
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
AlCl3-graphite intercalation compounds as negative electrode materials for lithium-ion capacitors†
Yamato
Haniu
 a,
Hiroki
Nara
*bc,
Seongki
Ahn
d,
Toshiyuki
Momma
*ab,
Wataru
Sugimoto
e and
Tetsuya
Osaka
b
a,
Hiroki
Nara
*bc,
Seongki
Ahn
d,
Toshiyuki
Momma
*ab,
Wataru
Sugimoto
e and
Tetsuya
Osaka
b
aFaculty of Science and Engineering, Waseda University, 3-4-1, Okubo, Shinjuku-ku, Tokyo 169-8555, Japan. E-mail: momma@waseda.jp; Fax: +81-3-3205-2074; Tel: +81-3-5286-8537
bResearch Organization for Nano and Life Innovation, Waseda University, 513, Wasedatsurumakicho, Shinjuku-ku, Tokyo 162-0041, Japan. E-mail: h-nara@aoni.waseda.jp; Tel: +81-3-6265-9937
cJST-ERATO Yamauchi Materials Space-Tectonics Project, Kagami Memorial Research Institute for Materials Science and Technology, Waseda University, 2-8-26 Nishiwaseda, Shinjuku, Tokyo 169-0051, Japan
dDepartment of New Energy and Mining Engineering, Sangji University, 83 Sangjidae-gil, Wonju-si, Gangwon-do, Republic of Korea
eFaculty of Textile Science and Technology, Shinshu University, 3-15-1 Tokida, Ueda, Nagano, 386-8567, Japan
First published on 26th November 2021
Abstract
Lithium-ion capacitors (LICs) are energy storage devices that bridge the gap between electric double-layer capacitors and lithium-ion batteries (LIBs). A typical LIC cell is composed of a capacitor-type positive electrode and a battery-type negative electrode. The most common negative electrode material, graphite, suffers from low rate capability and cyclability due to the sluggish kinetics of the Li+ intercalation/de-intercalation process. In this work, metal chloride-pillared graphite, which has recently attracted attention as high-rate LIB anodes, is applied as the negative electrode for LICs for the first time to overcome this drawback. It is shown that AlCl3-graphite intercalation compounds (AlCl3-GICs) with a wide interlayer spacing benefit faster Li+ diffusion. The low molecular weight and conversion reaction of the AlCl3 pillar further enhance the specific capacity per mass. An optimized LIC cell composed of an AlCl3-GIC negative electrode and activated carbon as the positive electrode exhibited higher energy and power densities compared to LICs using graphite as the negative electrode, and displayed stable cycling performance with 85% capacity retention after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 charge/discharge cycles. The AlCl3-GICs synthesized in this work displayed improved electrochemical performances and have the potential to replace the graphite electrode in conventional LICs.
000 charge/discharge cycles. The AlCl3-GICs synthesized in this work displayed improved electrochemical performances and have the potential to replace the graphite electrode in conventional LICs.
Introduction
The recent increase in demand for efficient use of clean energy and the reduction of CO2 emissions has expanded requirements for energy storage devices. There are many kinds of energy storage devices and each has unique characteristics and suitable applications. Electric double-layer capacitors (EDLCs), also known as supercapacitors, are characterized by high power performance and cycle stability, while lithium-ion batteries (LIBs) exhibit high energy density.1–5 The reaction in EDLCs is non-faradaic and ions adsorb at the electrode surface to form an electric double layer. On the other hand, in a LIB cell, the electric charge carrier, Li+, is stored in the electrode bulk material by a faradaic reaction. Because of different reaction mechanisms, i.e. surface confined versus bulk reactions, there is a large performance gap between EDLCs and LIBs.6 Lithium-ion capacitors (LICs) have been developed to bridge the gap between EDLCs and LIBs. LICs can be considered as capacitor-battery hybrid cells where a capacitive material is used as the positive electrode and a battery-type material is utilized as the negative electrode.6–8 LICs are expected to be applied in applications where the combination of high energy densities and long cycle life is required. Typical LIC negative electrode materials are carbon-based materials such as graphite,8–10 hard carbon,11–13 and carbon nanotubes.10,14 Layered oxides, such as Li4Ti5O12,8,15,16 Nb2O5,17,18 and Li3VO519,20 have also been studied as alternatives. Graphite is the most popular negative electrode material because of its stable cycling performance, low potential plateau, and low environmental impact. However, graphite negative electrodes suffer from limited rate capability, owing to the sluggish solid phase diffusion of Li+ ions, and a low Li+ ion capacity.7,9 Metal-chloride graphite intercalation compounds (MClx-GICs) such as FeCl3-GICs, which have been reported as LIB negative electrodes,21–23 are attractive alternatives to graphite. FeCl3-GICs have a structure consisting of graphite as the host material and FeCl3 as the guest material. FeCl3 exists between graphene layers resulting in an increased interlayer spacing that enables rapid Li+ ion diffusion. In addition, FeCl3 has a higher theoretical capacity (approximately 500 mA h g−1) than graphite (372 mA h g−1) owing to the conversion reaction expressed in eqn (1). | (1) |
Due to these features, FeCl3-GICs have shown potential as negative electrode materials with improved rate capability and capacity for LIBs. In addition, low-graphitized carbon is reported to be favorable for better electrochemical performance.24 The above features are attractive for LIC negative electrodes that require fast kinetics to counter the rapid positive electrode reactions. To the best of our knowledge, MClx-GICs have not been studied as negative electrodes for LICs.
In this study, we report the properties of AlCl3-GICs, which are derived from carbon with a low graphitization degree, as a negative electrode for LICs. Notably, AlCl3 can vaporize at relatively low temperatures and has a low molecular weight compared to other metal chlorides. Therefore, the synthesis of AlCl3-GICs is possible under moderate conditions25,26 requiring low energy consumption. Fig. 1 illustrates the increase in the interlayer spacing due to the presence of pillar-molecules (Fig. 1b) compared to the spacing without pillar-molecules (Fig. 1a). After the introduction of the AlCl3 pillar, accelerated diffusion is expected due to the expanded interlayer. In addition, pillars with lower molecular weight are advantageous in terms of mass-specific capacity. The conversion reaction (2) is as follows:
 | (2) |
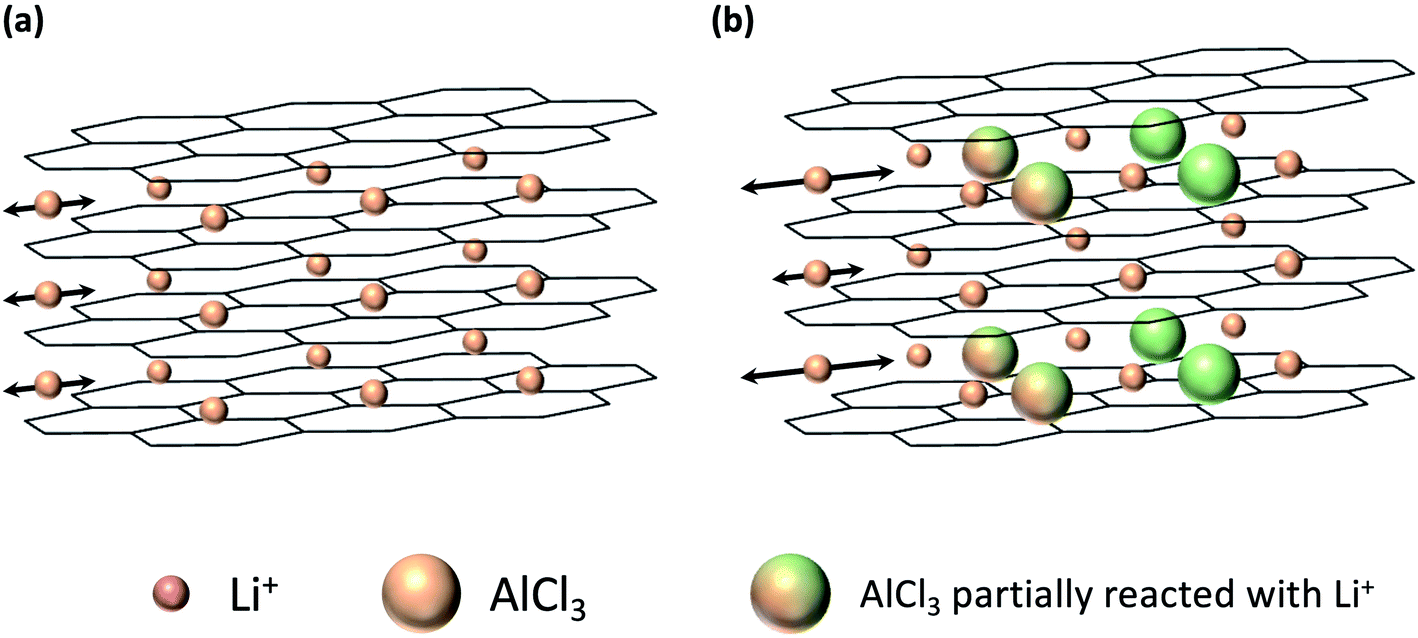 | ||
| Fig. 1 Illustration of (a) graphite and (b) AlCl3-GIC negative electrodes. In the AlCl3-GIC negative electrode, Li+ diffusion into the graphene layers was facilitated by the increased interlayer spacing due to the presence of pillar-molecules. | ||
Thus, AlCl3-GICs are projected as new LIC negative electrodes displaying high rates and capacities. This study describes the synthesis and characterization of AlCl3-GICs and their electrochemical evaluation as a new LIC negative electrode displaying high rates and capacities including LIC full cell characterization. The results reveal the advantageous Li+ storage properties of AlCl3-GICs as negative electrodes for LIC cells with improved performance.
