
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The onset of copper-ion mobility and the electronic transition in the kesterite Cu2ZnGeSe4†
Panagiotis
Mangelis
 a,
Paz
Vaqueiro
a,
Ronald I.
Smith
b and
Anthony V.
Powell
*a
a,
Paz
Vaqueiro
a,
Ronald I.
Smith
b and
Anthony V.
Powell
*a
aDepartment of Chemistry, University of Reading, Whiteknights, Reading RG6 6DX, England, UK. E-mail: a.v.powell@reading.ac.uk
bISIS Pulsed Neutron and Muon Source, Rutherford Appleton Laboratory, Harwell Campus, Didcot, OX11 0QX, UK
First published on 23rd November 2021
Abstract
Kesterite-related phases have attracted considerable interest as earth-abundant photovoltaic and thermoelectric materials. For the kesterite Cu2ZnGeSe4, we have established a direct link between anomalies in the temperature dependence of transport properties and an order-disorder transition. Powder neutron diffraction as a function of temperature reveals an order-disorder transition at 473 K, involving disordering of copper and zinc cations over three crystallographic positions. Vacancies are simultaneously created on the copper-ion sub-lattice, indicative of the concomitant onset of copper-ion mobility. Differential scanning calorimetry data show a weak thermal signature in this temperature region, typical of a second-order phase transition, which is consistent with the absence of anomalies in the temperature dependence of the unit cell volume. The partial melting of the copper-ion sub-lattice induces a transition in the electrical-transport properties. The changes in electrical resistivity and Seebeck coefficient suggest this involves a transition from a conventional, activated semiconductor, to a degenerate semiconductor. The entry of an increasing fraction of the copper-ion sub-lattice into a liquid-like state is reflected in a reduction in thermal conductivity above 473 K. The order-disorder phase transition identified here has consequences for the performance of photovoltaic and thermoelectric devices based on kesterites.
Introduction
The diamond-like quaternary chalcogenides of general formula A2BCQ4 where A, B, C are elements from groups 11, 12 and 14 respectively and Q = S or Se, have attracted particular interest in recent years as materials for environmentally-friendly energy applications, including photovoltaic devices,1–5 intermediate-temperature thermoelectrics6–12 and photocatalysts for solar water splitting.13 These materials exhibit promising photovoltaic and thermoelectric properties and, being composed of Earth-abundant elements, offer considerable advantages in terms of device manufacturing cost. For example, Dong et al.,14 have demonstrated that the copper-rich phase Cu2.2Zn0.8SnSe4 exhibits a maximum thermoelectric figure-of-merit, ZT = 0.86 at 800 K. Similarly, high-performance thin-film solar cells fabricated from quaternary chalcogenides, Cu2ZnSn(S,Se)4, exhibit efficiencies of more than 12%.15,16 However, the efficiency is limited by band gap fluctuations which lead to low open-circuit voltages. Computational and experimental investigations suggest this behaviour is related to the degree of Cu/Zn disorder within the structure.17,18 These materials also exhibit low thermal conductivities, giving rise to potential applications in thermoelectric devices, which can also be related to the underlying structure.Materials of general formula A2BCQ4 adopt structures that can be considered as ordered derivatives of zinc blende or wurtzite. Each anion is coordinated by two A+ cations and one each of B2+ and C4+ cations. The different bonding properties of these cations leads to a range of anion–cation distances and a distortion away from ideal tetrahedral geometry.19 It has been suggested that this may contribute to enhancing the phonon scattering, leading to low lattice thermal conductivities.20
Despite more than 40 years of structural investigations, the detailed structural description of many of the phases in this family of materials remains a matter of some debate. The majority of reports describe the compounds at room temperature as adopting either the stannite (I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2m) or kesterite (I) zinc-blende-related forms (Fig. 1). In the ideal (fully ordered) kesterite structure, A and B cations are ordered over the 2c and 2d Wyckoff sites respectively in planes perpendicular to the c axis at z = 0.25, 0.75. The remaining A cations are located in planes at z = 0, 0.5 where together with the C cations they form an ordered array over the 2a and 2b sites. In the stannite structure, A cations exclusively occupy the four-fold (4d) site in the plane located at z = 0.25, 0.75, whereas B and C cations are ordered over layers located at z = 0, 0.5. The principal difference therefore is that in the kesterite structure A cations (Cu in the material discussed here) are located in each of the cation layers in the unit cell, whereas in the stannite structure they are present only in those layers located at z = 0.25 and z = 0.75.
2m) or kesterite (I) zinc-blende-related forms (Fig. 1). In the ideal (fully ordered) kesterite structure, A and B cations are ordered over the 2c and 2d Wyckoff sites respectively in planes perpendicular to the c axis at z = 0.25, 0.75. The remaining A cations are located in planes at z = 0, 0.5 where together with the C cations they form an ordered array over the 2a and 2b sites. In the stannite structure, A cations exclusively occupy the four-fold (4d) site in the plane located at z = 0.25, 0.75, whereas B and C cations are ordered over layers located at z = 0, 0.5. The principal difference therefore is that in the kesterite structure A cations (Cu in the material discussed here) are located in each of the cation layers in the unit cell, whereas in the stannite structure they are present only in those layers located at z = 0.25 and z = 0.75.
 | ||
| Fig. 1 Comparison of (a) the fully-ordered kesterite (I) and (b) the stannite (I2m) structures of the quaternary A2BCQ4 phases. In the kesterite form, A-cations are located in each of the cation layers, whereas in the stannite structure, they are confined to the cation layers at z = 0.25, 0.75. A, B and C cations are denoted by red, blue and black spheres respectively and yellow spheres denote Q anions. | ||
The similarity of the X-ray diffraction patterns of these two structural modifications, together with the presence of elements with similar atomic numbers, introduces ambiguities into the correct description of the ground-state structure of these materials. The structural characterization of Cu2ZnGeQ4 (Q = S, Se) in particular has been a contentious issue. While several reports suggest these phases adopt the stannite structure12,21,22 first-principles calculations reveal that both Cu2ZnGeS4 and Cu2ZnGeSe4 compounds are more stable in the kesterite form.23 Polarized Raman spectroscopy on single crystals24 suggests that Cu2ZnGeSe4 crystallizes in the kesterite structure, I, while Zeier et al. have described the series Cu2ZnGeSe4−xSx (0 ≤ x ≤ 4), including the end-member (x = 0) phase, in the space group I2m, consistent with a stannite-type structure.25 Using average neutron scattering length analysis, a recent neutron diffraction study concluded that Cu2ZnGeSe4 crystallizes in the kesterite structure and that while there is disorder in the cation planes at z = 0.25 and 0.75, there is an off-stoichiometry defect corresponding to a copper rich phase.26 Similarly our own recent work27 using a combination of powder neutron diffraction and Density Functional Theory (DFT) simulations demonstrated that materials of general formula, A2ZnCQ4 (A = Cu, Ag; C = Sn, Ge; Q = S, Se) all adopt the lower energy kesterite structure, albeit with differing degrees of disorder of A and Zn cations, the extent of which depends on the identity of both the A and C cations.
