. | ||
Meanwhile, SN38 release kinetics study was executed to evaluate the release behavior of SNP and Glu-SNP (Fig. 2E). SNP and Glu-SNP released a negligible amount of SN38 on Day 1. On Day 14, the release amount of total SN38 from SNP and Glu-SNP increased to 34.67 ± 3.54% and 33.00 ± 2.20%, respectively, which indicated that our prodrug design and nanoformulation strategy enabled SNP and Glu-SNP with a remarkably sustained-release property, further inhibited the premature release of chemotherapeutics (SN38) from nanoparticles during blood circulation, ultimately enhancing intratumoral drug accumulation. Moreover, it was reported that abundant esterase in the cancer cells played an important role in the intracellular release of chemotherapeutics.36 Herein, 50 U mL−1 esterase was added into the nanoparticle solutions to mimic the intracellular esterase-rich environment. Notably, the release amount of SN38 from SNP and Glu-SNP increased to 8.11 ± 1.12% and 8.00 ± 1.56% on Day 1; while the amount was determined as 67.32 ± 1.78% and 72.19 ± 3.02% on Day 14, respectively, which was significantly higher than the amount of released SN38 from SNP or Glu-SNP without esterase. These data supported a favorable quick intracellular release of SN38, which probably enhance the antitumor efficiency of SNP and Glu-SNP. These data demonstrated that there existed limited differences in morphology, release kinetics and other physicochemical properties of SNP and Glu-SNP.
3.3 In vitro antitumor efficiency of SN38, SNP and Glu-SNP
Surface modification of nanoparticles was a common way to enhance intracellular drug accumulation and further improve antitumor efficiency.25,37 CCK-8 assay was carried out to compare the cytotoxicity of SN38, SNP and Glu-SNP in the MKN45 and SGC-7901 cells (Fig. 3A and Table 1). IC50 of SN38, SNP and Glu-SNP in MKN45 cells were 68.36 ± 3.826, 167.7 ± 9.321 and 125.8 ± 8.684 nM; while in SGC-7901 cells, IC50 were 63.48 ± 5.936, 208.8 ± 20.34 and 154.3 ± 16.33 nM, respectively. IC50 of Glu-SNP was lower than SNP in both tested cell lines, which indicated that the decoration of glucose enhanced in vitro antitumor efficacy of Glu-SNP. EdU incorporation assay was further conducted to compare differences in the ability of SN38, SNP and Glu-SNP to inhibit the proliferation of cancer cells (Fig. 3B and C). EdU, as a thymine analog, can insert into replicating DNA and label cells in proliferation. As shown in Fig. 3B and C, SN38, SNP and Glu-SNP all potently inhibited proliferation of MKN45 and SGC-7901 cells. Glu-SNP exhibited comparable inhibition of proliferation to SN38, which was more efficient than SNP. | ||
| Fig. 3 In vitro antitumor evaluation of SN38, SNP and Glu-SNP. (A) In vitro cytotoxicity analysis of SN38, SNP and Glu-SNP against gastric cancer cells MKN45 and SGC-7901. MKN45 and SGC-7901 cells were treated with SN38, SNP and Glu-SNP for 72 h at serial dilutions. At the end of exposure, cell viabilities were determined by CCK-8 assay. (B and C) Proliferation of MKN45 and SGC-7901 cells after 24 h treatment with SN38, SNP and Glu-SNP at an SN38 concentration of 20 nM quantified by EdU assay. The cell nuclei were stained with Hoechst 33342 (blue) and only proliferating cells were stained with EdU (red). Scale bars = 100 μm. The data are presented as the mean ± SD for n > 5 regions with 1500–2000 total cells. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001 and ‘ns’ represents p > 0.05. | ||
| Drug formulation | IC50 (nM) | |
|---|---|---|
| MKN45 | SGC-7901 | |
| SN38 | 68.36 ± 3.826 | 63.48 ± 5.936 |
| SNP | 167.7 ± 9.321 | 208.8 ± 20.34 |
| Glu-SNP | 125.8 ± 8.684 | 154.3 ± 16.33 |
3.4 Mode of action of SN38-loaded nanoparticles to induce gastric cell death