Experimental
The synthesis of AlCl3-GICs was conducted following literature procedures.25,26 Graphite (0.1 g, CGB-20, a graphitization degree of 84.8%, Nippon Graphite, Japan) and AlCl3 (1 g, Tokyo Kasei, Japan) powders were used as starting materials. The powders were mixed with 0.15 g N-chlorosuccinimide (Tokyo Kasei, Japan) as a chlorinating agent in an Erlenmeyer flask under an Ar atmosphere. The container with the mixed powder was then placed in a vacuum for 1 hour and heated to different temperatures (115, 135, 150, and 200 °C) for 24 hours. The obtained powder was then washed with deionized water to remove unreacted starting materials and water-soluble by-products and vacuum dried overnight. The obtained AlCl3-GICs were labeled according to their heating temperatures (115, 135, 150, and 200 °C).Surface morphologies and elemental mapping images were obtained via field emission-scanning electron microscopy (FE-SEM, SU8200, Hitachi High-tech). Crystal structures were characterized by X-ray diffraction (XRD, RINT-Ultima III, Rigaku) with CuKα radiation (λ = 1.542 Å). X-ray fluorescence (XRF, ZSX-Primus2, Rigaku) measurement was performed using RhKα radiation. Raman microprobe spectroscopy (InVia Reflex, Renishaw) was performed with a 532 nm excitation wavelength. The specific surface area of the products was calculated from N2 gas adsorption/desorption data (BELSORP-mini II, MicrotracBEL).
All electrodes were fabricated via the slurry method. In the negative electrode, the ratio of the active material, acetylene black (AB, Denka, Japan), and polyvinylidene difluoride (PVdF, Sigma-Aldrich, US) was 80:10:10 wt%. The loading amount of the active material was 0.4–0.5 mg cm−2. The active material was AlCl3-GICs (115, 135, 150, and 200) or graphite for comparison. The mixture was dispersed in N-methyl-2-pyrrolidone (Kanto Chemical, Japan). The slurry was coated on Cu foil and vacuum dried for 24 hours. The positive electrode was fabricated similarly using a slurry of activated carbon (AC, YP-50F Kuraray, Japan), AB, and PVdF in a 90:5:5 wt% ratio coated on Al foil. Half-cell measurements were conducted with the prepared negative electrodes applied as the working electrode in 2032-type coin cells with Li metal foil as the counter electrode. A glass fiber filter soaked with 1 M LiPF6 in ethylene carbonate/diethylene carbonate (1:1 volume ratio) was used as the electrolyte. Cyclic voltammetry and galvanostatic charge/discharge tests were conducted with the half cells between 0.01 and 3.0 V. Full-cell device testing was conducted with a LIC cell composed of the AlCl3-GIC (115) negative electrode and AC positive electrode balanced so that the electrode capacity ratios were 3:1, 5:1, and 7:1 in coin cells with the same electrolyte as that used in the half-cells. A LIC cell with a graphite negative electrode and an AC positive electrode with an electrode capacity ratio of 5:1 was used as a reference cell.
For pre-lithiation of the negative electrodes, five galvanostatic charge/discharge cycles were conducted and then the state of charge (SoC) was adjusted to allow the negative electrodes in the LIC cells to operate in a deep SoC range. Cyclic voltammetry and galvanostatic charge/discharge tests were performed between 2.2 and 3.8 V using a potentiostat and a galvanostat (TOSCAT, Toyo System; VSP300, Biologic). To calculate the energy densities, E [W h kg−1], and power densities, P [W kg−1], of the LIC full cells, the following equations were used:
 | (3) |
| P [W kg−1] = E/t | (4) |
Results and discussion
Structural characterization
Vapor-phase AlCl3 is reported to behave as a guest of GICs in a Cl2 gas atmosphere and for AlCl3-GIC.25 In this study, AlCl3 powder was vaporized by heating in a container filled with Cl2 gas derived from N-chlorosuccinimide.Fig. 2a depicts the XRD patterns of the samples (full scale XRD patterns are shown in Fig. S1†). All AlCl3-GICs (115, 135, 150, and 200) displayed peaks at approximately 26.5° and 27.5°. The peaks at 26.5° and 27.5° correspond to the graphite (002) plane and the GIC (00l) plane, respectively. The absence of higher-order (00l) peaks for AlCl3-GICs indicates that it has a less ordered structure compared to graphite. The GIC (00l) plane is derived from the AlCl3 intercalated interlayer (003) of stage 1, (004) of stage 2, or (005) of stage 3.25 These results reflect the intercalation of AlCl3 between the layers of the graphitic structure, although some pristine interlayers were retained. This irregular intercalation of AlCl3 is caused by the low graphitization degree of GCB-20. As previously reported, the graphitization degree affects the MClx-GIC structure.27 The increased interlayer spacing due to AlCl3 intercalation was reported in the literature to be 0.954 nm.26 When comparing the GIC (00l) and graphite (002) plane intensity ratios, the ratios of the AlCl3-GICs (200 and 150) were higher than those of the AlCl3-GICs (135 and 115). This reflects the degree of structural changes caused by AlCl3 intercalation. This intercalation of AlCl3 between the layers of the graphitic structure was suggested based on a comparison of the electrochemical behavior of graphite and AlCl3-GICs in a propylene carbonate (PC)-based electrolyte (Fig. S2†). As previously reported,28 the cyclic voltammogram of graphite only shows the cathodic peak derived from the co-intercalation of a Li+-PC complex. The co-intercalation of PC-solvated Li+ destroys the graphitic structure, resulting in the lack of de-intercalation of Li+. In contrast, cyclic voltammograms of AlCl3-GICs (115 and 200) showed both cathodic and anodic peaks owing to the enlarged interlayer spacing caused by the AlCl3 insertion that enabled PC-solvated Li+ to intercalate into the graphite layer without destroying the graphitic structure.