Cu2ZnGeSe4 is a p-type semiconductor. Previous studies have shown that hole doping through the partial replacement of zinc by copper in Cu2+xZn1−xGeSe4 (0 ≤ x ≤ 0.1), enhances the thermoelectric properties.12 The introduction of holes results in a significant reduction in electrical resistivity, due to an increase in the charge-carrier concentration. The composition x = 0.075 exhibits the maximum ZT = 0.45 at 670 K. A similar improvement in thermoelectric performance is achieved through the substitution of Ge by In, where the maximum value of ZT = 0.33 is obtained for Cu2ZnGe0.925In0.075Se4 at 673 K.22 In both investigations, the temperature dependence of the electrical-transport properties suggests the occurrence of a semiconductor-to-metal transition in the region 450 ≤ T/K ≤ 470. The origin of this transition has not been identified. Here, we report an investigation of the detailed structure of Cu2ZnGeSe4 by powder neutron diffraction that reveals the electronic transition is accompanied by structural changes involving a phase transition and the partial delocalisation of copper cations.
Experimental
A polycrystalline sample of Cu2ZnGeSe4 was prepared by high temperature synthesis. An appropriate mixture of elemental copper (Sigma-Aldrich, powder, 99.999%), zinc (Sigma-Aldrich, powder, <150 μm, 99.995%), germanium (Sigma-Aldrich, powder, ≥99.999%) and selenium (Sigma-Aldrich, pellets, <5 mm, ≥99.99%) was ground thoroughly using an agate pestle and mortar. The resulting powder was sealed into an evacuated (10−4 mbar) fused silica tube. The mixture was subjected to two 48 hour firings at 650 °C, with an intermediate grinding, before being heated for a third time at 800 °C for 96 hours. Heating and cooling rates of 2 °C min−1 were used throughout.The initial structural characterization of Cu2ZnGeSe4 was carried out by powder X-ray diffraction using a Bruker D8 Advance diffractometer (Ge-monochromated Cu Kα1, λ = 1.5406 Å and a LynxEye linear detector). Powder neutron diffraction data28 were collected using the POLARIS diffractometer29 at the ISIS facility, Rutherford Appleton Laboratory. The powdered sample was sealed at 10−4 mbar into a high-purity, fused-silica ampoule, which was placed in an 8 mm diameter vanadium can. This was mounted inside a furnace for the collection of data over the temperature range 300 ≤ T/K ≤ 973. Rietveld refinements were performed with the GSAS software package30 using data from banks centred at 2θ = 146.72° (Bank 5) and 92.59° (Bank 4). The background parameters were refined using a reciprocal interpolation function and the peak shape modelled using a pseudo-Voigt function.
Analysis of the thermal stability of Cu2ZnGeSe4 in the temperature range 290 ≤ T/K ≤ 773 was carried out through DSC measurements performed on a TA Instruments SDT Q600. Measurements were made on ca. 20 mg of powder placed in an alumina pan under a flow of nitrogen.
In order to conduct electrical-transport property measurements ca. 1.8 g of Cu2ZnGeSe4 was hot-pressed (45 min at 65 bar, 600 °C) under a nitrogen atmosphere, using equipment constructed in-house. The density of the resulting pellet was determined as ca. 99% of the crystallographic value by the Archimedes method, using an Adam PW184 balance. Electrical resistivity and Seebeck coefficient measurements were conducted over the temperature range 300 ≤ T/K ≤ 655 using a Linseis LSR3-800. The measurements were carried out under a partial pressure (50 mbar) of helium, with a temperature gradient of 50 K for the determination of the Seebeck coefficient and a current of 100 mA for the resistivity data. Thermal diffusivity data were collected using a Netzsch LFA 447 Nanoflash instrument. Data were collected in 25 K increments over the temperature range 323 ≤ T/K ≤ 575 on 13 mm diameter pellets that had been first coated with graphite. The Dulong–Petit law was used to provide a value for the heat capacity required to calculate the thermal conductivity.
Results and discussion
Powder X-ray diffraction data confirm the product of the high-temperature synthesis is a single phase. Rietveld refinement using powder X-ray diffraction data does not unambiguously identify the superstructure and allow discrimination between structures described in the space groups I2m and I, due to the almost identical X-ray scattering factors of Cu+ and Zn2+ cations and the similarity of the diffraction patterns arising from the two structure types. The initial Rietveld refinements using room-temperature powder X-ray diffraction data were therefore conducted for both the stannite and kesterite structure types in order to provide lattice parameters and initial structural models for subsequent analysis of powder neutron diffraction data.
Powder neutron diffraction provides the necessary contrast between constituent cations in Cu2ZnGeSe4 (b(Cu) = 7.7, b(Zn) = 5.7, b(Ge) = 8.2 fm). Weighted residuals (I: Rwp, (Bank 5) = 1.5%, Rwp, (Bank 4) = 1.6%; I2m: Rwp (Bank 5) = 2.4%, Rwp (Bank 4) = 3.6%) arising from Rietveld refinement using powder neutron diffraction data collected close to room temperature, indicate that the crystal structure is better described in the kesterite structure, space group I, in agreement with our earlier investigation using a combination of powder neutron diffraction and DFT simulations.27 A similar conclusion has been reached for the related sulphide, Cu2ZnSnS4 on the basis of 65Cu NMR data,31 which exhibits peaks due to the presence of copper cations in two crystallographically-distinct sites, as occurs in the kesterite structure. The stannite structure possesses a single copper site.
The thermal vibration parameters of all atoms were constrained to be equal to reduce problems of correlation with the atomic coordinates and site occupancy factors. Site occupancy factors were initially constrained so that all sites remain fully occupied. In the space group I, the site occupancy factors associated with the cation sites show partial disordering of copper and zinc cations at 2c and 2d sites, within the planes located at z = 0.25 and 0.75, consistent with previous neutron studies.26,27 At 323 K, the 2c position is occupied by ca. 71(4)% of Cu and ca. 29(4)% of Zn cations: the occupancies being reversed at the 2d site. By contrast the neutron diffraction data are consistent with complete ordering of copper and germanium cations at the 2a and 2b sites respectively. Rietveld refinement provides no evidence for vacancy formation at any of the 2a, 2b and 8g sites and site occupancy factors were therefore fixed at unity for these sites. Final observed, calculated and difference powder neutron diffraction profiles for Cu2ZnGeSe4 at 323 K are presented in Fig. 2(a), while the corresponding refined parameters are presented in Table 1.
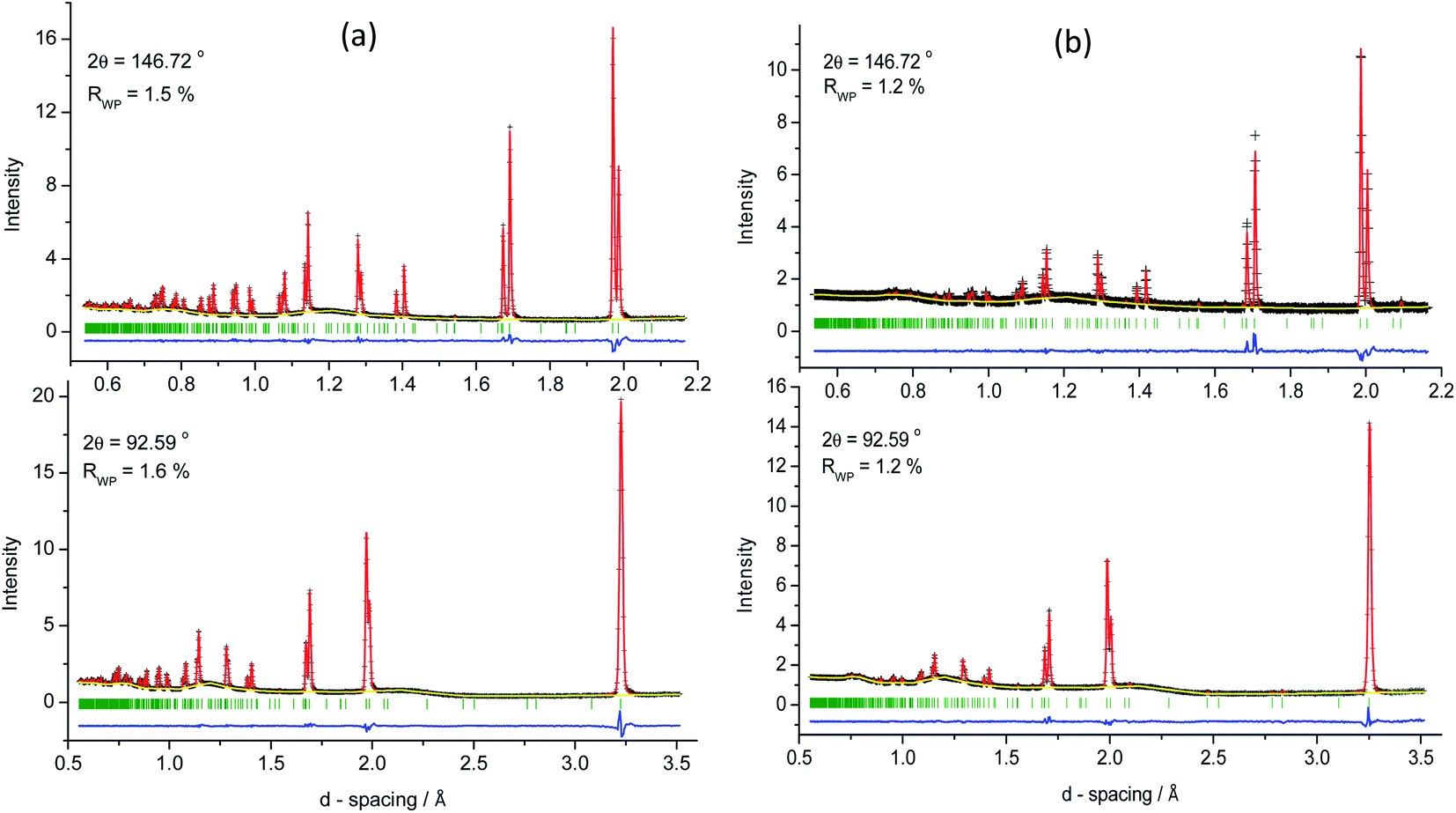 | ||
| Fig. 2 Final observed (black crosses), calculated (red solid lines) and difference (lower blue line) profiles from Rietveld refinement for Cu2ZnGeSe4 in the space group I using powder neutron diffraction data collected at (a) 323 K and (b) 973 K. Data are shown for the back-scattering and 90° detector banks. Reflection positions are marked by vertical lines. | ||
| Temperature/K | |||||
|---|---|---|---|---|---|
|
a Space group I. Cu(1) on 2a (0, 0, 0); Ge(1) on 2b (0, 0, ½); Cu(2)/Zn(2) on 2c (0, ½, ¼) and 2d (0, ½, ¾); Se(1) on 8g (x, y, z). All site occupancy factors (SOFs) are 1.0 unless otherwise stated.
b Site occupancy factors of Cu/Zn cations at the 2(d) site are the reverse of the values at the 2c site.
|
|||||
| 323 | 450 | 473 | 973 | ||
| a/Å | 5.61309(2) | 5.62256(2) | 5.62372(2) | 5.66470(3) | |
| c/Å | 11.05630(7) | 11.07134(7) | 11.07476(7) | 11.1348(1) | |
| V/Å3 | 348.348(3) | 349.999(4) | 350.274(4) | 357.274(5) | |
| c/2a | 0.98486(8) | 0.98455(8) | 0.98465(8) | 0.9828(1) | |
| 2a site | SOF(Cu) | 1.0(–) | 1.0(–) | 0.965(4) | 0.648(5) |
| 2c siteb | SOF(Cu), SOF(Zn) | 0.71(4), 0.29(4) | 0.71(4), 0.29(4) | 0.487(2), 0.50(–) | 0.377(3), 0.50(–) |
| 8g site | x, y, z | 0.2448(3), 0.2578(2), 0.1230(1) | 0.2458(5), 0.2597(2), 0.1225(1) | 0.2500(3), 0.2606(2), 0.1228(2) | 0.2598(3), 0.2599(4), 0.1278(2) |
| U iso/Å2 | 1.345(2) | 1.954(3) | 2.063(3) | 4.541(7) | |
| R wp/% | 2θ = 146.7°, 2θ = 92.6° | 1.5, 1.6 | 1.4, 1.5 | 1.3, 1.5 | 1.2, 1.2 |
| χ 2 | 3.54 | 2.99 | 2.81 | 2.06 | |
Our electrical transport property data (Fig. 3) show similar features in their temperature dependence to those observed by Zeier and co-workers.12,25 The ρ(T) behaviour in the range 325 ≤ T/K ≤ 520 is indicative of semiconducting behaviour, with the increase in gradient at ca. 400 K, suggestive of a change in conduction mechanism. Above 510 K, dρ/dT changes sign and the resistivity thereafter increases with increasing temperature. The positive sign of dρ/dT is the behaviour associated with metallic conduction, leading to previous suggestions that the anomaly in transport properties is associated with an insulator to metal transition.12 The Seebeck coefficient is positive at all temperatures in the range investigated, consistent with p-type conductivity, in which holes are the dominant charge carriers. There are complementary features in S(T). Initially, the Seebeck coefficient increases on heating, before reaching a maximum value at 400 K; the temperature at which the change in gradient of ρ(T) is observed, before decreasing on further heating to 500 K. Above this temperature the Seebeck coefficient increases linearly with temperature, which is also characteristic of metallic conduction.