SN38 acts as a typical DNA topoisomerase I inhibitor to induce cell cycle arrest and death. The effects of SN38-induced DNA damages on cell cycle distribution were studied in MKN45 cells (Fig. 4A and B). After treated with SN38, SNP and Glu-SNP for 24 h, an obvious arrest in S phase was observed. More SN38- and Glu-SNP-treated MKN45 cells accumulated in S phase than SNP-treated MKN45. The cell apoptosis-inducing capability of SN38, SNP and Glu-SNP were also evaluated by Annexin V-FITC/PI double staining and western blot assay. As shown in Fig. 4C and D, SN38 and Glu-SNP induced comparable apoptotic proportion at the same SN38 concentration, which was higher than that of SNP. The sum of the early and late apoptotic cell proportion in SN38 and Glu-SNP reached 30.6 ± 2.33% and 27.2 ± 0.82%, respectively; by contrast, SNP induced 18.6 ± 4.28% apoptotic cells in total. Results of western blot assay were consistent with flow cytometer analysis (Fig. 4E). The elevated expression of cleaved-PARP and cleaved-caspase 3 protein indicated a strong apoptosis-inducing capability of both SNP and Glu-SNP. These consistent results suggested Glu-SNP was able to induce a higher proportion of apoptotic cells than SNP.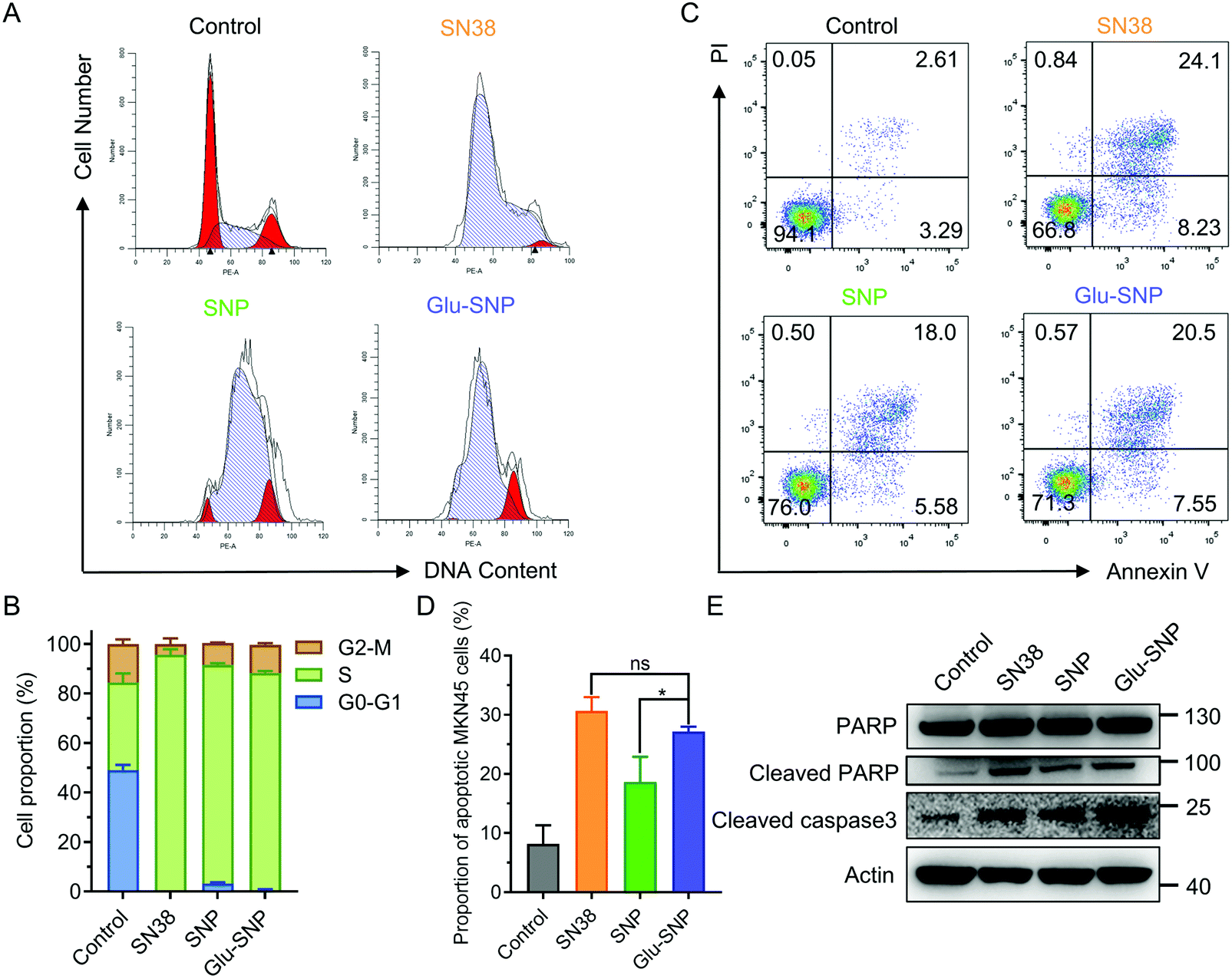 | ||
| Fig. 4 Cell cycle arrest and apoptotic analysis of SN38, SNP and Glu-SNP. (A and B) Cell cycle distribution of MKN45 cells detected by flow cytometry after 24 h treatment with SN38, SNP and Glu-SNP at an SN38 concentration of 20 nM. Cells were stained with propidium iodide (PI) and analyzed by flow cytometry. The data are presented as the mean ± SD (n = 3). (C and D) Apoptotic analysis of MKN45 cells using flow cytometry. Cells were treated with SN38, SNP and Glu-SNP at an SN38 concentration of 200 nM for 36 h. After exposure, cells were harvested and stained with Annexin V-FITC and propidium iodide (PI) for a following flow cytometry analysis. The apoptosis rates were detected by a flow cytometer. The data are presented as the mean ± SD (n = 3). (E) Western blot analysis of apoptotic protein (Cleaved-PARP, Cleaved-caspase 3) expression. MKN45 cells were treated with SN38, SNP and Glu-SNP at an SN38 concentration of 100 nM for 48 h. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001 and ‘ns’ represents p > 0.05. | ||
An in vitro cytotoxicity study process was executed in addition to verify the advantage of gastric cancer-targeted SN38 delivery. As illustrated in Fig. 5A, cells were incubated with SNP or Glu-SNP for 24 h and then the medium was replaced with fresh medium. After incubation for an additional 24 h, cell viability was accessed by CCK-8 assay. In MKN45 and SGC-7901 cells, targetable Glu-SNP exhibited more potent to inhibit cell activity than SNP (Fig. 5B). Comparing with the results in Fig. 3A, differences in cytotoxicity between Glu-SNP and SNP against gastric cancer cells were enlarged when the incubation was shorten from 72 h to 24 h, which might attribute to a nanoparticles “over-saturation” status.25,37 After incubation with Glu-SNP or SNP for 72 h, enough nanoparticles probably have been uptaken by cancer cells via multiple ways, which further smoothed out the advantage of targeted Glu-SNP in cellular uptake. Regarding the normal human gastric epithelial cell line GES-1, there existed limited difference in the cytotoxicity between SNP and Glu-SNP. To further analyze the role of GLUT1 in the gastric cancer-targeted SN38 delivery, WZB-117, a typical GLUT1 inhibitor, was applied to block GLUT1 and added 6 h before SNP and Glu-SNP administration. Notably, differences in the cytotoxicity between SNP and Glu-SNP against gastric cancer cells were almost erased (Fig. 5C and Fig. S4, ESI†). Moreover, we found that WZB-117 sensitized both MKN45 and SGC-7901 cells to SNP and Glu-SNP, which indicated a potential synergetic effect of WZB-117 and SN38. Results of cell cycle analysis, apoptotic assessment and CCK-8 assays indicated Glu-SNP was more potent than SNP in the in vitro antitumor efficiency.
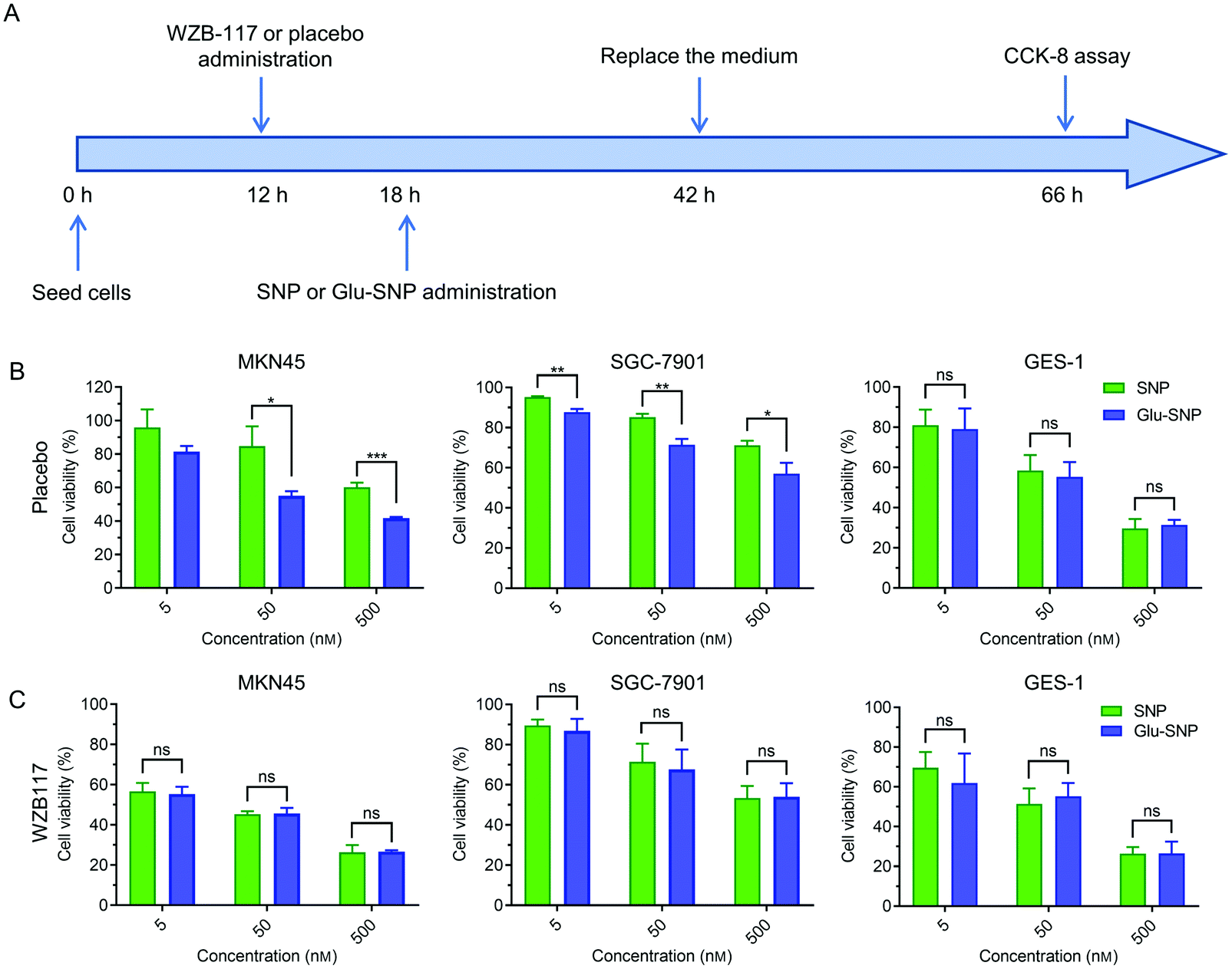 | ||
| Fig. 5 In vitro cytotoxicity evaluation of SNP and Glu-SNP for a short-time incubation. (A) Schematic illustration of in vitro cytotoxicity evaluation of SNP and Glu-SNP for a short-time incubation. Cells were pretreated with GLUT1 inhibitor WZB-117 or placebo for 6 h, followed by a 24 h culture with SNP and Glu-SNP. After exposure, the medium was replaced with fresh medium and cells were incubated for an additional 24 h. Finally, cell viability was assessed by the CCK-8 assay. (B) In vitro cytotoxicity of SNP and Glu-SNP against MKN45, SGC-7901 and GES-1 cells pretreated with placebo. (C) In vitro cytotoxicity of SNP and Glu-SNP against MKN45, SGC-7901 and GES-1 cells pretreated with 10 μM WZB-117 for 6 h. The data are presented as the mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001 and ‘ns’ represents p > 0.05. | ||
3.5 Cell-specific recognition and enhanced uptake of Glu-SNP
The cell-specific recognition and intracellular uptake of nanoparticles were monitored by confocal laser scanning microscopy (CLSM) and flow cytometer. To track intracellular uptake of nanoparticles, Cy5.5, a NIR fluorescence probe, was conjugated to PLA and assembled with PLA-SN38 and DSPE-PEG2000/DSPE-PEG2000-Glu to form Cy5.5@SNP and Cy5.5@Glu-SNP. Firstly, we testified whether the glucose decoration could enhance the cell-specific uptake of nanoparticles. The gastric cancer cells (MKN45) and normal stomach epithelial cells (GES-1) were treated with Cy5.5@SNP and Cy5.5@Glu-SNP for the same time intervals (1 h, 3 h and 6 h), and then observed upon CLSM. In MKN45 cells, a significantly stronger red fluorescence signal derived from Cy5.5 was detected following 1 h and 3 h treatment with Cy5.5@Glu-SNP compared with Cy5.5@SNP post-treatment group (Fig. 6A and B). At 6 h post-treatment, the variety between two nanomedicines weakened, which might attribute to the high concentration of Cy5.5-labeled nanoparticles in the medium and the cell uptake approaching the saturation point. Results of flow cytometer analysis were consistent with CLSM observation (Fig. 6C and Fig. S5, ESI†). However, both CLSM observation and flow cytometer analysis displayed negligible differences between two nanomedicines in GES-1 cells (Fig. 6D and E), which may be attributed to the relatively low expression of GLUT1 in normal stomach epithelial cells. The differences in the cellular uptake of SN38-loaded nanoparticles might account for enhancement in the in vitro cytotoxicity of Glu-SNP. | ||