 | ||
| Fig. 2 (a) XRD patterns, (b) micro-Raman spectra and (c) enlarged Raman spectra of AlCl3-GICs synthesized at different temperatures and labelled according to the heating temperature (115, 135, 150, and 200). Data for graphite are also shown for reference. | ||
Micro-Raman spectroscopy was used to determine the graphitic structure. The Raman spectra of the AlCl3-GICs and graphite are shown in Fig. 2b and c. In all the spectra shown in Fig. 2b, the G and D bands of the graphitic structure were detected at approximately 1575 cm−1 and 1350 cm−1, respectively. The ratios of the D and G bands, i.e., the D/G values, of graphite and the AlCl3-GICs (115, 135, 150, and 200) were 0.257, 0.251, 0.324, 0.324, and 0.478, respectively. This demonstrates that products with higher heat-treatment temperatures had a greater number of graphitic structural defects. Enlarged views of the Raman spectra in the vicinity of the G band are shown in Fig. 2c. The Raman shift of the peaks of graphite and AlCl3-GICs (115, 135, 150, and 200) were 1572, 1574, 1578, 1580, and 1581 cm−1, respectively. In previous reports, the wavenumber of the G bands of metal chloride-GICs such as FeCl3-GICs shifted to higher values due to electron donation from the graphite host to the guest metal chlorides.21,22 In this study, the same observation was made for each of the AlCl3-GICs. Therefore, the intercalation of AlCl3 is believed to have occurred in all the products. Products with higher heat-treatment temperatures displayed larger G band shifts, which suggests that larger amounts of AlCl3 were intercalated into the AlCl3-GIC graphitic structure at higher heat-treatment temperatures.
From these results, the degree of structural change caused by the intercalation of AlCl3 decreased in the order 200 > 150 > 135 > 115.
The surface morphologies of graphite and the AlCl3-GICs observed via FE-SEM are shown in Fig. 3. In the FE-SEM images of graphite shown in Fig. 3a and the AlCl3-GICs (115, 135, 150, and 200) shown in Fig. 3b–e, stacked-layer structures were widely observed. Moreover, clear micro-scale structural differences between graphite and the AlCl3-GICs could be seen. The structures of the AlCl3-GICs appeared to be slightly rougher with thin stacked layer units. In contrast, graphite appeared as a highly ordered structure with stacked units composed of thicker layers. The thinner layers are believed to be formed by the micro-scale exfoliation of graphite layers due to the intercalation of AlCl3.
 | ||
| Fig. 3 FE-SEM images of (a) graphite and (b)–(e) AlCl3-GICs (115, 135, 150, and 200). | ||
The composition of AlCl3-GICs was obtained through elemental mapping with SEM-EDX analysis. The mapping images of AlCl3-GICs (115, 135, 150, and 200) are shown in Fig. 4a–d. C, Al, and Cl were uniformly distributed in all the AlCl3-GICs. The atomic ratio of Cl to Al was approximately three in all the products in agreement with the presence of AlCl3. The AlCl3 content in the AlCl3-GICs (115, 135, 150, and 200) was estimated to be approximately 8, 11, 16, and 32 wt% via quantitative analysis. The trend in the AlCl3 content corresponded to the observations from the XRD and Raman patterns. For a detailed analysis, XRF was conducted on AlCl3-GIC (115), which had the lowest AlCl3 content. The AlCl3 content of AlCl3-GIC (115) based on XRF was 15.3 wt.%, indicating that all the AlCl3-GICs synthesized in this study contained at least 15.3 wt% AlCl3. Additionally, based on the results of the XRF analysis, the atomic ratio of Al/Cl was 1:2.62.
 | ||
| Fig. 4 (a)–(d) SEM-EDX elemental mapping images of AlCl3-GICs (115, 135, 150, and 200). | ||
Due to the morphological changes mentioned above, surface area changes were expected. The results of N2 gas adsorption/desorption measurements shown in Fig. 5 confirm this. The AlCl3-GICs displayed higher gas adsorption than graphite. The specific surface areas of the AlCl3-GICs (115, 135, 150, and 200) and graphite were calculated to be8.47, 7.71, 7.14, 6.02, and 5.60 m2 g−1, respectively. Although the difference in specific surface areas may not be significant, the higher surface areas displayed by the AlCl3-GICs may be due to an increase in both the edge and basal plane areas as confirmed by SEM images. AlCl3-GIC (115) had the highest specific surface area among all the AlCl3-GICs, while AlCl3-GIC (200) had the lowest. This trend appeared to be related to the molecular weights of carbon and AlCl3. Products with higher contents of AlCl3, which has a higher molecular weight than carbon, displayed lower specific surface areas.
 | ||
| Fig. 5 Nitrogen adsorption/desorption isotherms of graphite and AlCl3-GICs (115, 135, 150, and 200). | ||
Half cell studies
Electrochemical characterization experiments of different negative electrodes were performed via cyclic voltammetry and galvanostatic charge/discharge tests in the voltage range of 0.01 to 3.0 V in a half-cell configuration and the results are shown in Fig. 6. Cyclic voltammograms of the initial and second cycle at 0.2 mV s−1 are shown in Fig. S3† and 6a, respectively. Reductive currents with voltages higher than 0.2 V derived from the initial irreversible reaction were detected, with especially large currents detected in the AlCl3-GICs. Because the voltage of Li+ intercalation in graphite is lower than 0.2 V, the irreversible current was believed to be derived from other reactions, namely solid electrolyte interphase formation and the insertion of solvated Li+, which was promoted by the high surface area and increased interlayer spacing.21,29,30 In the second cycle of the AlCl3-GICs, sets of reversible peaks in the voltage range of 0–0.2 V and above 0.2 V appeared. The peaks between 0 and 0.2 V were clearly observable and were attributed to the insertion reaction of Li+ into graphite. The increasing parts of the anodic peaks of the AlCl3-GICs which are associated with the deintercalation of Li+ shifted to lower voltages compared to the voltage of the peak of pristine graphite, indicating a lower overvoltage for the reaction with Li+. The peaks above 0.2 V displayed a lower intensity and are attributed to the conversion reactions of AlCl3 in the interlayer spaces. | ||
| Fig. 6 (a) Cyclic voltammograms, (b) charging/discharge curves (c) at 0.1 A g−1 and (c) at 2.0 A g−1, respectively. (d) Discharge capacities at various current densities between 0.1 and 5.0 A g−1, and (e) discharge capacities of graphite and the AlCl3-GICs (115, 135, 150, and 200) cycled at 0.2 A g−1. | ||
XPS analysis of Al 2p and Cl 2p in the lithiated and de-lithiated states supports the conversion reaction (Fig. S4†). The Al 2p peak at 73 eV in the charged state is attributed to the presence of the Al metal, while the peak at a higher binding energy of ∼75 eV can be attributed to the presence of AlCl3.31 The Cl 2p peak at 198.5 eV is attributed to the presence of LiCl.32,33 Furthermore, the Cl 2p peak shifted to a higher binding energy, implying metal chloride formation. The Cl 2p peaks of metal chlorides other than the alkaline metal chlorides are known to be observed at a higher binding energy than LiCl. It has been reported that FeCl3 conversion occurs at 0.2 V. In addition, the peaks may also be due to Li+ absorption in the pores as observed in hard carbon and surface capacitance.13,21,22,34 Fig. S5† and 6b show the charge/discharge curves of the initial and second cycle at 0.1 A g−1, respectively. These curves reflect almost the same characteristics as the cyclic voltammograms, i.e., a large initial irreversible capacity and the existence of reactions in various voltage ranges. Taking AlCl3-GIC (115) as an example, the total discharge capacity was 435 mA h g−1. The curve was divided into two parts at 0.2 V. The capacity below 0.2 V, derived from the graphitic structure, was 306 mA h g−1 based on the discharge curve. By subtraction, the capacity above 0.2 V, unique to AlCl3-GICs and not found in pristine graphite, was calculated to be 128 mA h g−1. Even if the AlCl3 in AlCl3-GIC (115) (15.3 wt%) had completely reacted with the Li+, the calculated theoretical value would have only been 87 mA h g−1, using 571 mA h g−1 as the theoretical capacity of AlCl3 conversion. Thus, the increase in capacity of the AlCl3-GICs was caused not only by the reaction of AlCl3 but also by the other effects mentioned above, such as Li+ absorption in the pores as observed in hard carbon and the surface adsorption of numerous ions due to structural changes. The decrease in the discharge overvoltage was also confirmed from the curves and is believed to have led to the high-rate performance. The discharge capacities during rate performance tests at 0.1, 0.2, 0.5, 1.0, 2.0, and 5.0, and then again at 0.1 A g−1 are shown in Fig. 6d. In the low current density region, the AlCl3-GICs (115 and 135) displayed a higher capacity than graphite. Moreover, all the AlCl3-GICs displayed an improved capacity in the high current density region. These results are evidence of improved rate performance and higher capacity compared to graphite. AlCl3-GIC (115) displayed the highest performance of the AlCl3-GICs, while AlCl3-GIC (200) displayed the lowest. For AlCl3-GIC (115), even under severe conditions such as 2.0 and 5.0 A g−1 corresponding to 4.8 and 12 C-rates, capacities of 105 and 53 mA h g−1, respectively, were delivered. To examine the behaviors under high-rate conditions, charge/discharge curves at 2.0 A g−1 are shown in Fig. 6c. Factors that enhance the reactions with Li+, which occur in the AlCl3-GICs but not in graphite, can increase the capacity. This is attributed to the higher surface area and improved Li+ diffusion due to the increased interlayer spacing. The AlCl3-GICs (115, 135, 150, and 200) displayed superior electrochemical performances in the order 115 < 135 < 150 < 200. There are two possible reasons for this trend. The first is related to the contact area between the edge plane of the active material and the electrolyte. The surface area of active materials containing more AlCl3 was found to be lower than expected. This may lead to a decrease in the surface area of the edge plane and a decrease in Li+ transfer. The second cause is related to the Li+ diffusibility inside the active material. A wider interlayer spacing may contribute to a higher rate of Li+ diffusion.
Cycling performance was evaluated by repeating charge/discharge tests between 0.01 and 3.0 V at 0.2 A g−1. The obtained capacities for each cycle are plotted in Fig. 6e. AlCl3-GICs displayed small capacity decays during the 400 cycles. During the initial cycles, an increase in capacity was observed which can be attributed to some type of activation process. Structural changes during cycling should increase the supply of Li+ to AlCl3, which would increase the number of active sites in pores, thereby changing the charge storage mechanism. The complicated behavior associated with the capacity change may be attributed to the surface area (8.47, 7.71, 7.14, and 6.02 m2 g−1) and AlCl3 content (8, 11, 16, and 32%) of the AlCl3-GICs (115, 135, 150, and 200).
LIC full cell studies
LIC cell performance was evaluated with coin-type cells consisting of AlCl3-GIC (115) as the negative electrode and activated carbon as the positive electrode. AlCl3-GIC (115) was chosen as it showed the best performance among all the AlCl3-GICs based on half-cell data. AlCl3-GIC (115) negative electrodes were pre-lithiated before cell fabrication. Three test cells with different electrode capacity ratios, i.e., negative:positive = 3:1, 5:1, and 7:1, were fabricated. Cyclic voltammetry and galvanostatic charge/discharge tests were conducted in the voltage range of 2.2–3.8 V. The cyclic voltammograms in Fig. S6† display rectangular shape with no clear redox peaks. In addition, linear voltage changes over time were observed in the charge/discharge curves at 0.1 A (g-AC)−1 (Fig. 7a). The above two behaviors correspond to the theoretical nature of hybrid capacitors.7,8 Rate performance tests were conducted at 0.1–20 A (g-AC)−1, and the measured discharge capacities are shown in Fig. 7b. The cell with an electrode capacity ratio of 7:1 displayed the highest capacity per gram of AC at all current densities. In this case, the capacity per gram of AC was determined to be proportional to the operating voltage range of the positive electrode. The cell with the negative electrode with the narrowest operating voltage range displayed the highest capacity of ∼34 mA h (g-AC)−1 at low current densities. Consequently, the ability to retain high capacity at high current densities can be used as an index to evaluate rate performance. At high current densities, LICs with negative:positive capacity ratios of 7:1 and 5:1 displayed the highest and the second highest capacities, thus, the order of rate capability from the highest to lowest was 7:1 > 5:1 > 3:1. The order was affected by the width of the operating voltage range of the negative electrode. During charge/discharge in cells with high-rate performances, the negative electrodes, which displayed lower rate capabilities than the positive electrodes, operated in a narrower SoC range.
 | ||
| Fig. 7 (a) Charge/discharge curves, (b) discharge capacities at various current densities between 0.1 and 20 A g−1, (c) discharge capacities cycled at 1.0 A g−1 of LICs containing AlCl3-GIC (115) negative electrodes after pre-lithiation and AC positive electrodes with electrode capacity ratios of 3:1, 5:1, and 7:1, and (d) Ragone plots of LICs containing AlCl3-GICs, the LIC containing graphite, and reported LICs containing other insertion-type negative electrodes such as Li4Ti5O12,15,16 Nb2O5,17 and Li3VO5.20 | ||
Cycling performance tests of the LIC full cells were conducted at 1.0 A (g-AC)−1. Fig. S7† and 7c show capacities and capacity retentions after each cycle for over 10000 cycles. The LIC cells effectively operated without a steep capacity decay over long periods of cycling and the retentions of the LIC cells after 10000 cycles with negative:positive capacity ratios of 7:1, 5:1, and 3:1 were 85.9, 72.8, and 53.4%, respectively. Cycling performance appeared to be related to the operating SoC range of the negative electrode, and the rate capability mentioned above.
Ragone plots of the LIC cells are shown in Fig. 7d. All energy and power densities were based on the total mass of the active materials (negative electrode and positive electrode). The maximum energy densities of the cells (AlCl3-GIC (115)//AC cell (3:1, 5:1, and 7:1)) were 71, 69, and 58 W h kg−1 at 222, 208, and 187 W kg−1, respectively. The cells retained energy densities of 20, 25, and 40 W h kg−1 at their highest power densities of 9.1, 9.8, and 15.9 kW kg−1, respectively. The forced use of a wider SoC range for the negative electrode led to a higher maximum energy density, while a narrower SoC range resulted in a higher power performance. At the same capacity ratio, the energy densities at each power density and the maximum power density of AlCl3-GIC (115)//AC were higher than those of graphite//AC. Thus, improvements in power and energy densities by using AlCl3-GIC negative electrodes were demonstrated. Compared to the LIC cells using insertion-type negative electrodes reported previously, such as Li4Ti5O12,15,16 Nb2O5,17 and Li3VO5,20 the energy densities demonstrated in this study were higher, especially at high power densities. Based on these results, the excellent performance and great prospects of AlCl3-GICs as a new LIC negative electrode are evident.
Conclusions
AlCl3-graphite intercalation compounds (GICs) were synthesized and studied as potential negative electrodes for lithium-ion capacitors. Four different AlCl3-GICs (115, 135, 150, and 200) were synthesized by reacting AlCl3 vapor with graphite at four heating temperatures (115, 135, 150, and 200 °C). Intercalation of AlCl3 was confirmed by various analytical methods, and a higher content of AlCl3 was obtained in products when heated at higher temperatures. AlCl3-GICs, especially AlCl3-GIC (115), displayed higher rate capabilities and capacities than graphite. AlCl3-GICs also displayed stable cycling operation during half-cell measurements. Discharge capacities of 105 and 53 mA h g−1 were recorded for the AlCl3-GIC (115) negative electrode at 2.0 and 5.0 A g−1, which corresponds to C-rates of 4.8 and 12, respectively. Graphite retained less capacity under the same conditions. The AlCl3-GIC (115) negative electrode was applied in LIC full cells using activated carbon (AC) as the positive electrode. Three different capacity-balanced cells were constructed with negative:positive capacity ratios of 3:1, 5:1, and 7:1. Capacity retention after 10000 cycles was 53.4, 72.8, and 85.9% for AlCl3-GIC (115)//AC cells with 3:1, 5:1, and 7:1 capacity ratios, respectively, showing the long-term stability of cells with oversized negative electrodes. For the three cells, the maximum energy densities were 71, 69, and 58 W h kg−1. The cells retained energy densities of 20, 25, and 40 W h kg−1 at their highest power densities of 9.1, 9.8, and 15.9 kW kg−1, respectively. The AlCl3-GIC (115)//AC cell displayed higher energy and power densities than the graphite//AC cell under the same cell conditions. The improved energy densities, especially at high power densities, are among the highest reported values for LICs with insertion-type negative electrodes.
Author contributions
Yamato Haniu: conceptualization, investigation, data curation, visualization, and writing – original draft preparation. Hiroki Nara: conceptualization, visualization, writing – original draft preparation, and writing – review & editing. Seongki Ahn: conceptualization and writing – review & editing. Toshiyuki Momma: funding acquisition, project administration, and supervision. Wataru Sugimoto: funding acquisition and writing – review & editing. Tetsuya Osaka: supervision and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Advanced Low Carbon Technology Research and Development Program of the Japan Science and Technology Agency (JST-ALCA, JPMJAL1008, and JPMJAL1301).References
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS.
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 CrossRef.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 RSC.
- J. Ronsmans and B. Lalande, in Electrical Systems for Aircraft, Railway and Ship Propulsion, ESARS, IEEE Computer Society, 2015, vol. 2015-May, p. 1 Search PubMed.
- S. Ahn, Y. Haniu, H. Nara, T. Momma, W. Sugimoto and T. Osaka, J. Electrochem. Soc., 2020, 167, 040519 CrossRef CAS.
- K. Naoi, S. Ishimoto, J. I. Miyamoto and W. Naoi, Energy Environ. Sci., 2012, 5, 9363–9373 RSC.
- S. R. Sivakkumar and A. G. Pandolfo, Electrochim. Acta, 2012, 65, 280–287 CrossRef CAS.
- M. Cai, X. Sun, W. Chen, Z. Qiu, L. Chen, X. Li, J. Wang, Z. Liu and Y. Nie, J. Mater. Sci., 2018, 53, 749–758 CrossRef CAS.
- J. Jiang, Y. Zhang, Z. Li, Y. An, Q. Zhu, Y. Xu, S. Zang, H. Dou and X. Zhang, J. Colloid Interface Sci., 2020, 567, 75–83 CrossRef CAS PubMed.
- W. J. Cao and J. P. Zheng, J. Power Sources, 2012, 213, 180–185 CrossRef CAS.
- J. Ni, Y. Huang and L. Gao, J. Power Sources, 2013, 223, 306–311 CrossRef CAS.
- M. Cai, X. Sun, Y. Nie, W. Chen, Z. Qiu, L. Chen, Z. Liu and H. Tang, Nano, 2017, 12(4), 1750051 CrossRef CAS.
- S. Dsoke, B. Fuchs, E. Gucciardi and M. Wohlfahrt-Mehrens, J. Power Sources, 2015, 282, 385–393 CrossRef CAS.
- Y. Liu, W. Wang, J. Chen, X. Li, Q. Cheng and G. Wang, J. Energy Chem., 2020, 50, 344–350 CrossRef.
- E. Lim, C. Jo, H. Kim, M. H. Kim, Y. Mun, J. Chun, Y. Ye, J. Hwang, K. S. Ha, K. C. Roh, S. Yoon and J. Lee, ACS Nano, 2015, 9, 7497–7505 CrossRef CAS PubMed.
- X. Qu, Y. Liu, B. Li, B. Xing, G. Huang, H. Zhao, Z. Jiang, C. Zhang, S. W. Hong and Y. Cao, J. Mater. Sci., 2020, 55, 13062–13074 CrossRef CAS.
- H. Li, X. Liu, T. Zhai, D. Li and H. Zhou, Adv. Energy Mater., 2013, 3, 428–432 CrossRef CAS.
- M. Zhang, X. Zhang, Z. Liu, H. Peng and G. Wang, Ionics, 2020, 26, 4129–4140 CrossRef CAS.
- X. Qi, J. Qu, H. bin Zhang, D. Yang, Y. Yu, C. Chi and Z. Z. Yu, J. Mater. Chem. A, 2015, 3, 15498–15504 RSC.
- Y. Sun, F. Han, C. Zhang, F. Zhang, D. Zhou, H. Liu, C. Fan, X. Li and J. Liu, Energy Technol., 2019, 7, 1801091 CrossRef.
- Z. Li, C. Zhang, F. Han, F. Wang, F. Zhang, W. Shen, C. Ye, X. Li and J. Liu, J. Mater. Chem. A, 2020, 8, 2430–2438 RSC.
- Y. Sun, F. Han, C. Zhang, F. Zhang, D. Zhou, H. Liu, C. Fan, X. Li and J. Liu, Energy Technol., 2019, 7, 1–9 Search PubMed.
- H. Shioyama, TANSO, 1994, 1994, 35–37 CrossRef.
- K. Nagai, H. Kurata, S. Isoda and T. Kobayashi, Synth. Met., 1992, 48, 99–109 CrossRef CAS.
- Y. Sun, F. Han, C. Zhang, F. Zhang, D. Zhou, H. Liu, C. Fan, X. Li and J. Liu, Energy Technol., 2019, 7, 1801091 CrossRef.
- S. K. Jeong, M. Inaba, Y. Iriyama, T. Abe and Z. Ogumi, J. Power Sources, 2008, 175, 540–546 CrossRef CAS.
- R. Mogi, M. Inaba, S.-K. Jeong, Y. Iriyama, T. Abe and Z. Ogumi, J. Electrochem. Soc., 2002, 149, A1578 CrossRef CAS.
- T. Abe, H. Fukuda, Y. Iriyama and Z. Ogumi, J. Electrochem. Soc., 2004, 151, A1120 CrossRef CAS.
- G. E. Mcguire, G. K. Schweitzer and T. A. Carlson, Inorg. Chem., 1973, 12, 2450–2453 CrossRef CAS.
- W. E. Morgan, J. R. van Wazer and W. J. Stec, J. Am. Chem. Soc., 2002, 95, 751–755 CrossRef.
- J. P. Contour, A. Salesse, M. Froment, M. Garreau, J. Thevenin and D. Warin, J. Microsc. Spectrosc. Electron., 1979, 4, 483–491 CAS.
- L. Wang, Y. Zhu, C. Guo, X. Zhu, J. Liang and Y. Qian, ChemSusChem, 2014, 7, 87–91 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08378c |
| This journal is © The Royal Society of Chemistry 2021 |