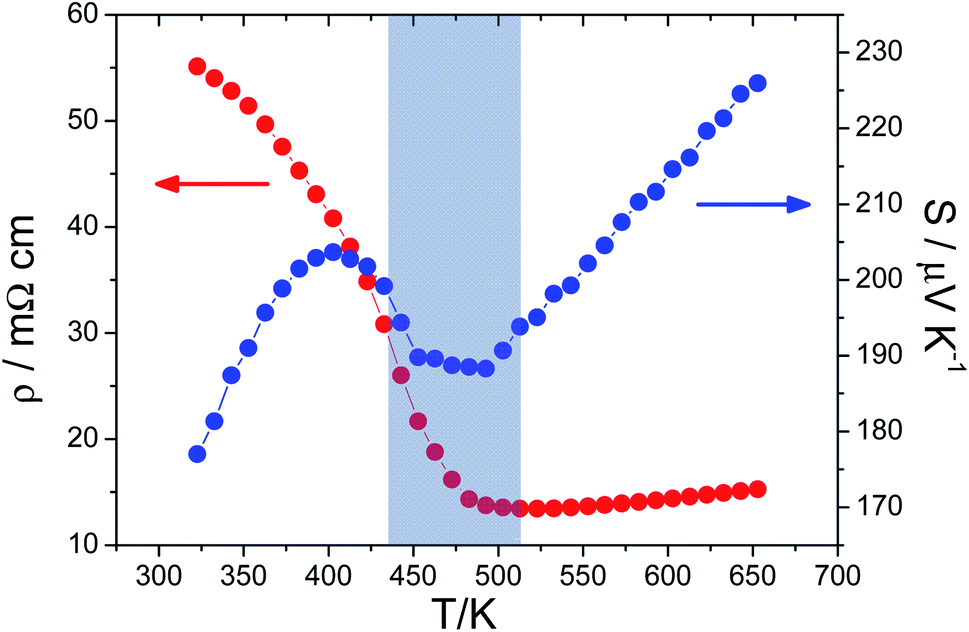 | ||
| Fig. 3 Electrical resistivity (ρ) and Seebeck coefficient (S) data for Cu2ZnGeSe4 as a function of temperature. The shading highlights the temperature region in which the changes in electrical-transport properties occur. | ||
Rietveld analysis of powder neutron diffraction data collected as a function of temperature were initiated in the kesterite structure (space group I) using atomic coordinates determined at 323 K for the initial structural model. Refinement was performed sequentially with the refined structure at a given temperature being used as the initial structural model for the next highest temperature. Final observed, calculated and difference powder neutron diffraction profiles from refinement using data collected at 973 K, are presented in Fig. 2(b). Selected fitted powder neutron diffraction profiles using data collected at intermediate temperatures are provided as ESI.† Analysis reveals that data are well described by the kesterite structure over the temperature range 300 ≤ T/K ≤ 973, with goodness of fit parameters in the range 1.2 ≤ Rwp/% ≤ 1.6 and 2.1 ≤ χ2 ≤ 3.5. Refined parameters for selected temperatures are presented in Table 1. The data provide no evidence of a structural transition to a stannite-type structure on heating, nor is there any indication of the formation of secondary phases through exsolution.
The unit-cell parameters increase smoothly with temperature (Fig. 4), with no evidence of a discontinuity in the region 440 ≤ T/K ≤ 512, where changes in transport properties and anomalies in the first derivative of the heat flow are observed (vide infra). Thermal expansion results in an increase of unit-cell volume (by ca. 2.6%, at 973 K relative to that at 323 K). The absence of any discontinuities in the temperature dependence of the unit-cell volume, suggest that the anomalies in the electrical-transport properties are associated with a second-order phase transition.
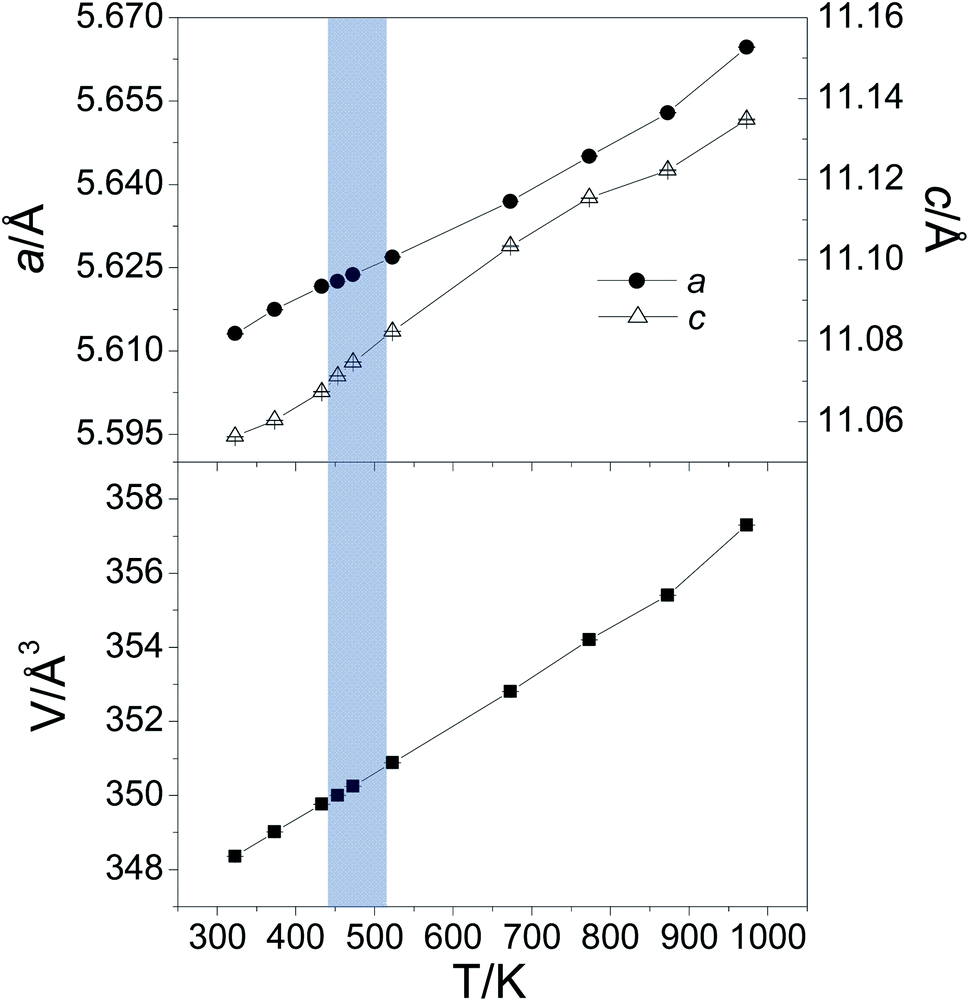 | ||
| Fig. 4 Refined unit-cell parameters (upper graph) and unit-cell volume (lower graph) of Cu2ZnGeSe4 determined from Rietveld analysis of powder neutron diffraction data collected in the temperature range 323 ≤ T/K ≤ 973. The shading indicates the temperature region in which changes in electrical-transport properties are observed. | ||
Following refinement of lattice and atomic positional parameters, site occupancy factors were introduced as variables into structural refinements using data collected as a function of temperature, in order to explore temperature-induced changes in the nature and degree of cation disorder over all (Cu/Zn, Cu/Ge, Zn/Ge) cation sites. Initially, the sum of site occupancy factors at each site was constrained to unity, to maintain full occupancy of a site. At temperatures up to 453 K, the degree of Cu/Zn disorder over the 2c and 2d sites remains effectively unchanged from that determined at 323 K, while full ordering is retained at the remaining cation sites (2a and 2b) (Fig. 5). Relaxing the constraint on full occupancy does not lead to the indication of vacancy formation at any of the cation or anion sites below 473 K. Therefore, the kesterite structure with partial disorder of Cu and Zn cations over the 2c/2d sites is maintained at temperatures below 473 K.
 | ||
| Fig. 5 The temperature dependence of the copper site-occupancy factors associated with the 2c, 2d and 2a crystallographic sites in Cu2ZnGeSe4 determined from Rietveld analysis of powder neutron diffraction data collected over the temperature range 300 ≤ T/K ≤ 973. The shading indicates the temperature region in which changes in electrical-transport properties are observed and the location of each of the sites is indicated on the right. | ||
At 473 K, the site occupancy factor associated with the 2c and 2d positions changes abruptly (Fig. 5) towards the value of 0.5, which would correspond to complete disorder of Cu and Zn cations in the z = 0.25 and 0.75 planes. However, the goodness-of-fit parameters (Rwp, χ2) from Rietveld refinement at 473 K increase from the values obtained at lower temperatures. This led us to remove the constraint on full site occupancy at these sites, to allow for vacancy formation, in addition to Cu/Zn disorder. This improves the goodness-of-fit parameters and results in deviations of the site-occupancy factor from the value of 0.5 associated with copper cations at the 2c and 2d sites. Furthermore, there is a reduction of the site occupancy factor of the 2a site that is fully occupied by copper at lower temperatures. These changes in site occupancy factors reveal that vacancies are created on the copper-ion sub-lattice, simultaneously with the onset of Cu/Zn disorder. By contrast, germanium (2b site) and zinc (2c and 2d sites) cations retain their full site occupancy, consistent with the original stoichiometry, on heating above 453 K. Structure refinement with powder neutron diffraction data collected over the temperature range 473 ≤ T/K ≤ 973 reveals a progressive increase in the number of copper vacancies at the 2a, 2c and 2d sites (Fig. 5). In this temperature range, the structure is therefore best described as a defective disordered-kesterite type, in which there is incomplete occupancy of the 2a site by copper cations, with the 2c and 2d positions half-occupied by zinc cations and the balance comprising copper cations and vacancies. The 2b and 8g sites remain fully occupied by germanium cations and selenium anions up to 973 K.
Rietveld analysis indicates that the total of the copper vacancies at these three sites at 973 K represents ca. 30% of the total copper content, corresponding to a composition of Cu1.40(1)ZnGeSe4 at this temperature. Examination of calculated difference Fourier maps reveals the presence of residual nuclear density at multiple sites within the unit cell. Attempts to refine the coordinates and occupancies of the interstitial copper cations, from the position of the maxima in the residual nuclear density, were unsuccessful owing to the smearing out of the copper distribution over numerous sites. This suggests that cations are not localised at a particular crystallographic position and is indicative of appreciable ion mobility at temperatures above 473 K.
The creation of interstitial copper ions is supported by 63Cu MAS-NMR data,25 which at room temperature show at least two peaks arising from copper that merge into one broad feature above 450 K. Although the low-temperature data were interpreted in terms of a stannite structure, containing a single copper site, together with interstitial copper species to account for the second peak, the MAS-NMR data are consistent with the results of the analysis of powder neutron diffraction data reported here, which gives rise to three distinct copper sites in the kesterite structure at low temperature, owing to Cu/Zn disorder. The merging into a single feature at high temperatures may then be associated with the onset of copper ion mobility leading to a superionic phase transition, as proposed by Zeier et al.25 However, as demonstrated by the analysis of powder neutron diffraction data, only partial melting of the copper-ion sub-lattice occurs above 473 K, with an increasing proportion of the copper ions becoming delocalised on heating.
The phase transition that occurs in the region of the anomalies in the electron-transport properties leads to retention of the principal structural features of the low-temperature kesterite structure. In particular, the anion sub-lattice is retained, while the 2b cation sites remain fully occupied by germanium cations, and zinc cations remain located entirely on the 2c and 2d sites, albeit with a change in their distribution. The phase transition is associated with partial melting of the copper-ion sub-lattice only, which would be consistent with the phase transition being second order. Thermal analysis data support this view. Other workers report a very weak feature at 450 K in DSC data, while the data for the material reported here (Fig. 6) reveal only a change in slope of heat flow in the temperature region where the anomalies in electron transport properties are observed. This is more clearly evidenced in the behaviour of the first-derivative of the heat flow, which show several features in the temperature region 450 ≤ T/K ≤ 512. The weak thermal signature is consistent with minimal structural change having occurred through the temperature region where both the resistivity and Seebeck coefficient data indicate significant changes in the electron-transport properties. The absence of a discontinuity in the temperature dependence of unit-cell volume of Cu2ZnGeSe4 shows parallels with class III solid electrolytes in the classification scheme proposed by O'Keefe32 for superionic transitions. This would also be consistent with the weak thermal signature, since in class III transitions anomalies in specific heat may extend over a very wide range of temperatures.33
 | ||
| Fig. 6 DSC data in the temperature range 375 ≤ T/K ≤ 575 collected for Cu2ZnGeSe4 under flowing nitrogen. Changes in slope in the heat flow as function of temperature are more apparent in the behaviour of the first derivative (right-hand axis). The shading indicates the temperature region in which changes in electrical-transport properties are observed. | ||
A marked rise in Seebeck coefficient that introduces a local maximum into S(T), as is the case here (Fig. 3), has been observed prior to the onset of a second-order phase transition to a superionic state in Cu2Se.34,35 Mahan36 has suggested that this arises from a temperature-dependent broadening of the energy gap through critical fluctuations, whereas Liu et al.,34 have proposed the origin lies in structural fluctuations leading to rising local states and an increase in the entropy of charge carriers, giving rise to a power law dependence to the Seebeck coefficient as the superionic transition is approached. However, Kang et al.37 have demonstrated that in the case of Cu2Se the transition is first order in nature. Based on an analysis of the phase diagram, they conclude that the additional contribution to the Seebeck coefficient, prior to the melting of the copper sub-lattice, may be accounted for by an extended region in which a mixture of two phases evolves, necessarily implying a discontinuous phase transition. However, in the present case, the weak thermal signature, retention of the principal structural features and only partial melting of the copper-ion sub-lattice indicate that the transition is unlikely to be first order. Consequently, the increase in Seebeck coefficient prior to the phase transition is more likely to be associated with structural fluctuations as the phase transition is approached.
It has been proposed12 that the marked change in electron-transport properties in the region 450 ≤ T/K ≤ 500, signifies a transition from non-metallic to metallic conduction. While the temperature dependence of the resistivity (ρ ∝ T) above this temperature is that expected for a metallic system, the magnitude of the resistivity (14–15 mΩ cm) is more appropriate to a semiconductor. Similarly, while the S ∝ T dependence of the Seebeck coefficient is that expected for a metal, the magnitude of the Seebeck coefficient (S ≈ 190–225 μV K−1) is more characteristic of semiconducting behaviour. The relatively high values of the resistivity and Seebeck coefficient, coupled with the metal-like temperature dependence, is indicative of a degenerate semiconductor in which there is a high (and temperature-independent) concentration of charge carriers. This is consistent with the Hall measurement data of Zeier et al.,38 which show that the carrier concentration increases on heating to ca. 500 K, above which temperature it attains a constant value of ca 1020 cm−3. This high carrier concentration is commensurate with degenerate semiconducting behaviour.
Below the phase transition the conduction is due to intrinsic defects, given the relatively large band gap of 1.63 eV.39 The positive sign of the Seebeck coefficient indicates that these defects result in the creation of holes, which serve as the dominant charge carriers at ambient temperature. As has been suggested by Song et al.40 these are likely to arise from slight deviations from the ideal stoichiometry. Calculations41 for the analogous tin-containing phase, Cu2ZnSnSe4, suggest that after CuZn anti-site defects, copper vacancies (VCu) have the lowest vacancy formation energy, and can form spontaneously during processing. Similar conclusions regarding the spontaneous formation of VCu defects have been arrived at from calculations on the related phase Cu2FeSnSe4.40 Even a small number of copper vacancies would result in a relatively high hole carrier concentration: the reported carrier concentration, nH ≈ 1018 cm−3 at 300 K, requiring < 0.01% of ionized copper vacancies. As the temperature is increased, VCu defects are progressively ionized resulting in an increase in hole concentration with temperature, as is observed experimentally38 and a corresponding reduction in resistivity. Although the conduction process below 473 K is thermally activated, efforts to fit the data using an expression for either an Arrhenius or a variable-range-hopping conduction mechanism were unsuccessful, suggesting the mechanism is more complex, while the change in gradient of ρ(T) at ca. 400 K, may be indicative of a change in the balance between competing mechanisms.
Above the onset of copper-ion delocalisation at T ≥ 473 K, both the resistivity and Seebeck coefficient have contributions from electronic and ionic conductivities, preventing detailed interpretation of the data. The movement of copper from its regular crystallographic position to interstitial sites creates additional vacancies on the copper sub-lattice at the 2a, 2c and 2d positions, according to:
| CuCu + Vi → VCu + Cui |
The formation of defects will impact on carrier mobility as the periodicity of the crystal structure is perturbed, while interstitial copper ions serve as additional electron-scattering centres. The increase in resistivity with increasing temperature may be associated with the increase in the number of interstitial copper ions. In addition, new defect electronic states are created within the band gap. Ionization of the VCu defects would create additional holes in the valence band and ionization of the interstitial copper cations (Cui), electrons. Providing both types of defect state are shallow, for every hole that is produced by ionization of VCu, an electron is generated by ionization of Cui, resulting in no net increase in carrier concentration above ca. 500 K, consistent with the experimental data of Zeier et al.12,38 Further support for this is provided by the observation that the hole concentration, determined from Hall measurements at T < 500 K, is more than an order of magnitude lower than would be generated by the number of copper vacancies determined by powder neutron diffraction at high temperatures.
The impact of the onset of copper ion mobility on heating above 473 K is also evident in the thermal conductivity data (Fig. 7). The moderately high electrical resistivity results in a low electronic contribution (κel), of the order of 3–10% (determined using the Wiedemann–Franz Law) at temperatures above 473 K, to the total thermal conductivity (κ). The total thermal conductivity is therefore dominated by the lattice contribution (κL). Between 300 K and 473 K, κL decreases slowly with heating. Above 473 K, the decrease in κL with increasing temperature is more marked, and the value of 1.38 W m−1 K−1 attained at 573 K is ca. 30% lower than that at 473 K. This significant decrease in thermal conductivity on heating above 473 K may be associated with the formation of copper interstitials. This is similar to the behaviour observed in the binary copper chalcogenides, Cu2−δQ (Q = S, Se), in which copper migration leads to a liquid-like copper sub-lattice. The exceptionally low lattice thermal conductivity of such phases (κL < 0.6 W m−1 K−1) at high temperature has been attributed to the liquid-like character of the copper sub-lattice and the materials have been described as exhibiting phonon-liquid electron-crystal (PLEC) behaviour, reflecting the fact that the electron-transport properties are determined principally by the anion sub-lattice and thermal transport by the disordered copper sub-lattice. The origin of the low lattice thermal conductivity has been attributed to the softness of the shear modes, which reduces the transverse phonon velocity and the average speed of sound in the liquid like state.42 Although in Cu2ZnGeSe4, only partial melting of the copper sub-lattice occurs, the difference Fourier map indicates that the copper ions are not located at specific interstitial sites. This high degree of disorder of interstitial copper ions confers liquid-like characteristics, analogous to the situation in Cu2−δQ, providing a mechanism for the observed reduction in κL.
 | ||
| Fig. 7 Total (κ), electrical (κe) and lattice (κL) thermal conductivity of Cu2ZnGeSe4 over the temperature range 325 ≤ T/K ≤ 575.38 The shading indicates the temperature region in which changes in electrical-transport properties are observed. | ||
Conclusions
In conclusion, we have demonstrated by analysis of powder neutron diffraction data collected as a function of temperature that Cu2ZnGeSe4 adopts a kesterite-type structure at ambient temperature in which there is partial disordering of copper and zinc cations over crystallographic sites in layers at z = ¼ and z = ¾ along the c-axis of the unit cell. The p-type semiconduction arising from intrinsic copper vacancies gives rise to a relatively large electrical resistivity and Seebeck coefficient at room temperature. A decrease in resistivity and increase in Seebeck coefficient are observed on heating as the copper vacancies are progressively ionized, increasing the hole concentration. On heating, the extent of CuZn antisite disorder, remains effectively constant to 473 K. Above this temperature Cu and Zn cations become fully disordered. However, this disorder is accompanied by partial melting of the copper-ion sub-lattice, which results in the formation of copper interstitials and a significant number of defects on the copper sub-lattice. The number of these defects increases with increasing temperature until at 973 K, ca. 30% of the copper ions are displaced from their regular crystallographic sites. Analysis of difference Fourier maps suggests the interstitials are not located at specific cation sites, consistent with entry of a fraction of the copper cations into a liquid-like state, analogous to that observed in the binary copper chalcogenides at high temperature. The phase transition appears to be second order, as evidenced by the absence of anomalies in the temperature dependence of the unit-cell volume and the appearance of a local maximum in S(T) prior to the onset of copper delocalization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank EPSRC (award ref. 1367504) and the University of Reading for financial support for P. M., and for access to the Chemical Analysis Facility for powder X-ray diffraction and thermal analysis measurements. The authors also thank the STFC (Science and Technology Facilities Council) for the neutron beam time allocation at the ISIS Facility.References
- S. Chen, X. G. Gong, A. Walsh and S.-H. Wei, Crystal and Electronic Band Structure of Cu2ZnSnX4 (X = S and Se) Photovoltaic Absorbers: First-principles Insights, Appl. Phys. Lett., 2009, 94, 041903 CrossRef.
- S. Delbos, Kësterite Thin Films for Photovoltaics: a Review, EPJ Photovoltaics, 2012, 3, 35004 CrossRef CAS.
- Q. Guo, G. M. Ford, W.-C. Yang, C. J. Hages, H. W. Hillhouse and R. Agrawal, Enhancing the Performance of CZTSSe Solar Cells with Ge Alloying, Sol. Energy Mater. Sol. Cells, 2012, 105, 132–136 CrossRef CAS.
- S. Nakamura, T. Maeda and T. Wada, Phase Stability and Electronic Structure of In-Free Photovoltaic Materials: Cu2ZnSiSe4, Cu2ZnGeSe4, and Cu2ZnSnSe4, Jpn. J. Appl. Phys., 2010, 49, 121203 CrossRef.
- T. K. Todorov, K. B. Reuter and D. B. Mitzi, High-Efficiency Solar Cell with Earth-Abundant Liquid-Processed Absorber, Adv. Mater., 2010, 22, E156–E159 CrossRef CAS PubMed.
- C. P. Heinrich, T. W. Day, W. G. Zeier, G. J. Snyder and W. Tremel, Effect of Isovalent Substitution on the Thermoelectric Properties of the Cu2ZnGeSe4–xSx Series of Solid Solutions, J. Am. Chem. Soc., 2014, 136, 442–448 CrossRef CAS PubMed.
- F. S. Liu, B. Wang, W. Q. Ao, Y. Li and J. Q. Li, Crystal structure and Thermoelectric Properties of Cu2Cd1−xZnxSnSe4 Solid Solutions, Intermetallics, 2014, 55, 15–21 CrossRef CAS.
- M.-L. Liu, F.-Q. Huang, L.-D. Chen and I.-W. Chen, A Wide-band-gap p-type Thermoelectric Material Based on Quaternary Chalcogenides of Cu2ZnSnQ4 (Q = S, Se), Appl. Phys. Lett., 2009, 94, 202103 CrossRef.
- C. Sevik and T. Çağın, Assessment of Thermoelectric Performance of Cu2ZnSnX4, X = S, Se, and Te, Appl. Phys. Lett., 2009, 95, 112105 CrossRef.
- C. Sevik and T. Çağın, Ab initio Study of Thermoelectric Transport Properties of Pure and Doped Quaternary Compounds, Phys. Rev. B, 2010, 82, 045202 CrossRef.
- K. Wei, L. Beauchemin, H. Wang, W. D. Porter, J. Martin and G. S. Nolas, Enhanced Thermoelectric Properties of Cu2ZnSnSe4 with Ga-doping, J. Alloys Compd., 2015, 650, 844–847 CrossRef CAS.
- W. G. Zeier, A. LaLonde, Z. M. Gibbs, C. P. Heinrich, M. Panthöfer, G. J. Snyder and W. Tremel, Influence of a Nano Phase Segregation on the Thermoelectric Properties of the p-Type Doped Stannite Compound Cu2+xZn1–xGeSe4, J. Am. Chem. Soc., 2012, 134, 7147–7154 CrossRef CAS PubMed.
- I. Tsuji, Y. Shimodaira, H. Kato, H. Kobayashi and A. Kudo, Novel Stannite-type Complex Sulfide Photocatalysts AI2–Zn–AIV–S4 (AI = Cu and Ag; AIV = Sn and Ge) for Hydrogen Evolution under Visible-Light Irradiation, Chem. Mater., 2010, 22, 1402–1409 CrossRef CAS.
- Y. Dong, H. Wang and G. S. Nolas, Synthesis and Thermoelectric Properties of Cu Excess Cu2ZnSnSe4, Phys. Status Solidi RRL, 2014, 8, 61–64 CrossRef CAS.
- J. Kim, H. Hiroi, T. K. Todorov, O. Gunawan, M. Kuwahara, T. Gokmen, D. Nair, M. Hopstaken, B. Shin, Y. S. Lee, W. Wang, H. Sugimoto and D. B. Mitzi, High Efficiency Cu2ZnSn(S,Se)4 Solar Cells by Applying a Double In2S3/CdS Emitter, Adv. Mater., 2014, 26, 7427–7431 CrossRef CAS PubMed.
- W. Wang, M. T. Winkler, O. Gunawan, T. Gokmen, T. K. Todorov, Y. Zhu and D. B. Mitzi, Device Characteristics of CZTSSe Thin-Film Solar Cells with 12.6% Efficiency, Adv. Energy Mater., 2014, 4, 1301465 CrossRef.
- J. J. S. Scragg, L. Choubrac, A. Lafond, T. Ericson and C. Platzer-Björkman, A Low-temperature Order-disorder Transition in Cu2ZnSnS4 Thin Films, Appl. Phys. Lett., 2014, 104, 041911 CrossRef.
- J. J. S. Scragg, J. K. Larsen, M. Kumar, C. Persson, J. Sendler, S. Siebentritt and C. Platzer Björkman, Cu–Zn Disorder and Band Gap Fluctuations in Cu2ZnSn(S,Se)4: Theoretical and Experimental Investigations, Phys. Status Solidi B, 2016, 253, 247–254, DOI:10.1002/pssb.201552530.
- X. Y. Shi, F. Q. Huang, M. L. Liu and L. D. Chen, Thermoelectric Properties of Tetrahedrally Bonded Wide-gap Stannite compounds Cu2ZnSn1−xInxSe4, Appl. Phys. Lett., 2009, 94, 122103 CrossRef.
- J. M. Skelton, A. J. Jackson, M. Dimitrievska, S. K. Wallace and A. Walsh, Vibrational Spectra and Lattice Thermal Conductivity of Kesterite-structured Cu2ZnSnS4 and Cu2ZnSnSe4, APL Mater., 2015, 3, 041102 CrossRef.
- W. G. Zeier, H. Zhu, Z. M. Gibbs, G. Ceder, W. Tremel and G. J. Snyder, Band Convergence in the Non-cubic Chalcopyrite Compounds Cu2MGeSe4, J. Mater. Chem. C, 2014, 2, 10189–10194 RSC.
- R. Chetty, A. Bali, O. E. Femi, K. Chattopadhyay and R. C. Mallik, Thermoelectric Properties of In-Doped Cu2ZnGeSe4, J. Electron. Mater., 2016, 45, 1625–1632 CrossRef CAS.
- S. Chen, A. Walsh, Y. Luo, J.-H. Yang, X. G. Gong and S.-H. Wei, Wurtzite-derived Polytypes of Kesterite and Stannite Quaternary Chalcogenide Semiconductors, Phys. Rev. B, 2010, 82, 195203 CrossRef.
- M. Guc, S. Levcenko, V. Izquierdo-Roca, X. Fontané, E. Arushanov and A. Pérez-Rodríguez, Polarized Raman scattering Analysis of Cu2ZnSnSe4 and Cu2ZnGeSe4 Single Crystals, J. Appl. Phys., 2013, 114, 193514 CrossRef.
- W. G. Zeier, C. P. Heinrich, T. Day, C. Panithipongwut, G. Kieslich, G. Brunklaus, G. J. Snyder and W. Tremel, Bond Strength Dependent Superionic Phase Transformation in the Solid Solution Series Cu2ZnGeSe4−xSx, J. Mater. Chem. A, 2014, 2, 1790–1794 RSC.
- G. Gurieva, D. M. Többens, M. Y. Valakh and S. Schorr, Cu–Zn Disorder in Cu2ZnGeSe4: A Complementary Neutron Diffraction and Raman Spectroscopy Study, J. Phys. Chem. Solids, 2016, 99, 100–104 CrossRef CAS.
- P. Mangelis, A. Aziz, I. de Silva, R. Grau-Crespo, P. Vaqueiro and A. V. Powell, Understanding the Origin of Disorder in Kesterite-type Chalcogenides A2ZnBQ4 (A = Cu, Ag; B = Sn, Ge; Q = S, Se): The Influence of Inter-layer Interactions, Phys. Chem. Chem. Phys., 2019, 21, 19311–19317 RSC.
- A. V. Powell, Structural Changes through the Semiconductor to Metal Transition in Cu2ZnGeSe4 STFC ISIS Neutron and Muon Source, 2015, DOI:10.5286/ISIS.E.RB1620098.
- R. I. Smith, S. Hull, M. G. Tucker, H. Y. Playford, D. J. McPhail, S. P. Waller and S. T. Norberg, The upgraded Polaris powder diffractometer at the ISIS neutron source, Rev. Sci. Instrum., 2019, 90, 115101 CrossRef CAS PubMed.
- A. C. Larson and R. B. von DreeleLos Alamos National Laboratory Report, LAUR, 1994, pp. 86–784 Search PubMed.
- L. Choubrac, M. Paris, A. Lafond, C. Guillot-Deudon, A. Rocquefeite and S. Jobic, Multinuclear (67Zn, 119Sn and 65Cu) NMR Spectroscopy – an Ideal Technique to Probe the Cationic Ordering in Cu2ZnSnS4 Photovoltaic Materials, Phys. Chem. Chem. Phys., 2013, 15, 10722–10725 RSC.
- M. O'Keefe and G. G. Hyde, The Solid Electrolyte Transition and Melting in Salts, Philos. Mag., 1976, 33, 219–224 CrossRef.
- T. Hibma, Gradual Solid Electrolyte Transition in (C5H5NH)Ag5I6, Phys. Rev. B, 1977, 15, 5797–5803 CrossRef CAS.
- H. Liu, X. Shi, M. Kirkham, H. Wang, Q. Li, C. Uher, W. Zhang and L. Chen, Structure-transformation-induced Abnormal Thermoelectric Properties in Semiconductor Copper Selenide, Mater. Lett., 2013, 93, 121–124 CrossRef CAS.
- D. R. Brown, T. Day, K. A. Borup, S. Christensen, B. B. Iversen and G. J. Snyder, Phase Transition Enhanced Thermoelectric Figure-of-merit in Copper Chalcogenides, APL Mater., 2013, 1, 052107 CrossRef.
- G. D. Mahan, The Seebeck Coefficient of Superionic Conductors, J. Appl. Phys., 2015, 117, 045101 CrossRef.
- S. D. Kang, S. A. Danilkin, U. Aydemir, M. Avdeev, A. Studer and G. J. Snyder, Apparent Critical Phenomena in the Superionic Phase Transition of Cu2−xSe, New J. Phys., 2016, 18, 013024 CrossRef.
- W. G. Zeier, Y. Pei, G. Pomrehn, T. Day, N. Heinz, C. P. Heinrich, G. J. Snyder and W. Tremel, Phonon Scattering Through a Local Anisotropic Structural Disorder in the Thermoelectric Solid Solution Cu2Zn1−xFexGeSe4, J. Am. Chem. Soc., 2013, 135, 726–732 CrossRef CAS PubMed.
- H. Matsushita, T. Maeda, A. Katsui and T. Takizawa, Thermal Analysis and Synthesis from the Melts of Cu-based Quaternary Compounds Cu–III–IV–VI4 and Cu2–II–IVVI4 (II = Zn, Cd; III = Ga, In; IV = Ge, Sn; VI = Se), J. Cryst. Growth, 2000, 208, 416–422 CrossRef CAS.
- Q. Song, P. Qiu, H. Chen, K. Zhao, M. Guan, Y. Zhou, T.-R. Wei, D. Ren, L. Xi, J. Yang, Z. Chen, X. Shi and L. Chen, Enhanced Carrier Mobility and Thermoelectric Performance in Cu2FeSnSe4 Diamond-like Compound via Manipulating the Intrinsic Lattice Defects, Mater. Today Phys., 2018, 7, 45–53 CrossRef.
- S. Chen, A. Walsh, X.-G. Gong and S.-H. Wei, Classification of lattice defects in the kesterite Cu2ZnSnS4 and Cu2ZnSnSe4 Earth-abundant Solar Cell Absorbers, Adv. Mater., 2013, 25, 1522–1539 CrossRef CAS PubMed.
- Y. He, T. Day, T. Zhang, H. Liu, X. Shi, L. Chen and G. J. Snyder, High Thermoelectric Performance in Non-Toxic Earth-Abundant Copper Sulfide, Adv. Mater., 2014, 26, 3974–3978 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08642a |
| This journal is © The Royal Society of Chemistry 2021 |