
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineered protein cages for selective heparin encapsulation†
Salla
Välimäki‡
a,
Qing
Liu‡
a,
Lise
Schoonen
 b,
Daan F. M.
Vervoort
c,
Nonappa
d,
Veikko
Linko
ad,
Roeland J. M.
Nolte
b,
Jan C. M.
van Hest
bc and
Mauri A.
Kostiainen
*ad
b,
Daan F. M.
Vervoort
c,
Nonappa
d,
Veikko
Linko
ad,
Roeland J. M.
Nolte
b,
Jan C. M.
van Hest
bc and
Mauri A.
Kostiainen
*ad
aDepartment of Bioproducts and Biosystems, Aalto University, P.O. Box 16100, Aalto FI-00076, Espoo, Finland. E-mail: mauri.kostiainen@aalto.fi
bInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, Nijmegen 6525 AJ, The Netherlands
cDepartment of Bio-Organic Chemistry, Eindhoven University of Technology, Institute of Complex Molecular Systems (ICMS), Het Kranenveld 14, Eindhoven 5600 MB, The Netherlands
dHYBER Centre, Department of Applied Physics, Aalto University, Aalto FI-00076, Finland
First published on 17th December 2020
Abstract
A heparin-specific binding peptide was conjugated to a cowpea chlorotic mottle virus (CCMV) capsid protein, which was subsequently allowed to encapsulate heparin and form capsid-like protein cages. The encapsulation is specific and the capsid-heparin assemblies display negligible hemolytic activity, indicating proper blood compatibility and promising possibilities for heparin antidote applications.
Heparin (Fig. 1a) is a highly anionic polysaccharide belonging to the family of glycosaminoglycans and is widely used as an anticoagulant in surgical practices and thrombotic events.1 The anticoagulant activity is based on its ability to bind and subsequently activate antithrombin-III, which leads to the inactivation of vital coagulation cascade substances such as thrombin and factor Xa.2 For safe clinical operation, heparin requires an antidote, which can counteract the anticoagulant effect and suppress potential side effects, if needed.3 Protamine sulfate is a small arginine-rich cationic protein that binds heparin via electrostatic interactions, and is commonly used for this purpose.2,4 This interaction is electrostatic in nature, but nonspecific, and protamine sulfate can cause multiple adverse effects.5 This has initiated diverse attempts to develop safer heparin antidotes, such as cationic polymers and small molecules, which all largely rely on the electrostatic binding with heparin.6–12 Although efficient binding can be achieved, cationic platforms often lack specificity to heparin and usually exhibit high cytotoxicity. Furthermore, the complexes formed with heparin are often large and ill-defined aggregates. Therefore, more sophisticated alternatives that would transform heparin into small and well-defined particles, which are biologically inert, are needed.9,12
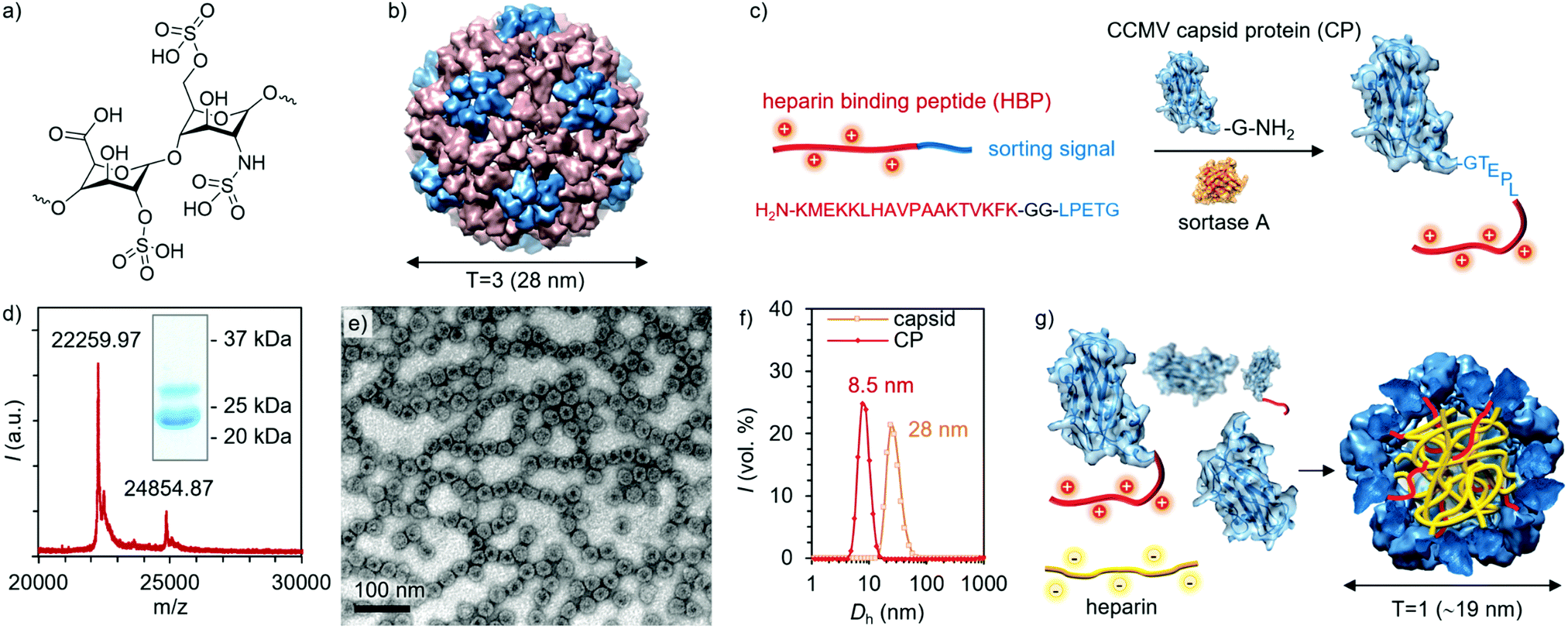 | ||
| Fig. 1 (a) Structure of the major repeating unit of heparin. (b) Drawing of the CCMV capsid (T = 3) structure. (c) Illustration of the Sortase A mediated coupling of HBP to CP. (d) MALDI-TOF spectrum and SDS-PAGE (inset) of HBP modified CPs. (e) TEM image of empty capsids formed by HBP-modified CPs. (f) DLS measurements (volume-weighted size distributions) of the empty capsid and free CP. (g) Schematic representation of heparin (yellow chain) encapsulation by a mixture of CP and HBP modified CP to form T = 1 structures. | ||
Virus-like particles (VLPs) are protein assemblies derived from wild-type viruses. They lack the viral genome but have similar well-defined capsid structures as native virus particles, and have therefore been explored for biomedical and nano-technological applications.13–17 Indeed, their most appealing features are the accurate sizes and shapes, which are combined with a relatively high stability and versatile modification potential.18 The removal of the viral genome also generates a highly confined cavity, which can be harnessed to compartmentalize other nanoscale substances.19–21 Cowpea chlorotic mottle virus (CCMV) capsid is one of the most studied VLPs as they can be produced in high yields22 and undergo pH and ionic strength-dependent swelling and assembly,23,24 which have therefore been intensively utilized in cargo-loading.25
In more detail, CCMV is a plant-infecting icosahedral virus consisting of 180 capsid proteins (CPs) in its native T = 3 form, leading to a capsid with an outer diameter of 28 nm and an inner cavity diameter of 18 nm (Fig. 1b).26 Its T = 1 and pseudo T = 2 forms consist of 60 and 120 capsid proteins and have diameters of roughly 18 and 22 nm, respectively.27 Each CP is formed from 190 amino acids with its N-terminus located on the interior of the capsid. CCMV can be modified in multiple different ways, for example, by functionalizing carboxylic acids or amines on the exterior of the capsid.28,29 In addition, the CCMV interior may be altered to enable enhanced cargo loading and encapsulation through electrostatic interactions.30,31 To obtain a highly defined modification of the capsid interior, an appealing approach is to engineer the N-termini, which provides site- and number-specificity. For this purpose, an enzymatic method involving a reaction catalyzed by the enzyme Sortase A (SrtA) has been developed.32 This benign methodology was first utilized to label the CCMV capsids with a fluorescent tag, but due to the generic nature of the strategy, a wide range of cargos loaded in VLPs can be envisioned.33,34
So far, only the exteriors of virions have been employed to display polycationic motifs (poly-Arg) for heparin-binding.35–38 However, multiple mutations may yield unstable virions and lead to nonspecific electrostatic interactions with heparin. In this work, we report on the conjugation of a heparin-binding peptide (HBP), i.e. a heparin-specific sequence derived from the fibroblast growth factor having a dissociation constant of ∼134 pM with heparin, to the interior of the CCMV capsid.39 The binding and packing of heparin into the protein cage was studied with dynamic light scattering (DLS), transmission electron microscopy (TEM), and fast protein liquid chromatography (FPLC). Assemblies were not observed in the presence of two other glycosaminoglycan analogues, demonstrating high selectivity towards heparin. Moreover, no hemolytic activity was detected, implying proper biocompatibility.
The particular HBP was selected based on its high selectivity and affinity to heparin, thus minimizing off-target binding even in complex biological environments.12,39 Conjugation of HBP to the CP N-terminus was realized with the SrtA-based method, as illustrated in Fig. 1c. SrtA catalyzes the cleavage of the HBP C-terminal sorting signal (LPETG) after threonine and the subsequent ligation with an N-terminal glycine in the CP. The detailed coupling procedure and characterization of the intermediate conjugates can be found in the ESI† (Fig. S1–S6, ESI†). The final product was characterized by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometry. Based on the SDS-PAGE and MALDI-TOF analysis, approximately 25% of the CPs were successfully conjugated with HBP (Fig. 1d and Fig. S7, ESI†), which corresponds on average to 45 modified CPs per one T = 3 CCMV capsid. Initially, TEM imaging was employed to investigate the morphology of the assemblies formed by HBP modified CPs alone. Negatively stained spherical VLPs were observed under the TEM microscope, having a diameter of ∼28 nm, which corresponds well with the expected dimension of the T = 3 icosahedral capsids (Fig. 1e and Fig. S9a, ESI†). The presence of dark regions in the capsid cores indicates that the capsids are empty as the stain is able to enter the voids.40 Empty CCMV capsids were then disassembled by dialysis against Tris–HCl buffer (50 mM, pH = 7.4). Disassembly was confirmed by TEM and DLS measurements, which showed a size shift from 28 nm to 8–9 nm (Fig. 1f and Fig. S9b, ESI†). The obtained CP solution was then mixed with heparin in different quantities yielding heparin-loaded capsids (Fig. 1g).
As observed in Fig. 2a–c and Fig. S12, ESI,† the heparin-loaded capsid assemblies formed with three different heparin to protein mass ratios (ρ = 0.02, 0.22, and 0.44) all had a roughly spherical morphology and a smaller diameter than the empty T = 3 capsid (28 nm). For instance, TEM images indicated an average size of 18.6 nm at ρ = 0.02 (Fig. 2d). Similar diameters were observed with the ρ = 0.22 (18.6 nm) and 0.44 (20.2 nm) samples (Fig. 2d), matching well with the dimension of T = 1 capsids. The preferential formation of T = 1 capsids has been observed previously, i.e. when highly negatively charged synthetic polymers40–43 and nucleic acids were encapsulated in CCMV VLPs.44,45 The particle size has been shown to depend on the molecular weight of the encapsulated compound, and commonly higher molecular weight compounds promote the formation of larger capsids.40,45 Unlike empty capsids, the heparin-loaded assemblies did not show dark cores (Fig. 2e), demonstrating that the capsid voids were filled and successfully loaded with heparin molecules. Cryo-TEM images of the heparin-loaded capsids further verified the spherical morphology and ∼18 nm size of the complexes and confirmed that the observed structures are not a result of a drying effect.
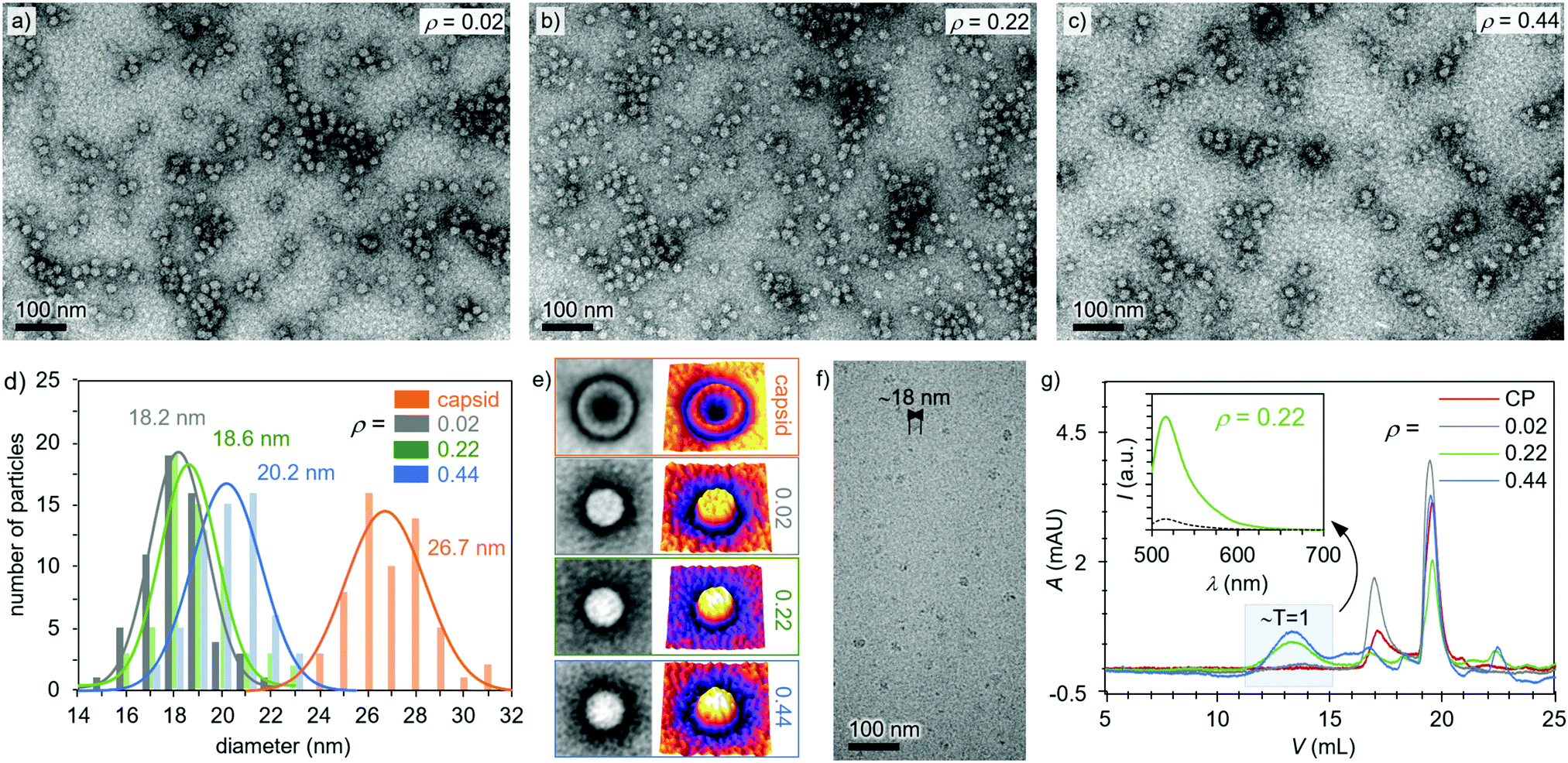 | ||
| Fig. 2 TEM images of heparin-loaded capsids at (a) ρ = 0.02, (b) ρ = 0.22, (c) ρ = 0.44. (d) Analysis of the dimensions of the complexes (ρ = 0.02–0.44) and capsids before disassembly (ρ = 0), as derived from the TEM images. Solid curves show Gaussian fits. (e) Averaged (n = 40) particle images (size 50 nm × 50 nm), as taken from panels a–c (left) and their 3D intensity profiles (right). (f) Cryo-TEM image of heparin-loaded capsids at ρ = 0.16. (g) FPLC chromatogram (280 nm) of free CPs and heparin-loaded capsids (ρ = 0.02–0.44). Free CPs elute at 19.6 mL and 17.1 mL, and T = 1 heparin-loaded capsids elute at 13.9 mL. Inset graph shows the fluorescence emission spectrum (excitation at 485 nm) of the heparin-loaded capsids (ρ = 0.22, green line) and pure heparin-FITC (black dashed line) collected at the 13.9 mL. | ||
The effect of the amount of heparin on capsid formation was quantified with fluorescein (FITC) moieties according to a previously published method.46 Successful heparin-FITC conjugation was confirmed with FPLC and nuclear magnetic resonance (NMR) spectroscopy (Fig. S8, ESI†). Fig. 2g shows that the free CPs were eluting at 19.6 mL and 17.1 mL. Comparison of the peak areas revealed that 71% of protein was eluting at the higher elution volume (19.6 mL, Fig. S11, ESI†), which corresponds to the protein dimers. These values are well in line with previously published work conducted under similar conditions where the CP dimers were eluting at 18.5 mL.42 In the present studies, another peak at 17.1 mL, corresponding to slightly larger capsomers, was also observed.47 The assembled capsids were expected to elute at lower volumes, and for the ρ = 0.02 sample, a minor broad peak appeared around 13.9 mL (Fig. 2g), which corresponds to T = 1 capsids.42 When heparin concentration was increased to ρ = 0.22, the peak fraction at 13.9 mL increased from 6% to 37%, showing the formation of more T = 1 capsids. However, further addition of heparin (ρ = 0.44) did not significantly increase the fraction of T = 1 capsids. Instead it enhanced the amount of the intermediate particles as observed by the larger peak area between 17.1 mL and 13.9 mL. Additionally, unbound heparin (peak at 22.4 mL) started to appear (Fig. S12d, ESI†), indicating that the excess of heparin did not induce further formation of capsids. This finding is supported by previous studies, which have shown that capsid formation depends on the amount of the encapsulated compound and that high cargo ratios limit capsid formation.41,42 Fractions corresponding to the T = 1 capsids (13 mL) were collected and investigated with the help of fluorescence spectroscopy measurements. A clear emission spectrum from the fluorescein-conjugated heparin was observed for all the heparin-loaded samples, while that for pure heparin-FITC was neglectable, demonstrating that the encapsulations had been successful (Fig. 2g inset and Fig. S13, ESI†).
The selectivity of the heparin encapsulation was evaluated qualitatively with DLS. Studies aimed at encapsulating other negatively charged target molecules were conducted with two glycosaminoglycan analogues, i.e. hyaluronic acid and chondroitin sulfate as well as bovine serum albumin. No size change was observed when these compounds were added to CCMV CPs (ρ = 0.02–0.22), indicating that capsids or other higher-order structures are not formed (Fig. 3a). However, adding heparin in similar mass ratio clearly induced T = 1 capsid formation, which is visible as a peak between the CP dimers and T = 3 capsid (Fig. 3b and Fig. S14, ESI†). Adding heparin to the preassembled T = 3 capsid did not result in any changes in the size distribution indicating that heparin is not bound to the exterior of the capsid. Such observation is expected since the empty capsid remains assembled only at pH 5 in the presence of high electrolyte concentration (500 mM NaCl and 10 mM MgCl2), which efficiently screens electrostatic interactions.
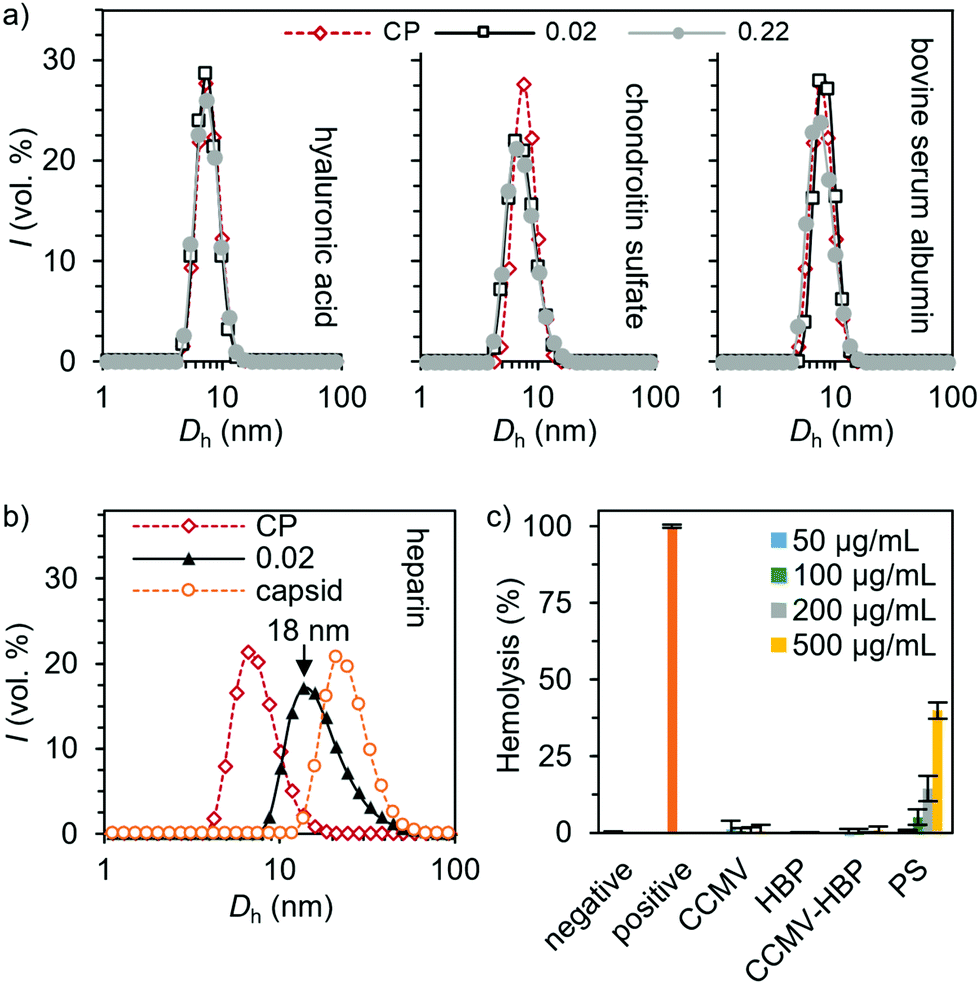 | ||
| Fig. 3 Selectivity of the capsid assembly induced by (a) hyaluronic acid, chondroitin sulfate, bovine serum albumin and (b) heparin, as measured by DLS. (c) Hemolytic activity of the unmodified and HBP-conjugated CPs. Protamine sulfate (PS) was tested for comparison. Solutions of 1× phosphate-buffered saline (PBS) and 1% Triton X-100 were used as the negative and positive controls, respectively. Measurements were performed using triplicate samples, and the averaged results with standard deviation are presented. | ||
Furthermore, we found negligible hemolytic activity of the CPs up to 0.5 mg mL−1 concentration on red blood cells (RBCs) (Fig. 3c), which is in sharp contrast to the effect of commonly employed protamine sulfate, thus providing a promising strategy in heparin neutralization.
In conclusion, we have shown that CCMV capsid proteins modified with a heparin-binding peptide exhibit specific heparin binding at physiological pH's. Co-assembly with heparin yielded T = 1 capsids with diameters ranging between 18–20 nm according to the DLS measurements and TEM characterization. FPLC results further confirmed the capsid formation and revealed a heparin concentration-limiting effect on capsid formation. The binding of heparin was proved to be specific as no capsid formation was induced with two other glycosaminoglycan analogs. Moreover, hemolysis assays demonstrated a better biocompatibility than the commercial heparin antidote protamine sulfate. These results indicate that VLPs are promising materials for heparin binding, especially when the binding is further optimized for physiological conditions and after thorough hematologic studies have been conducted.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We gratefully thank the Academy of Finland (projects 308578, 303804, 267497), the Jane and Aatos Erkko Foundation, the Sigrid Jusélius Foundation and the Emil Aaltonen Foundation for financial support. We acknowledge the provision of facilities at OtaNano – Nanomicroscopy Center (Aalto-NMC) and Bioeconomy Infrastructure. Support was also received from the European Research Council (ERC Advanced Grant 740295 ENCOPOL to R. J. M. N.) and from the Dutch Ministry of Education, Culture and Science (Gravity Program 024.001.035 to R. J. M. N. and J. C. M. v. H.).References
- B. Casu, A. Naggi and G. Torri, Carbohydr. Res., 2015, 403, 60–68 CrossRef CAS.
- J. Hirsh, T. E. Warkentin, S. G. Shaughnessy, S. S. Anand, J. L. Halperin, R. Raschke, C. Granger, E. M. Ohman and J. E. Dalen, Chest, 2001, 119, 64S–94S CrossRef CAS.
- B. Girolami and A. Girolami, Semin. Thromb. Hemostasis, 2006, 32, 803–809 CrossRef CAS.
- R. Balhorn, Genome Biol., 2007, 8, 227 CrossRef.
- J. C. Horrow, Anesth. Analg., 1985, 64, 348–361 CrossRef CAS.
- S. M. Bromfield, E. Wilde and D. K. Smith, Chem. Soc. Rev., 2013, 42, 9184–9195 RSC.
- M. Schuksz, M. M. Fuster, J. R. Brown, B. E. Crawford, D. P. Ditto, R. Lawrence, C. A. Glass, L. Wang, Y. Tor and J. D. Esko, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13075–13080 CrossRef CAS.
- S. Välimäki, A. Khakalo, A. Ora, L.-S. Johansson, O. J. Rojas and M. A. Kostiainen, Biomacromolecules, 2016, 17, 2891–2900 CrossRef.
- M. T. Kalathottukaren, C. A. Haynes and J. N. Kizhakkedathu, Drug Delivery Transl. Res., 2018, 8, 928–944 CrossRef.
- V. M. P. Vieira, V. Liljeström, P. Posocco, E. Laurini, S. Pricl, M. A. Kostiainen and D. K. Smith, J. Mater. Chem. B, 2017, 5, 341–347 RSC.
- S. Välimäki, N. K. Beyeh, V. Linko, R. H. A. Ras and M. A. Kostiainen, Nanoscale, 2018, 10, 14022–14030 RSC.
- Q. Liu, S. Välimäki, A. Shaukat, B. Shen, V. Linko and M. A. Kostiainen, ACS Omega, 2019, 4, 21891–21899 CrossRef CAS.
- Y. Ma, R. J. M. Nolte and J. J. L. M. Cornelissen, Adv. Drug Delivery Rev., 2012, 64, 811–825 CrossRef CAS.
- P. Singh, M. J. Gonzalez and M. Manchester, Drug Dev. Res., 2006, 67, 23–41 CrossRef CAS.
- V. Liljeström, A. Ora, J. Hassinen, H. T. Rekola, M. Heilala Nonappa, V. Hynninen, J. J. Joensuu, R. H. A. Ras, P. Törmä, O. Ikkala and M. A. Kostiainen, Nat. Commun., 2017, 8, 671 CrossRef.
- M. Rother, M. G. Nussbaumer, K. Rengglic and N. Bruns, Chem. Soc. Rev., 2016, 45, 6213 RSC.
- J. G. Heddle, S. Chakraborti and K. Iwasaki, Curr. Opin. Struct. Biol., 2017, 43, 148 CrossRef CAS.
- M. Young, D. Willits, M. Uchida and T. Douglas, Annu. Rev. Phytopathol., 2008, 46, 361–384 CrossRef CAS.
- W. M. Aumiller, M. Uchida and T. Douglas, Chem. Soc. Rev., 2018, 47, 3433–3469 RSC.
- B. Wörsdörfer, K. J. Woycechowsky and D. Hilvert, Science, 2011, 331, 589–592 CrossRef.
- A. Korpi, E. Anaya-Plaza, S. Välimäki and M. A. Kostiainen, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12, e1578 CAS.
- J.-P. Michel, M. Gingery and L. Lavelle, J. Virol. Methods, 2004, 122, 195–198 CrossRef CAS.
- L. Lavelle, J.-P. Michel and M. J. Gingery, J. Virol. Methods, 2007, 146, 311–316 CrossRef CAS.
- L. Lavelle, M. Gingery, M. Phillips, W. M. Gelbart, C. M. Knobler, R. D. Cadena-Nava, J. R. Vega-Acosta, L. A. Pinedo-Torres and J. Ruiz-Garcia, J. Phys. Chem. B, 2009, 113, 3813–3819 CrossRef CAS.
- J. G. Heddle, S. Chakraborti and K. Iwasaki, Curr. Opin. Struct. Biol., 2017, 43, 148–155 CrossRef CAS.
- J. A. Speir, S. Munshi, G. Wang, T. S. Baker and J. E. Johnson, Structure, 1995, 3, 63–78 CrossRef CAS.
- J. Tang, J. M. Johnson, K. A. Dryden, M. J. Young, A. Zlotnick and J. E. Johnson, J. Struct. Biol., 2006, 154, 59–67 CrossRef CAS.
- E. Gillitzer, D. Willits, M. Young and T. Douglas, Chem. Commun., 2002, 2390–2391 RSC.
- E. Gillitzer, P. Suci, M. Young and T. Douglas, Small, 2006, 2, 962–966 CrossRef CAS.
- I. J. Minten, L. J. A. Hendriks, R. J. M. Nolte and J. J. L. M. Cornelissen, J. Am. Chem. Soc., 2009, 131, 17771–17773 CrossRef CAS.
- P. A. Suei, Z. Varpness, E. Gillitzer, T. Douglas and M. Young, Langmuir, 2007, 23, 12280–12286 CrossRef.
- L. Schoonen, J. Pille, A. Borrmann, R. J. M. Nolte and J. C. M. van Hest, Bioconjugate Chem., 2015, 26, 2429–2434 CrossRef CAS.
- L. Schoonen, S. Maassen, R. J. M. Nolte and J. C. M. van Hest, Biomacromolecules, 2017, 18, 3492–3497 CrossRef CAS.
- L. Schoonen, R. J. M. Nolte and J. C. M. van Hest, Nanoscale, 2016, 8, 14467–14472 RSC.
- A. K. Udit, C. Everett, A. J. Gale, J. Reiber Kyle, M. Ozkan and M. G. Finn, ChemBioChem, 2009, 10, 503–510 CrossRef CAS.
- A. J. Gale, D. J. Elias, P. M. Averell, P. S. Teirstein, M. Buck, S. D. Brown, Z. Polonskaya, A. K. Udit and M. G. Finn, Thromb. Res., 2011, 128, e9–e13 CrossRef CAS.
- H. Y. Cheong, M. Groner, K. Hong, B. Lynch, W. R. Hollingsworth, Z. Polonskaya, J.-K. Rhee, M. M. Baksh, M. G. Finn, A. J. Gale and A. K. Udit, Biomacromolecules, 2017, 18, 4113 CrossRef CAS.
- J. M. Choi, V. Bourassa, K. Hong, M. Shoga, E. Y. Lim, A. Park, K. Apaydin and A. K. Udit, Mol. Pharmaceutics, 2018, 15, 2997 CrossRef CAS.
- M. Kan, F. Wang, J. Xu, J. Crabb, J. Hou and W. McKeehan, Science, 1993, 259, 1918–1921 CrossRef CAS.
- Y. Hu, R. Zandi, A. Anavitarte, C. M. Knobler and W. M. Gelbart, Biophys. J., 2008, 94, 1428–1436 CrossRef CAS.
- F. D. Sikkema, M. Comellas-Aragonès, R. G. Fokkink, B. J. M. Verduin, J. J. L. M. Cornelissen and R. J. M. Nolte, Org. Biomol. Chem., 2006, 5, 54–57 RSC.
- M. Brasch and J. J. L. M. Cornelissen, Chem. Commun., 2012, 48, 1446–1448 RSC.
- J. B. Bancroft, E. Hiebert and C. E. Bracker, Virology, 1969, 39, 924–930 CrossRef CAS.
- S. J. Maassen, M. V. de Ruiter, S. Lindhoud and J. J. L. M. Cornelissen, Chem. – Eur. J., 2018, 24, 7456–7463 CrossRef CAS.
- R. D. Cadena-Nava, M. Comas-Garcia, R. F. Garmann, A. L. N. Rao, C. M. Knobler and W. M. Gelbart, J. Virol., 2012, 86, 3318–3326 CrossRef CAS.
- Q. Li, L. Ye, A. Zhang and Z. Feng, Carbohydr. Polym., 2019, 211, 370–379 CrossRef CAS.
- A. Liu, M. Verwegen, M. V. De Ruiter, S. J. Maassen, C. H. Traulsen and J. J. L. M. Cornelissen, J. Phys. Chem. B, 2016, 120, 6352–6357 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Viral capsid protein preparation, purification and characterization methods. Details of TEM, DLS, FPLC, NMR, fluorescence spectroscopy and hemolysis assay. Additional TEM, FPLC and DLS data. See DOI: 10.1039/d0tb02541k |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |