
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-healing and mechanical performance of dynamic glycol chitosan hydrogel nanocomposites†
Spyridon
Efstathiou
 a,
Alan M.
Wemyss
ab,
Georgios
Patias
a,
Lucas
Al-Shok
a,
Maria
Grypioti
a,
Despina
Coursari
a,
Congkai
Ma
a,
Christophe J.
Atkins
a,
Ataulla
Shegiwal
a,
Chaoying
Wan
b and
David M.
Haddleton
*a
a,
Alan M.
Wemyss
ab,
Georgios
Patias
a,
Lucas
Al-Shok
a,
Maria
Grypioti
a,
Despina
Coursari
a,
Congkai
Ma
a,
Christophe J.
Atkins
a,
Ataulla
Shegiwal
a,
Chaoying
Wan
b and
David M.
Haddleton
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, CV4 7AL, Coventry, UK. E-mail: d.m.haddleton@warwick.ac.uk
bInternational Institute for Nanocomposites Manufacturing (IINM), WMG, University of Warwick, CV4 7AL, UK
First published on 8th December 2020
Abstract
The application of functional self-healing and mechanically robust hydrogels in bioengineering, drug delivery, soft robotics, etc., is continuously growing. However, fabricating hydrogels that simultaneously possess good mechanical and self-healing properties remains a challenge. Developing robust hydrogel formulations for the encapsulation and release of hydrophobic substances is a major challenge especially in some pharmaceutical treatments where the many of drugs show incompatibility with the hydrophilic hydrogel matrices. Schiff base hydrogels have been developed using a benzaldehyde multifunctional amphiphilic polyacrylamide crosslinker in conjunction with glycol chitosan. The polymeric crosslinker was synthesized by a two-step reaction using aqueous Cu-RDRP to give an ABA telechelic copolymer of N,N-dimethyl acrylamide (DMAc) and N-hydroxyethyl acrylamide (HEAm) from a bifunctional PEG. The polymer was then modified by post functionalization leading to a multifunctional benzaldehyde crosslinker that was shown to be capable of self-assembly into aggregates in aqueous media serving as a possible candidate for the entrapment of hydrophobic substances. Aqueous solutions of the crosslinker spontaneously formed hydrogels when mixed with glycol chitosan due to the in situ formation of imine bonds. Hydrogels were characterized while additional comparisons were made with a commonly used bifunctional PEG crosslinker. The effect of introducing partially reduced graphene oxide (GO) nanosheets was also examined and led to enhancements in both mechanical properties (2.0 fold increase in modulus and 1.4 fold increase in strain) and self-healing efficiencies (>99% from 60% by rheology) relative to the pristine polymer hydrogels.
Introduction
Recently, there has been growing interest in developing materials that are able to intrinsically self-heal after damage to recover their properties.1,2 Despite the wide ranging application of hydrogels in fields such as biomedicine,3 tissue engineering,4,5 and batteries,6 amongst others, their soft characteristics makes them susceptible to damage, such as macro- or microscale cracks that can be caused by mechanical or chemical factors.7,8 To address these limitations, self-healing hydrogels have been developed that have the ability to extend the service life of these materials.9,10 However, a common issue that is observed in such systems, is that hydrogels with good self-healing abilities often lack high mechanical properties, therefore limiting their application window. Conversely, stiffer hydrogels can show less efficient self-healing properties as they frequently lack chain mobility and network flowability, which restricts the generation of appropriate “mobile phase” to autonomously cover and heal damaged areas.11,12 Thus, there is a desire to develop hydrogels that possess both good mechanical and self-healing properties.An additional challenge that most clinically related hydrogel systems face, is the difficulty to encapsulate and potentially release hydrophobic substances of biological interest at the appropriate time and place, a consequence of the high hydrophilicity of the polymer matrix.13 A common way that deals with this limitation is the introduction of hydrophobic domains by choosing hydrophobic monomers or crosslinkers, the use of thermoresponsive polymer blocks (usually triblock ABA copolymers) that can undergo sol–gel transitions from room to physiological temperature and finally the introduction of nanoparticles which can either take part on the crosslinking or get adsorbed inside the material. In 2015, Appel et al.14 prepared self-assembled biocompatible injectable hydrogels arising from the interactions between poly(ethylene)glycol-b-poly(lactic acid) particles and a derivative of cellulose. Hydrogels could afford release of both hydrophilic and hydrophobic molecules promising for controlled drug delivery applications. In another paradigm, Qu et al.15 developed antibacterial wound dressing adhesive hydrogels prepared by mixing quaternized chitosan and a benzaldehyde-terminated crosslinker of Pluronic®F127 able to form micelles of various diameters depending on the amount of incorporated curcumin. Hydrogels showed good mechanical and self-healing properties while their adhesiveness and antibacterial characteristics made them promising for wound healing applications.
A common method used to enhance both mechanical and self-healing characteristics of materials involves the incorporation of nanoparticles to form nanocomposites. Nanofillers can offer superior properties to materials when embedded inside their network due to their high surface area and nanoscale interactions with the polymer components.16,17 Specifically, their use in the development of new self-healing materials has proven fruitful as they can often lead to tough hydrogels with good self-healing abilities. Some commonly used nanofillers are silica,18 carbon nanotubes (CNTs),19,20 and graphene oxide (GO).21,22 Recently, graphene-based nanocomposites have attracted research interest due to their exceptional mechanical, electrical, optical and thermal properties, which lead to high-end materials.23 In 2017, Pan et al.24 reported new nanocomposite hydrogels based on poly(acrylamide-co-(dimethylamino)ethylacrylatemethochloride)/GO that successfully demonstrated excellent self-healing efficiencies (>99%), stretchability and relatively good mechanical properties (Young's modulus: ∼1.1 MPa). Addition of various concentrations of GO in combination with different monomer ratios seemed to effectively tune the mechanical properties, making them good candidates for practical applications. Furthermore, Jing et al.25 utilized GO as a reinforcing agent for mussel inspired conductive hydrogels with a chitosan skeleton. GO was partially reduced from chitosan and polydopamine resulting into conductive hydrogels with fast self-healing ability and good adhesiveness for tissue engineering applications.
Hydrogels can be generally described as water-swollen three dimensional (3D) networks that may possess self-healing properties depending on the type (physical or chemical) and degree of crosslinking. In order to introduce intrinsic self-healing ability into hydrogels, dynamic covalent bonds are often used to crosslink the network since they can reversibly form and break under certain conditions, acting as either permanent covalent bonds or reversible non-covalent physical ones at an equilibrium. Some common examples of such bonds include, disulfide bonds,26 imine bonds,27–29 Diels–Alder adducts,30 and acylhydrazone bonds.31 More specifically, imine based hydrogels which arise from in situ Schiff base reactions between aldehydes and amines, have attracted attention in the field of smart biocompatible hydrogels as they can undergo fast bond exchange between other imines and amines with no requirement for additional catalysts and with tuneable sol–gel transitions.29,32
Many of the reported imine based hydrogels utilize chitosan or glycol chitosan (the water soluble analogue) as the main source of primary amines due to their low toxicity as well as excellent biocompatibility and biodegradability in certain circumstances.33,34 For example, in 2016 Zhu et al.35 reported the effective incorporation and release of colistin from non-toxic self-healing hydrogels prepared from glycol chitosan and a bifunctional benzaldehyde poly(ethylene glycol) crosslinker. Disk diffusion assays showed that pristine hydrogels had no inhibitory activity against bacteria while incorporation and release of colistin revealed antimicrobial activity with potential applications as localized wound healing gels. The same bifunctional crosslinker was also used by Qu et al.36 to develop pH-responsive and self-healing N-carboxyethyl chitosan hydrogels as potential candidates for drug delivery vehicles. Hydrogels showed good cytocompatibility while exhibited a pH depended degradation suitable for tumor therapy and release of drugs like doxorubicin. A number of these studies use small benzaldehyde crosslinkers, such as bi-, tri-, or tetra-functional PEGs, however, fewer reports exist on the use of high molecular weight crosslinkers with higher degrees of functionality.37–39 In one example, Cao et al.,39 used anionic polymerization to synthesize a multi-benzaldehyde functionalized PEG analogue crosslinker to form injectable hydrogels after in situ reaction with glycol chitosan. Hydrogels showed controlled gelation times and long term degradation profiles important for the encapsulation of chondrocytes for cartilage tissue applications. However, the three-step process that was followed for the synthesis of the crosslinker was laborious while the self-healing properties of the hydrogels were not assessed.
Controlled radical polymerization techniques have been widely used for the synthesis of advanced materials with narrow molecular weight distributions (MWDs) and controlled molecular weight and architectures.40–42 Among them, copper-mediated reversible deactivation radical polymerization (Cu-RDRP) has been established as a versatile method for the preparation of well-defined polymers with a broad monomer and solvent compatibility.43–46 Specifically, aqueous Cu-RDRP based on the rapid disproportionation of Cu(I)Br/Me6TREN at low temperatures, is an efficient, simple and fast radical polymerization technique that can offer excellent control over molecular weight and well defined water-soluble polymers at various architectures in <10 min.47–50
In this present study we introduce a faster way to access well-defined amphiphilic benzaldehyde modified crosslinkers using as precursors ABA telechelic copolymers from a simple, environmental and fast polymerization technique in water. Dynamic hydrogel systems are then formulated utilizing the water soluble glycol chitosan (GC) as a source of amine functionality. More specifically, hydrogels were formed from the in situ Schiff base reaction between the amines of glycol chitosan and a novel ABA telechelic crosslinker with multiple pendent benzaldehyde groups, p(DMAcx-co-CHOy)-b-PEG2000-b-p(DMAcx-co-CHOy). A narrow dispersity crosslinker was desired in order to have greater control over the functionality and molecular weight. For the crosslinker synthesis, a two-step procedure was followed, initially using aqueous Cu-RDRP to produce the telechelic copolymer p(DMAcx-co-HEAmy)-b-PEG2000-b-p(DMAcx-co-HEAmy) followed by post-polymerization modification by carbodiimide esterification to attach 4-formyl benzoic acid moieties into the polymer chains. An important difference of our multi-functional crosslinker compared to lower functionality benzaldehyde ones is its ability to self-assemble in water opening future possibilities for drug delivery of more hydrophobic drugs with higher loading capabilities. However, this current study focuses on the evaluation of the materials properties and their comparison with other similar systems. Rheological assessment of the hydrogels demonstrated self-healing properties due to the reversibility and dynamic exchange of imine bonds while the results were also compared with those obtained from a bifunctional PEG one. At a final stage, an attempt to enhance the mechanical properties of the gels was made through reinforcement with GO nanofillers. Our idea was to introduce nanofillers as partially reduced nanocomposites from glycol chitosan. For that reason, a mild technique for the reduction of GO was followed leading to GC/GO suspensions at two different concentrations of GO (0.5 and 2.0 wt%). Characterization of GC/GO nanocomposites via various analytical techniques proved the partial reduction of GO nanosheets while their incorporation in the hydrogels was assessed showing certain enhancements on the self-healing ability (>99% recovery) and the mechanical properties compared to the pristine hydrogels.
Results and discussion
Synthesis and characterization of ABA telechelic copolymer crosslinker
Inspired by previously reported benzaldehyde-terminated PEG crosslinkers,35,39,51 we wanted to investigate the effect of a high benzaldehyde functionality crosslinker on the properties of imine based hydrogels. A prerequisite for the design of the multi-functional crosslinker was to be structurally comparable with a benzaldehyde-terminated PEG crosslinker while simultaneously being able to form assemblies in water due to our desire to investigate this effect on the hydrogel properties. Forming an ABA telechelic copolymer structure seemed to serve both purposes, as it would be quite similar with the structure of a bifunctional PEG crosslinker, making the comparison feasible, while the choice of right chemistry would enable self-assembly in water working as potential nanocarrier for hydrophobic substances in the future.14,52–54 Thus, a narrow dispersity ABA telechelic copolymer crosslinker with a predetermined number of benzaldehyde groups in the A blocks and PEG as the B block was synthesized in a two-step process. Choosing PEG as the B block didn’t only resemble the bifunctional PEG crosslinker but PEG moieties are well known for their antifouling and antimicrobial properties important when non-specific interactions with proteins or cells are needed.55–57 In addition, the high mobility and flexibility of the PEG chains would be expected to be advantageous for the formation of the hydrogels increasing the diffusivity and mobility of the multi-functional crosslinker throughout the matrix something important for the self-healing characteristics.Firstly, a bifunctional PEG initiator was used to synthesize a copolymer of N,N-dimethyl acrylamide (DMAc) and N-hydroxyethyl acrylamide (HEAm) from aqueous Cu-RDRP, while in the second step the Steglich esterification via N,N′-dicyclohexylcarbodiimide (DCC) chemistry was used to couple 4-formyl benzoic acid to the free hydroxyl groups of HEAm (Fig. 1a). Our choice to copolymerize with DMAc (a highly water soluble monomer) arose from our previous attempts to solely synthesize the A blocks out of benzaldehyde groups leading to undesired limited micellar stability and precipitation of the crosslinker in water.
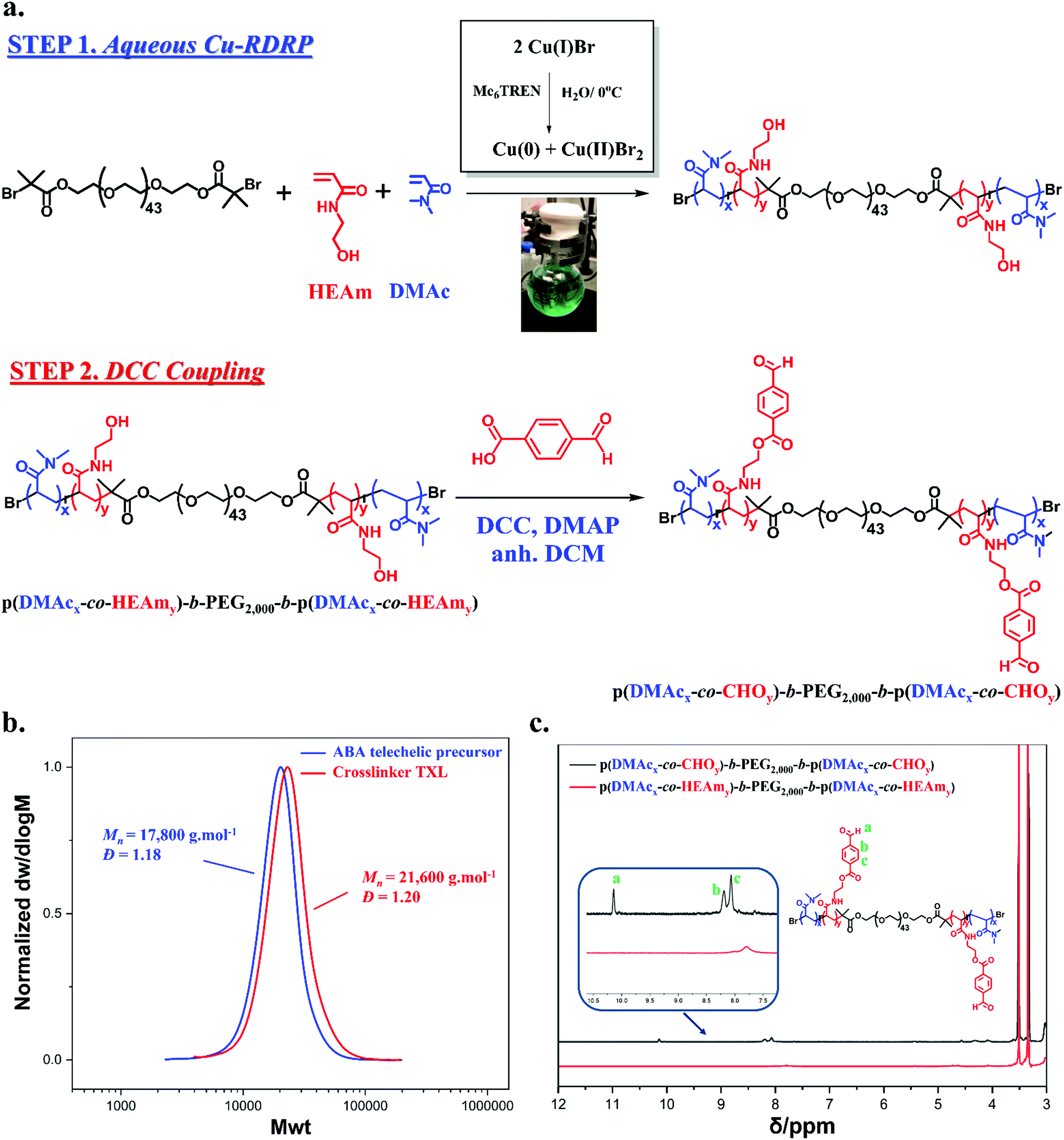 | ||
| Fig. 1 (a) Schematic of the synthesis of the ABA telechelic copolymer by aqueous Cu-RDRP and subsequent modification to crosslinker C. (b) Molecular weight distribution traces of the ABA telechelic copolymer before and after modification (DMF SEC). (c) 1H-NMR (500 MHz, DMSO-d6) spectrum of synthesized crosslinker highlighting the incorporation of benzaldehyde moieties in the polymer chains. | ||
The bifunctional water soluble initiator was synthesized from α,ω-dihydroxy polyethylene glycol (PEG, Mn = 2000 g mol−1) and characterized by NMR, FT-IR and MALDI-ToF MS (Fig. S1–S4, ESI†). This initiator was then used for the copolymerization of DMAc and HEAm by aqueous Cu-RDRP to form the telechelic p(DMAcx-co-HEAmy)-b-PEG2000-b-p(DMAcx-co-HEAmy). The reaction was conducted using a reagent ratio of [Cu(I)Br]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Me6TREN] = [0.4]:[0.4]. Polymerization reached high conversions in 30 min (>95% conversion for both monomers calculated by 1H-NMR; see Fig. S20, ESI†) while targeting a degree of polymerization of DPn,total = 80 at a molar ratio of DMAc to HEAm = 88:12 to ensure the solubility of the ABA crosslinker in water after the DCC coupling step. As shown in Table 1, SEC analysis of the telechelic copolymer gave a narrow molecular weight distribution (Đ = 1.18), however, a difference between the values of Mn,th and Mn,SEC was observed. This discrepancy was attributed to the calibration of the instrument with commercial PMMA calibration standards. Further molecular weight analysis was achieved by 1H-NMR through integration of the –CH2 groups (δ = 3.45–3.63 ppm), incorporated through the initiator (a, Fig. S5 and S11, ESI†), against the proton of the –OH groups (δ = 4.54–4.86 ppm) (j, Fig. S5 and S11, ESI†) to calculate DPn,p(HEAM) and those of –CH3 (δ = 2.66–3.10 ppm) to calculate DPn,p(DMAc) (m, Fig. S5 and S11, ESI†). 1H-NMR calculations gave a molecular weight close to the theoretical value (Mn,NMR = 9800 g mol−1). Assignment of the 1H-NMR and 13C-NMR of the polymer showed the desired product had been synthesized while DOSY NMR provided additional evidence for the copolymerization of the two monomers due to the overlapping of their diffusion coefficients (Fig. S7, ESI†).
:[Me6TREN] = [0.4]:[0.4]. Polymerization reached high conversions in 30 min (>95% conversion for both monomers calculated by 1H-NMR; see Fig. S20, ESI†) while targeting a degree of polymerization of DPn,total = 80 at a molar ratio of DMAc to HEAm = 88:12 to ensure the solubility of the ABA crosslinker in water after the DCC coupling step. As shown in Table 1, SEC analysis of the telechelic copolymer gave a narrow molecular weight distribution (Đ = 1.18), however, a difference between the values of Mn,th and Mn,SEC was observed. This discrepancy was attributed to the calibration of the instrument with commercial PMMA calibration standards. Further molecular weight analysis was achieved by 1H-NMR through integration of the –CH2 groups (δ = 3.45–3.63 ppm), incorporated through the initiator (a, Fig. S5 and S11, ESI†), against the proton of the –OH groups (δ = 4.54–4.86 ppm) (j, Fig. S5 and S11, ESI†) to calculate DPn,p(HEAM) and those of –CH3 (δ = 2.66–3.10 ppm) to calculate DPn,p(DMAc) (m, Fig. S5 and S11, ESI†). 1H-NMR calculations gave a molecular weight close to the theoretical value (Mn,NMR = 9800 g mol−1). Assignment of the 1H-NMR and 13C-NMR of the polymer showed the desired product had been synthesized while DOSY NMR provided additional evidence for the copolymerization of the two monomers due to the overlapping of their diffusion coefficients (Fig. S7, ESI†).
:[Cu(I)]:[Me6TREN] = [1]:[0.4]:[0.4] targeting a DPn,total = 80 at a molar ratio of DMAc to HEAm = 88:12
| Entry | M n,SEC (g mol−1) | M n,th (g mol−1) | M n,NMR (g mol−1) | Đ | DPp(DMAc),NMR | DPp(HEAm),NMR | DPp(CHO),NMR |
|---|---|---|---|---|---|---|---|
| a Determined by SEC analysis in DMF compared to PMMA narrow molecular weight standards. b Calculated for 95% conversion of DMAc and 97.5% conversion of HEAm. | |||||||
| Copolymer | 17800 |
10000b |
9800 | 1.18 | ∼73 | ∼10 | — |
| TXL | 21600 |
12000b |
11400 |
1.20 | ∼67 | — | ∼10 |
| Bi-PEG4000 | 9500 | 4260 | 4260 | 1.07 | — | — | ∼2 |
N,N′-Dicyclohexylcarbodiimide (DCC coupling) was then used to esterify the free hydroxyl groups of HEAm with 4-formyl benzoic acid. Consequently, a narrow dispersity p(DMAcx-co-CHOy)-b-PEG2000-b-p(DMAcx-co-CHOy) crosslinker was synthesized bearing a defined number of benzaldehyde groups, determined by the initial molar ratios of the monomers in a statistical fashion. The final crosslinker retained its narrow dispersity after modification (Đ = 1.20), with a small shift to higher molecular weights according to SEC (Table 1 and Fig. 1b). The incorporation of benzaldehyde groups was confirmed by both 1H and 13C-NMR from the presence of the characteristic aromatic protons at δ = 8.05 and 8.18 ppm and the aldehyde proton at δ = 10.13 ppm (Fig. 1c and Fig. S8, ESI†). Additional DOSY data confirmed the functionalization of the copolymer with aromatic groups as well as the UV250nm trace of the SEC (Fig. S13, ESI†). The number of functional groups was estimated by 1H-NMR by the integration of the –CH2 groups (δ = 3.45–3.63 ppm), incorporated through the initiator, against the aromatic protons –CH– at δ = 8.05 and 8.18 ppm (Fig. S12, ESI†). As a result, a high molecular weight crosslinker with ∼10 benzaldehyde groups per chain was synthesized (Mn,NMR = 11400 g mol−1) referred to as “TXL”.
In addition, a lower molecular weight PEG4000 based crosslinker was also synthesized bearing on average two benzaldehyde groups per polymer chain (Bi-PEG4000). Similar crosslinkers have been previously utilized to prepare self-healing hydrogels after mixing with aqueous solutions of glycol chitosan,35,58,59 hence it was used as a comparison model for our telechelic copolymer crosslinker. DCC coupling was again used to esterify the terminal hydroxyl ends of PEG4000. The crosslinker was efficiently synthesized as confirmed by 1H-NMR, 13C-NMR (Fig. S17a, b and S18, ESI†) and by MALDI-ToF MS (Fig. S16c, ESI†) with complete disappearance of the terminal hydroxyl groups after modification (>99% modification) and a narrow dispersity (Đ = 1.07) according to SEC (Table 1). The incorporation of benzaldehyde groups was additionally confirmed by the UV280nm trace of the SEC which overlapped with the RI signal (Fig. S16b, ESI†).
Formation and characterization of GC/TXL(x) hydrogels
We first investigated the formation and properties of dynamic hydrogels by mixing the synthesized crosslinker TXL with glycol chitosan (GC). GC/TXL(x) (where x denotes the molar ratio –CHO/–NH2) hydrogels were formed by homogeneously mixing aqueous solutions of GC with the ABA telechelic crosslinker at room temperature and pH = 7.4. Transparent hydrogels were observed after few minutes of mixing (Fig. 2a), arising from the reaction between the amine groups of the GC backbone and the aldehyde groups of the crosslinker to form a reversible imine bond network in the material. FT-IR was used to monitor the progress of the in situ crosslinking reaction. The magnitude of the band at 1723 cm−1, attributed to the carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the benzaldehyde, was reduced after mixing, indicating the occurrence of a Schiff base reaction between the aldehyde and the amine groups of glycol chitosan (Fig. 2c).
O) of the benzaldehyde, was reduced after mixing, indicating the occurrence of a Schiff base reaction between the aldehyde and the amine groups of glycol chitosan (Fig. 2c).
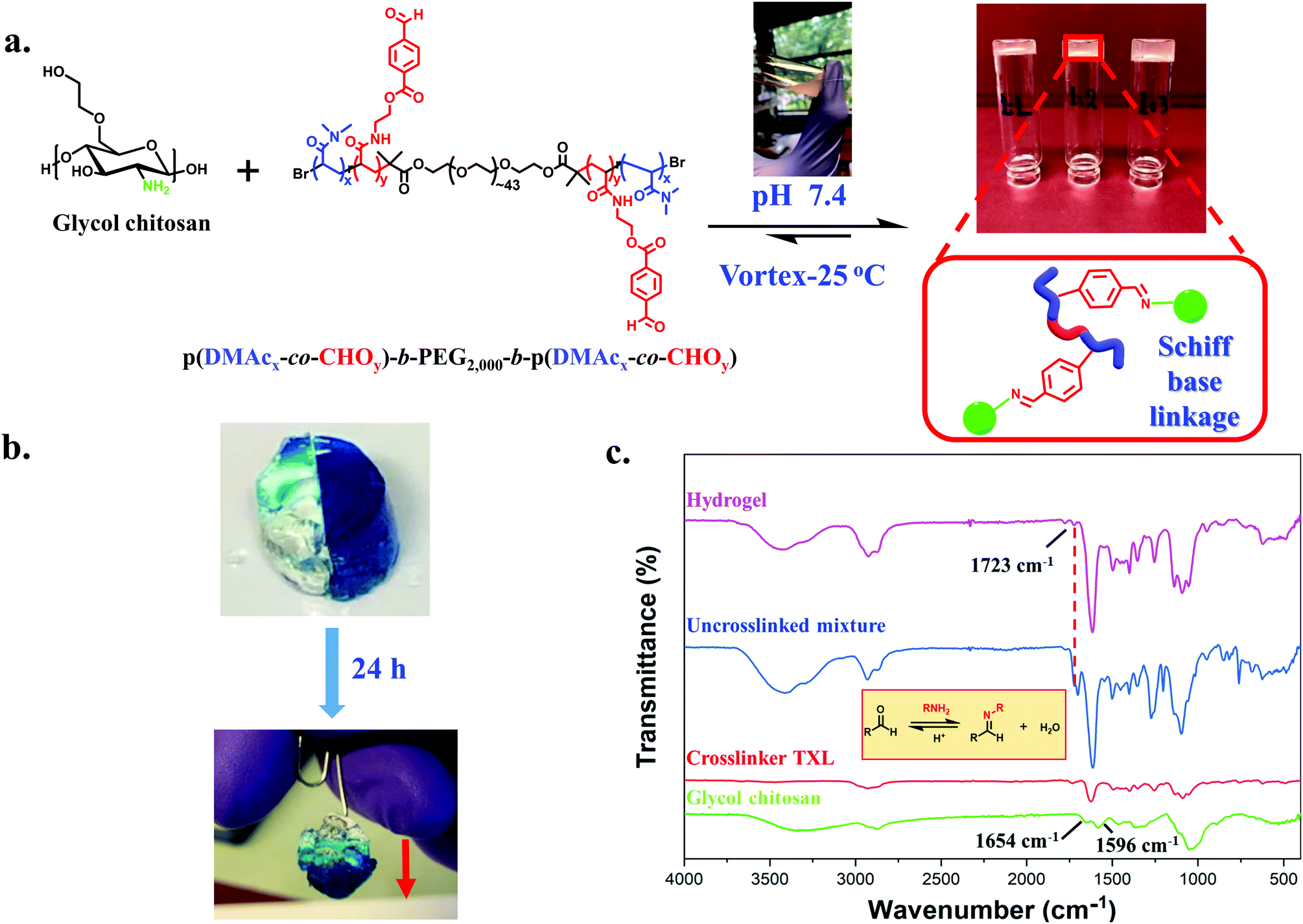 | ||
| Fig. 2 (a) Formation of hydrogels via Schiff base linkages after mixing GC with TXL crosslinker. (b) Photographs illustrating the combination of two separate peaces of gels and the healed gel after 24 h. (c) Combined FT-IR spectrums of GC, TXL crosslinker, uncrosslinked hydrogel mixture and formed hydrogel proving the formation of imine bonds. | ||
Hydrogels with different molar ratios (–CHO/–NH2) and solid contents (wt%) were formed to assess the impact of the crosslinker on their gelation kinetics and viscoelastic properties. As a relatively high molecular weight polymer (Mn,NMR = 11400 g mol−1), compared to frequently used smaller crosslinkers, the chains were expected to have slower diffusion inside the hydrogel matrix that could affect both crosslinking and dynamic exchange of bonds, which are important for directing the self-healing characteristics.4,60,61
In order to assess the gelation process of the systems, gelation kinetics were conducted by rheology at 25 °C using a time sweep mode (Fig. S22, ESI†). A kinetic study indicated that gelation time was reduced by increasing the molar ratio of (–CHO/–NH2), as more free benzaldehyde groups were introduced to the mixture resulting in faster crosslinking and solidification. More specifically, the formulation with a molar ratio of 0.03 (GC/TXL(0.03)) gelled within ∼5–6 min while GC/TXL(0.32) in less than ∼10 s, as indicated by the crossover point of the storage (G′) and loss moduli (G′). The time-dependence of gelation was also demonstrated by the trends of the phase angle values (tanδ = G′′/G′) which in every case came in good agreement with the G′ and G′′ curves. At small time intervals, essentially prior gelation, tanδ values were higher than 1 indicative of viscous materials. However, after gelation (tanδ = 1) a descending profile was observed with tanδ values approaching 0, characteristic of perfectly formed elastic materials. In the case of GC/TXL(0.32), tanδ values were early enough below 1 due to fast gelation kinetics.
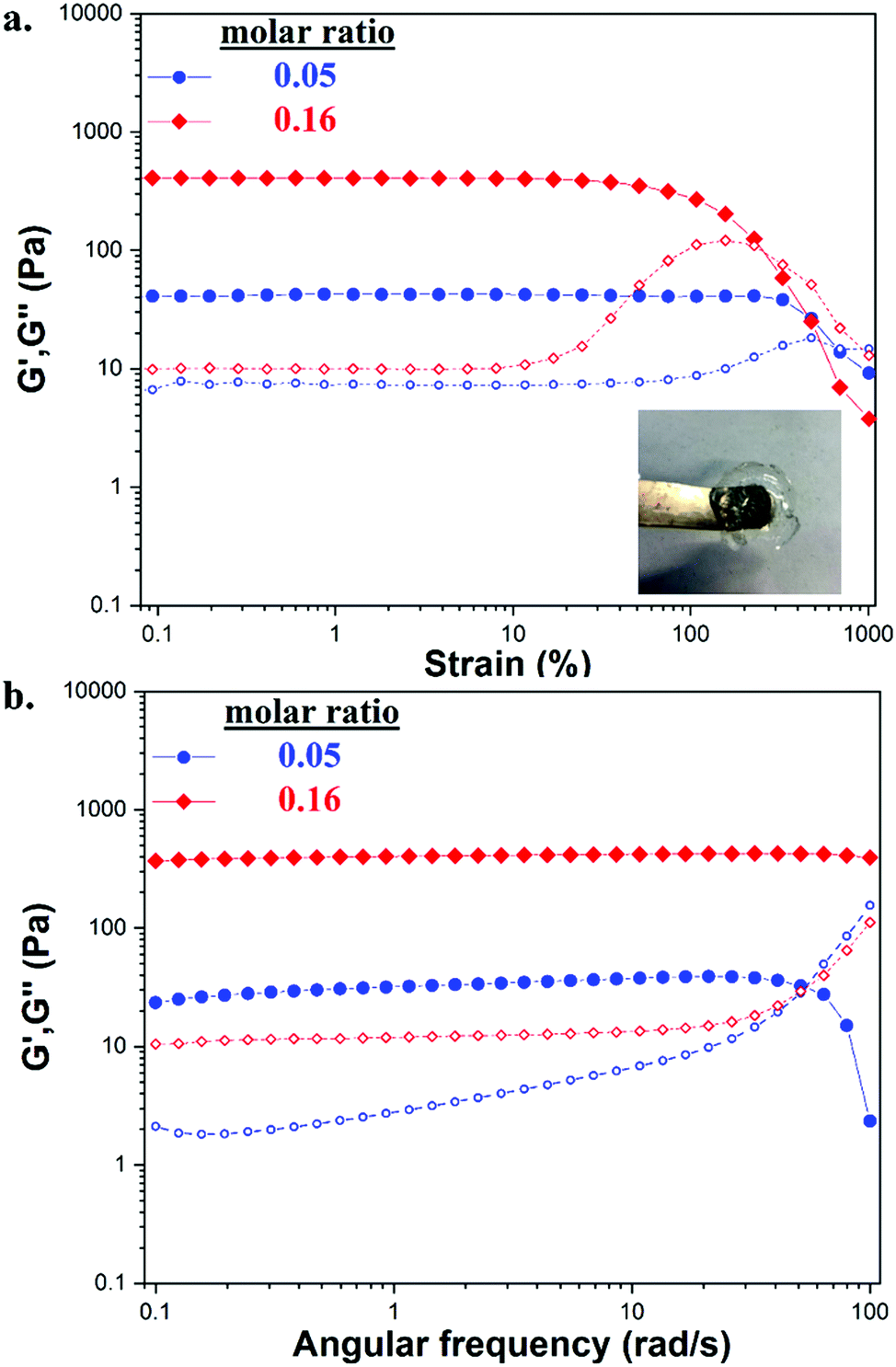
The strength and viscoelastic properties of the hydrogels, using crosslinker TXL, were evaluated by oscillatory rheology at 25 °C by monitoring the progress of their storage (G′) and loss (G′′) moduli. Amplitude sweeps were performed at three different molar ratios (0.05, 0.16 and 0.32), at a constant frequency of ω = 10 rad s−1 to find the linear viscoelastic regions (LVE) (Fig. 3a). Gels formed with molar ratios >0.32 were found to be too brittle to record data, thus measurements were limited to the three stated molar ratios. The crossover points of G′ and G′′ indicated the strain (%) at which the materials begin to flow. Gels tended to lose their elasticity at lower strains (%) by increasing the amount of crosslinker, demonstrating that stiffer materials were not able to withstand high levels of strain. A strain at break of 250% was found for GC/TXL(0.05) formulations which dropped at 116% when testing a slightly higher ratio, GC/TXL(0.16), and finally reaching a value of 70% for the formulations with the highest molar ratio, GC/TXL(0.32). Despite these differences, G′ values remained constant between γ = 0.1–10% indicating a useful strain region to examine the frequency dependent properties.
 | ||
| Fig. 3 (a) Amplitude sweep experiments of GC/TXL(x) hydrogels at 25 °C and at a constant frequency of ω = 10 rad s−1 for three molar ratios (0.05, 0.16, and 0.32). (b) Frequency sweep experiments of GC/TXL(x) hydrogels at 25 °C and at a constant strain of 5.0% for three molar ratios (0.05, 0.16, and 0.32). In all occasions, storage modulus (G′, filled symbols) and loss modulus (G′′, plane symbols). | ||
Frequency sweep experiments were conducted at 25 °C in the angular frequency range of ω = 0.5–100 rad s−1 while keeping a constant strain of γ = 5.0%, as indicated from the measured LVE regions (Fig. 3b). The G′ values for all examined molar ratios seemed to be independent of frequency changes. Hydrogels with higher concentrations of crosslinker demonstrated higher G′ values suggesting that a more rigid crosslinked network was formed due to an increase in the crosslinking density. The lowest G′ value, 119 ± 15 Pa, was obtained from the hydrogel with 0.05 molar ratio. When the molar ratio was 0.16, G′ increased at 231 ± 53 Pa while at 0.32 the hydrogel reached a value of 460 ± 72 Pa. The dynamic character of this system was observed by following the independence of the G′′ versus the frequency alternations, a characteristic that ‘static’ covalent networks do not possess.63,64 The G′ values of the same materials were also compared against their tanδ values during the frequency sweep process, Fig. S29a (ESI†). An increase in the crosslinker content resulted to smaller tanδ values indicating the formation of more elastic materials with lower dumping properties under applied load. In any case, tanδ values remained relative constant during the frequency tests being below 0.2 while showing a weak increase at higher frequencies.
The viscoelastic properties of GC/TXL(x) hydrogels were then compared with those prepared using the synthesized Bi-PEG4000 crosslinker, GC/Bi-PEG4000(x) at 25 °C. Comparisons between the two types of gels were made in terms of same aldehyde to amine ratio (–CHO/–NH2), as shown in Table 2. Initially, amplitude sweep experiments were performed at a constant frequency of ω = 10 rad s−1 to examine the LVE regions (Fig. 4a). GC/Bi-PEG4000(0.05) were found to have lower G′ values and withstand significantly higher strains prior to flow (690%), when compared to GC/TXL(0.05) hydrogels. An increase in the molar ratio for GC/Bi-PEG4000(0.16) gels resulted in a lowering of the crossover point to 230%, however, this was still higher than the values recorded from the GC/TXL(0.16) gels, formed using the multifunctional crosslinker at the same (–CHO/–NH2) molar ratio.
| Entry | Molar ratio (–CHO/–NH2)a | Volume ratio (GC/C(x)) | Solid content (wt%) | Storage modulus (G′) (Pa) | Loss modulus (G′′) (Pa) | Crossover point (γ %) |
|---|---|---|---|---|---|---|
| a The free amine groups were quantified by the ninhydrin assay.62 There are 2.82 ± 0.5 mmol of free amine groups in 1 g of glycol chitosan (see assay data in Fig. S21 and Table S3, ESI). | ||||||
| GC/TXL(0.05) | 0.05 | 10 | 3.2 | 119 ± 15 | 9.7 ± 15 | 250 |
| GC/TXL(0.16) | 0.16 | 3.3 | 3.5 | 213 ± 53 | 6.3 ± 2.2 | 116 |
| GC/TXL(0.32) | 0.32 | 1.6 | 3.8 | 460 ± 72 | 35 ± 13 | 70 |
| GC/Bi-PEG4000(0.05) | 0.05 | 5.3 | 3.3 | 34 ± 9.0 | 2.7 ± 0.8 | 690 |
| GC/Bi-PEG4000(0.16) | 0.16 | 1.7 | 3.7 | 403 ± 84 | 5.4 ± 0.4 | 230 |
 | ||
| Fig. 4 (a) Amplitude sweep experiments of Bi-PEG4000 hydrogels at 25 °C and at a constant frequency of ω = 10 rad s−1 for two molar ratios (0.05 and 0.16). (b) Frequency sweep experiments of Bi-PEG4000 hydrogels at 25 °C and at a constant strain of 5.0% for two molar ratios (0.05 and 0.16). In all occasions, storage modulus (G′, filled symbols) and loss modulus (G′′, plane symbols). | ||
Frequency sweep experiments in the angular frequency range of ω = 0.1–100 rad s−1 and at a constant strain of γ = 5.0% (Fig. 4b), showed that the GC/Bi-PEG4000(0.05) gels had significantly lower G′ values (34 ± 9.0 Pa) compared to the GC/TXL(0.05) formulations. Although the two formulations contained same concentration of aldehyde and amine functionalities, the benzaldehyde groups analogy per polymer crosslinker chain was different (i.e. 2 benzaldehydes per Bi-PEG4000 chains and ∼10 per TXL) potentially causing this G′ variation at low molar ratios. In contrast, a ∼2.0 fold increase in the G′ value (403 ± 84 Pa) was observed for the GC/Bi-PEG4000(0.16) formulations compared to the GC/TXL(0.16) gels which was unexpected based on the previous results. Data could not be recorded from GC/Bi-PEG4000(0.32) gels, as they were too brittle.
These results show that the rate of G′ increase was dramatically faster with increasing Bi-PEG4000 crosslinker, relative to increasing TXL. A possible reason for this could be the faster diffusion of the lower molecular weight Bi-PEG4000, allowing it to better distribute throughout the GC matrix prior to the formation of imines in contrast to the expected slower diffusion of TXL. However, the lower G′ values could also be attributed to the self-assembly properties of TXL in water which was a major difference to the Bi-PEG4000. As previously discussed, TXL crosslinker is expected to self-assemble in aqueous media due to its amphiphilic character after functionalization with benzaldehyde. The aromatic moieties introduce hydrophobicity, while the hydrophilic PEG block (B) and DMAc moieties would organize the polymer chains into assemblies limiting the interactions of the amine groups with the aldehydes hidden in the core.
To further elaborate this, a second ABA crosslinker (TXL′) with lower degree of functionality (DPp(CHO),NMR = ∼3 per polymer chain) was synthesized and compared with TXL (Fig. S14 and Table S2, ESI†). Our goal was to demonstrate that lowering the benzaldehyde functionality (less amphiphilic) can lead to smaller or no assemblies thus increasing the G′ values of the hydrogels due to more available interactions with the amines. To investigate that, transmission electron microscopy (TEM) was initially employed showing micellar structures of ∼350 nm when dispersing TXL crosslinker in water and much smaller ones of ∼30 nm for TXL′ (Fig. S15a and b, ESI†). Hydrogels were then casted at a molar ratio (–CHO/–NH2) of 0.05 using crosslinker TXL′, GC/TXL′(0.05). The amplitude and frequency sweeps of these hydrogels (Fig. S23a and b, ESI†) showed an increase in the G′ values (from 119 ± 15 to 375 ± 13 Pa) compared to GC/TXL(0.05) formulations proving a possible correlation between the way the crosslinker assembles and its interactions with the amines which eventually affect the crosslinking density. The above experiment also proved us that we can tune the hydrogel properties by changing the degree of functionality of the crosslinker which is useful according to a future application. The self-healing properties of GC/TXL(0.05) are exhibited in Fig. 2b. A number of GC/TXL(0.05) hydrogels were coloured using Brilliant blue dye while others were left transparent. Two hydrogel samples of different colours were cut in half and recombined by applying a small force by hand and then leaving at room temperature. After 24 h, the two pieces merged due to the dynamic exchange of imine bonds, restoring the cracked surface and combining the two pieces into one gel which could be lifted without tearing apart under gravitational force. The macroscopic self-healing time of the system was relatively long showing no expected differences due to the use of a higher functionality crosslinker something attributed to the slow mobility of the chains. The same process was repeated but this time a small droplet of 10−4 M NaOH (pH = 10) was added between the two fractured surfaces. As shown from the images (Fig. S24, ESI†), in just 5 h the hydrogels rapidly healed and a visible wet surface was observed, where the increase of pH caused base assisted imine hydrolysis, thereby lowering the crosslinking density. In contrast, gels having no NaOH did not self-heal within 5 h. Attempts to prepare hydrogels at more basic conditions led to no gelation, most likely due to a lack of protons required to catalyse Schiff base reactions.
Formation and characterization of GC/TXL(x)–GO(y) dynamic nanocomposite hydrogels
Chitosan has been previously reported to interact with the surface of GO, under certain conditions, reducing it in stages depending on the concentration and the pH of the medium.65–67 Our main goal was to partially reduce GO, keeping a balance between its reduced form and surface oxygen functionality. A fully reduced GO could be expected to have low aqueous dispersibility and limited interactions with the network components consequence of the loss of majority of its oxygen functionality crucial for the formation of non-covalent bonds and aqueous stability.68 In contrast, a partially reduced GO by GC, in our case, would potentially strengthen the hydrogel network as the free amine groups of the GC would be both interacting with the GO surface and the synthesized ABA crosslinker (through dynamic bonds) causing possible enhancements in the mechanical properties of the materials.To ensure the partial reduction of GO, we avoided very strong reducing agents such as ascorbic acid or hydrazine. Instead, we mixed glycol chitosan with graphene oxide under milled conditions in order to induce interactions, which could be either electrostatic forces and hydrogen bonding or covalent bonds between the functional groups of GC (amines and hydroxyls) and those found on the surface of GO (epoxides, carboxyls and hydroxyls) (Fig. 6a). Thinking back to our network, the preparation of hydrogels occurred at a pH = 7.4 which was also a suitable medium to prepare stable dispersions of GO in water (ζ < −50 mV, Konkena et al.68). GC and GO were mixed for 72 h at 37 °C to form dispersed GC/GO(y) (y being the wt% of GO added in the mixture) nanocomposites. GO was added at two concentrations, 0.5 and 2.0 wt%, and then the nanocomposite products were dried and further characterized.
UV-Vis absorption spectra of the isolated GC/GO(0.5) and GC/GO(2.0) nanocomposites were obtained (Fig. 5a). Pristine GO showed an absorption peak at 230 nm attributed to π → π* transitions of CC bonds and a shoulder peak at 300 nm arising from the n → π* transitions of the carbonyl groups CO located on its surface. For both GC/GO(0.5) and GC/GO(2.0), the absorption peaks of CC bonds showed a bathochromic shift to higher wavelengths (252 and 242 nm respectively) indicating restoration of conjugation due to removal of oxygen containing groups by GC hence leading to lower energy transitions.69–71 The carbonyl groups CO peak at 300 nm, remained visible after 72 h indicating that oxygen functional groups were still present on the surface of GO even after partial reduction from GC.
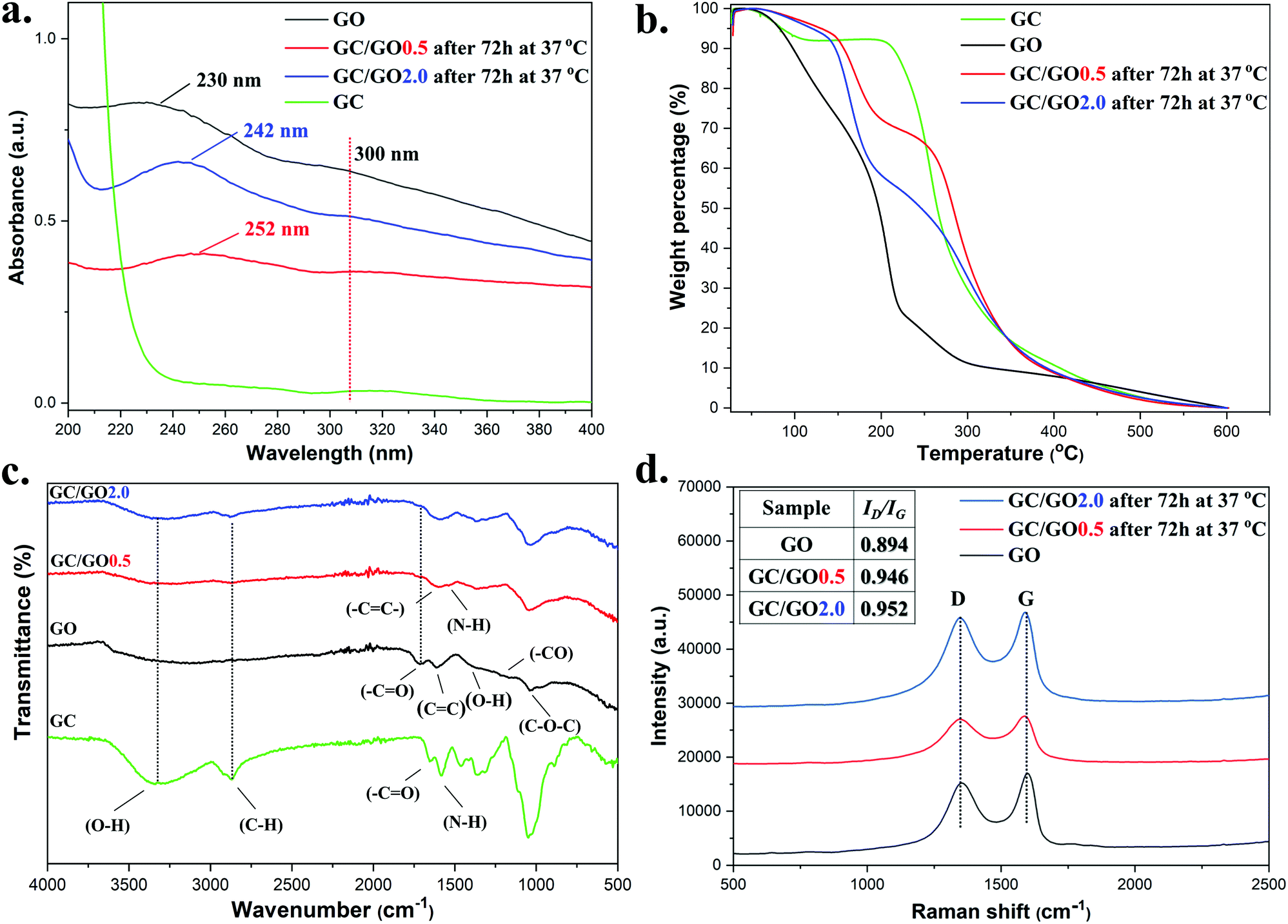 | ||
| Fig. 5 (a) UV-vis absorption spectra of GC/GO(0.5) and GC/GO(2.0) after 72 h at 37 °C, GO and GC at 0.1 mg mL−1. (b) TGA of GC/GO(0.5) and GC/GO(2.0) after 72 h at 37 °C, GO and GC. (c) FT-IR spectra of GC/GO(0.5) and GC/GO(2.0) after 72 h at 37 °C, GO and GC and (d) Raman spectra of GC/GO(0.5) and GC/GO(2.0) after 72 h at 37 °C and GO. | ||
Thermogravimetric analysis (TGA) was used to characterize the GC/GO(y) materials (Fig. 5b). For comparison, the thermogravimetric profile of GC was obtained showing a 5.0% mass loss at ∼83 °C, attributed to adsorbed water, and a rapid 56% mass loss at ∼258 °C due to GC's backbone degradation.72 The thermal profile of GO contained more degradation stages beginning with a 14% mass loss at ∼100 °C due to adsorbed water. Then, a rapid 23% mass loss at the range of 158–230 °C was observed attributed to the removal of oxygen-containing groups releasing CO and CO2, following a 6.0% mass loss between 250–314 °C from the further removal of oxygen-containing groups. Compared with GO, the mass loss in this temperature range dropped from 23% to 13% proving that GC/GO(0.5) lost part of its oxygen functionality due to the chemical interaction with GC and the restoration of the π conjugated system. A further mass loss of 32% at ∼284 °C was finally attributed to glycol chitosan and the remainder of the carbon skeleton of GO. GC/GO(2.0) also showed a mass loss at the range of 61–230 °C which was slightly smaller (22.5%) than that of GO, indicating the presence of more oxygen containing functional groups than GC/GO(0.5), in agreement with the UV-vis data. First derivative graphs of all thermal curves are provided in Fig. S26 (ESI†).
Fig. 5c illustrates the FT-IR spectra of GC, GO and prepared GC/GO(0.5) and GC/GO(2.0) nanocomposites. For GC the broad band at 3345 cm−1 was mainly attributed to –OH stretching vibrations while the peak at 2868 cm−1 was attributed to C–H stretching. At 1581 cm−1, the N–H bending was observed together with the C–N stretching at 1318 cm−1. For GO, the peaks at 1720, 1205, 1394 and 1042 cm−1 correspond to the –COOH, C–O epoxy, C–OH and C–O–C groups respectively that existed on the surface of the GO nanosheets. In addition the peak at 1613 cm−1 was caused by CC proving the existence of some sp2 character. Similarly to previous reported literature,66,73,74 for both spectra of GC/GO(0.5) and GC/GO(2.0) nanocomposites, the carbonyl groups (CO) located on the surface of GO nanosheets at 1720 cm−1, showed a significant reduction in intensity indicating loss of oxygen functionality from the surface of the GO nanosheets through possible reaction with part of the free amine groups of GC. The C–H bond stretch of GC at 2868 cm−1 was detected at 2876 cm−1 giving a strong indication of the existence of GC on the surface of the GO nanosheets since repeated washings with water ensured the complete removal of the high water-soluble GC prior analysis. The formation of hydrogen bonding was evidenced by the downshift of the –NH groups of GC from 1581 cm−1 to 1538 cm−1 and 1542 cm−1 for GC/GO(0.5) and GC/GO(2.0) respectively, giving strong indication of hydrogen bonding interactions. In addition, the appearance of a slightly stronger band between 3000 and 3500 cm−1 was attributed to O–H stretching demonstrating a small downshifting at 3300 and 3310 cm−1 for GC/GO(0.5) and GC/GO(2.0) indicating once more hydrogen bond interactions.
Raman spectroscopy was also employed to characterize the electronic properties of the isolated nanosheets as often carried for graphene and graphite composites.75 From the Raman spectra (Fig. 5d), two characteristic absorption bands, D and G, appeared at the frequency range between 1000–2000 cm−1, as previously reported.74,76 The D band at 1347 cm−1 showed the A1g symmetry mode caused by the GO edges while the G band at 1589 cm−1 described the E2g mode of sp2 carbon atoms. Usually, the intensity ratio between the D and G bands (ID/IG) gives a good indication of the conjugation state of GO. An increase in the intensity ratio indicates smaller sp2 in-plane domains, hence restoration of the conjugated network due to reduction of the GO. According to the spectrum of pristine GO, the ID/IG was calculated equal to 0.894. However, treatment of GO with GC at 37 °C increased this value to 0.946 and 0.952 for GC/GO(0.5) and GC/GO(2.0), respectively, indicating a partial reduction of the GO nanosheets from GC at pH 7.4.
Our next step was to examine the formation of GC/TXL(x)–GO(y) hydrogels by mixing the GC/GO(y) suspensions with the synthesized ABA telechelic crosslinker (Fig. 6a). Every solution appeared to have low viscosity prior to mixing, while a sol–gel transition was observed immediately after blending, leading to black opaque hydrogels, Fig. 6b. It should be noted that when higher amounts of GO were used (>2.0 wt%) no gelation occurred either due to low availability of free amine groups or due to high steric effects that reduced interactions. To confirm the formation of hydrogels through imine bonds in the presence of GO, an experiment was conducted by dissolving a piece of gel in acetic acid (pH = 3). The sudden drop of pH fully dissolved the hydrogel after a period of 1 h, where the protonation of the amine groups led to the dissociation of the crosslinked network (Fig. S28, ESI†).
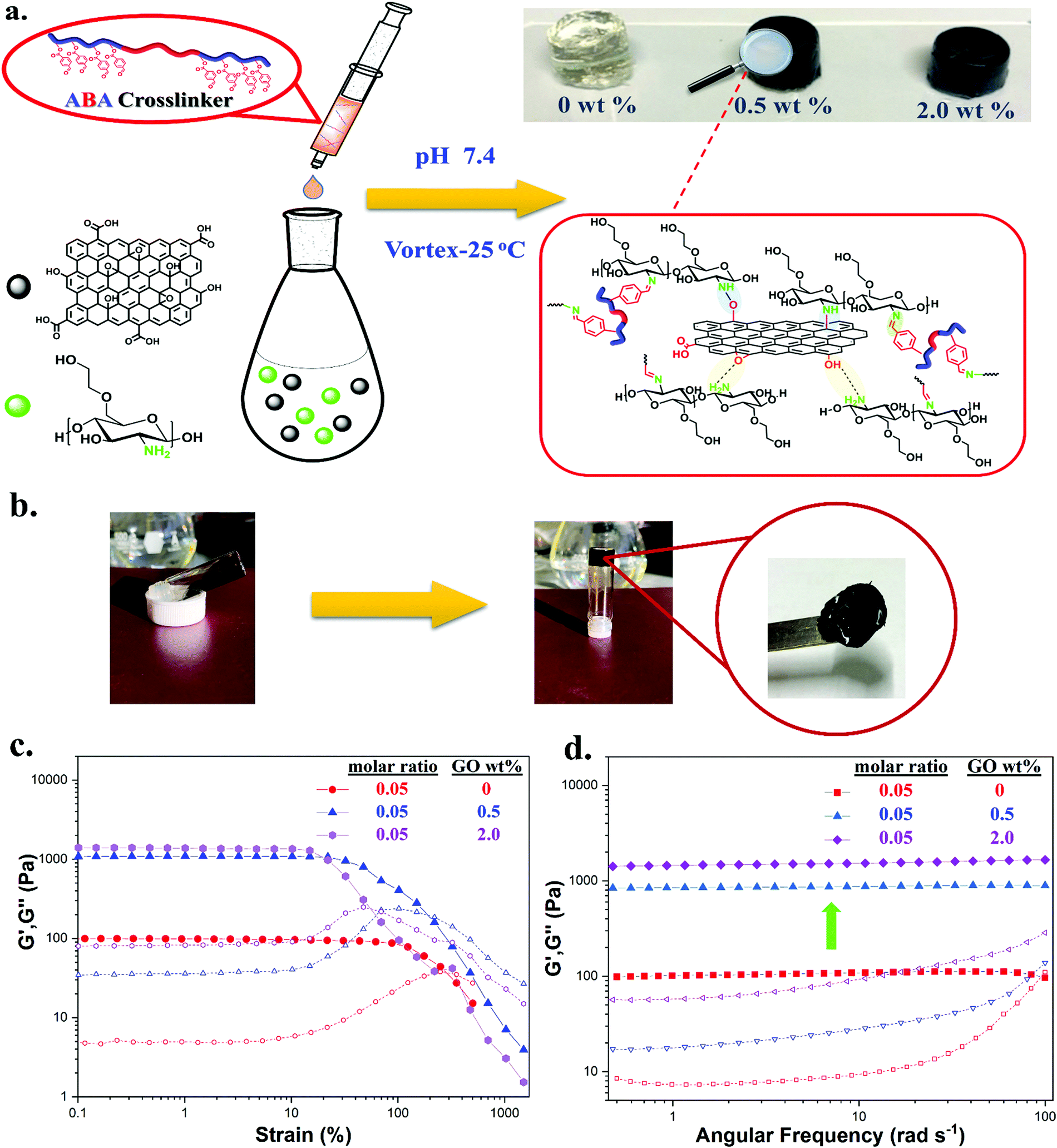 | ||
| Fig. 6 (a) Schematic illustration of the formation of free standing GC/TXL(x)–GO(y) hydrogels at a pH of 7.4. (b) Vial inversion test that shows the sol–gel transition after mixing with the appropriate amount of TXL crosslinker. (c) Amplitude sweep experiment comparisons between GC/TXL(0.05), GC/TXL(0.05)–GO(0.5) and GC/TXL(0.05)–GO(2.0) at 25 °C and at a constant frequency of 10 rad s−1. (d) Frequency sweep experiment comparisons between GC/TXL(0.05), GC/TXL(0.05)–GO(0.5) and GC/TXL(0.05)–GO(2.0) at 25 °C at a constant strain of 5.0%. For all occasions, storage modulus (G′, filled symbols) and loss modulus (G′′, plane symbols). | ||
Oscillatory sweep experiments were conducted at a constant frequency of ω = 10 rad s−1 to examine the strain dependent rheological properties of the nanocomposite hydrogels. To investigate the effect of GO, a molar ratio (between –CHO/–NH2) of 0.05 was chosen. Hydrogels withstood less strain (%) after the addition of GO compared to the pristine hydrogels of the same molar ratio. Addition of 0.5 wt% GO in GC/TXL(0.05) gels decreased their crossover point to 220% while addition of 2.0 wt% dropped it to 56%, indicating that the presence of GO makes the chains dislocate more easily from each other. Frequency sweep experiments, showed a definite increase in the crosslinking degree of the network as depicted by the G′ values. More specifically, after addition of 0.5 wt% of GO, a ∼7.0 fold rise of G′ was observed (851 ± 42 Pa) while 2.0 wt% of GO gave a ∼12 fold increase reaching the value of 1450 ± 230 Pa. Hydrogen bonds and covalent interactions between GC and GO possibly increased the junctions and crosslinking points of the network leading to higher crosslinking densities (Fig. 6a). To further understand this, the crosslinking density values, N, of GC/TXL(0.05)–GO(y) hydrogels were determined based on the James and Guth rubber elasticity theory described in eqn (1):24,77,78
| N = RT/Ge | (1) |
The tanδ values of the GC/TXL(0.05)–GO(y) nanocomposite materials gave additional insights on their energy dissipation when compared with the GC/TXL(0.05) formulations during frequency sweep oscillations, Fig. S29b (ESI†). Results indicated that addition of GO caused a small reduction on the tanδ profiles proving the more elastic response of these materials in contrast to their pristine analogue. This could be explained from the restriction of movement of the polymer chains due to the appearance of the graphene nanosheets that made the hydrogel nanocomposite store the applied load instead of dissipating it.
Evaluation of mechanical and self-healing properties
Mechanical properties were evaluated by uniaxial compression testing. The crosslinker content regulated the mechanical performance of the gels, although over a fairly narrow range (Table S4, ESI† and Fig. 7a). Hydrogels were considered soft due to their low compression modulus values at 10% deformation. By comparing the stiffness of GC/TXL(0.05) and GC/TXL(0.16) gels, no significant differences were observed due to their similar modulus values (∼23.0 ± 3.0 and ∼19.9 ± 2.6 kPa respectively). On the other hand, the stiffness of GC/TXL(0.32) gels increased by 1.2 folds (∼27.7 kPa), coming in to agreement with their viscoelastic properties (highest G′ value between the three examined pristine gels). The crosslink density between GC/TXL(0.05) and GC/TXL(0.16) although different, did not seem to alter the stiffness of the materials in contrast to GC/TXL(0.32) gels which appeared slightly stiffer due to a higher concentration of aldehyde groups in relation to amines. Regarding the strength of the pristine hydrogels, both GC/TXL(0.16) and GC/TXL(0.32) seemed to have similar strain (%) at break values (∼38.9 and ∼40.2% respectively) which were slightly higher than those of GC/TXL(0.05) (∼33.2%), showing that the higher crosslink density strengthens the gels.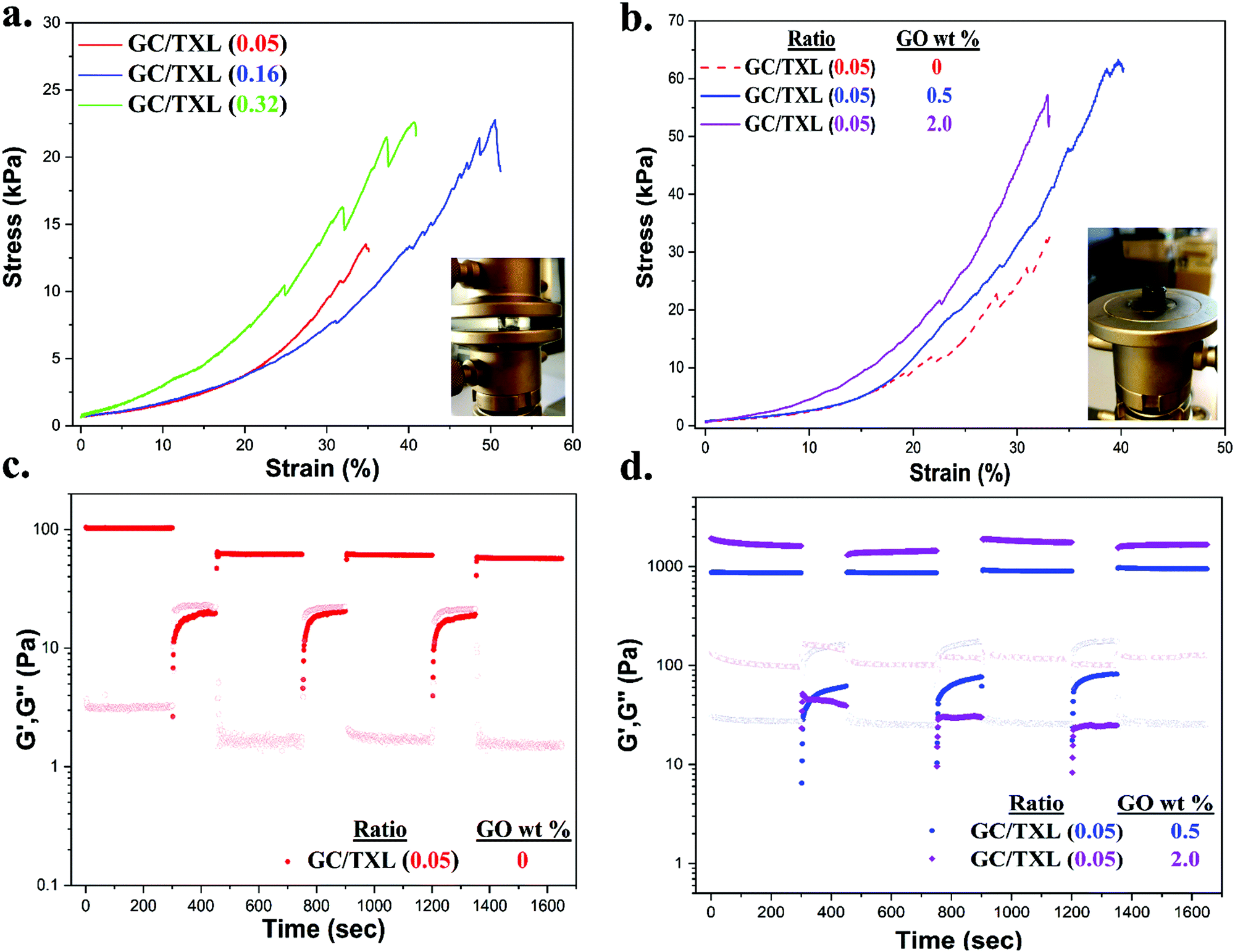 | ||
| Fig. 7 Compression tests on (a) GC/TXL(x) hydrogels at the molar ratios of 0.05, 0.16 and 0.32. (b) GC/TXL(0.05) hydrogels containing 0, 0.5 and 2.0 wt% of GO. Alternate strain sweep experiments (between 1.0% and 350% at 25 °C) for (c) GC/TXL(0.05) hydrogels containing no GO (d) GC/TXL(0.05) hydrogels containing 0.5 and 2.0 wt% of GO. For all occasions, storage modulus (G′, filled symbols) and loss modulus (G′′, fade symbols). | ||
With the addition of GO the stiffness and strength of the hydrogels increased. Compression testing on GC/TXL(0.05)–GO(0.5) gels showed a 1.5 fold increase compared to the pristine GC/TXL(0.05) analogues (∼35 kPa), while GC/TXL(0.05)–GO(2.0) exhibited a further raise in its modulus to ∼48.7 kPa (∼2.1 fold increase) (Fig. 7b). A similar trend was observed by addition of GO in GC/TXL(0.16) formulations, showing the same 1.5 fold increase in modulus for GC/TXL(0.16)–GO(0.5) (∼28.9 kPa) and ∼2.0 fold increase for GC/TXL(0.16)–GO(2.0) (∼38.8 kPa) (Fig. S30a, ESI†).
In the presence of GO, hydrogels became stronger, handling strains of ∼33.8% for GC/TXL(0.05)–GO(0.5) and ∼35.0% for GC/TXL(0.05)–GO(2.0). GC/TXL(0.16) hydrogels were found to be the strongest, withstanding strains of ∼54% when 2.0 wt% GO was present. GC/TXL(0.32)–GO(0.5) hydrogels appeared to have quite close modulus values with the other GO containing gels, with a higher strain at break comparable to GC/TXL(0.05)–GO(0.5) (∼32%) (Fig. S27b, ESI†).
The self-healing behaviour of both pristine and reinforced hydrogels was evaluated by rheology. Hydrogels were subjected in continuous step change strain sweeps for a total of seven cycles at a constant frequency of 10 rad s−1 (Fig. 7c and d). Firstly the GC/TXL(0.05) hydrogels were analyzed. At low strains (1.0% for 300 s), G′ values exceeded the G′′ confirming the formation of a stable network. When higher shear strains were applied (350% for 150 s), a sudden drop occurred for both G′ and G′′ until crossover, which indicated disruption of the network and loss of viscoelasticity (sol state). After returning to low strains (1.0%), the G′ value increased back, though stabilized at ∼60 Pa showing a 60% recovery from the initial value of ∼100 Pa. In the following cycles, these gels continued to show approximately a 60% recovery of their G′ with respect to the pristine material. Similar step strain experiments were applied to GC/TXL(0.16) hydrogels. After a period of high shear strains, its G′ value recovered to ∼170 Pa showing a self-healing efficiency of 85% (Fig. S25, ESI†). Interestingly, despite the minor descending trend of G′ during the following cycles, a ∼70% recovery was observed after seven cycles, which was higher than the one achieved for the GC/TXL(0.05) gels due to the greater availability of aldehyde groups.
The self-healing properties of GC/Bi-PEG4000 hydrogels were also investigated at the molar ratios of 0.05 and 0.16 (Fig. S19a and b, ESI†). The GC/Bi-PEG4000(0.05) hydrogels showed a ∼88% recovery after repeated cycles of low (1.0%) and high (350%) shear strain, demonstrating their good self-healing ability which was slightly higher than the hydrogels from crosslinker TXL at same molar ratio. In contrast, results seemed to be quite different in the case of 0.16 hydrogels as they demonstrated very low recoveries after high shear strain periods, which was not detected in the GC/TXL(0.16) analogues. More specifically, after the first cycle, gels demonstrated a ∼22% recovery that continued descending for the rest of the cycles reaching a final low recovery of ∼6% after 7 cycles. Hence, despite their higher G′ values, GC/Bi-PEG4000(0.16) seemed to suffer more irreversible damage in contrast to GC/TXL(0.16).
In order to investigate the microscopic effect of GO on the self-healing properties, GC/TXL(0.05) gels containing 0.5 and 2.0 wt% of GO were subjected to continuous step change strain sweeps and their self-healing characteristics were compared with the pristine ones. As illustrated from the rheological data (Fig. 7d), in both cases at low strain (1.0%) the G′ values overcame G′′ creating a plateau, indicating free standing hydrogels. When a higher shear strain (350%) was applied, a sudden decrease of the G′ and G′′ values occurred until G′′ exceeded G′ due to transition into a sol state. With the immediate application of 1.0% strain, both hydrogels had the ability to fully recover to their initial properties without any network loses (>99% self-healing efficiency) with reproducible recovery through all the examined cycles. In order to evaluate the self-healing efficiency of the nanocomposite hydrogels at a macroscopic level, cylindrical samples were casted and cut in half with a blade. Then, the cut surfaces were recombined and let self-heal overnight at room temperature. The healed gels were self-supported similarly to their pristine analogues hence analyzed by compression testing (Fig. S31 and Table S5, ESI†). The self-healing efficiency (SH%) of each hydrogel was determined from the compressive modulus at 10% strain. Both GC/TXL(0.05)–GO(0.5) and GC/TXL(0.05)–GO(2.0) formulations appeared to have lower modulus values after 24 h of healing with a calculated efficiency of ∼50% taking into consideration the modulus values of their intact analogues. Results indicated that addition of GO managed to cause an average enhancement on the mechanical properties of these hydrogels while also increased the self-healing capabilities of the system at a microscopic level, offering fast recovery after periods of high strain while avoiding the loses that were observed in the GO free gels.
In vitro antibacterial evaluation of dynamic nanocomposite hydrogels
Antibacterial activity has been previously reported in injectable hydrogel wound dressings containing quaternarized chitosan and benzaldehyde modified crosslinkers.79,80 The antibacterial properties were attributed to the positive charged species, known to damage the bacteria walls, while imine based materials specifically containing aromatic functionality have also shown enhanced antimicrobial activities.81,82As previously mentioned, pristine hydrogels of GC/Bi-PEG4000 have shown no antimicrobial activity though working as potential carriers of colistin for wound healing applications with good biocompatibility.35 To similarly evaluate the antimicrobial activity of our nanocomposite hydrogels, a disk diffusion assay was conducted against E. coli (Gram-negative bacteria) strains. More specifically, a 5 wt% solution of TXL crosslinker in PBS pH = 7.4, GC/TXL(0.05)–GO(0.5) and GC/TXL(0.05)–GO(2.0) hydrogels were place on an agar plate (three replicates) containing the bacterial solution following by incubation for 24 h at 37 °C (Fig. S32, ESI†). The antibacterial activity is usually evaluated from the formation of a zone of inhibition (a free bacteria zone) and the bigger is the zone the better the activity. In our case, none of the tested samples showed any visible zone of inhibition coming in agreement with the GC/Bi-PEG4000 hydrogels which similarly showed no antimicrobial activity.
Conclusions
In summary, we have synthesized and characterized a well-defined narrow dispersity ABA amphiphilic crosslinker p(DMAcx-co-CHOy)-b-PEG2000-b-p(DMAcx-co-CHOy) with multiple benzaldehyde functionality using a relatively simple and fast aqueous Cu-RDRP polymerization technique followed by DCC post-modification. Aqueous solutions of this crosslinker spontaneously formed hydrogels when mixed with GC through the in situ formation of imine bonds while gelation times were effectively tuned by varying the amounts of crosslinker between 10 s to 6 min. Despite its relatively high molecular weight, hydrogels demonstrated self-healing properties with various recoveries depending on the molar ratio of (–CHO/–NH2), as shown by rheological analysis. Both rheology and mechanical characterization showed relatively average properties compared to previously reported systems attributing that to the limited interactions of the crosslinker with the amines of GC a possible result of the formation of large micelles (∼350 nm). A direct comparison was further made with a bifunctional PEG4000 crosslinker which seemingly led to higher G′ values at same molar ratios with our synthesized crosslinker. To further investigate that behaviour, a lower functionality well-defined ABA benzaldehyde crosslinker was synthesized leading to enhanced rheological characteristics helping us to realize that by tuning the amphiphilic character of the crosslinker we can gain access to imine based gels with various properties. The average properties of the pristine hydrogels were attempted to be enhanced by introducing predetermined amounts of partially reduced GO in the hydrogels matrix. Results illustrated an effective tuning of mechanical properties (maximum modulus: 48.7 kPa and maximum strain at break: 54.3%) and an enhancement of the microscopic self-healing (>99% efficiency) characteristics of the nanocomposite gels compared to their pristine formulations. However, their macroscopic self-healing differed demonstrating healing efficiencies of ∼50% compared to their intact analogues attributing that to the slow diffusion properties of our crosslinker not allowing the materials to return 100% back to their initial properties. Finally, similarly to GC/Bi-PEG4000 formulations our nanocomposite hydrogels showed no antimicrobial behaviour against E. coli strains hence they can be examined as high loading hydrogel platforms for wound healing applications where hydrophobic drug loading is desired.We believe that the proposed synthesis of the crosslinker could serve as a platform for the facile incorporation of different families of monomers in the near future e.g. acrylates and acrylamides with thermoresponsive and pH-responsive properties leading to responsive gels. In addition, despite the average characteristics of the materials, the high functionality of our crosslinker together with its self-assembly characteristics have the potential for increased loading capabilities of various amine based hydrophobic substances with biological interest.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Warwick Polymer Characterisation Research and Technology Platform (RTP) for all the provided equipment and specifically Dr Daniel Lester and James S. Town for their help and support. We would also like to thank Andrea Dsouza for performing the antibacterial assays. Finally we are grateful for the studentship funding provided by Unilever (SE, MG), Lubrizol (GP, CJA), DSM (AS) and Syngenta (LS) as well as the University of Warwick and the EPSRC Centre for Doctoral Training in Molecular Analytical Science.Notes and references
- A. M. Wemyss, C. Bowen, C. Plesse, C. Vancaeyzeele, G. T. M. Nguyen, F. Vidal and C. Wan, Mater. Sci. Eng., R, 2020, 141, 100561 CrossRef.
- S. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CrossRef.
- S. Uman, A. Dhand and J. A. Burdick, J. Appl. Polym. Sci., 2020, 137, 48668 CrossRef.
- S. Talebian, M. Mehrali, N. Taebnia, C. P. Pennisi, F. B. Kadumudi, J. Foroughi, M. Hasany, M. Nikkhah, M. Akbari, G. Orive and A. Dolatshahi-Pirouz, Adv. Sci., 2019, 6, 1801664 CrossRef PubMed.
- M. Liu, X. Zeng, C. Ma, H. Yi, Z. Ali, X. Mou, S. Li, Y. Deng and N. He, Bone Res., 2017, 5, 17014 CrossRef PubMed.
- Y. Guo, J. Bae, F. Zhao and G. Yu, Trends Chem., 2019, 1, 335–348 CrossRef.
- M. D. Hager, P. Greil, C. Leyens, S. van der Zwaag and U. S. Schubert, Adv. Mater., 2010, 22, 5424–5430 CrossRef PubMed.
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef.
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrínyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- D. L. Taylor and M. in het Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef PubMed.
- M. M. Perera and N. Ayres, Polym. Chem., 2020, 11, 1410–1423 RSC.
- R. P. Wool, Soft Matter, 2008, 4, 400–418 RSC.
- E. Larrañeta, S. Stewart, M. Ervine, R. Al-Kasasbeh and R. F. Donnelly, J. Funct. Biomater., 2018, 9, 13 CrossRef.
- E. A. Appel, M. W. Tibbitt, M. J. Webber, B. A. Mattix, O. Veiseh and R. Langer, Nat. Commun., 2015, 6, 6295 CrossRef.
- J. Qu, X. Zhao, Y. Liang, T. Zhang, P. X. Ma and B. Guo, Biomaterials, 2018, 183, 185–199 CrossRef.
- R. A. Hule and D. J. Pochan, MRS Bull., 2007, 32, 354–358 CrossRef.
- D. Das, T. Kar and P. K. Das, Soft Matter, 2012, 8, 2348–2365 RSC.
- A. K. Gaharwar, C. Rivera, C. J. Wu, B. K. Chan and G. Schmidt, Mater. Sci. Eng., C, 2013, 33, 1800–1807 CrossRef.
- S. Dai, X. Zhou, S. Wang, J. Ding and N. Yuan, Nanoscale, 2018, 10, 19360–19366 RSC.
- Z. Deng, T. Hu, Q. Lei, J. He, P. X. Ma and B. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 6796–6808 CrossRef PubMed.
- A. Ma, G. Wang, Z. Yang, L. Bai, H. Chen, W. Wang, H. Yang, D. Wei and L. Yang, Chem. Eng. J., 2020, 385, 123962 CrossRef.
- Y. Zhang, M. Zhang, H. Jiang, J. Shi, F. Li, Y. Xia, G. Zhang and H. Li, Carbohydr. Polym., 2017, 177, 116–125 CrossRef.
- Z. Deng, H. Wang, P. X. Ma and B. Guo, Nanoscale, 2020, 12, 1224–1246 RSC.
- C. Pan, L. Liu, Q. Chen, Q. Zhang and G. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 38052–38061 CrossRef.
- X. Jing, H.-Y. Mi, B. N. Napiwocki, X.-F. Peng and L.-S. Turng, Carbon, 2017, 125, 557–570 CrossRef.
- H. Yu, Y. Wang, H. Yang, K. Peng and X. Zhang, J. Mater. Chem. B, 2017, 5, 4121–4127 RSC.
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef PubMed.
- X. Han, X. Meng, Z. Wu, Z. Wu and X. Qi, Mater. Sci. Eng., C, 2018, 93, 1064–1072 CrossRef CAS PubMed.
- R. Bui and M. A. Brook, Polymer, 2019, 160, 282–290 CrossRef CAS.
- Z. Wei, J. H. Yang, X. J. Du, F. Xu, M. Zrinyi, Y. Osada, F. Li and Y. M. Chen, Macromol. Rapid Commun., 2013, 34, 1464–1470 CrossRef CAS.
- G. Deng, C. Tang, F. Li, H. Jiang and Y. Chen, Macromolecules, 2010, 43, 1191–1194 CrossRef CAS.
- J. Xu, Y. Liu and S. H. Hsu, Molecules, 2019, 24, 3005 CrossRef CAS.
- C. Ding, L. Zhao, F. Liu, J. Cheng, J. Gu, S. Dan, C. Liu, X. Qu and Z. Yang, Biomacromolecules, 2010, 11, 1043–1051 CrossRef CAS.
- T.-C. Tseng, L. Tao, F.-Y. Hsieh, Y. Wei, I.-M. Chiu and S.-h. Hsu, Adv. Mater., 2015, 27, 3518–3524 CrossRef CAS PubMed.
- C. Zhu, J. Zhao, K. Kempe, P. Wilson, J. Wang, T. Velkov, J. Li, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Macromol. Biosci., 2017, 17, 1600320 CrossRef PubMed.
- J. Qu, X. Zhao, P. X. Ma and B. Guo, Acta Biomater., 2017, 58, 168–180 CrossRef CAS.
- M. Wu, J. Chen, W. Huang, B. Yan, Q. Peng, J. Liu, L. Chen and H. Zeng, Biomacromolecules, 2020, 21, 2409–2420 CrossRef CAS.
- L. Li, B. Yan, J. Yang, L. Chen and H. Zeng, Adv. Mater., 2015, 27, 1294–1299 CrossRef CAS PubMed.
- L. Cao, B. Cao, C. Lu, G. Wang, L. Yu and J. Ding, J. Mater. Chem. B, 2015, 3, 1268–1280 RSC.
- K. Matyjaszewski and J. Spanswick, Mater. Today, 2005, 8, 26–33 CrossRef CAS.
- R. Whitfield, A. Anastasaki, V. Nikolaou, G. R. Jones, N. G. Engelis, E. H. Discekici, C. Fleischmann, J. Willenbacher, C. J. Hawker and D. M. Haddleton, J. Am. Chem. Soc., 2017, 139, 1003–1010 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- J. A. Burns, C. Houben, A. Anastasaki, C. Waldron, A. A. Lapkin and D. M. Haddleton, Polym. Chem., 2013, 4, 4809–4813 RSC.
- A. Anastasaki, A. J. Haddleton, Q. Zhang, A. Simula, M. Droesbeke, P. Wilson and D. M. Haddleton, Macromol. Rapid Commun., 2014, 35, 965–970 CrossRef CAS.
- C. Ma, T. Han, M. Kang, E. Liarou, A. M. Wemyss, S. Efstathiou, B. Z. Tang and D. Haddleton, ACS Macro Lett., 2020, 9, 769–775 CrossRef CAS.
- E. Liarou, M. Staniforth, J. S. Town, A. Marathianos, M. Grypioti, Y. Li, Y. Chang, S. Efstathiou, E. Hancox, A. M. Wemyss, P. Wilson, B. A. Jones, M. Aljuaid, V. G. Stavros and D. M. Haddleton, Eur. Polym. J., 2020, 123, 109388 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2016, 116, 835–877 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002–1026 RSC.
- F. Alsubaie, E. Liarou, V. Nikolaou, P. Wilson and D. M. Haddleton, Eur. Polym. J., 2019, 114, 326–331 CrossRef CAS.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS PubMed.
- W. Huang, Y. Wang, Y. Chen, Y. Zhao, Q. Zhang, X. Zheng, L. Chen and L. Zhang, Adv. Healthcare Mater., 2016, 5, 2813–2822 CrossRef CAS.
- J. Zhu, Y. Jiang, H. Liang and W. Jiang, J. Phys. Chem. B, 2005, 109, 8619–8625 CrossRef CAS PubMed.
- M. D. Baumann, C. E. Kang, J. C. Stanwick, Y. Wang, H. Kim, Y. Lapitsky and M. S. Shoichet, J. Controlled Release, 2009, 138, 205–213 CrossRef CAS.
- M. Xie, H. Han, Y. Kong, J. Li and Y. Zhang, Acta Polym. Sin., 2009, 9, 345–351 CrossRef.
- T. Ekblad, G. Bergström, T. Ederth, S. L. Conlan, R. Mutton, A. S. Clare, S. Wang, Y. Liu, Q. Zhao, F. D’Souza, G. T. Donnelly, P. R. Willemsen, M. E. Pettitt, M. E. Callow, J. A. Callow and B. Liedberg, Biomacromolecules, 2008, 9, 2775–2783 CrossRef CAS.
- S. Lowe, N. M. O'Brien-Simpson and L. A. Connal, Polym. Chem., 2015, 6, 198–212 RSC.
- Q. Chen, S. Yu, D. Zhang, W. Zhang, H. Zhang, J. Zou, Z. Mao, Y. Yuan, C. Gao and R. Liu, J. Am. Chem. Soc., 2019, 141, 16772–16780 CrossRef CAS PubMed.
- D. Gupta, A. Gangwar, K. Jyoti, V. G. S. Sainaga Jyothi, R. K. Sodhi, N. K. Mehra, S. B. Singh and J. Madan, Colloids Surf., B, 2020, 194, 111171 CrossRef CAS PubMed.
- Y. Zhang, B. Yang, X. Zhang, L. Xu, L. Tao, S. Li and Y. Wei, Chem. Commun., 2012, 48, 9305–9307 RSC.
- R. Wool and K. Oconnor, J. Appl. Phys., 1981, 52, 5953–5963 CrossRef CAS.
- Z. Wei, J. Hai, J. Zhou, F. Xu, M. Zrinyi, P. Dussault, Y. Osada and Y. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- E. Curotto and F. Aros, Anal. Biochem., 1993, 211, 240–241 CrossRef CAS.
- Y. Zhang, C. Fu, Y. Li, K. Wang, X. Wang, Y. Wei and L. Tao, Polym. Chem., 2017, 8, 537–544 RSC.
- L. Zhao, L. Niu, H.-Z. Liang, H. Tan, C. Liu and F. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 37563–37574 CrossRef CAS PubMed.
- R. Justin and B. Chen, J. Mater. Chem. B, 2014, 2, 3759–3770 RSC.
- T. Zhou, X. Qi, H. Bai and Q. Fu, RSC Adv., 2016, 6, 34153–34158 RSC.
- K. Kosowska, P. Domalik-Pyzik, M. Nocuń and J. Chłopek, Mater. Chem. Phys., 2018, 216, 28–36 CrossRef CAS.
- B. Konkena and S. Vasudevan, J. Phys. Chem. Lett., 2012, 3, 867–872 CrossRef CAS.
- K. Liu, J.-J. Zhang, F.-F. Cheng, T.-T. Zheng, C. Wang and J.-J. Zhu, J. Mater. Chem., 2011, 21, 12034–12040 RSC.
- R. Justin and B. Chen, Mater. Sci. Eng., C, 2014, 34, 50–53 CrossRef CAS PubMed.
- Z. Xu, H. Gao and H. Guoxin, Carbon, 2011, 49, 4731–4738 CrossRef CAS.
- I. Charhouf, A. Bennamara, A. Abdelmjid, A. Chenite, J. Zhu and M. Berrada, Int. J. Sci. Basic Appl. Res., 2014, 16, 336–348 Search PubMed.
- J. Nath, A. Chowdhury and S. K. Dolui, Adv. Polym. Technol., 2018, 37, 3665–3679 CrossRef CAS.
- B. Haghighi and M. A. Tabrizi, RSC Adv., 2013, 3, 13365–13371 RSC.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS.
- H. Ding, S. Zhang, J.-T. Chen, X.-P. Hu, Z.-F. Du, Y.-X. Qiu and D.-L. Zhao, Thin Solid Films, 2015, 584, 29–36 CrossRef CAS.
- Z. Tang, F. Chen, Q. Chen, L. Zhu, X. Yan, H. Chen, B. Ren, J. Yang, G. Qin and J. Zheng, Polym. Chem., 2017, 8, 4659–4672 RSC.
- L. R. G. Treloar, The Physics of Rubber Elasticity, Oxford University Press, Oxford, United Kingdom, 2005 Search PubMed.
- M. S. Mohy Eldin, E. A. Soliman, A. I. Hashem and T. M. Tamer, Adv. Polym. Technol., 2012, 31, 414–428 CrossRef CAS.
- X. Zhao, H. Wu, B. Guo, R. Dong, Y. Qiu and P. X. Ma, Biomaterials, 2017, 122, 34–47 CrossRef CAS.
- C. M. da Silva, D. L. da Silva, L. V. Modolo, R. B. Alves, M. A. de Resende, C. V. B. Martins and Â. de Fátima, J. Adv. Res., 2011, 2, 1–8 CrossRef.
- L. Xia, Y.-F. Xia, L.-R. Huang, X. Xiao, H.-Y. Lou, T.-J. Liu, W.-D. Pan and H. Luo, Eur. J. Med. Chem., 2015, 97, 83–93 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Includes all synthetic procedures and additional characterization data. See DOI: 10.1039/d0tb02390f |
| This journal is © The Royal Society of Chemistry 2021 |