| Fig. 6 Cellular uptake evaluation of Cy5.5 labeled SNP (Cy5.5@SNP) and Glu-SNP (Cy5.5@Glu-SNP) in gastric cancer cell line MKN45 and normal gastric epithelial cell line GES-1. (A and B) Representative confocal laser scanning microscopy (CLSM) images and quantification of Cy5.5 labeled nanoparticles taken up by MKN45 cells for different time intervals (1 h, 3 h and 6 h). (C) Representative flow cytometric analysis of Cy5.5 labeled nanoparticles taken up by MKN45 cells for different time intervals (1 h, 3 h and 6 h) (n = 3). (D and E) Representative CLSM images and quantification of Cy5.5 labeled nanoparticles taken up by GES-1 cells for different time intervals (1 h, 3 h and 6 h). (F) Representative flow cytometric analysis of Cy5.5 labeled nanoparticles taken up by GES-1 cells for different time intervals (1 h, 3 h and 6 h) (n = 3). The quantification in CLSM images is executed via Image J software for at least 6 regions. In CLSM images, cell nuclei stained with Hoechst 33342 are shown in blue, and Cy5.5 labeled nanoparticles are shown in red. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001 and ‘ns’ represents p > 0.05. | ||
Furthermore, we hypothesized that glucose-decorated nanoparticles facilitate the intracellular uptake of gastric cancer cells in a GLUT1-dependent manner. To test this assumption, 10 μM WZB-117 was added to block GLUT1.38–40 Although there remained differences in the intracellular accumulation of nanoparticles after 1 h incubation, WZB-117 generally attenuated the enhanced cellular uptake of Cy5.5@Glu-SNP (Fig. 7A–C). Extra glucose was also added into the medium to compete with Glucose-decorated nanoparticles. Both CLSM observation and flow cytometer analysis indicated that there existed limited difference in the cellular uptake of Cy5.5@SNP and Cy5.5@Glu-SNP when the cells were pretreated with high glucose medium (Fig. 7D–F). Moreover, glucose in the medium was deprived to mimic the intratumoral physiological environment and to further explore the role of GLUT1 in the enhancement of cellular uptake of Cy5.5@Glu-SNP. Notably, compared with incubation in the normal medium, the difference in the cellular uptake of Cy5.5@SNP and Cy5.5@Glu-SNP was enlarged after 3 h and 6 h treatment possibly owing to the activity of unoccupied GLUT1 (Fig. 7G–I). All these results demonstrated that glucose decoration enabled Glu-SNP an enhanced cellular uptake in gastric cancer cells, which was in some way attributed to the overexpression of GLUT1.
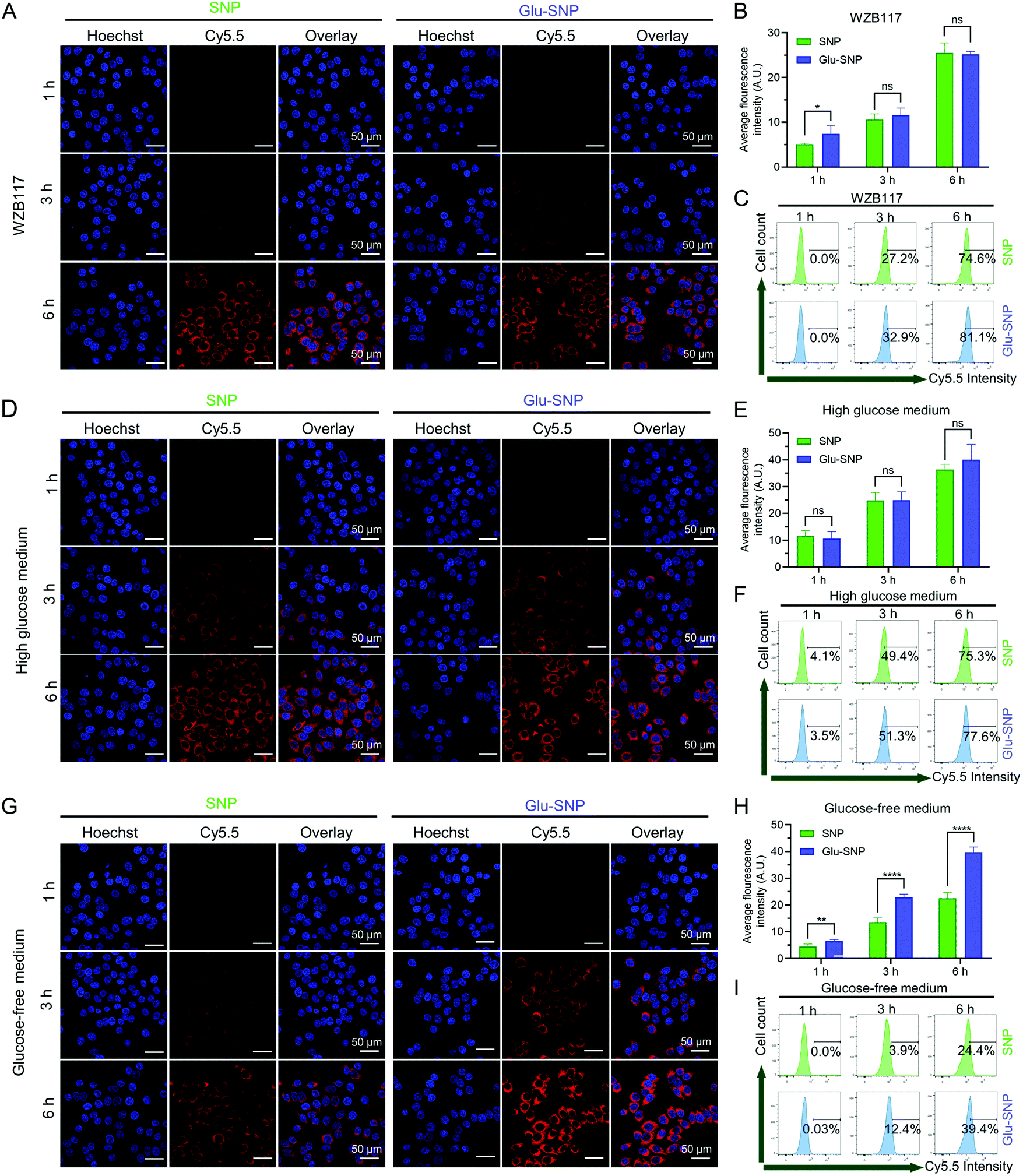 | ||
| Fig. 7 Cellular uptake evaluation of Cy5.5@SNP and Cy5.5@Glu-SNP in gastric cancer cell line MKN45 in different culture mediums. (A and B) Representative CLSM images and quantification of Cy5.5 labeled nanoparticles taken up by MKN45 cells pretreated with 10 μM WZB-117 for 3 h. (C) Representative flow cytometric analysis of Cy5.5 labeled nanoparticles taken up by MKN45 cells pretreated with 10 μM WZB-117 for 3 h (n = 3). (D and E) Representative CLSM images and quantification of Cy5.5 labeled nanoparticles taken up by MKN45 cells pretreated with high glucose medium (25 mM) for 3 h. (F) Representative flow cytometric analysis of Cy5.5 labeled nanoparticles taken up by MKN45 cells pretreated with high glucose medium (25 mM) for 3 h (n = 3). (G and H) Representative CLSM images and quantification of Cy5.5 labeled nanoparticles taken up by MKN45 cells pretreated with the glucose-free medium for 3 h. (I) Representative flow cytometric analysis of Cy5.5 labeled nanoparticles taken up by MKN45 cells pretreated with the glucose-free medium for 3 h (n = 3). The quantification in CLSM images is executed via Image J software for at least 6 regions. In CLSM images, cell nuclei stained with Hoechst 33342 are shown in blue, and Cy5.5 labeled nanoparticles are shown in red. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001 and ‘ns’ represents p > 0.05. | ||
3.6 In vivo distribution of SN38-loaded nanoparticles in an MKN45 xenograft nude mouse model
Encouraged by the excellent in vitro antitumor efficiency and enhanced cellular uptake of Glu-SNP, we further analyzed whether glucose decoration could increase the intratumoral accumulation of SN38-loaded nanoparticles in the MKN45 xenograft nude mouse model. After a single intravenous injection of Cy5.5@SNP or Cy5.5@Glu-SNP, in vivo imaging system was applied to visualize biodistribution of nanomedicines at different timepoints. The fluorescence signal of Cy5.5@Glu-SNP in tumor sites was significantly higher than Cy5.5@SNP (Fig. 8A), indicating a favorable intratumoral accumulation of Glu-SNP. After 48 h post-administration, mice were sacrificed and tumors and major organs were isolated for ex vivo imaging analysis immediately. As shown in Fig. 8B, Cy5.5 mainly concentrated in the kidney, liver and spleen in Cy5.5@SNP-treated mice. By contrast, in Cy5.5@Glu-SNP-treated mice, fluorescence signal detected in tumor sites was higher and a large number of nanoparticles still accumulated in the kidney, liver and spleen possibly owing to the metabolism effect of kidney and liver. Quantification of fluorescence intensity in different excised organs also verified an enhanced intratumoral accumulation of Glu-SNP (Fig. 8C). Results of in vivo distribution assay demonstrated that glucose decoration endowed Glu-SNP with increased targeting capability toward gastric cancer. | ||
| Fig. 8 Biodistribution of Cy5.5@SNP and Cy5.5@Glu-SNP in an MKN45 xenograft nude mouse model. (A) In vivo real-time imaging of MKN45 xenograft nude mice after a single intravenous injection of Cy5.5@SNP and Cy5.5@Glu-SNP at a Cy5.5 dose of 20 μg per mouse. (B) Ex vivo fluorescence images of the heart, lung, liver, kidney, spleen and tumor of Cy5.5@SNP- and Cy5.5@Glu-SNP-treated mice at 48 h post-injection. (C) Relative signal of Cy5.5 in the excised organs at 48 h post-injection (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001 and ‘ns’ represents p > 0.05. | ||
3.7 In vivo antitumor and biosafety evaluation in an MKN45 xenograft nude mouse model
MKN45 xenograft nude mouse model was established for in vivo antitumor evaluation of SNP and Glu-SNP, we’ve constructed. Irinotecan (CPT-11), the front-line drug against advanced gastric cancer, could be conversed to active metabolite SN38 with the help of carboxylesterases in the liver and was set as a positive control in our study. Mice in different treatment groups were administrated with saline, CPT-11, SNP and Glu-SNP, respectively, every other day for three times in total. Tumor volume and weight of mice were monitored every day. As shown in Fig. 9A–C, both SNP and Glu-SNP exhibited outstanding ability to inhibit tumor growth, with a tumor-growth inhibition rate (TIR%) of 91.5 ± 1.49% and 93.9 ± 1.08%, respectively, in terms of tumor weight at the end of the experiment, significantly higher than that of CPT-11 (20.6 ± 13.2%). And body weight monitorization displayed negligible variation between different groups (Fig. 9D). H&E and IHC analysis was executed to further evaluate in vivo antitumor efficiency of SNP and Glu-SNP (Fig. 9E–G). Impressively, H&E staining of the tumor samples, excised from Glu-SNP-treated mice, exhibited numerous apoptotic cells with extensive vacuolization and nucleus shrinkage (black arrow in Fig. 9E). As for comparisons, H&E staining of tumor sections, from both saline- and CPT-11-treated mice, showed tightly-packed cancer cells. There existed much less apoptotic cells in the SNP group, compared with apoptotic cell proportion in the Glu-SNP group. Results of Ki67 and TUNEL immunohistochemistry assays were highly consistent with that of H&E staining. There were fewer cells in proliferation (Ki67 positive rate) and more apoptotic cells (Tunel positive rate) in the Glu-SNP group than those in the SNP group, which also supported that Glu-SNP was more efficient for therapeutics against MKN45 xenograft nude mouse model. | ||
| Fig. 9 The antitumor activity of SNP and Glu-SNP in an MKN45 xenograft nude mouse model. (A) The tumor growth curves after intravenous injection of saline, CPT-11 (15 mg kg−1), SNP and Glu-SNP (10 mg kg−1, at SN38 equivalent dose) according to a q2 × 3 regimen. Black arrows indicate drug injections. (B) The average tumor weight of the excised tumor in each group at the experimental endpoint (n = 6). (C) Images of excised tumors of mice in each group at the experimental endpoint. (D) Body weight variation in the tumor-bearing mice during the experimental period (n = 6). (E) Representative images of H&E staining, Ki67 immunohistochemistry and TUNEL histopathology analysis of tumors in each group. Scale bars = 100 μm. (F and G) Quantitative analysis of Ki67 immunohistochemistry and TUNEL histopathology positive cells. The data are presented as the mean ± SD for at least 3 regions. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001 and ‘ns’ represents p > 0.05. | ||
Myelosuppression (mainly leukopenia and thrombocytopenia) and gastrointestinal toxicity were reported to be the prominent side effects of CPT-11 and SN38. Hence, major organs, including liver, stomach and colon, were excised and subjected for H&E histological analysis at the end of in vivo antitumor evaluation (Fig. 10A). H&E histological analysis didn’t exhibit obvious systemic damages in all the groups. Meanwhile, blood samples of mice in the different groups at the end of in vivo antitumor evaluation were also collected (Fig. 10B). Blood routine and blood biochemical examinations are performed to explore whether CPT-11 and SN38-loaded nanoparticles (SNP and Glu-SNP) could induce myelosuppression and liver and kidney damages. Except elevated neutrophil percentage and decreased lymphocyte percentage in the CPT-11 group, all the other blood routine and blood biochemical parameters were within a normal range, which indicated CPT-11 and SN38-loaded nanoparticles (SNP and Glu-SNP) at current therapeutic dosages didn’t cause severe or permanent myelosuppression and liver and kidney damages. And transient continuous injections of CPT-11 thrice might induce a mild inflammation, possibly accounting for changes in neutrophil and lymphocyte percentages.
 | ||
| Fig. 10 In vivo biosafety evaluation of SNP and Glu-SNP. (A) Representative H&E staining of hearts, livers, spleens, lungs and kidneys excised from nude mice in the different groups (n = 6 for each group). Scale bars = 100 μm. (B) Whole-cell counts, liver and kidney toxicity analysis of different therapies. The data are represented as the mean ± SD (n = 6). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001 and ‘ns’ represents p > 0.05. RBC: red boold cell; HGB: hemoglobin; HCT: hematocrit; PLT: platelet count; WBC: white boold cell; NE%: neutrophil percentage; LY%: lymphocyte percentage; AST: aspartate aminotransferase; ALT: alanine aminotransferase; BUN: blood urea nitrogen; CRE: creatinine. | ||
4. Conclusion
In this study, we have conducted TCGA data analysis and a convictive tissue array analysis to verify the overexpression of GLUT1 on both mRNA and protein level in gastric cancers. Furthermore, the “Sweet Tooth”-oriented SN38 prodrug delivery nanoplatform (Glu-SNP) for targeted gastric cancer therapy was established through prodrug strategy and nanoformulation. Both SNP and Glu-SNP possessed appropriate particle sizes, superior long-term stability and sustained drug release profiles. Glucose decoration of nanoparticles resulted in an enhancement in the cytotoxicity via GLUT1-specific intracellular accumulation in vitro relative to the undecorated counterparts. Glucose competition (WZB-117 and high glucose concentration) and glucose deprivation experiments provided strong evidence that Glu-SNP enhanced the intracellular accumulation and cytotoxicity in a GLUT1-mediated manner. Moreover, owing to the proper exploitation of the EPR effect and active targeting of glucose decoration, Glu-SNP displayed a favorable intratumoral accumulation and avoided the possible off-target effects. In an MKN45 xenograft nude mouse model, Glu-SNP exhibited an outstanding tumor growth inhibition capability and biosafety. Collectively, motivated by the “Sweet Tooth” of gastric cancer, we developed a glucose-decorated nanoplatform that combines prodrug strategy with nanoformulation. This devised nanoplatform is an easy approach, reproducible and scalable. Hence, this approach could also be used in reformulation of other potential antitumor agents with poor pharmacokinetics or bioavailability to improve the targeting ability and therapeutic efficacy.Author contributions
L. W., X. X., N. D., S. X., conceived and designed the experiments; N. D., S. X., S. Z., Q. Y., W. C., Z. Z. performed the experiments; L. X., S. L., S. X., Z. L. analyzed the data; N. D., S. X. wrote the manuscript; L. W., X. X., M. X. made revisions to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (82072623, 82073229 and 81802348). The authors express deepest thanks to Prof. Hangxiang Wang for providing helpful suggestions. Shengjun Xu appreciate the help of Jiali Zhang and Rong Su in the flow cytometer analysis.References
- M. G. V. Heiden, L. C. Cantley and C. B. Thompson, Science, 2009, 324, 1029–1033 CrossRef PubMed.
- T. Bui and C. B. Thompson, Cancer Cell, 2006, 9, 419–420 CrossRef CAS PubMed.
- M. Y. Fong and S. E. Wang, Cell Cycle, 2015, 14, 1768–1769 CrossRef CAS PubMed.
- V. Ganapathy, M. Thangaraju and P. D. Prasad, Pharmacol. Ther., 2009, 121, 29–40 CrossRef CAS PubMed.
- J.-w. Kim and C. V. Dang, Cancer Res., 2006, 66, 8927–8930 CrossRef CAS PubMed.
- J. W. Fletcher, B. Djulbegovic, H. P. Soares, B. A. Siegel, V. J. Lowe, G. H. Lyman, R. E. Coleman, R. Wahl, J. C. Paschold, N. Avrill, L. H. Einhorn, W. W. Suh, D. Samson'O, D. Delbekell, M. Gorman and A. F. Shields, J. Nucl. Med., 2008, 49, 480–508 CrossRef PubMed.
- R. L. Wahl, H. Jacene, Y. Kasamon and M. A. Lodge, J. Nucl. Med., 2009, 50, 122S–150S CrossRef CAS PubMed.
- T. Amann and C. Hellerbrand, Expert Opin. Ther. Targets, 2009, 13, 1411–1427 CrossRef CAS PubMed.
- B. Y. Shim, J.-H. Jung, K.-M. Lee, H.-J. Kim, S. H. Hong, S. H. Kim, D. S. Sun and H.-M. Cho, Int. J. Colorectal Dis., 2013, 28, 375–383 CrossRef PubMed.
- T. Kawamura, T. Kusakabe, T. Sugino, K. Watanabe, T. Fukuda, A. Nashimoto, K. Honma and T. Suzuki, Cancer, 2001, 92, 634–641 CrossRef CAS PubMed.
- J. Wang, W. Xu, B. Wang, G. Lin, Y. Wei, M. Abudurexiti, W. Zhu, C. Liu, X. Qin, B. Dai, F. Wan, H. Zhang, Y. Zhu and D. Ye, Cancer Lett., 2020, 485, 45–55 CrossRef CAS PubMed.
- F.-Q. Zhao and A. F. Keating, Curr. Genomics, 2007, 8, 113–128 CrossRef CAS PubMed.
- D. Deng, C. Xu, P. Sun, J. Wu, C. Yan, M. Hu and N. Yan, Nature, 2014, 510, 121–125 CrossRef CAS PubMed.
- Z. Wan, X. Li, R. Sun, Y. Li, X. Wang, X. Li, L. Rong, Z. Shi and J. Bao, J. Mol. Struct., 2015, 1101, 57–65 CrossRef CAS.
- Y. Liu, W. Zhang, Y. Cao, Y. Liu, S. Bergmeier and X. Chen, Cancer Lett., 2010, 298, 176–185 CrossRef CAS PubMed.
- Y.-S. Lin, R. Tungpradit, S. Sinchaikul, F.-M. An, D.-Z. Liu, S. Phutrakul and S.-T. Chen, J. Med. Chem., 2008, 51, 7428–7441 CrossRef CAS PubMed.
- T. Mandai, H. Okumoto, T. Oshitari, K. Nakanishi, K. Mikuni, K. J. Hara, K. Z. Hara, W. Iwatani, T. Amano, K. Nakamura and Y. Tsuchiya, Heterocycles, 2001, 54, 561–566 CrossRef CAS.
- D.-Z. Liu, S. Sinchaikul, P. Vasu, G. Reddy, M.-Y. Chang and S.-T. Chen, Bioorg. Med. Chem. Lett., 2007, 17, 617–620 CrossRef CAS PubMed.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- N. C. C. Network, NCCN clinical practice guidelines in oncology, (accessed October 14, 2020).
- B. Glimelius, K. Ekstrom, K. Hoffman, W. Graf, P. O. Sjoden, U. Haglund, C. Svensson, L. K. Enander, T. Linne, H. Sellstrom and R. Heuman, Ann. Oncol., 1997, 8, 163–168 CrossRef CAS PubMed.
- A. D. Wagner, N. L. X. Syn, M. Moehler, W. Grothe, W. P. Yong, B.-C. Tai, J. Ho and S. Unverzagt, Cochrane Database Syst. Rev., 2017, 8, 1465–1858 Search PubMed.
- H. A. Schloesser, U. Drebber, A. Urbanski, S. Haase, C. Baltin, F. Berlth, S. Neiss, M. von Bergwelt-Baildon, U. K. Fetzner, U. Warnecke-Eberz, E. Bollschweiler, A. H. Hoelscher, S. P. Moenig and H. Alakus, Gastric Cancer, 2017, 20, 83–91 CrossRef CAS PubMed.
- P. Huang, L. Bao, C. Zhang, J. Lin, T. Luo, D. Yang, M. He, Z. Li, G. Gao, B. Gao, S. Fu and D. Cui, Biomaterials, 2011, 32, 9796–9809 CrossRef CAS PubMed.
- L. Xu, S. Xu, H. Wang, J. Zhang, Z. Chen, L. Pan, J. Wang, X. Wei, H. Xie, L. Zhou, S. Zheng and X. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 3229–3240 CrossRef CAS PubMed.
- E. Fernandes, D. Ferreira, A. Peixoto, R. Freitas, M. Relvas-Santos, C. Palmeira, G. Martins, A. Barros, L. L. Santos, B. Sarmento and J. A. Ferreira, Int. J. Pharm., 2019, 570, 118646 CrossRef CAS PubMed.
- E. C. Calvaresi and P. J. Hergenrother, Chem. Sci., 2013, 4, 2319–2333 RSC.
- A. Bukchin, G. Pascual-Pasto, M. Cuadrado-Vilanova, H. Castillo-Ecija, C. Monterrubio, N. G. Olaciregui, M. Vila-Ubach, L. Ordeix, J. Mora, A. M. Carcaboso and A. Sosnik, J. Controlled Release, 2018, 276, 59–71 CrossRef CAS PubMed.
- S. Jin, Z. Du, H. Guo, H. Zhang, F. Ren and P. Wang, Int. J. Mol. Sci., 2019, 20, 697 CrossRef CAS PubMed.
- H. Wang, L. Zhou, K. Xie, J. Wu, P. Song, H. Xie, L. Zhou, J. Liu, X. Xu, Y. Shen and S. Zheng, Theranostics, 2018, 8, 3949–3963 CrossRef CAS PubMed.
- S. Ling, J. Li, Q. Shan, H. Dai, D. Lu, X. Wen, P. Song, H. Xie, L. Zhou, J. Liu, X. Xu and S. Zheng, Mol. Oncol., 2017, 11, 682–695 CrossRef CAS PubMed.
- J. Wan, Y. Qiao, X. Chen, J. Wu, L. Zhou, J. Zhang, S. Fang and H. Wang, Adv. Funct. Mater., 2018, 28, 1804229 CrossRef.
- T. Amann, U. Maegdefrau, A. Hartmann, A. Agaimy, J. Marienhagen, T. S. Weiss, O. Stoeltzing, C. Warnecke, J. Schoelmerich, P. J. Oefner, M. Kreutz, A. K. Bosserhoff and C. Hellerbrand, Am. J. Pathol., 2009, 174, 1544–1552 CrossRef CAS PubMed.
- C.-H. Chan, C.-F. Li, W.-L. Yang, Y. Gao, S.-W. Lee, Z. Feng, H.-Y. Huang, K. K. C. Tsai, L. G. Flores, Y. Shao, J. D. Hazle, D. Yu, W. Wei, D. Sarbassov, M.-C. Hung, K. I. Nakayama and H.-K. Lin, Cell, 2012, 149, 1098–1111 CrossRef CAS PubMed.
- G. G. Chabot, Clin. Pharmacokinet., 1997, 33, 245–259 CrossRef CAS PubMed.
- N. Qiu, X. Liu, Y. Zhong, Z. Zhou, Y. Piao, L. Miao, Q. Zhang, J. Tang, L. Huang and Y. Shen, Adv. Mater., 2016, 28, 10613–10622 CrossRef CAS PubMed.
- L. Wu, F. Zhang, X. Chen, J. Wan, Y. Wang, T. Li and H. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 3327–3340 CrossRef CAS PubMed.
- F. Zhao, J. Ming, Y. Zhou and L. Fan, Cancer Chemother. Pharmacol., 2016, 77, 963–972 CrossRef CAS PubMed.
- O. A. Ojelabi, K. P. Lloyd, A. H. Simon, J. K. De Zutter and A. Carruthers, J. Biol. Chem., 2016, 291, 26762–26772 CrossRef CAS PubMed.
- K. Shibuya, M. Okada, S. Suzuki, M. Seino, S. Seino, H. Takeda and C. Kitanaka, Oncotarget, 2015, 6, 651–661 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb02787a |
| ‡ Ning Ding and Shengjun Xu contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |