
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFibronectin adsorption on oxygen plasma-treated polyurethane surfaces modulates endothelial cell response†
Ruben
Daum‡
ab,
Ivana
Mrsic‡
c,
Johanna
Hutterer
 c,
Achim
Junginger
c,
Svenja
Hinderer
ab,
Alfred J.
Meixner
cd,
Günter
Gauglitz
c,
Thomas
Chassé
cd and
Katja
Schenke-Layland
*abef
c,
Achim
Junginger
c,
Svenja
Hinderer
ab,
Alfred J.
Meixner
cd,
Günter
Gauglitz
c,
Thomas
Chassé
cd and
Katja
Schenke-Layland
*abef
aNMI Natural and Medical Sciences, Institute at the University of Tübingen, Markwiesenstr. 55, 72770 Reutlingen, Germany Web: http://www.nmi.de/en
bDepartment of Women's Health, Research Institute for Women's Health, Eberhard Karls University Tübingen, Silcherstr. 7/1, 72076 Tübingen, Germany. E-mail: katja.schenke-layland@uni-tuebingen.de; Tel: +49-707-1298-5205 Web: http://www.schenke-layland-lab.de
cInstitute of Physical and Theoretical Chemistry, Eberhard Karls University Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany
dCenter for Light–Matter Interaction, Sensors & Analytics (LISA+) at the University of Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany
eCluster of Excellence iFIT (EXC 2180) “Image-Guided and Functionally Instructed Tumor Therapies”, Eberhard Karls University Tübingen, 72076 Tübingen, Germany
fDepartment of Medicine/Cardiology, Cardiovascular Research Laboratories, David Geffen School of Medicine at UCLA, Los Angeles, CA, 90095, USA
First published on 18th January 2021
Abstract
Fibronectin coating increases implant biocompatibility by enhancing surface endothelialization via integrin-mediated binding. Surface properties determine the fibronectin orientation and conformation, dictating which ligands are presented, and therefore altering the bioactivity of an implant surface. In this study, polyurethane was treated with oxygen plasma, which allowed for a simultaneous modification of the surface chemistry and topography to modulate fibronectin adsorption. By varying the parameters of the treatment, human plasma fibronectin adsorbed on the surfaces in different conformations, orientations, and binding affinities, which was investigated by atomic force microscopy, fluorescence microscopy, monoclonal and polyclonal antibody staining and reflectometric interference spectroscopy. Apart from the most hydrophilic rough surfaces, the adsorbed fibronectin showed a lower binding affinity and less conformational change on the more hydrophilic surfaces. A large amount of exposed fibronectin–cell binding was detected on the rough treated and the smooth untreated surfaces. Primary isolated human umbilical vein and human microvascular endothelial cells showed a significantly higher cell adherence on the absorbed fibronectin with a low binding affinity and low conformational changes. Significant differences in the formation of mature focal adhesions and the reorganization of F-actin were identified on the rough treated and the smooth untreated surfaces. Our data suggest that oxygen plasma treatment is a reliable technique for the modulation of fibronectin adsorption in order to adjust fibronectin bioactivity and impact cell responses to implant surfaces.
1. Introduction
The interaction between cardiovascular implants and cells in the human body is highly influenced by the layer of blood proteins initially adsorbed onto the synthetic material surfaces.1 In order to trigger specific reactions in the body, an implant material can be functionalized with bioactive substances. An attractive candidate that has been reported to support endothelialization of the surface is fibronectin (FN), a well-studied glycoprotein of the extracellular matrix (ECM).2–4 However, the influence of the surface properties on the deposition and thus bioactivity of FN is often neglected. FN fulfils a wide range of biological functions as it interacts with other ECM molecules and growth factors, binds to cell surface receptors and forms fibrils through intermolecular interactions.5,6 It is a 440 kDa macromolecule that is secreted as a dimer connected by two disulfide S–S bonds at the C-terminus. The two peptide chains consist of several repeating domains (FNI, FNII and FNIII) that can associate with each other, resulting in a globular folding structure of FN.7 Moreover, FN is very flexible and can change its conformation depending on the environment.8In vivo, FN is synthesized, for example, by hepatocytes and secreted into the blood plasma in an inactive globular conformation.9 The soluble plasma FN binds poorly to many cell types. In early wound-healing responses; however, it undergoes conformational changes by interacting with fibrin and platelets.10 During this finely tuned unfolding process of the protein, cell- and FN-binding sides are exposed, which leads to further stages of tissue repair.10 The folding of FN adsorbed on biomaterial surfaces has been shown to be influenced by surface properties such as wettability,11 surface chemistry,12 and roughness.13 In recent years, several studies have investigated the influence of modified substrates on the conformation of FN and its effect on cell behavior.14–17 Based on this knowledge, research now aims to specifically control cell attachment, proliferation, and differentiation through substrate-dependent changes.18–20 Cells interact with FN mainly via the α5β1 integrin, which binds to the RGD loop on FNIII10 and the neighboring PHSRN sequence in FNIII9.21–23 This interaction induces integrin clustering and the formation of focal adhesion complexes.10 These protein complexes are dynamic structures that form mechanical links between FN and the cell cytoskeleton, and mediate the transduction of signaling events, which in turn affects the growth, differentiation, adhesion, and motility of the cell.9 At the same time, the cell assembles FN into a fibrillar network through an integrin-dependent mechanism.24 All these processes are strongly influenced by the conformation and the adhesive force of the adsorbed FN, and ultimately determine cell behavior.20In this study, we investigated the adsorption behavior of FN and subsequently the endothelial cell–surface interaction through the fine-tuning of the surface wettability with oxygen plasma treatment of polyurethane (PU). The selected material is a commercially available PU of the Pellethane® series, which is being currently studied in cardiovascular devices.3 The effects of oxygen plasma on the wettability of Pellethane® was examined in a recent study over a wide range of plasma treatment parameters.25 Using this method allows the incorporation of oxygen-rich functional groups onto the material surface changing the chemical composition.26 Simultaneously to functionalization, the competing plasma etching of low molecular contaminations and upper polymer surface layers is modifying the surface topography and therefore material roughness.27,28 The objective of this study was a defined modification of the surface chemistry and roughness of PU in order to modulate the adsorption behavior and conformation of FN, which affects its bioactivity and subsequently influences endothelial cell responses, potentially providing surfaces for various cardiovascular applications.
2. Materials & methods
2.1 Sample preparation
Plasma surface modifications of the polymer films were performed in a FEMTO low pressure plasma system (Diener electronic, Ebhausen, Germany) consisting of a borosilicate glass round vacuum and a low-frequency (40 kHz) generator using oxygen plasma. A previous study investigated the plasma modification of PU according to a statistical experimental design.25 Based on this study, to achieve varied modifications with few plasma parameters, the process time and pressure were varied between 0.2–3.0 minutes and 0.2–0.8 mbar (Table 1). The applicable power was kept constant at 100 W.
2.2 Characterization of the surfaces
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500, F3648-100UL, Sigma-Aldrich, St. Louis, USA) and a monoclonal anti-FN antibody, which is described to bind against the flexible linker between the 9th and 10th type III repeat of FN33 (HFN7.1, 1:200, ABIN284417, antibodies-online, Aachen, Germany). After washing once with PBS, the samples were incubated with goat anti-rabbit IgG-Alexa Fluor® 546 and goat anti-mouse IgG-Alexa FluorΔ 488 (both 1:250, Thermo Fisher Scientific) for 45 minutes in the dark.34 Images were acquired by using a Zeiss fluorescence microscope (Cell Observer, Carl Zeiss AG, Oberkochen, Germany). The focal plane was adjusted manually for each image.
:500, F3648-100UL, Sigma-Aldrich, St. Louis, USA) and a monoclonal anti-FN antibody, which is described to bind against the flexible linker between the 9th and 10th type III repeat of FN33 (HFN7.1, 1:200, ABIN284417, antibodies-online, Aachen, Germany). After washing once with PBS, the samples were incubated with goat anti-rabbit IgG-Alexa Fluor® 546 and goat anti-mouse IgG-Alexa FluorΔ 488 (both 1:250, Thermo Fisher Scientific) for 45 minutes in the dark.34 Images were acquired by using a Zeiss fluorescence microscope (Cell Observer, Carl Zeiss AG, Oberkochen, Germany). The focal plane was adjusted manually for each image.
2.3 Cell culture
For cell–material studies, human primary-isolated microvascular endothelial cells (HMVECs) were isolated from the foreskin biopsies under the ethics approval no. 495/2018BO2 (IRB, University Hospital Tübingen) by enzymatic digestion with dispase and trypsin as previously described.35 Cells were cultured in endothelial cell growth medium MV (C-22020, PromoCell, Heidelberg, Germany) and used between passages 2 and 4. Human umbilical vein endothelial cells (HUVECs, C-12205, PromoCell, Heidelberg, Germany) were cultured in endothelial cell growth medium (C-22010, PromoCell) and used between passages 4 and 6.36 For cell–material–interaction experiments, 400 cells mm−2 were seeded on the FN-adsorbed surfaces and incubated for 24 hours at 37 °C and 5% CO2.2.4 Immunocytochemistry
After cell culture experiments, the cell-seeded PU samples were washed with PBS− and fixed with 4% (w/w) paraformaldehyde in PBS for 10 minutes. In order to reduce nonspecific binding, the cells were incubated with 2% (w/w) goat block solution for 30 minutes. Cells were incubated with anti-FN (1:200, ab2413, Abcam, Cambridge, UK), anti-CD31 (PECAM-1; 1:100, sc-71872, Santa Cruz, USA), anti-von Willebrand factor (1:200, A0082, DAKO, Glostrup, Denmark), anti-focal adhesion kinase (FAK; 1:100, PA5-17591, Thermo Fisher scientific, Waltham, USA), anti-VE-Cadherin (1:250, HPA004726, Sigma-Aldrich, St. Louis, USA) and anti-vinculin (1:500, MAP3574, Milipore, Burlington, USA) over night at 4 °C. One primary antibody of the same species was used per sample. After washing with PBS, samples were incubated with goat anti-mouse IgG-Alexa Fluor® 488, goat anti-rabbit IgG-Alexa Fluor® 546 (both 1:250, Thermo Fisher Scientific) and Alexa Fluor® 647 Phalloidin (1:1000, A22283, Thermo Fisher Scientific) for 45 minutes in the dark. Finally, nuclei were counterstained with DAPI (10236276001, Roche, Basel, Switzerland). Images were observed by using a Zeiss fluorescence microscope (Cell Observer, Carl Zeiss AG, Oberkochen, Germany).
2.5 Image analysis
Polyclonal and monoclonal anti-FN IF staining was quantified by measuring the relative pixel intensity (RPI) of the immunofluorescence images. Counting the DAPI-stained cell nuclei per area determined the number of adherent cells. The formation of focal adhesions was quantified by counting the vinculin and FAK foci per cell. To assess VE-cadherin and PECAM-1 expression as well as F-actin organization, the area within a defined fluorescence intensity threshold was measured and normalized to the cell number. All images were analyzed using ImageJ.372.6 Statistical analysis
Results are presented as mean ± standard deviation. For statistical analysis, a one-way ANOVA/Fisher's Least Significant Difference test was performed using Origin 2018 (OriginLab, Northampton, MA, USA). Probability values of 95%, 99% and 99.9% were used to determine significance.3. Results
3.1 Analysis of the surface chemistry and its influence on FN adsorption
XPS, AFM and contact angle measurements showed that surface roughness affects the hydrophilic character of the polymers and FN adsorption behavior (Fig. 1). According to the XPS data (Fig. 1a, Fig. S1, ESI†), the chemical composition of the sample surfaces was separable into plasma-incorporated functional groups and PU-specific functionalities. The plasma-generated −OH functional groups and the polymers C–O–C species peaks are situated on identical binding energies, and therefore could not be adequately separated within the functional group content calculations. From the XPS data analysis it was discernible that samples A and B had a higher –COOH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O content than all other samples, while the C, D and E surfaces had a high content of –OH and C–O–C functionalities (Fig. 1a). The untreated control surface F had no new plasma-created species. In terms of surface roughness, the untreated sample F showed a smooth surface that was only little impacted by most plasma treatments (Fig. 1b). Only surface A, treated for a longer time under low oxygen pressure, exhibited a clearly higher surface roughness compared to the other samples. (Fig. 1b). This effect was also seen in the water contact angle measurements of the series (Fig. 1c). The untreated F surface had the highest contact angle with 71.1° ± 1.6°. In contrast, the surfaces C, D and E, which were similar in chemical composition and roughness, were only slightly more hydrophilic with similar contact angle values ranging around 50° (C: 48.2° ± 0.6° versus D: 50.8° ± 1.7°, p < 0.05; C versus E: 54.3° ± 1.0°, p < 0.001; D versus E, p < 0.05). Since the roughness was less impacted by these plasma treatments, the biggest contribution to the hydrophilization of these surfaces came from the introduced hydroxyl groups. While the –COOH and CO surface coverage of the samples A and B was very similar, the contact angle of surface A (17.5° ± 1.9°) was half as high compared with surface B (33.4° ± 2.7°). The major contribution to this additional lowering of the contact angle of the longer plasma-treated sample came from the increased surface roughness, which was twice as high as every other sample (A: 1.45 nm versus B: 0.5 nm, C: 0.61 nm, D: 0.63 nm, E: 0.5 nm and F: 0.39 nm).
O content than all other samples, while the C, D and E surfaces had a high content of –OH and C–O–C functionalities (Fig. 1a). The untreated control surface F had no new plasma-created species. In terms of surface roughness, the untreated sample F showed a smooth surface that was only little impacted by most plasma treatments (Fig. 1b). Only surface A, treated for a longer time under low oxygen pressure, exhibited a clearly higher surface roughness compared to the other samples. (Fig. 1b). This effect was also seen in the water contact angle measurements of the series (Fig. 1c). The untreated F surface had the highest contact angle with 71.1° ± 1.6°. In contrast, the surfaces C, D and E, which were similar in chemical composition and roughness, were only slightly more hydrophilic with similar contact angle values ranging around 50° (C: 48.2° ± 0.6° versus D: 50.8° ± 1.7°, p < 0.05; C versus E: 54.3° ± 1.0°, p < 0.001; D versus E, p < 0.05). Since the roughness was less impacted by these plasma treatments, the biggest contribution to the hydrophilization of these surfaces came from the introduced hydroxyl groups. While the –COOH and CO surface coverage of the samples A and B was very similar, the contact angle of surface A (17.5° ± 1.9°) was half as high compared with surface B (33.4° ± 2.7°). The major contribution to this additional lowering of the contact angle of the longer plasma-treated sample came from the increased surface roughness, which was twice as high as every other sample (A: 1.45 nm versus B: 0.5 nm, C: 0.61 nm, D: 0.63 nm, E: 0.5 nm and F: 0.39 nm).
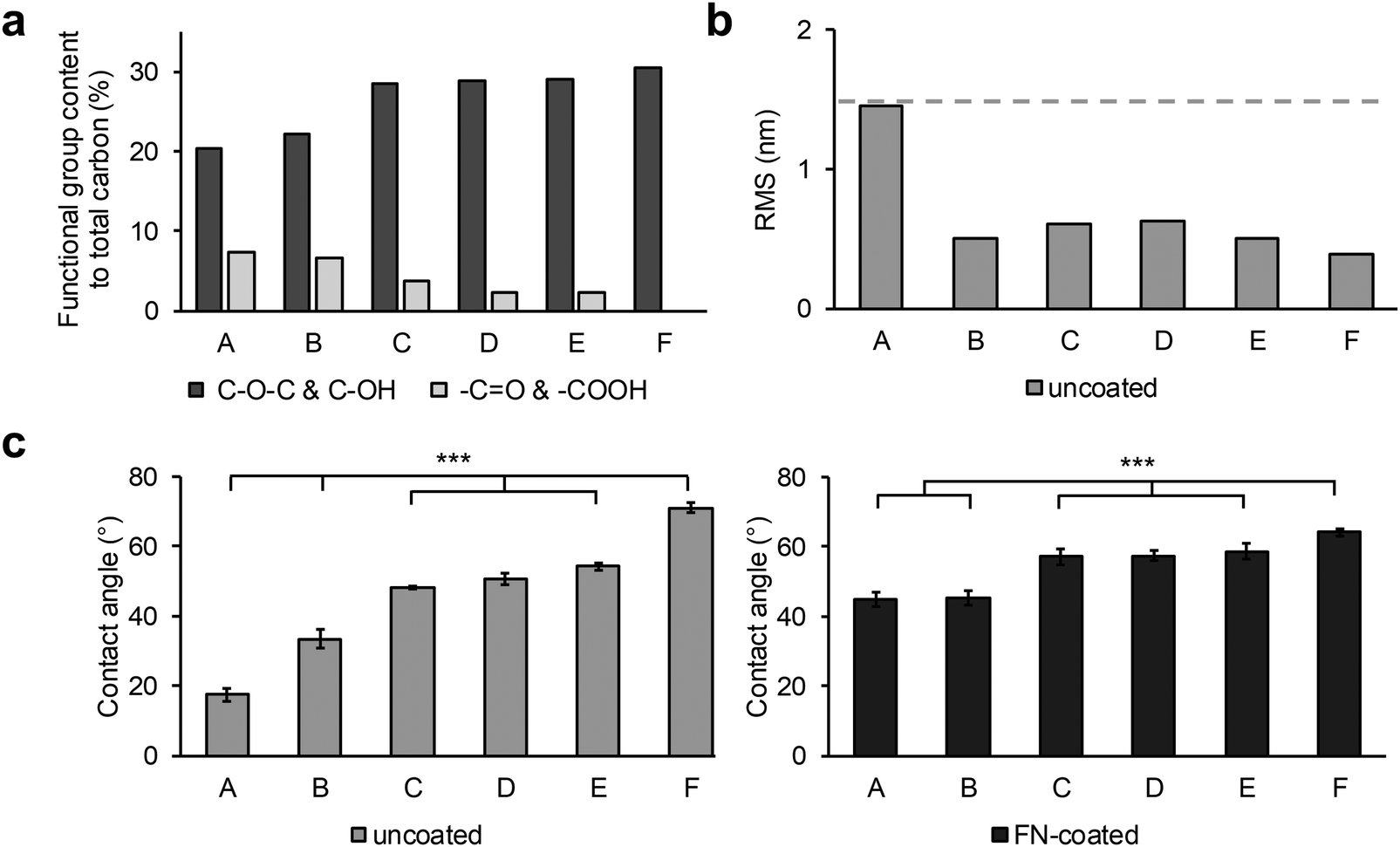 | ||
| Fig. 1 Investigation of plasma modifications on the PU surfaces and adsorption of fibronectin. (a) XPS obtained chemical composition of plasma-induced functional groups on the polymer surface before FN coating. (b) Qualitative roughness analysis r(RMS) of the polymer surfaces after plasma treatment. The dash line represents the maximum roughness value. (c) Contact angle analysis of plasma-treated and untreated surfaces before (left, “uncoated”, one-way ANOVA, n = 3, p*** < 0.001) and after FN coating (right, “FN-coated”, one-way ANOVA, n = 3, p*** < 0.001). (d) AFM topography images of sample surfaces after FN coating. Scale bar equals 200 nm. | ||
The water contact angles and r(RMS) were also measured after FN coating of the samples (Fig. 1c, “FN-coated”, Fig. 2a). As with the previous contact angle series, surface F (64.2° ± 1.1°) was the least hydrophilic with the lowest change in the contact angle after FN coating (Table S2, ESI†). Samples C, D, and E had a contact angle of around 57° (C: 57.2° ± 2.3°, D: 57.4° ± 1.5°, E: 58.7° ± 2.1°, not significant). Unlike surface F, the A (45.0° ± 2.0°) and B (45.3° ± 2.0°) surfaces showed an increase in contact angle with the same result at about 45°. The AFM images, recorded after FN coating, resulted in visible topographic changes and increase in roughness compared with their uncoated counterparts only for the control sample F (Fig. 1d). From the height contrast images, and extracted height profiles of the images, no difference between the plasma treated FN-coated surfaces was visible (Fig. 2). The protein domains and the polymer background were indistinguishable, as was the case with phase AFM imaging (Fig. S4 and S5, ESI†). Through the plasma treatment of the samples, the FN proteins lost their material contrast with the polymer. The untreated surface (sample F) had a noticeable increased roughness after the protein coating, from approximately 0.4 to 3.1 nm, which was due to topographical changes caused by the protein network rather than modifications in the PU layer. This network was partially up to 10 nm high with differently sized pores ranging from 200 nm to 500 nm in diameter.
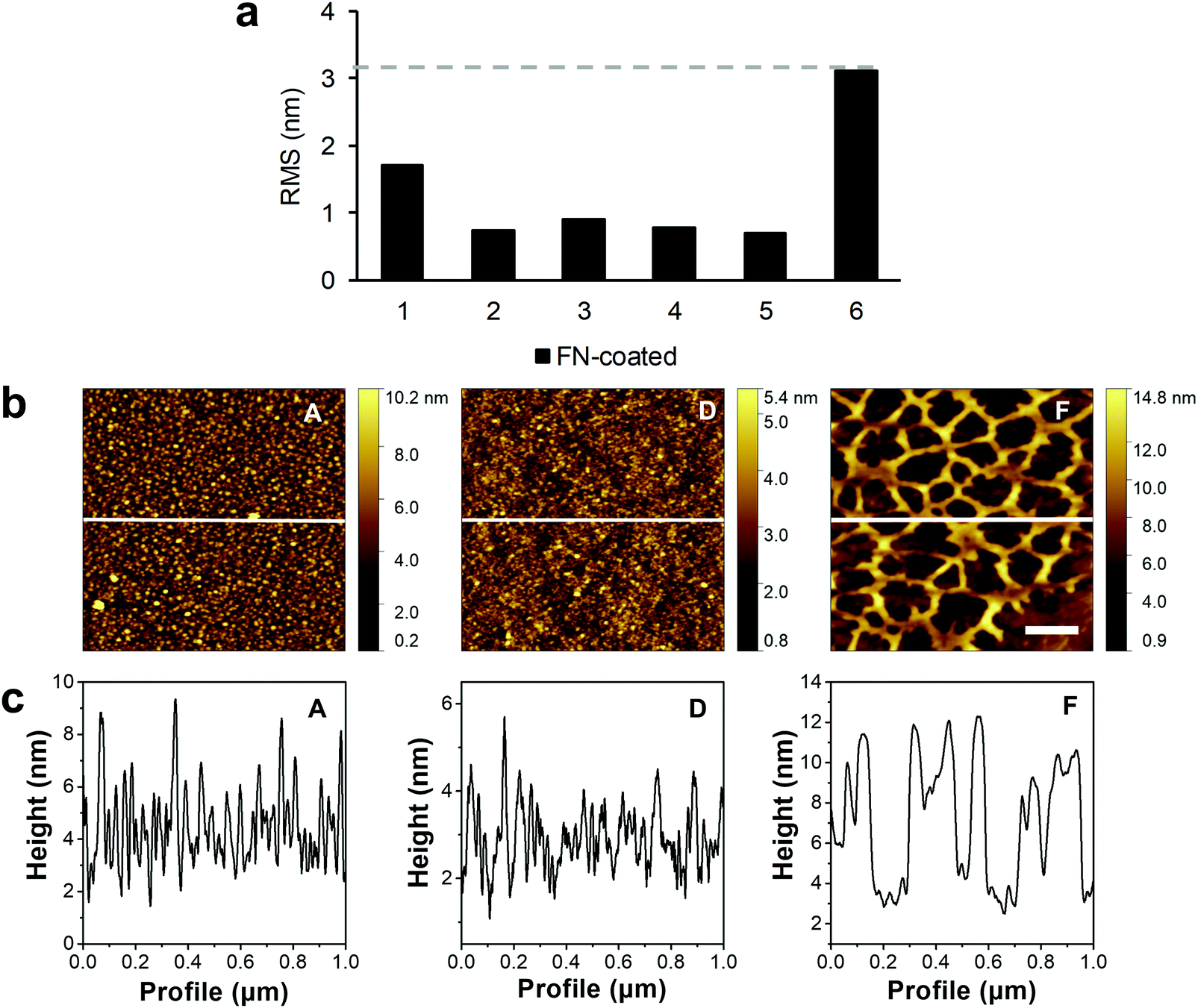 | ||
| Fig. 2 Qualitative analysis of the polymer surface topography after protein coating. (a) r(RMS) results of the AFM measurements of the sample control and plasma-treated surfaces after FN coating. The dash line represents the highest r(RMS) value of the series. (b) AFM height contrast images of the FN-coated surfaces. The white line shows the profile extraction region. The scale bar equals 200 nm. (c) Extracted line profiles from the AFM images showing the structure of the surface. | ||
In order to study protein adsorption kinetics, the adsorption of FN on the oxygen plasma-treated PU surfaces was monitored by RIfS. An exemplary binding curve is depicted in Fig. 3a showing the baseline (with PBS), followed by the association of FN (with the FN solution), starting at the time point 0 s, and the dissociation (rinsing with PBS) of FN from 2000 s to 4000 s. The recorded binding curves (Fig. S2, ESI†) showed a different adsorption behavior of FN on the surfaces. These differences were evaluated by calculating the respective association rate constant ka and dissociation rate constant kd using a basic kinetic model (Fig. 3b and c). FN showed on all surfaces small kd values in the magnitude of 10−3 s−1. On surface F, FN exhibited the highest kd value compared to all other investigated surfaces, indicating an easier desorption of FN from surface F. The calculated ka values showed faster rates of the forward adsorption process on surface A, E and F in comparison to the surfaces B, C and D. The larger error of the fit of the calculated rate constants on the surfaces A, E and F in respect to the surfaces B, C and D demonstrated a larger discrepancy from the assumed first order kinetic model on the surfaces A, E and F.
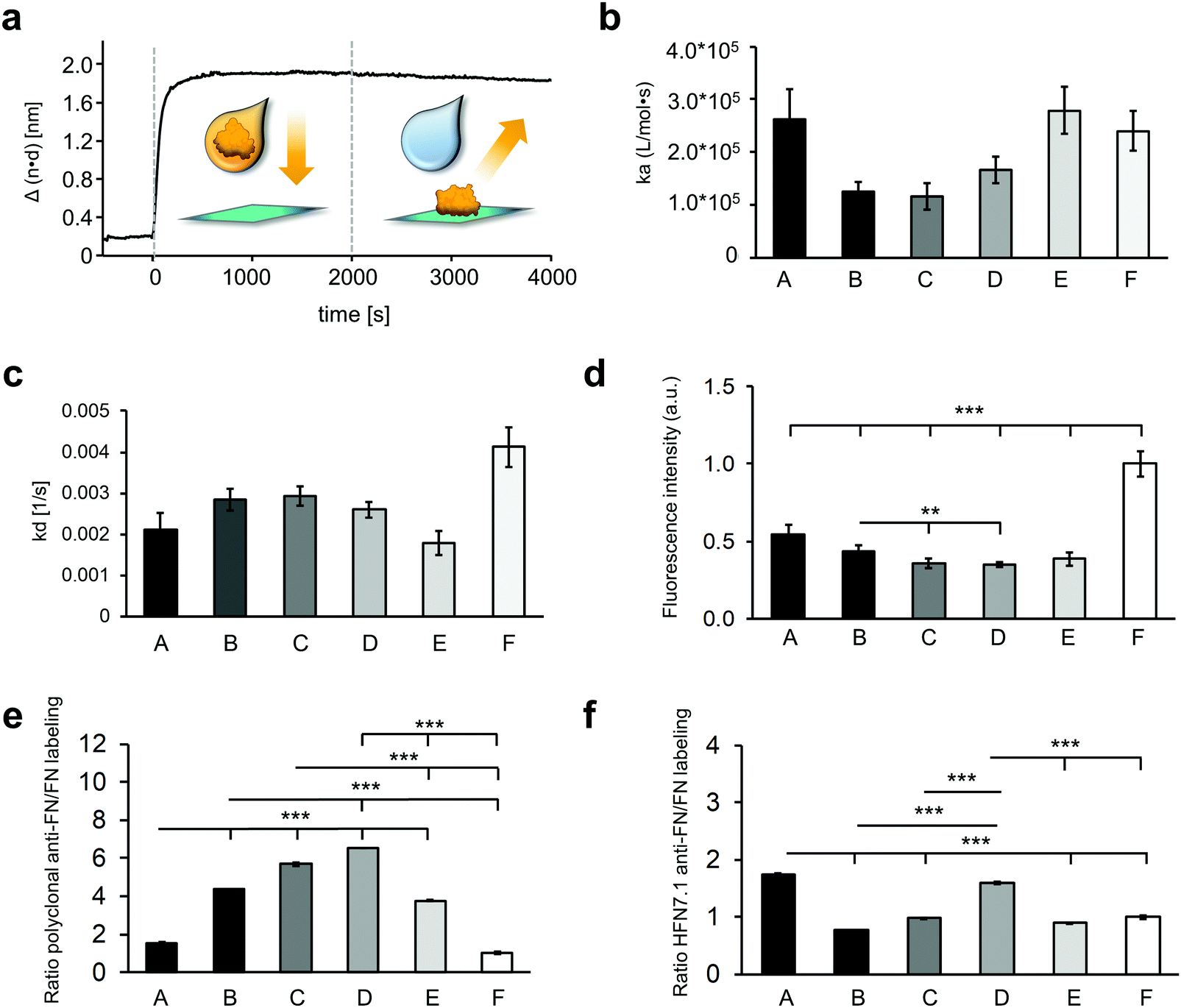 | ||
| Fig. 3 The adsorbed fibronectin varies in adsorption kinetics, amount and conformation on different hydrophilic surfaces. (a) Exemplary binding curve of 10 μg ml−1 FN on surface F. Association (with the FN solution) of FN starting at time point 0 s and dissociation (with PBS) of FN from 2000 s to 4000 s. Calculated association rate constants (ka) (b) and calculated dissociation rate constants (kd) (c) of FN adsorption. Error bars depict the error of the fit. (d) Fluorescence intensity of labeled FN that was coated on the different surfaces. Data is shown relative to surface F. (e) Ratio of the polyclonal anti-FN immunofluorescence staining and the labeled FN. (f) Ratio of the monoclonal anti-FN immunofluorescence staining targeting the cell binding domain and the labeled FN. One-way ANOVA, n = 3, **p < 0.01, ***p < 0.001; RPI = relative pixel intensity. | ||
The relative amount of the total adsorbed FN on the different plasma-treated samples was determined indirectly by comparing the fluorescence intensity of DY-490 labeled FN after laser excitation (Fig. 3d). As the degree of labeling is consistent throughout the FN molecules, the amount of fluorescence intensity is directly linked to the amount of FN molecules on the surface. While sample F showed the highest fluorescence signal and therefore the highest amount of FN (1.00 ± 0.08), sample A revealed about half of it (0.54 ± 0.06). Samples C, D and E showed the same fluorescence intensity, which was significantly lower than sample A (A versus C: 0.36 ± 0.03, p < 0.001; A versus D: 0.35 ± 0.02, p < 0.001; A versus E: 0.39 ± 0.04, p < 0.001), sample B (B: 0.43 ± 0.04 versus C, p < 0.01; B versus D, p < 0.01), and sample F (F versus B, p < 0.001; F versus C, p < 0.001; F versus D, p < 0.001), which indicated fewer FN amounts on these surfaces.
The orientation and conformation of the adsorbed FN was further investigated with IF staining using a polyclonal antibody against the full-length FN (Fig. 3e and Fig. S6a, ESI†). The ratio of the anti-FN staining to the amount of adsorbed FN, determined by the fluorescence-labeled FN, indicates conformational and orientation changes of the protein. Comparing the polyclonal anti-FN/FN labeling ratios of the different surfaces, a similar result was found on the surfaces C and D. Compared to them, the ratios on the A, B, E and F surfaces were significantly lower (A: 1.54 ± 0.05 versus C: 5.68 ± 0.07, A versus D: 6.51 ± 0.01, B: 4.35 ± 0.01 versus C, B versus D, E: 3.77 ± 0.05 versus C, E versus D, F: 1.00 ± 0.06 versus C, F versus D, all p < 0.001). The FN coating on the A and F surfaces even showed a significantly lower value compared with the B and E surfaces (all p < 0.001). Additionally, the adsorbed FN was stained with a monoclonal antibody (Fig. S6b, ESI†), whose epitope maps to a segment between the 9th and 10th type III repeat (HFN7.1), indicating the availability of the cell binding domain.38 The ratio of the monoclonal antibody to the amount of adsorbed FN showed significantly higher signals on the A and D surfaces compared with the surfaces B, C, E and F (A: 1.74 ± 0.01 versus B: 0.77 ± 0.00, A versus C: 0.98 ± 0.01, A versus E: 0.89 ± 0.01, A versus F: 1.00 ± 0.03, D: 1.60 ± 0.02 versus B, D versus C, D versus E, D versus F, all p < 0.001), suggesting a more exposed cell binding domain of the FN on these surfaces (Fig. 3f).
3.2 Investigation of the endothelial cell–material–interaction on the FN-coated surfaces
The investigation of the endothelial cell–material–interaction on the different surfaces was of particular interest as it provides information to predict the behavior of the cells depending on surface properties and FN deposition. For this purpose, HUVECs and HMVECs were utilized.First, the number of adherent HUVECs and HMVECs was investigated 24 hours after seeding showing a rather similar distribution of adherent cells on the different surfaces (Fig. 4a and b). However, on surfaces A and E, significantly less adherent HUVECs and HMVECs were found when compared with the surfaces B and D (HUVECs: A: 0.97 ± 0.01 versus B: 1.16 ± 0.04, p < 0.01 and D: 1.15 ± 0.05, p < 0.05; E: 0.95 ± 0.10 versus B and D, p < 0.01; HMVECs: A: 0.87 ± 0.04 versus B: 1.04 ± 0.05 and D: 1.05 ± 0.07, p < 0.001; E: 0.92 ± 0. 60 versus B, p < 0.001 and D, p < 0.01, cell count was normalized to surface F). In addition, the HUVECs showed a significantly increased cell count on surface C compared to surface A and E (C: 1.11 ± 0.13 versus A and E, p < 0.05). For the HUVECs, but not for the HMVECs, significantly fewer adherent cells were found on surface F compared to surfaces B and D (F: 1.00 ± 0.09 versus B and D, p < 0.05). Both cell types showed no significant differences in the number of adherent cells between the B, C and D surfaces, and between the A and E samples.
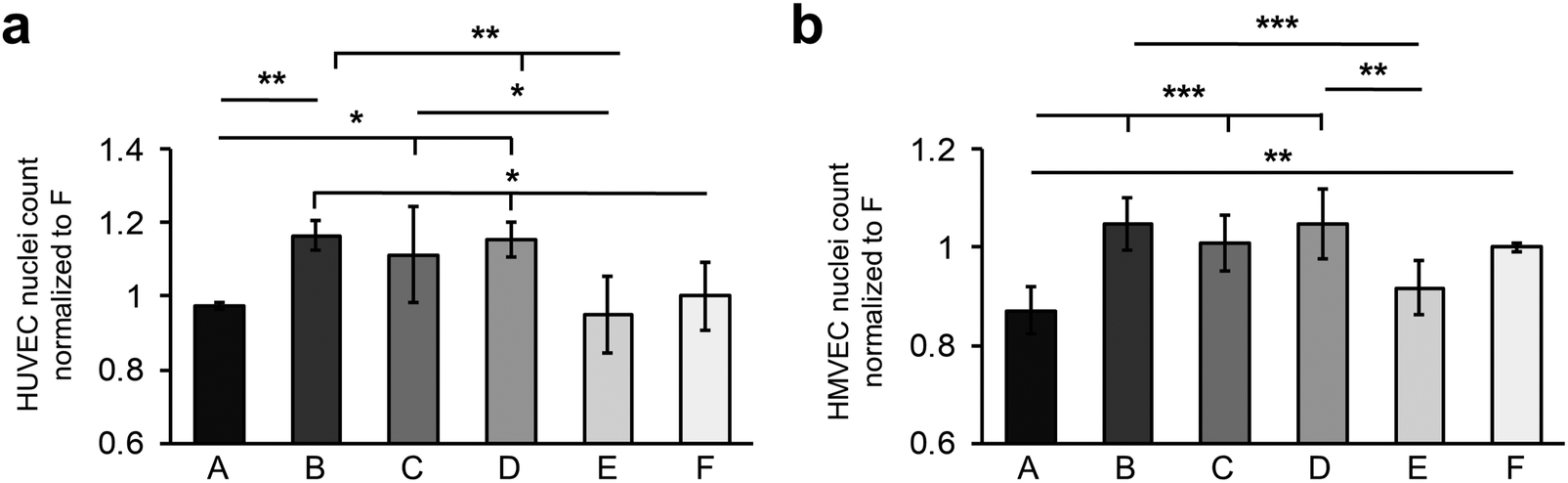 | ||
| Fig. 4 Fibronectin coating on different hydrophilic surfaces impacts the number of adherent cells. The number of adherent (a) HUVECs and (b) HMVECs 24 h after cell seeding. Data is shown as change relative to F. One-way ANOVA, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001. | ||
The different surfaces did not appear to influence the size of the two cell types, although the HUVECs tended to be smaller on the surfaces B and C compared to the other samples, and larger on surfaces C, D and E (Fig. S7, ESI†).
The interaction of HUVECs and HMVECs with the pre-adsorbed FN layer led to a FN reorganization, which was demonstrated by anti-FN IF staining (Fig. 5a and Fig. S8, ESI†). Both cell types showed fibrillar structures on the A surface in the area of the cells (Fig. 5a, white arrow). In addition, FN reorganization appeared on all surfaces as dark areas around the cells, most notably in the C and D samples (Fig. 5a, white dotted arrow).
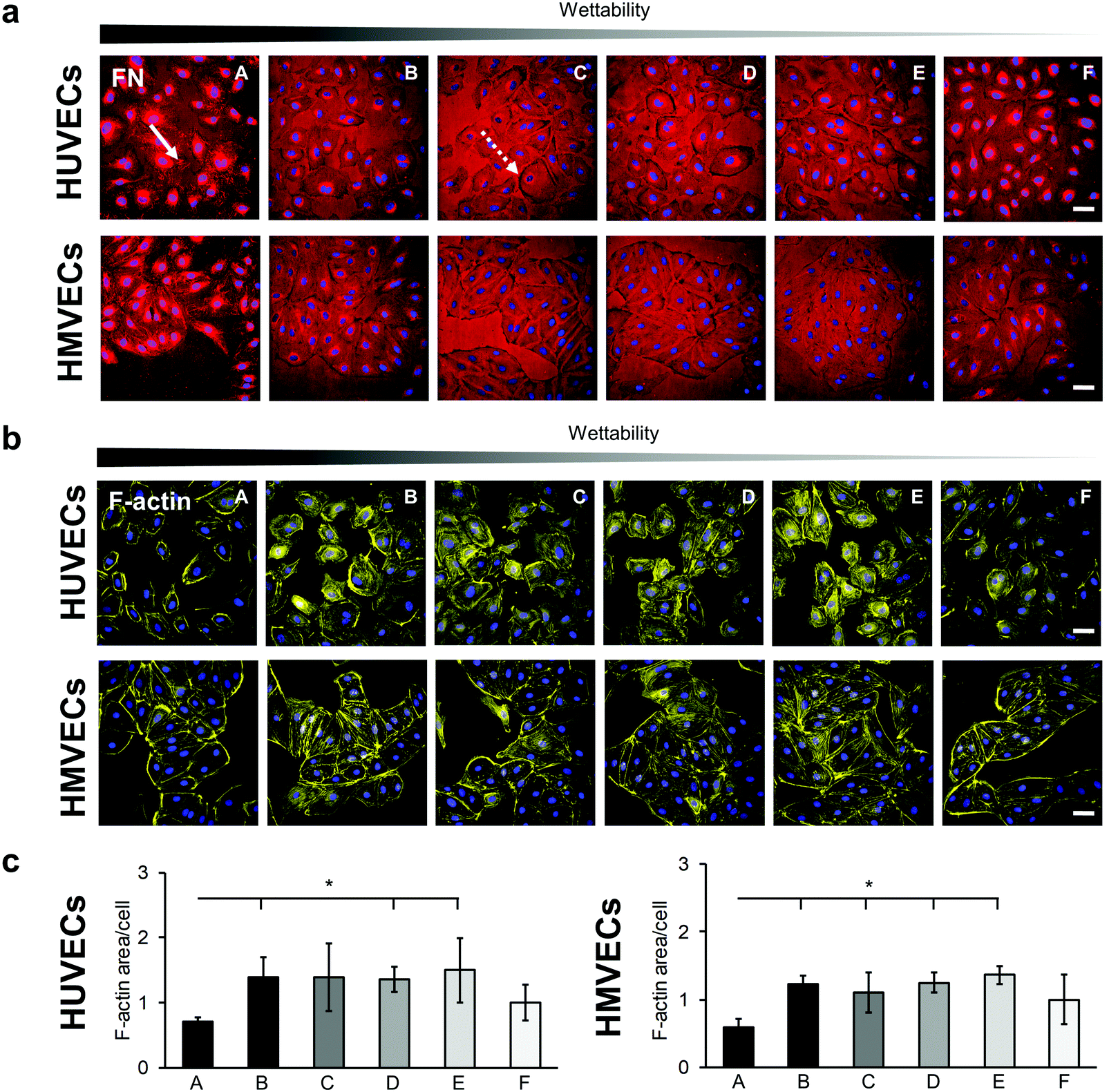 | ||
| Fig. 5 Cell–fibronectin-interaction and F-actin reorganization in HUVECs and HMVECs. (a) Anti-FN immunofluorescence staining shows the interaction of HUVECs and HMVECs with the adsorbed FN, which is indicated by fibrillar structures (white arrow) and dark areas (white dotted arrow). Scale bars equal 100 μm. (b) Peripheral F-actin bundles are formed in HUVECs and HMVECs that are seeded on A and F surfaces. Scale bars equal 100 μm. (c) Semi-quantitative area analysis of F-actin distribution in HUVECs and HMVECs. Data is shown relative to F. One-way ANOVA, n = 3, *p < 0.05. | ||
The cytoskeleton was examined by using F-actin staining, as its distribution provides information about the barrier function of an endothelial cell.39 A distinct reorganization of the F-actin was observed on the different surfaces (Fig. 5 and Fig. S9, ESI†). For both cell types, peripheral ventral stress fibers were found on the A and F surfaces with more prominent F-actin bundles on the A surface (Fig. 5b). In contrast, in the cells on the samples B, C, D and E, the stress fibers were distributed over the entire cell. A semi-quantitative analysis of the F-actin-stained areas revealed a significantly lower amount of stress fibers in the cells cultured on the A surfaces compared with cells grown on surfaces B, D and E for both cell types (HUVECs: A: 0.71 ± 0.06 versus B: 1.39 ± 0.31, D: 1.35 ± 0.19 and E: 1.50 ± 0.50, p < 0.05; HMVECs: A: 0.60 ± 0.12 versus B: 1.23 ± 0.12, D: 1.25 ± 0.15 and E: 1.36 ± 0.13, p < 0.05). In addition, the HMVECs showed a significantly lower signal on surface A compared with surface C (A versus C: 1.10 ± 0.30, p < 0.05; Fig. 5c).
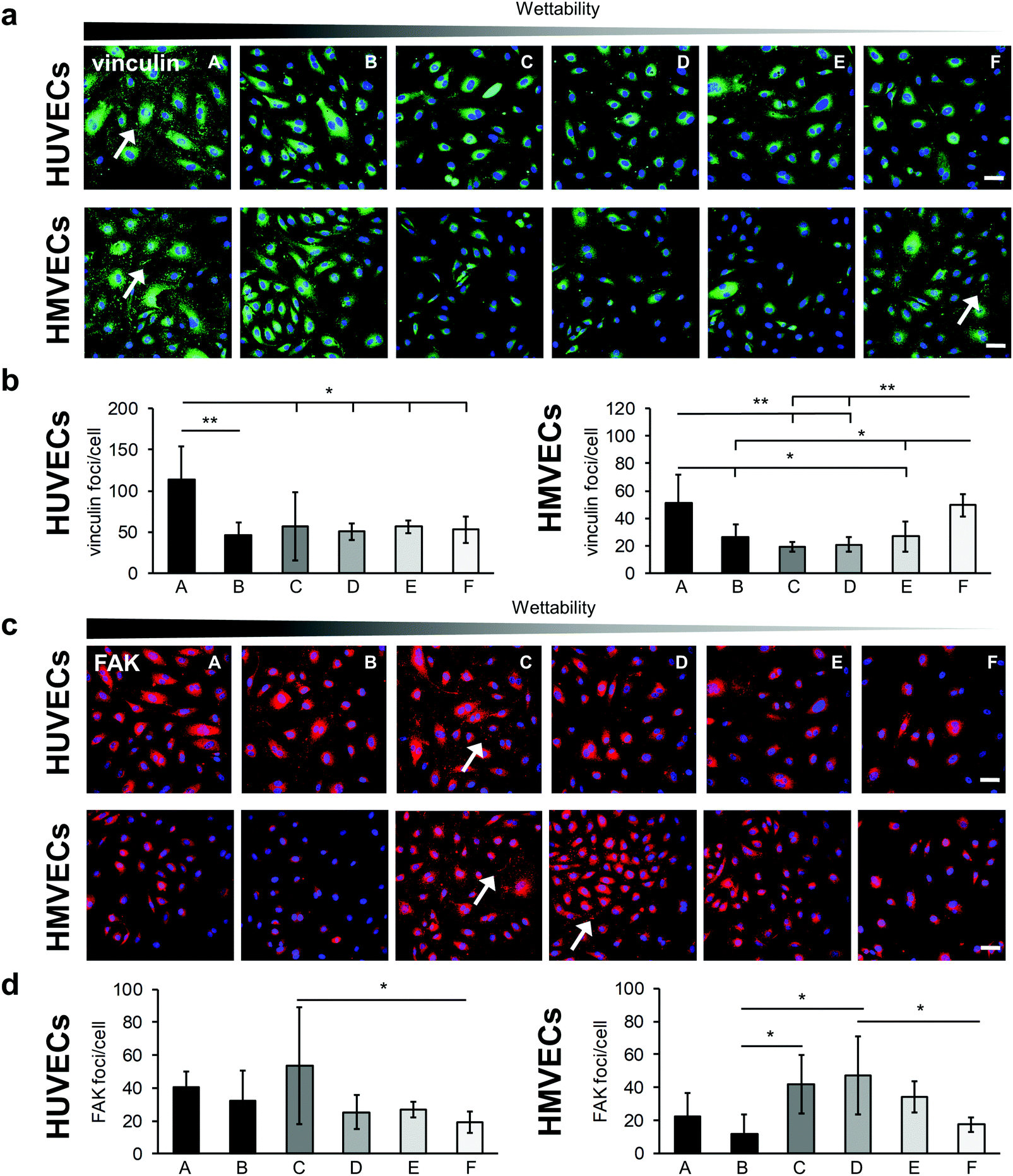
The integrin–mediated cell adhesion is associated with the formation of focal adhesions.40 Vinculin, a scaffolding protein that is involved in the mechanical regulation of focal adhesions, was studied by statistically analyzing the number of vinculin foci per cell (Fig. 6).41 Both cell types showed a significantly higher number of vinculin foci per cells on surface A compared with the other surfaces (HUVECs: A: 113 ± 40 versus B: 46 ± 16, p < 0.01; A versus C: 57 ± 41, D: 51 ± 10, E: 56 ± 7 and F: 53 ± 16, p < 0.05; HMVECs: A: 51 ± 21 versus B: 26 ± 9 and E: 27 ± 11, p < 0.05; A versus C: 19 ± 4 and D: 21 ± 5, p < 0.01), with the exception of surface F (49 ± 8) where a similar number of vinculin foci were counted in the HMVECs (Fig. 6a and b). For both cell types, the average number of foci per cell was similar for surfaces B, C, D and E.
 | ||
| Fig. 6 The formation of focal adhesion complexes influenced by fibronectin coating on different hydrophilic surfaces. (a) Anti-vinculin immunofluorescence-stained HUVECs and HMVECs on the FN-coated surfaces. Vinculin foci are indicated by white arrows. Scale bars equal 100 μm. (b) Semi-quantitative analysis of the anti-vinculin staining for HUVECs (left) and HMVECs (right). Data is shown relative to surface F. (c) FAK-stained HUVECs and HMVECs seeded on the different hydrophilic FN-coated surfaces. FAK foci are indicated by white arrows. Scale bars equal 100 μm. (d) Semi-quantitative analysis of the anti-FAK staining for HUVECs (left) and HMVECs (right). Data is shown relative to surface F. One-way ANOVA, n = 3, *p < 0.05, **p < 0.01. | ||
Furthermore, the FAK, which plays a critical role in integrin–mediated signal transduction, was examined (Fig. 6).42 For the HUVECs, the number of FAK foci per cell tended to be higher on the very hydrophilic surfaces (A: 40 ± 10, B: 32 ± 18 and C: 54 ± 35) than on the less hydrophilic surfaces (D: 25 ± 10, E: 27 ± 5 and F: 20 ± 7). Due to a high variation within the experiments, statistically significant more FAK foci per cells were only found on the C surface compared with the surface F (C: 54 ± 35 versus F: 20 ± 7, p < 0.05) (Fig. 6c and d). The HMVECs showed a slightly different behavior. They inclined to form less FAK foci on the very hydrophilic surfaces compared with moderately hydrophilic surfaces. In detail, on surface B, significantly fewer FAK foci per cell were found compared to surfaces C and D (B: 12 ± 12 versus C: 42 ± 18 and D: 47 ± 24, p < 0.05). Both cell types tended to form few FAK foci on the F surface; however, in the case of HMVECs the number of FAK foci per cell was significantly lower compared to surface D (D: 47 ± 24 versus F: 17 ± 4, p < 0.05).
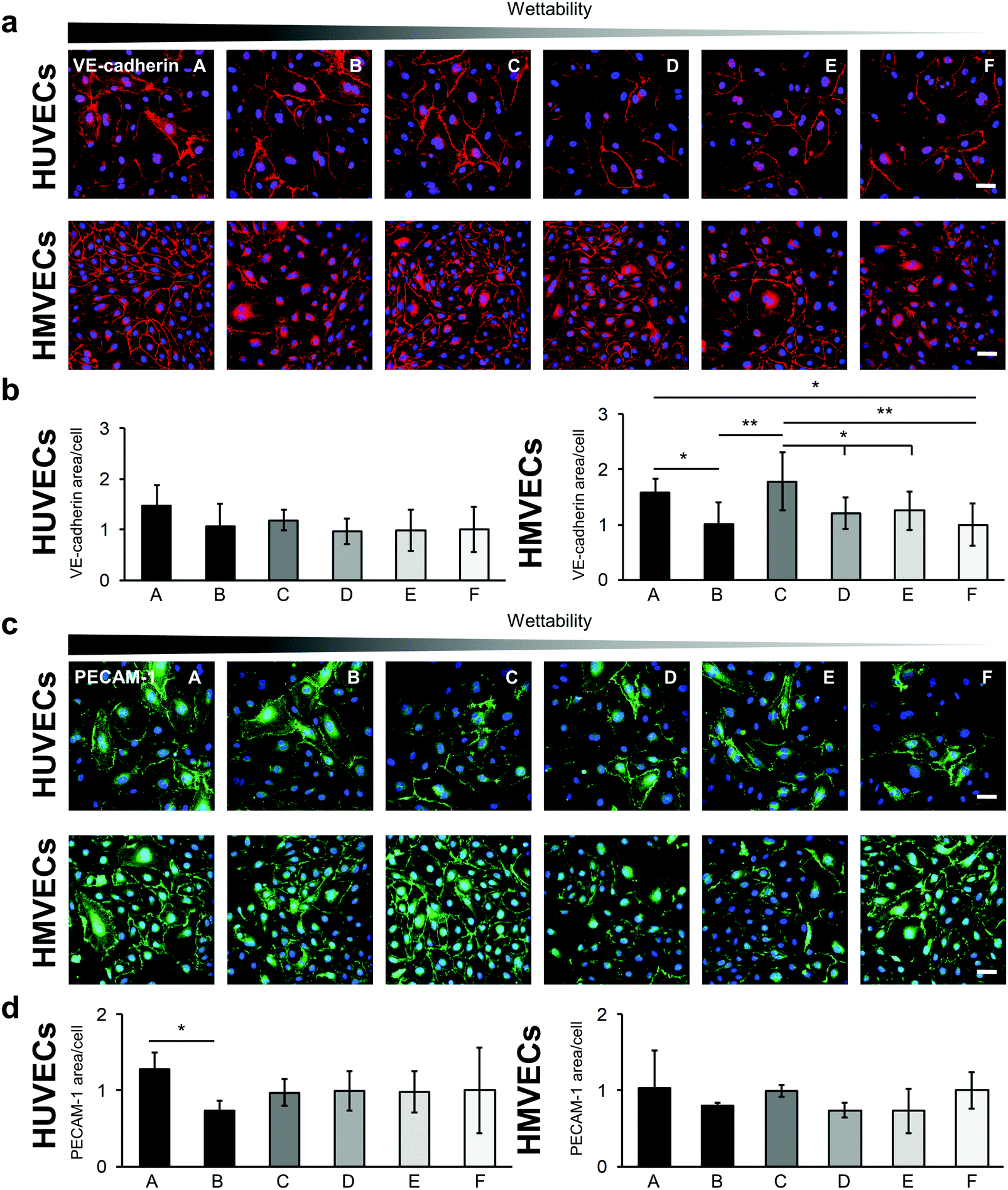
Cell–cell interactions were investigated by studying the expression of the endothelial cell type-specific markers VE-cadherin and PECAM-1 (Fig. 7). Both proteins are located on the surface of endothelial cells and account for a large portion of the endothelial cell–to–cell junctions that contribute to the maintenance of the endothelial permeability barrier.43 Expression levels were evaluated by the IF-stained area per cell (Fig. 7b and d). The VE-cadherin staining showed similar expression levels for the HUVECs on all surfaces. Significant differences were found for the HMVECs (Fig. 7b and Fig. S10, ESI†). In detail, a higher VE-cadherin signal was detected on the A surface compared with surfaces B and F (A: 1.59 ± 0.24 versus B: 1.02 ± 0.38 and F: 1.00 ± 0.39, p < 0.05). In addition, the cells on the C surface revealed a higher expression level compared with the B, D, E and F surfaces (C: 1.78 ± 0.52 versus D: 1.21 ± 0.28 and E: 1.30 ± 0.34, p < 0.05 and versus B and F, p < 0.01).
 | ||
| Fig. 7 Endothelial cell–to–cell junction variation on the different fibronectin-coated surfaces for both HUVECs and HMVECs. (a) VE-cadherin immunofluorescence staining of HUVECs (upper row) and HMVECs (lower row) on the different hydrophilic surfaces coated with FN. Scale bars equal 100 μm. (b) Semi-quantitative analysis of the anti-VE-cadherin-stained area for HUVECs (left) and HMVECs (right). Data is shown relative to surface F. (c) PECAM-1 immunofluorescence staining of HUVECs and HMVECs on the FN-coated surfaces. Scale bars equal 100 μm. (d) Semi-quantitative analysis of the anti-PECAM-1-stained area for HUVECs (left) and HMVECs (right). Data is shown relative to surface F. One-way ANOVA, n = 3, *p < 0.05, **p < 0.01. | ||
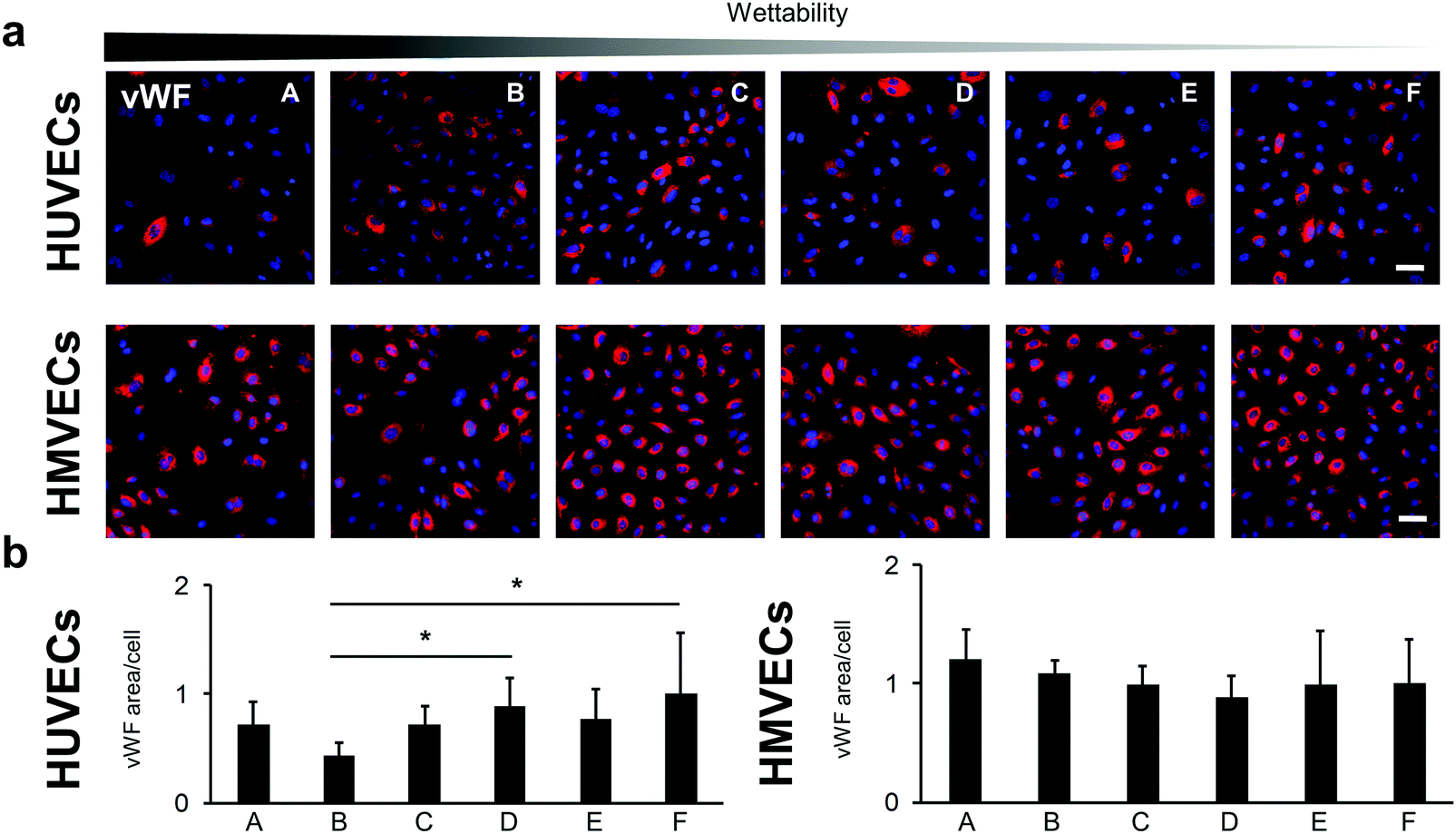
The PECAM-1 expression was the same for the HMVECs on all samples (Fig. 7c and d). For the HUVECs, less PECAM-1 was expressed in the cells on surface B compared with surface A. Interestingly, the HUVECs on surface B showed also a significantly lower expression of the endothelial cell marker von Willebrand factor (vWF) when compared with cells grown on surfaces D and F (B: 0.43 ± 0.12 versus D: 0.89 ± 0.26 and versus F: 1.00 ± 0.57, p < 0.05; Fig. 8).
 | ||
| Fig. 8 von Willebrand factor (vWF) expression in HUVECs and HMVECs cultured on the FN-coated surfaces. (a) Anti-vWF immunofluorescence staining of HUVECs and HMVECs. The scale bar equals 100 μm. (b) Semi-quantitative analysis of the anti-vWF-stained cells. Data is shown relative to surface F. One-way ANOVA, n = 3, *p < 0.05. | ||
4. Discussion
In the present study, oxygen plasma surface treatment was used to incorporate hydroxyl, carbonyl, carboxyl and other oxygen-rich groups onto PU surfaces in order to modify the polarity and chemical structure of the surface.44–49 During the plasma treatment, removal of material from the surface layer through plasma etching processes with UV irradiation and ion bombardment resulted in topographical changes.27,44,47 The impact of the plasma modification on the different surfaces was evaluated using AFM imaging, XPS analysis and contact angle measurements. The XPS and contact angle results showed that higher oxidized functional groups were incorporated with low oxygen pressure treatments, which led to a greater impact on PU surface hydrophilicity.25 From the contact angle measurements, specifically the low standard deviations of the samples, it can be concluded that the plasma treatment of the polymer surfaces had a high reproducibility. AFM measurements showed that an increased surface roughness contributed to lowering the contact angle, demonstrating a more hydrophilic surface.50 Especially with surface A, which has a similar chemical composition to surface B, the higher wettability can be attributed to surface roughness effects. This roughening of the sample topography was caused by the ablative nature of the oxygen plasma, which had a larger effect during the longer plasma treatment of sample A under low oxygen pressure conditions. FN-coated plasma-treated samples were difficult to distinguish using AFM topographic imaging; however, contact angle analysis showed differences between the plasma-treated samples. Only on the FN-coated control surface F, AFM images showed a fibrillar network that may be attributed to material-driven fibrillogenesis where FN molecules unfold and bind to each other.1,19,51RIfS was employed to investigate the conformation and orientation of FN on the samples. Here, the kinetic inspection of the binding curves revealed different rate constants for the chemical forward and reverse process, indicating different interaction sites on the reactants. Surface A and B had a similar chemical composition; however, there was a pronounced difference in the calculated ka on these surfaces, which could be attributed to the changed surface roughness and different reaction sites on the adsorbed FN. In contrast, the increase of ka from surface B to E was induced by the changes in the chemical composition of the surfaces and the corresponding different reaction sites on FN. In our study, we identified the lowest ka values for the surfaces B, C and D, and larger ka values for the more hydrophilic surface A as well as for the more hydrophobic surfaces E and F. This may be explained by the different influences of surface chemistry and surface roughness of the investigated PU surfaces on FN adsorption. A higher flexibility of FN on surface F in comparison to the other surfaces was found by a large ka value and a coincidental large kd value. Although FN appeared in a very stable fibrillary form on surface F, it exhibited a more dynamic interaction towards the surface. The oversimplification of the actual processes at the surface by the applied kinetic model is reflected in the calculated error accompanying the results.52 The error was particularly large on surface A, E and F, indicating that the discrepancy from the Langmuir adsorption model was pronounced on these surfaces. On surface F, the lateral interactions between the adsorbed proteins in the network demonstrated by the AFM measurements (Fig. 1 and 2) can cause the discrepancy from the adsorption model. It is also very plausible that FN underwent greater conformational changes on surfaces A, E and F during adsorption when compared with surfaces B, C and D. To test this hypothesis, the adsorption of FN was further examined with fluorescence-labeled FN as well as with polyclonal and monoclonal antibody IF staining.
The total amount of adsorbed FN was determined with fluorescence-labeled FN. Here, the measured fluorescence intensity of the covalently labeled FN is directly proportional to the amount of adsorbed FN. This is, because the fluorescence DY-490 FN labeling took place prior to the adsorption. Compared to the antibody staining, the fluorescence signal is not affected by the protein adsorption and the associated changes in protein conformation and orientation. As we had a mean number of 2.6 dye molecules per FN, we do not expect that our labeling impaired the function and adsorption behavior of FN. A study by Hoffmann et al. analyzed the influence of FITC (a dye that is chemically related to DY-490) when covalently binding it to FN. For three dye molecules per FN, they could not detect significant modifications to the structure and biological function compared with unlabeled FN.53 Especially for surface A and F, the amount of coated FN was significantly higher when compared with the other surfaces. While for surface A the surface roughness probably allowed an increased FN adsorption, the amount of adsorbed FN on surface F was influenced by the material-driven FN fibrillogenesis, where the FN molecules were probably adsorbed in several layers.54,55
The ratio of the amount of bound monoclonal and polyclonal anti-FN antibodies to the total amount of adsorbed fluorescence-labeled FN provides information about the conformation and orientation of the adsorbed protein. In our study, the individual surfaces showed different ratios, which indicates a varying adsorption behavior of FN on the surfaces (Fig. 3e and f). Since the polyclonal antibody binds to many different epitopes of FN, we hypothesize that the lower the antibody signal, the more the conformation of the protein is altered and thus fewer epitopes are present or accessible to the antibody. Interestingly, there seems to be a reciprocal correlation between the results of the polyclonal antibody staining and the ka values of the RIfS measurements (Fig. 3). Since the ka value indirectly indicates the strength of the interaction between protein and surface, a greater conformational change can be assumed with stronger FN-material interaction.56 Thus, the results of the polyclonal antibody support the observations of the RIfS measurements, which indicates a slight conformational change of the FN on the surfaces B, C and D, and a stronger FN unfolding and expansion on the surfaces E and F. This observation is consistent with the results of several studies that have described more drastic conformational changes on more hydrophobic surfaces.14,57 Interestingly, despite their hydrophilic properties, conformational changes of the protein also occurred on the surface A. We assume that the surface roughness caused by oxygen plasma treatment influenced the conformational change of the protein. Several studies have already described that surface roughness can lead to a widespread and rigid protein conformation.13,58
An additional indication of the conformational change of the adsorbed FN was provided by the results of the monoclonal HFN7.1 antibody, which binds near the cell binding domain consisting of the PHSRN and RGD integrin binding sites.33 In literature, an increased HFN7.1 antibody binding to unfolded FN has been described.14,59 In our study, especially samples A and D showed a high level of accessible cell binding domain indicating an extended FN conformation. However, our data suggest that the accessibility of the cell-binding-domain is not necessary and is solely associated with the conformational change, but also with the orientation of the protein. The FN adsorbed on surface D suggested only small conformational changes, but nevertheless showed a high availability of the cell binding domain. In contrast, the data for surface F indicates a more unfolded adsorbed protein, but with a low-exposed cell binding domain (Fig. 3b, e and f). From this we can conclude that depending on the surface properties, the adsorbed FN can take a more or less favorable conformation and orientation with respect to the accessibility of the cell binding domain, which ultimately influences its bioactivity. Since the surface chemistry and roughness of the surfaces C, D and E are similar, the difference in FN adsorption behavior on sample D could not be explained from the gathered information. Protein adsorption onto biomaterial surfaces is affected by many additional factors such as the stiffness, which have not been investigated.60 Methods such as fluorescence resonance energy transfer could be helpful to obtain further details about the conformation of the adsorbed FN.
Bridging these results to later applications, the key question of how these differences influence cell behavior was studied. For both HUVECs and HMVECs we found the same trend of differences in the number of adherent cells on the different surfaces. It should be noted that the cells were examined 24 hours after seeding. Thus, the cell count is not only determined by the number of initially attached cells but also by cell proliferation. Interestingly, our results show that the number of adherent cells does neither correlate with the amount of the exposed FN cell binding side nor with the wettability or polarity of the underlying surface. Rather, a correlation between the ka value of the RIfS measurements and the cell count is observed (Fig. 3 and 4). We hypothesize that due to the stronger interaction with the surface and the resulting conformational change of the FN, certain domains are exposed, which influence the cell cycle. In the literature it is described that binding to the first type III repeat or the heparin binding site of FN inhibits endothelial cell growth.61,62 It is also possible that a phenomenon described by Podestá et al. took place.63 In their study, the authors observed a decreased endothelial cell proliferation due to a reorganization of the cytoskeleton into actin filament bundles, which was induced by an increased density of focal contacts. Reduced cell growth associated with cytoskeletal reorganization may also be related to the CaMKK2/AMPK signaling pathway. It is described that the activation of AMPK leads to the maturation of contractile actomyosin bundles and causes cell cycle arrest.64–66 In our study, a reduced cell number was observed for both cell types on surfaces A and F, accompanied by the formation of actin bundles and an increased density of vinculin foci. This cytoskeletal organization indicates a resting endothelium, while stress fibers, as observed on surfaces B, C, D and E, occur when the endothelium is activated, a condition associated with cell proliferation and migration.67
During cell–FN interaction, which is mainly mediated by the integrins α5β1 and αvβ3, focal adhesions are formed.68 On surfaces A and F, we think that the more unfolded conformation of FN, associated with an increased presentation of the cell binding side, facilitates the formation of mature focal adhesions composed of vinculin and αvβ3.69–71 In addition, a stronger FN–material interaction, as found on surface A, causes proper transmission of force through the actin cytoskeleton, allowing the maturation of vinculin-associated focal adhesions.72
Probably due to the stronger adhesion of the FN to the surface A, the cells can also form FN fibrils and develop actin bundles.67 It should be noted that the increased surface roughness of sample A to the other plasma treated samples can also influence the cell behavior.73
The more compact form of the FN molecule, which is assumed to be found on surfaces B, C and D, is only available for binding to α5β1 and not to αvβ3.74 Interestingly, higher levels of FAK foci, which are linked to integrin β1, were usually found on these surfaces.75 The higher cell count and pronounced stress fibers in the cells on these surfaces can be attributed to FAK, as it enhances cell proliferation and cell motility by stimulating the formation of actin stress fibers and promoting the turnover of integrin-based adhesions.76–78 In addition, the potentially weaker FN-material interaction on these surfaces hinder the cells to form vinculin-rich mature focal adhesions. Instead, when the cells exert a traction force on the FN, they detach it from the surface, which is indicated by the dark areas on the anti-FN-stained surfaces79 (Fig. 4).
In our study, the expression of the endothelial cell markers vWF, VE-cadherin, and PECAM-1 was confirmed for both cell types. With regard to the formation of a functional endothelium on the FN-coated surfaces, the cell–to–cell junctions that establish the endothelial permeability barrier play an important role.43 The slightly increased VE-cadherin and PECAM-1 expression in both cell types on surface A and for the HMVECs on surface C may indicate a more advanced formation of a functional endothelium compared to the other samples.
5. Conclusion
In our study, we showed that oxygen plasma treatment of PU surfaces through minor parameter changes induced large variations in the affinity, conformation and orientation of adsorbed FN, which in turn, significantly influenced the endothelial cell–material and cell–cell interactions. Due to the many simultaneous effects of plasma treatment on the surface through the introduction of a variety of functional groups and changes in surface topography, a direct link between a single parameter to the bioactivity of adsorbed FN or on the cell behavior could not be established; however, it was demonstrated that oxygen plasma treatment is a reliable, fast and reproducible technique with a high application potential in the adjustment of FN bioactivity and subsequent endothelial cell response.Source of funding
This research was funded by the Deutsche Forschungsgemeinschaft (SCHE701/14-1 to K. S.-L.) and by the doctoral program of the University of Tübingen and Reutlingen University “Intelligente Prozess- und Materialentwicklung in der Biomateriomics (IPMB)” that is supported by the MWK Baden-Württemberg (to R. D., I. M., J. H. and A. J.).Conflicts of interest
None.Acknowledgements
The authors are grateful to Elke Nadler (University of Tübingen) for scientific advice and Julia Senz (University of Tübingen) for the contact angle measurements. We thank Shannon Lee Layland (University of Tübingen) for his valuable input and support with the final revisions. This research was funded by the Deutsche Forschungsgemeinschaft (SCHE701/14-1 to K.S.-L.) and by the joint doctoral program of the University of Tübingen and Reutlingen University “Intelligente Prozess- und Materialentwicklung in der Biomateriomics (IPMB)” that is financially supported by the MWK Baden-Württemberg (to R.D., I.M., J.H. and A.J.).References
- V. Llopis-Hernández, P. Rico, J. Ballester-Beltrán, D. Moratal and M. Salmerón-Sánchez, PLoS One, 2011, 6, e19610 CrossRef.
- J. M. Seeger and N. Klingman, J. Vasc. Surg., 1988, 8, 476–482 CrossRef CAS.
- W. S. Choi, Y. K. Joung, Y. Lee, J. W. Bae, H. K. Park, Y. H. Park, J.-C. Park and K. D. Park, ACS Appl. Mater. Interfaces, 2016, 8, 4336–4346 CrossRef CAS.
- R. Daum, D. Visser, C. Wild, L. Kutuzova, M. Schneider, G. Lorenz, M. Weiss, S. Hinderer, U. A. Stock, M. Seifert and K. Schenke-Layland, Cells, 2020, 9, 778 CrossRef CAS.
- J. Zhu and R. A. F. Clark, J. Invest. Dermatol., 2014, 134, 895–901 CrossRef CAS.
- B. Geiger, A. Bershadsky, R. Pankov and K. M. Yamada, Nat. Rev. Mol. Cell Biol., 2001, 2, 793–805 CrossRef CAS.
- T. P. Ugarova, C. Zamarron, Y. Veklich, R. D. Bowditch, M. H. Ginsberg, J. W. Weisel and E. F. Plow, Biochemistry, 1995, 34, 4457–4466 CrossRef CAS.
- R. Mezzenga and M. Mitsi, Biomacromolecules, 2019, 20, 55–72 CrossRef CAS.
- R. Pankov and K. M. Yamada, J. Cell Sci., 2002, 115, 3861–3863 CrossRef CAS.
- W. S. To and K. S. Midwood, Fibrog. Tissue Repair, 2011, 4, 21 CrossRef CAS.
- M. Cantini, P. Rico, D. Moratal and M. Salmerón-Sánchez, Soft Matter, 2012, 8, 5575–5584 RSC.
- P. Thevenot, W. Hu and L. Tang, Curr. Top. Med. Chem., 2008, 8, 270–280 CrossRef CAS.
- M. B. Hovgaard, K. Rechendorff, J. Chevallier, M. Foss and F. Besenbacher, J. Phys. Chem. B, 2008, 112, 8241–8249 CrossRef CAS.
- A. J. García, M. D. Vega and D. Boettiger, Mol. Biol. Cell, 1999, 10, 785–798 CrossRef.
- B. G. Keselowsky, D. M. Collard and A. J. García, J. Biomed. Mater. Res., Part A, 2003, 66, 247–259 CrossRef.
- A. J. Campillo-Fernández, R. E. Unger, K. Peters, S. Halstenberg, M. Santos, M. S. Sánchez, J. M. M. Dueñas, M. M. Pradas, J. L. G. Ribelles and C. J. Kirkpatrick, Tissue Eng., Part A, 2009, 15, 1331–1341 CrossRef.
- F. A. Vanterpool, M. Cantini, F. P. Seib and M. Salmerón-Sánchez, BioRes. Open Access, 2014, 3, 286–296 CrossRef CAS.
- E. Grigoriou, M. Cantini, M. J. Dalby, A. Petersen and M. Salmeron-Sanchez, Biomater. Sci., 2017, 5, 1326–1333 RSC.
- V. Llopis-Hernández, M. Cantini, C. González-García, Z. A. Cheng, J. Yang, P. M. Tsimbouri, A. J. García, M. J. Dalby and M. Salmerón-Sánchez, Sci. Adv., 2016, 2, E1600188 CrossRef.
- C. González-García, M. Cantini, J. Ballester-Beltrán, G. Altankov and M. Salmerón-Sánchez, Acta Biomater., 2018, 77, 74–84 CrossRef.
- K. M. Yamada and D. W. Kennedy, J. Cell Biol., 1984, 99, 29–36 CrossRef CAS.
- M. D. Pierschbacher, E. G. Hayman and E. Ruoslahti, J. Cell. Biochem., 1985, 28, 115–126 CrossRef CAS.
- S. I. Aota, M. Nomizu and K. M. Yamada, J. Biol. Chem., 1994, 269, 24756–24761 CrossRef CAS.
- P. Singh, C. Carraher and J. E. Schwarzbauer, Annu. Rev. Cell Dev. Biol., 2010, 26, 397–419 CrossRef CAS.
- I. Mrsic, T. Bäuerle, S. Ulitzsch, G. Lorenz, K. Rebner, A. Kandelbauer and T. Chassé, Appl. Surf. Sci., 2021, 536, 147782 CrossRef CAS.
- P. Alves, S. Pinto, H. C. de Sousa and M. H. Gil, J. Appl. Polym. Sci., 2011, 122, 2302–2308 CrossRef CAS.
- O. Mrad, J. Saunier, C. Aymes-Chodur, V. Rosilio, S. Bouttier, F. Agnely, P. Aubert, J. Vigneron, A. Etcheberry and N. Yagoubi, Microscopy and Microanalysis, Cambridge University Press, 2010, vol. 16, pp. 764–778 Search PubMed.
- M. Griffin, R. Palgrave, V. G. Baldovino-Medrano, P. E. Butler and D. M. Kalaskar, Int. J. Nanomed., 2018, 13, 6123–6141 CrossRef CAS.
- R. Hesse, T. Chassé, P. Streubel and R. Szargan, Surf. Interface Anal., 2004, 36, 1373–1383 CrossRef CAS.
- D. Nečas and P. Klapetek, Cent. Eur. J. Phys., 2012, 10, 181–188 Search PubMed.
- G. Kraus and G. Gauglitz, Fresenius' J. Anal. Chem., 1994, 349, 399–402 CrossRef CAS.
- D. J. O’Shannessy, M. Brigham-Burke, K. Karl Soneson, P. Hensley and I. Brooks, Anal. Biochem., 1993, 212, 457–468 CrossRef.
- R. Bowditch, C. Halloran, S. Aota, M. Obara, E. Plow, K. Yamada and M. Ginsberg, J. Biol. Chem., 1991, 266, 23323–23328 CrossRef CAS.
- E. Brauchle, J. Kasper, R. Daum, N. Schierbaum, C. Falch, A. Kirschniak, T. E. Schäffer and K. Schenke-Layland, Matrix Biol., 2018, 68–69, 180–193 CrossRef CAS.
- J. Pusch, M. Votteler, S. Göhler, J. Engl, M. Hampel, H. Walles and K. Schenke-Layland, Biomaterials, 2011, 32, 7469–7478 CrossRef CAS.
- A. Zbinden, M. Urbanczyk, S. L. Layland, L. Becker, J. Marzi, M. Bosch, P. Loskill, G. P. Duffy and K. Schenke-Layland, Tissue Eng., Part A DOI:10.1089/ten.tea.2020.0250.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS.
- J. Ballester-Beltrán, P. Rico, D. Moratal, W. Song, J. F. Mano and M. Salmerón-Sánchez, Soft Matter, 2011, 7, 10803–10811 RSC.
- N. V. Bogatcheva and A. D. Verin, Microvasc. Res., 2008, 76, 202–207 CrossRef CAS.
- C. Wu, Cell Adhes. Migr., 2007, 1, 13–18 Search PubMed.
- E. Spanjaard and J. De Rooij, Curr. Biol., 2013, 23 Search PubMed.
- X. Zhao and J. L. Guan, Adv. Drug Delivery Rev., 2011, 63, 610–615 CrossRef CAS.
- M. G. Lampugnani, Cold Spring Harbor Perspect. Med., 2012, 2, a006528 Search PubMed.
- J. Friedrich, The Plasma Chemistry of Polymer Surfaces, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012 Search PubMed.
- B. Mutel, M. Bigan and H. Vezin, Appl. Surf. Sci., 2004, 239, 25–35 CAS.
- L. Yang, J. Chen, Y. Guo and Z. Zhang, Appl. Surf. Sci., 2009, 255, 4446–4451 CrossRef CAS.
- F. Arefi-Khonsari, M. Tatoulian, F. Bretagnol, O. Bouloussa and F. Rondelez, Surf. Coatings Technol., 2005, 200, 14–20 CrossRef CAS.
- J. Lai, B. Sunderland, J. Xue, S. Yan, W. Zhao, M. Folkard, B. D. Michael and Y. Wang, Appl. Surf. Sci., 2006, 252, 3375–3379 CrossRef CAS.
- N. Inagaki, K. Narushim, N. Tuchida and K. Miyazaki, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 3727–3740 CrossRef CAS.
- R. N. Wenzel, J. Phys. Colloid Chem., 1949, 53, 1466–1467 CrossRef CAS.
- M. Salmerón-Sánchez, P. Rico, D. Moratal, T. T. Lee, J. E. Schwarzbauer and A. J. García, Biomaterials, 2011, 32, 2099–2105 CrossRef.
- R. A. Latour, J. Biomed. Mater. Res., Part A, 2015, 103, 949–958 CrossRef.
- C. Hoffmann, J. Leroy-Dudal, S. Patel, O. Gallet and E. Pauthe, Anal. Biochem., 2008, 372, 62–71 CrossRef CAS.
- D. Khang, S. Y. Kim, P. Liu-Snyder, G. T. R. Palmore, S. M. Durbin and T. J. Webster, Biomaterials, 2007, 28, 4756–4768 CrossRef CAS.
- Y. Zhang, C. Chai, X. S. Jiang, S. H. Teoh and K. W. Leong, Mater. Sci. Eng., C, 2007, 27, 213–219 CrossRef CAS.
- P. Roach, D. Farrar and C. C. Perry, J. Am. Chem. Soc., 2005, 127, 8168–8173 CrossRef CAS.
- V. Vadillo-Rodríguez, M. A. Pacha-Olivenza, M. L. Gónzalez-Martín, J. M. Bruque and A. M. Gallardo-Moreno, J. Biomed. Mater. Res., Part A, 2013, 101A, 1397–1404 CrossRef.
- M. S. Lord, B. G. Cousins, P. J. Doherty, J. M. Whitelock, A. Simmons, R. L. Williams and B. K. Milthorpe, Biomaterials, 2006, 27, 4856–4862 CrossRef CAS.
- D. Gagné, Y. D. Benoit, J. F. Groulx, P. H. Vachon and J. F. Beaulieu, BMC Mol. Cell Biol., 2020, 21, 14 CrossRef.
- N. B. Guerra, C. González-García, V. Llopis, J. C. Rodríguez-Hernández, D. Moratal, P. Rico and M. Salmerón-Sánchez, Soft Matter, 2010, 6, 4748–4755 RSC.
- A. Ambesi, R. M. Klein, K. M. Pumiglia and P. J. McKeown-Longo, Cancer Res., 2005, 65, 148–156 CAS.
- G. A. Homandberg, J. Kramer-Bjerke, D. Grant, G. Christianson and R. Eisenstein, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1986, 874, 61–71 CrossRef CAS.
- F. Podestá, T. Roth, F. Ferrara, E. Cagliero and M. Lorenzi, Diabetologia, 1997, 40, 879–886 CrossRef.
- L. Miranda, S. Carpentier, A. Platek, N. Hussain, M. A. Gueuning, D. Vertommen, Y. Ozkan, B. Sid, L. Hue, P. J. Courtoy, M. H. Rider and S. Horman, Biochem. Biophys. Res. Commun., 2010, 396, 656–661 CrossRef CAS.
- S. Fogarty, F. A. Ross, D. V. Ciruelos, A. Gray, G. J. Gowans and D. G. Hardie, Mol. Cancer Res., 2016, 14, 683–695 CrossRef CAS.
- S. Tojkander, K. Ciuba and P. Lappalainen, Cell Rep., 2018, 24, 11–19 CrossRef CAS.
- H. Schnittler, M. Taha, M. O. Schnittler, A. A. Taha, N. Lindemann and J. Seebach, Cell Tissue Res., 2014, 355, 529–543 CrossRef CAS.
- E. H. J. Danen, P. Sonneveld, C. Brakebusch, R. Fässler and A. Sonnenberg, J. Cell Biol., 2002, 159, 1071–1086 CrossRef CAS.
- J. L. Coll, A. Ben-Ze’ev, R. M. Ezzell, J. L. Rodríguez Fernández, H. Baribault, R. G. Oshima and E. D. Adamson, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9161–9165 CrossRef CAS.
- R. M. Saunders, M. R. Holt, L. Jennings, D. H. Sutton, I. L. Barsukov, A. Bobkov, R. C. Liddington, E. A. Adamson, G. A. Dunn and D. R. Critchley, Eur. J. Cell Biol., 2006, 85, 487–500 CrossRef CAS.
- V. Schaufler, H. Czichos-Medda, V. Hirschfeld-Warnecken, S. Neubauer, F. Rechenmacher, R. Medda, H. Kessler, B. Geiger, J. P. Spatz and E. A. Cavalcanti-Adam, Cell Adhes. Migr., 2016, 10, 505–515 CrossRef CAS.
- A. Sales, K. Ende, J. Diemer, A. R. Kyvik, J. Veciana, I. Ratera, R. Kemkemer, J. P. Spatz and J. Guasch, ACS Omega, 2019, 4, 1791–1800 CrossRef CAS.
- H. Amani, H. Arzaghi, M. Bayandori, A. S. Dezfuli, H. Pazoki-Toroudi, A. Shafiee and L. Moradi, Adv. Mater. Interfaces, 2019, 6, 1900572 CrossRef.
- S. Huveneers, H. Truong, R. Fässler, A. Sonnenberg and E. H. J. Danen, J. Cell Sci., 2008, 121, 2452–2462 CrossRef CAS.
- X. K. Zhao, Y. Cheng, M. Liang Cheng, L. Yu, M. Mu, H. Li, Y. Liu, B. Zhang, Y. Yao, H. Guo, R. Wang and Q. Zhang, Sci. Rep., 2016, 6, 1–12 CrossRef CAS.
- A. P. Gilmore and L. H. Romer, Mol. Biol. Cell, 1996, 7, 1209–1224 CAS.
- K. Katoh, PeerJ, 2017, 2017, e4063 Search PubMed.
- C. Lawson and D. D. Schlaepfer, Cell Adhes. Migr., 2012, 6, 302–306 CrossRef.
- D. K. Pettit, T. A. Horbett and A. S. Hoffman, J. Biomed. Mater. Res., 1992, 26, 1259–1275 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb02757j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |