
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic mechanism of spinel oxides for oxidative electrolyte decomposition in Mg rechargeable batteries†
Jonghyun
Han
 a,
Shunsuke
Yagi
*a,
Hirokazu
Takeuchi
b,
Masanobu
Nakayama
bc and
Tetsu
Ichitsubo
d
a,
Shunsuke
Yagi
*a,
Hirokazu
Takeuchi
b,
Masanobu
Nakayama
bc and
Tetsu
Ichitsubo
d
aInstitute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8505, Japan. E-mail: syagi@iis.u-tokyo.ac.jp
bDepartment of Advanced Ceramics, Nagoya Institute of Technology, Gokiso, Showa, Nagoya, Aichi 466-8555, Japan
cDepartment of Frontier Materials, Nagoya Institute of Technology, Gokiso, Showa, Nagoya, Aichi 466-8555, Japan
dInstitute for Materials Research, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan
First published on 22nd November 2021
Abstract
One of the primary drawbacks in the development of Mg rechargeable batteries is their low operating voltage. Although electrolytes with a wide potential window have been used for high-voltage Mg rechargeable batteries, these electrolytes undergo oxidative decomposition at the surface of the positive electrode active materials at relatively low potentials. Moreover, the overpotential and kinetics of oxidative electrolyte decomposition significantly depend on the transition metal ion in spinel oxides (e.g., MgMn2O4, MgFe2O4, or MgCo2O4) used as positive electrode active materials. Because the catalytic activities of spinel oxides for electrolyte decomposition are different, electrolyte decomposition can be effectively suppressed by using transition metal ions with high overpotential for electrolyte decomposition in target spinel oxides. However, the mechanism of the catalytic reaction has not yet been elucidated. Herein, we determined that the direct electron transfer from the electrolyte to the electrode was slow, whereas the electron transfer via the oxidation reaction of spinel oxides was fast. Furthermore, we used experimental data and calculations to demonstrate that the catalytic activity for oxidative electrolyte decomposition was correlated with the valence band maximum (VBM) of spinel oxides; that is, low VBMs were correlated with high overpotentials for oxidative electrolyte decomposition.
Introduction
Recently, the demand for rechargeable batteries with high energy density has increased because the markets for long-range electric vehicles and high-performance mobile devices have expanded. Li-ion batteries (LIBs) are currently used in a wide range of applications because of their remarkable energy density and cyclability. Owing to the low standard electrode potential (−3.04 V vs. SHE) and high theoretical specific capacity (2062 mA h cm−3 or 3861 mA h g−1) of Li metal, rechargeable batteries with a Li metal negative electrode should exhibit high energy density. However, Li metal forms dendrites, which are needle-like structures, on the negative electrode surface during charging, causing the capacity to decrease, short circuits, and even fires. Hence, graphite, with a relatively low specific capacity (372 mA h g−1), has been used as a LIB negative electrode. Conversely, the tendency of Mg metal to form dendrites is lower than that of Li. Furthermore, Mg presents the lowest standard electrode potential (−2.37 V vs. SHE) among all the metals that can be safely handled in air, and its theoretical specific capacity can reach 3833 mA h cm−3 or 2205 mA h g−1.1–4 Therefore, Mg rechargeable batteries (MRBs) featuring a Mg metal negative electrode are considered some of the most promising and safe next-generation batteries with high energy density.5,6Many studies on MRBs have been conducted since 2000 when Aurbach et al. fabricated a prototype MRB featuring Mg metal as the negative electrode, Chevrel-phase Mo6S8 as the positive electrode, and Mg organohaloaluminate salts (Mg(AlCl2BuEt)2) synthesized via the complexation of MgBu2, a Lewis base, and AlCl2Et, a Lewis acid, (Bu = butyl, Et = ethyl) in tetrahydrofuran (THF), as the electrolyte.7 Even though Mo6S8 can be used at room temperature because of the high mobility of Mg2+ ions in its structure and exhibits excellent cyclability, the redox potential in the prototype MRB was low (1.0–1.2 V vs. Mg/Mg2+) and its capacity (∼75 mA h g−1) was significantly lower than the theoretical capacity (122 mA h g−1). Therefore, other compounds with higher redox potentials and capacities than Mo6S8 have been considered as positive electrode active materials (Fig. 1).7–16 Typically, the redox potentials and capacities of oxides are higher than those of sulfides. We revealed that MgM2O4 (M = Cr, Mn, Fe, Co) and Co3O4 with an oxide based spinel-type structure allow the insertion of Mg2+ ions at high potentials (∼2–3 V vs. Mg/Mg2+) and exhibit large theoretical capacities (220–270 mA h g−1).14 Especially, the redox potential of MgCo2O4 was high ∼2.9 V vs. Mg/Mg2+, and its capacity was 200 mA h g−1 (theoretical capacity: 260 mA h g−1). However, the system was maintained at 150 °C because the Mg2+ ion mobility of oxides is lower than that of sulfides, hindering room-temperature operation.17–19 In addition, several spinel oxides with controlled structures and/or compositions, such as MgMn2O4/V,20 ZnMnO3,21 Mg4V5−xNixO12,22 and Mg1+yCo2−x−yMnxO4,23 have been studied as positive electrode active materials for MRBs, but their evaluations were conducted at high temperatures (>90 °C) to enhance the mobility of Mg2+ ions. Moreover, despite their high redox potentials and theoretical capacities, MRBs featuring spinel oxides as positive electrode active materials typically suffer from being sufficiently charged because of the lack of suitable electrolytes with high anodic stability at potentials of up to ∼4 V vs. Mg/Mg2+, leading to lower specific capacity and inferior cyclability.
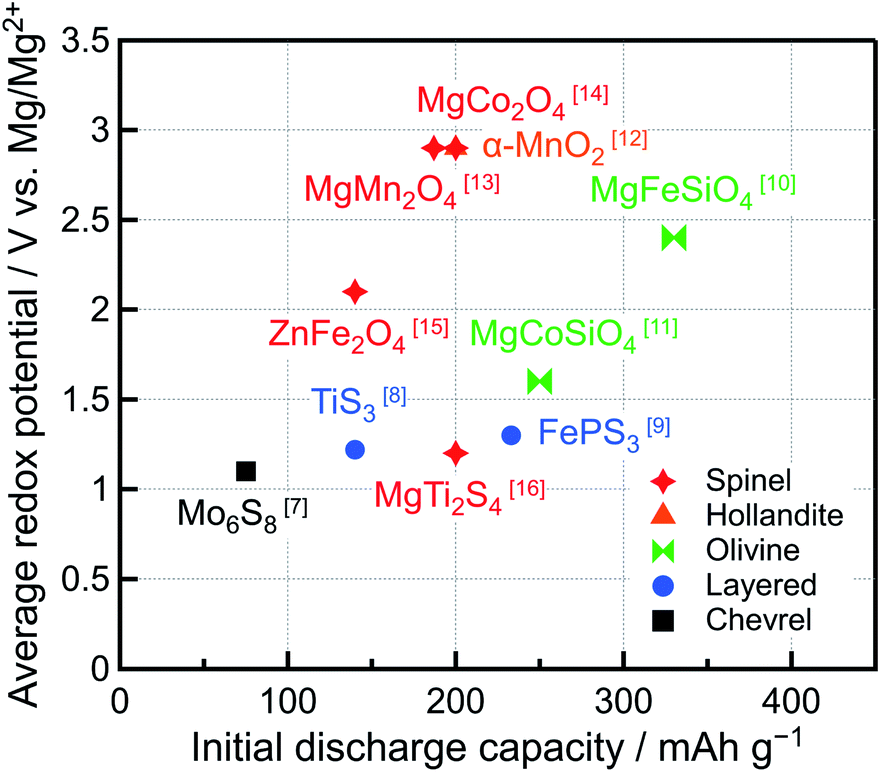 | ||
| Fig. 1 Average redox potentials and initial discharge capacities of reported positive electrode active materials for Mg rechargeable batteries. | ||
Table 1 lists MRB electrolytes with anodic stability at potentials higher than 2 V vs. Mg/Mg2+. Cl−-ion-containing electrolytes generally show a high coulombic efficiency (90–100%) of Mg deposition and dissolution. For example, 0.25 M Mg(AlCl2BuEt)2 in THF was reported to have ∼100% coulombic efficiency. However, the anodic stability was low (2.2 V vs. Mg/Mg2+). Although many Cl−-ion-containing electrolytes have been investigated, the anodic stability is not still sufficient. Furthermore, the corrosivity of Cl−-ion-containing electrolytes prevents the use of conventional and inexpensive current collectors and cell cases (e.g., Al, Ni, Cu, and stainless steel) used for LIBs. Hence, expensive Pt with high corrosion resistance was used as a current collector in many studies.24 Therefore, we focused on a bis(trifluoromethanesulfonyl)amide (TFSA)-based electrolyte, which is a Cl−-ion-free electrolyte.25 It was reported that this electrolyte inhibited electrode corrosion and its anodic stability was higher than 4.1 V vs. Mg/Mg2+ on an Al current collector; however, its coulombic efficiency was low (<60%) owing to the passivation of the Mg negative electrode in the presence of TFSA− ions. Passivation prevented Mg deposition/dissolution at room temperature and increased the overpotential for the Mg dissolution reaction during discharging.26–28 Although the anodic stability was reported to be higher than 4.1 V vs. Mg/Mg2+ on an Al current collector, the TFSA-based electrolyte was oxidatively decomposed at a potential over 3 V vs. Mg/Mg2+ when spinel oxides were used as positive electrode active materials.29 Therefore, the actual thermodynamic oxidation potential of this electrolyte is about 3 V vs. Mg/Mg2+ or less, and the reported high anodic limit of >4.1 V vs. Mg/Mg2+ was attributed to the high overpotential and sluggish kinetics of oxidative electrolyte decomposition on the Al current collector. In other words, spinel oxides have catalytic activities for anodic electrolyte decomposition. We have previously reported that oxidative electrolyte decomposition can be significantly suppressed by increasing the Fe ion content of Mg(Mn1−xFex)2O4 spinel oxides because Fe ions are less catalytically active than Mn ions.29 Changing the transition metal ion content of spinel oxides is a new method for improving the performance of positive electrode active materials of MRBs featuring conventional electrolytes.
| Type | Non-aqueous electrolytes | E ox/V vs. Mg/Mg2+ | Coulombic efficiency/% | Ref. |
|---|---|---|---|---|
| a E ox: oxidation potential; Bu: butyl; Et: ethyl; Ph: phenyl; HMDS: hexamethyldisilazane; Mes3B: tri(3,5-dimethylphenyl)borane; THF: tetrahydrofuran; DME: dimethoxyethane; G4: tetraethylene glycol dimethyl ether; Pyr1,3: N-methyl-N-propylpyrrolidinium. | ||||
| Halide-based | 0.25 M Mg(AlCl2BuEt)2 in THF | 2.2 (Pt) | ∼100 | 7, 30 and 31 |
| 0.4 M (PhMgCl)2–AlCl3 in THF | <3 (Pt) | 100 | 32 | |
| 0.5 M (HMDS-MgCl)3–AlCl3 in THF | 3.2 (Pt) | 90 | 33 | |
| 0.5 M (PhMgCl)2–Mes3B in THF | 3.5 (Pt) | 100 | 34 | |
| 0.25 M (MgCl2)2–AlCl3 in DME | 3.1 (Pt) | >99 | 35 | |
| Non-halide-based | 0.75 M Mg(CB11H12)2 in G4 | 3.8 (Al) | >99 | 36 and 37 |
| 0.5 M [Mg(G4)][TFSA]2 in [Pyr1,3][TFSA] | >4.1 (Al) | <60 | 25 and 38 | |
To the best of our knowledge, to date, no reports have been published on the effect of the transition metal ions of spinel oxides on the catalytic activity of spinel oxides for oxidative electrolyte decomposition. Therefore, in this study, we evaluated the catalytic mechanism using experimental and theoretical methods and demonstrated that oxidative electrolyte decomposition correlated with the location of the valence band maxima (VBMs) of spinel oxides and that of the highest occupied molecular orbital (HOMO) of the electrolyte.
Results and discussion
Oxidative decomposition behavior of the electrolyte
The electrochemical properties of a TFSA-based electrolyte were evaluated. The cyclic voltammogram of a cell featuring a Pt plate as the working electrode (WE), a Mg ribbon as the counter electrode (CE), Ag/Ag+ as the reference electrode (RE), and a 0.5 M [Mg(G4)][TFSA]2/[Pyr1,3][TFSA] solution as the electrolyte was obtained at 100 °C and a scan rate of 10 mV s−1 in the potential range of −3.6 to 1.4 V vs. Ag/Ag+ (Fig. 2a). The deposition/dissolution of Mg in the electrolyte was observed at ∼−2.6 V vs. Ag/Ag+. Subsequently, the measured potentials vs. Ag/Ag+ were converted into values vs. Mg/Mg2+ using the following equation: 0 V vs. Mg/Mg2+ = −2.6 V vs. Ag/Ag+, as previously described in the literature.25 The cyclic voltammograms of a cell featuring a blank electrode, which consisted of a mixture of Super P (SP) as the conductive agent, and polyvinylidene difluoride (PVdF) as the binder (SP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PVdF mass ratio of 9:1), on a Pt plate as the current collector were obtained to evaluate the anodic stability of the electrolyte in the potential range of 1–4 V vs. Mg/Mg2+ (Fig. 2b). The anodic current increased at potentials higher than 3.7 V vs. Mg/Mg2+ on the Pt plate, which was attributed to oxidative electrolyte decomposition.39,40 For comparison, the cyclic voltammetry (CV) of a cell featuring a blank electrode on an Al plate as the current collector was also measured in the wide potential range of 1–5 V vs. Mg/Mg2+ (Fig. 2c). Our results revealed that the electrolyte was oxidatively decomposed at potentials higher than 3.8 V vs. Mg/Mg2+ when the Al plate was used as the current collector. The anodic current was negligible at potentials lower than 3.8 V, indicating that the Al electrode did not dissolve even though the standard electrode potential of Al (−1.66 V vs. SHE) was only 0.7 V higher than that of Mg. In a preliminary experiment, we demonstrated that the TFSA-based electrolyte reinforces the passivation layer formed on the unpolished Al electrode (Fig. S1†), leading to the electrode stabilization over a wide potential range. In addition, Al anodic dissolution was observed at potentials higher than 2 V vs. Mg/Mg2+ when polishing the electrode surface, suggesting that the oxidized passivation layer originally formed on the Al is necessary for protecting the Al electrode. Therefore, in this study, an unpolished Al plate was used as the current collector owing to its low cost and high anodic stability.
:PVdF mass ratio of 9:1), on a Pt plate as the current collector were obtained to evaluate the anodic stability of the electrolyte in the potential range of 1–4 V vs. Mg/Mg2+ (Fig. 2b). The anodic current increased at potentials higher than 3.7 V vs. Mg/Mg2+ on the Pt plate, which was attributed to oxidative electrolyte decomposition.39,40 For comparison, the cyclic voltammetry (CV) of a cell featuring a blank electrode on an Al plate as the current collector was also measured in the wide potential range of 1–5 V vs. Mg/Mg2+ (Fig. 2c). Our results revealed that the electrolyte was oxidatively decomposed at potentials higher than 3.8 V vs. Mg/Mg2+ when the Al plate was used as the current collector. The anodic current was negligible at potentials lower than 3.8 V, indicating that the Al electrode did not dissolve even though the standard electrode potential of Al (−1.66 V vs. SHE) was only 0.7 V higher than that of Mg. In a preliminary experiment, we demonstrated that the TFSA-based electrolyte reinforces the passivation layer formed on the unpolished Al electrode (Fig. S1†), leading to the electrode stabilization over a wide potential range. In addition, Al anodic dissolution was observed at potentials higher than 2 V vs. Mg/Mg2+ when polishing the electrode surface, suggesting that the oxidized passivation layer originally formed on the Al is necessary for protecting the Al electrode. Therefore, in this study, an unpolished Al plate was used as the current collector owing to its low cost and high anodic stability.
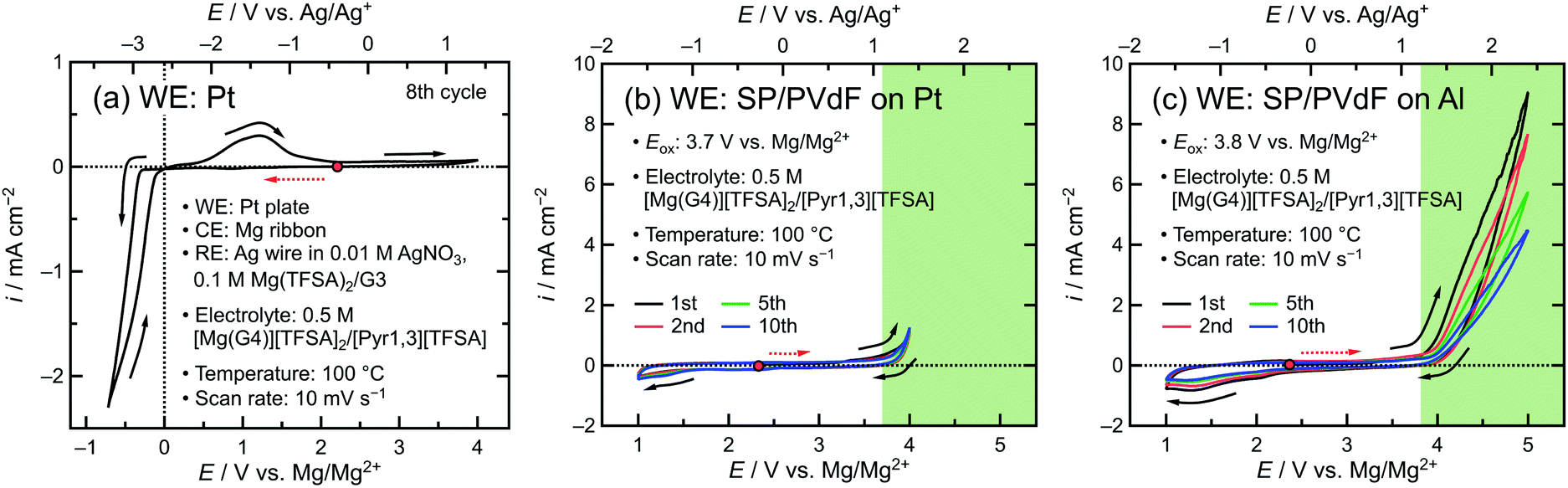 | ||
| Fig. 2 Cyclic voltammograms of cells featuring a 0.5 M [Mg(G4)][TFSA]2/[Pyr1,3][TFSA] solution as the electrolyte at 100 °C and a scan rate of 10 mV s−1. The cells comprised (a) a Pt plate, (b) a blank electrode on a Pt current collector, and (c) a blank electrode on an Al current collector as the working electrodes (WEs); a Mg ribbon as the counter electrode (CE); and a Ag wire in a 0.01 M AgNO3, 0.1 M Mg (TFSA)2/G3 solution as the reference electrode (RE). The electrolyte was decomposed at potentials higher than the oxidation potential (Eox), as indicated by the green areas in (b) and (c). The open circuit potentials and scan directions are illustrated using solid red circles and arrows, respectively. The dotted arrows indicate the initial potential scan direction. Here, G3, G4, TFSA, and Pyr1,3 denote triethyleneglycol dimethyl ether, tetraethylene glycol dimethyl ether, bis(trifluoromethanesulfonyl)amide, and N-methyl-N-propylpyrrolidinium, respectively. | ||
The cyclic voltammograms of MgM2O4 (M = Mn, Fe, and Co) spinel oxides on an Al plate as the positive electrode current collector measured in a 0.5 M [Mg(G4)][TFSA]2/[Pyr1,3][TFSA] solution at 100 °C are presented in Fig. 3a–c. It has been reported that according to the reaction described by eqn (1), the insertion of Mg2+ ions into MgM2O4 proceeded via a two-phase reaction between the MgM2O4 spinel and Mg2M2O4 rocksalt phases during discharging, as follows:14
| MgM2O4 + x(Mg2+ + 2e−) → (1 − x)MgM2O4 + xMg2M2O4 | (1) |
 | ||
| Fig. 3 Cyclic voltammograms of cells featuring (a) MgMn2O4, (b) MgFe2O4, and (c) MgCo2O4 as the working electrodes (WEs) in the cathodic direction and of cells featuring (d) MgMn2O4, (e) MgFe2O4, and (f) MgCo2O4 as the WEs in the anodic direction at 100 °C and a scan rate of 25 μV s−1. The electrolyte, counter electrode and reference electrode of the cells consisted of a 0.5 M [Mg(G4)][TFSA]2/[Pyr1,3][TFSA] solution, a Mg ribbon, and a Ag wire in 0.01 M AgNO3, 0.1 M Mg(TFSA)2/G3. The electrolyte decomposed above the oxidation potential (Eox), as indicated by the green rectangles. The open circuit potentials and scan directions are illustrated using the solid red circles and arrows, respectively; in particular, the dotted arrows indicate the initial potential scan direction. Here, G3, G4, TFSA, and Pyr1,3 denote triethyleneglycol dimethyl ether, tetraethylene glycol dimethyl ether, bis(trifluoromethanesulfonyl)amide, and N-methyl-N-propylpyrrolidinium, respectively. | ||
During charging, the reaction proceeds in the opposite direction. Hence, the cyclic voltammograms were obtained by scanning the potential in the cathodic direction from the open-circuit potential (OCP) in the range of 0.8–4 V vs. Mg/Mg2+ at a scan rate of 25 μV s−1. Two cathodic peaks are observed at 2.2 and 1.5 V vs. Mg/Mg2+ in the cyclic voltammogram of MgMn2O4 (inset of Fig. 3a). As shown in SXRD patterns for MgMn2O4 in Fig. S2a,† there is no phase transformation to rocksalt after the polarization at 1.85 V vs. Mg/Mg2+ (∼46 mA h g−1). Furthermore, only a small rocksalt peak was observed at around 13° after the polarization at 0.8 V vs. Mg/Mg2+ until the electric amount reached ∼188 mA h g−1 (theoretical capacity: 270 mA h g−1). These results suggest that the Mg2+ ions were inserted into MgMn2O4 mainly without the phase transition. It should be noted that MgCo2O4 showed the obvious phase transformation to rocksalt in Fig. S2c† as previously reported. In addition, there is a possibility of reductive electrolyte decomposition. Further study is required to clarify the emergence of the two peaks. An anodic peak, which was ascribed to the oxidation reaction of Mn ions, emerged at ∼2.75 V vs. Mg/Mg2+. The redox potential for each spinel oxide was determined as the midpoint between the onset potentials obtained using the cathodic and anodic currents. Therefore, the estimated redox potential of MgMn2O4 was ∼2.3 V vs. Mg/Mg2+. In contrast, a broad cathodic peak corresponding to the insertion of Mg2+ ions into spinel MgFe2O4 was observed at a potential lower than 2.3 V vs. Mg/Mg2+, and the cathodic peak at ∼1.2 V vs. Mg/Mg2+ was ascribed to the reduction of TFSA− ions (Fig. 3b). The anodic peak observed at ∼2.5 V vs. Mg/Mg2+ during the anodic scan was ascribed to the extraction of Mg2+ ions from rocksalt Mg2Fe2O4. The redox potential of MgFe2O4 was determined to be ∼2.1 V vs. Mg/Mg2+. Conversely, MgCo2O4 presented the highest redox potential of ∼2.4 V vs. Mg/Mg2+ among all the spinel oxides, and the distinct redox peaks corresponding to the Co2+/Co3+ valence change were observed at ∼1.6 and ∼3.2 V vs. Mg/Mg2+ (Fig. 3c).
The oxidation potentials (Eox) of the electrolyte were determined to be 3.05, 3.33, and 3.20 V vs. Mg/Mg2+ for MgMn2O4, MgFe2O4, and MgCo2O4 spinel oxides, respectively, based on the potentials at a current density of 0.5 mA g−1 during the cathodic sweep (Fig. 3a–c). The divalent transition metal ions in the corresponding rocksalt oxides also oxidized at these Eox values, resulting in residual rocksalt phases. These Eox values were lower than 3.8 V vs. Mg/Mg2+, which was the potential of the Al current collector in the absence of spinel oxides, suggesting that spinel oxides exhibited catalytic activity for oxidative electrolyte decomposition.
Therefore, next, the potential scan was initially conducted in the anodic direction from the OCP to evaluate the effect of the spinel oxides on oxidative electrolyte decomposition at the same scan rate of 25 μV s−1 (Fig. 3d–f). For MgMn2O4, the anodic current corresponding to oxidative electrolyte decomposition increased at potentials higher than 3.05 V vs. Mg/Mg2+ (Fig. 3d). The small cathodic current observed at a potential lower than 3 V vs. Mg/Mg2+ (inset of Fig. 3d) was related to the reduction of Mn4+ ions at higher potentials during electrolyte decomposition.14 However, no peaks ascribed to the Mn2+/Mn3+ redox reaction were observed even at potentials lower than 2 V vs. Mg/Mg2+, suggesting that electrolyte decomposition and/or decomposition products hindered the redox reaction. For MgFe2O4, electrolyte decomposition was significantly suppressed, and oxidative electrolyte decomposition occurred at potentials higher than 3.59 V vs. Mg/Mg2+ (Fig. 3e). The Fe2+/Fe3+ redox reaction obviously occurred, indicating that MgFe2O4 was not significantly affected by oxidative electrolyte decomposition. For MgCo2O4, oxidative electrolyte decomposition occurred at potentials higher than 3.27 V vs. Mg/Mg2+ (Fig. 3f). In contrast to MgMn2O4, for MgCo2O4, no cathodic current was observed at ∼3 V vs. Mg/Mg2+ during the cathodic scan, indicating that no Co4+ ions were formed. Similar to the cyclic voltammogram starting with a cathodic scan (Fig. 3c), the cyclic voltammogram starting with an anodic scan (Fig. 3f) presented a cathodic current at a potential lower than ∼2.5 V vs. Mg/Mg2+. However, the cathodic peak top was observed at a higher potential of ∼1.7 V vs. Mg/Mg2+ with a lower cathodic current in Fig. 3f compared to the cathodic peak top at ∼1.6 V vs. Mg/Mg2+ in Fig. 3c, suggesting that the electrolyte decomposition caused the passivation of the active materials and a decrease in the amount of MgCo2O4 participating in the reaction.
Band diagrams of spinel oxides
Before proposing a catalytic mechanism that can describe the dependence of oxidative electrolyte decomposition on the type of transition metal ions in the spinel oxides, the electronic states of the spinel oxides were determined using density functional theory (DFT) calculations. The density of states contour plots for the MgMn2O4, MgFe2O4, and MgCo2O4 slab models are presented in Fig. 4. The slab facet of the (100) surface with the lowest surface energy was selected for MgMn2O4 and MgFe2O4 (Table S1†). Owing to similarity, for MgCo2O4, the (100) surface, which is close to the most stable, was used instead of the (110) surface with the lowest surface energy. The slab models were constructed by stacking two unit cells to obtain more reliable results, where the slab thickness (>15 Å) is sufficiently larger than former studies on spinel-type LiMn2O4 (∼8 Å).41,42 | ||
| Fig. 4 Density of states (DOS) of the slab models of (a) MgMn2O4, (b) MgFe2O4, and (c) MgCo2O4 spinel oxides as a function of the energy level in the z-axis direction. | ||
The conduction band minima (CBMs), which were related to the reduction potentials of MgM2O4 (M = Mn, Fe, and Co) spinel oxides, were associated with the reduction of M3+ to M2+ upon receiving electrons. In contrast, the VBMs, which were related to the oxidation potentials of the spinel oxides, were associated with the M3+/M4+ oxidation reactions. The narrower the bandgap between the CBM and the VBM, the higher the electrical conductivity of the spinel oxide. Ultraviolet-visible spectroscopy revealed that the generalized gradient approximation (GGA)-calculated band gaps were comparable, yet smaller than the experimental values (Fig. S3†).43,44
Based on the DFT calculations (Fig. 4), we drew schematic band diagrams against the vacuum level for the MgMn2O4, MgFe2O4, and MgCo2O4 spinel oxides (Fig. 5).45 The VBMs of the spinel oxides were determined using the values at the center of the slab models. The CBMs of the spinel oxides decreased as follows: MgFe2O4 > MgMn2O4 > MgCo2O4, and these results were in agreement with the inverse trend in reduction potentials: MgFe2O4 (2.1 V vs. Mg/Mg2+) < MgMn2O4 (2.3 V vs. Mg/Mg2+) < MgCo2O4 (2.4 V vs. Mg/Mg2+) obtained from the cyclic voltammograms in Fig. 3a–c. In addition, because the spinel oxides did not undergo reduction upon immersion in the electrolyte, it was suggested that the HOMO of the electrolyte was lower than the CBMs of the spinel oxides. The specific position of the HOMO of the electrolyte is discussed in the following section.
 | ||
| Fig. 5 Schematic band diagram of the slab models of MgMn2O4, MgFe2O4, and MgCo2O4vs. the vacuum level (CB: conduction band, VB: valence band). | ||
Catalytic mechanism of oxidative electrolyte decomposition
The schematic of the catalytic mechanism of oxidative electrolytic decomposition is illustrated in Fig. 6. The experimentally demonstrated dependence of the Eox value of the electrolyte on the spinel oxides suggested that the direct charge transfer from the electrolyte to the electrode was slow. However, charge transfer through the temporary oxidation of the spinel oxides was promoted. Specifically, each spinel oxide was oxidized when the electronic energy of the electrode connected to the potentiostat became lower or the applied potential became higher than the VBM of the spinel oxide. The electron transfer from the electrolyte to the temporarily oxidized spinel oxides was immediately followed by charge compensation. Consequently, oxidative electrolyte decomposition proceeded based on the position of the VBM; the lower the VBM, the higher the Eox value of the electrolyte. According to the DFT calculations, the VBMs of the spinel oxides decrease as follows MgCo2O4 > MgMn2O4 > MgFe2O4 (Fig. 5), suggesting that MgFe2O4 exhibited the highest Eox value. The experimental results confirmed that MgFe2O4 presented the highest Eox value (3.59 V vs. Mg/Mg2+) among all the spinel oxides in this study (Fig. 3). However, the experimentally determined Eox value of the electrolyte on MgMn2O4 (3.05 V vs. Mg/Mg2+) was lower than that on MgCo2O4 (3.27 V vs. Mg/Mg2+), which contradicted the calculation results. The reason for this is explained below. | ||
| Fig. 6 Schematic energy level diagram of the electrolyte and positive electrode (PE) used to analyze oxidative electrolyte decomposition (HOMO: highest occupied molecular orbital, LUMO: lowest unoccupied molecular orbital, VBM: valence band maximum). Typically, if the electrolyte presents sufficiently high anodic stability, Mg2+ is extracted from spinel oxides for charge compensation during charging. Conversely, if the HOMO of the electrolyte is higher than the VBM of the cathode, the electrolyte is expected to be decomposed instead of undergoing Mg2+ extraction. | ||
In addition to the charge transfer from the electrolyte to the spinel oxides, Mg2+ extraction can promote the neutralization of the oxidized spinel oxides. Owing to the strong electrostatic attraction between Mg2+ and O2− ions, the extraction of Mg2+ ions from the spinel oxide except for chromate and manganate was hindered.14 A small cathodic current was observed at 3 V vs. Mg/Mg2+ only for the MgMn2O4 spinel oxide (Fig. 3d); however, the main reaction at higher potentials was oxidative electrolyte decomposition. Furthermore, we also confirmed that a small Mn3+/Mn4+ valence change occurred, as shown in Fig. S4.† Therefore, we hypothesized that the HOMO of the electrolyte was located just below the VBM of MgMn2O4. The extraction of Mg2+ ions from MgMn2O4 occurred when the electronic energy of the electrode became lower than the VBM upon charging. If the electronic energy of the electrode became lower than the HOMO of the electrolyte, electron transfer from the electrolyte occurred. Unlike MgMn2O4, MgCo2O4 did not undergo oxidation when the electronic energy of the electrode was intermediate between the VBM of the spinel oxide and the HOMO of the electrolyte because of the strong electrostatic attraction between Mg2+ and O2− ions. Therefore, the Eox value of MgMn2O4 was lower than that of MgCo2O4. Consequently, the Eox values of the electrolyte correlated with the positions of the VBMs of the positive electrode active materials unless the HOMO level of the electrolyte was low.
The overpotential and kinetics of oxidative electrolyte decomposition depended on the type of transition metal ions in the spinel oxides. In Fig. 3, the slopes of the anodic currents accompanying oxidative electrolyte decomposition increased in the same order as the Eox values: MgMn2O4 < MgCo2O4 < MgFe2O4. The linear correlation between the potential and logarithmic current density determined using the cyclic voltammograms in Fig. 3 is presented in Fig. 7. The Tafel slopes of MgMn2O4, MgCo2O4, and MgFe2O4 spinel oxides were 439, 377, and 97 mV dec−1, respectively (note that the order of the Tafel slopes is the opposite of the order of the corresponding slopes of the voltammograms). According to the Butler–Volmer equation, when charge transfer is the rate-limiting step, the current density increases exponentially with increasing polarization. In this case, the catalyst with the lowest overpotential should exhibit the largest current density at the same potential. However, the experimental data contradicted the electrochemical reaction given by the Butler–Volmer equation. Therefore, oxidative electrolytic decomposition was not rate-limited by charge transfer, and the overpotential and current slope should be evaluated separately when discussing catalytic activity.
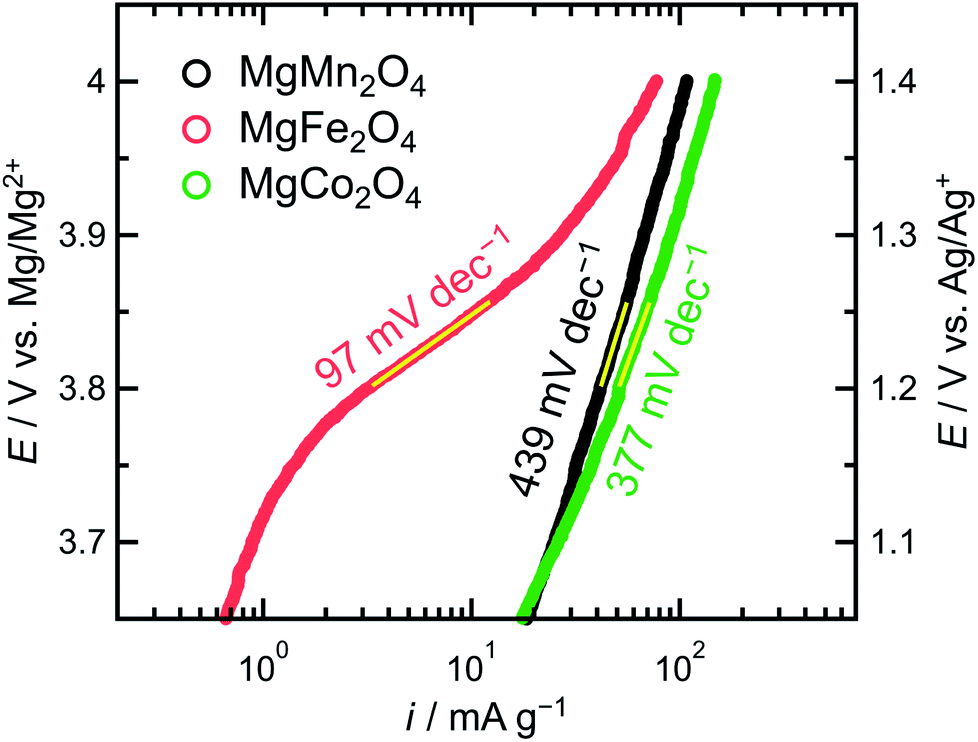 | ||
| Fig. 7 Tafel plots of the MgMn2O4, MgFe2O4, and MgCo2O4 spinel oxides. The Tafel slopes were determined in the potential range of 3.80–3.86 V vs. Mg/Mg2+. | ||
Experimental
Synthesis
Nanocrystalline MgM2O4 (M = Mn, Fe, and Co) powders were synthesized using an inverse co-precipitation method.46 Mg(NO3)2·6H2O (8 mmol) and Mn(NO3)2·6H2O, Fe(NO3)3·9H2O, or Co(NO3)2·6H2O (16 mmol) were dissolved in 100 mL of deionized water under stirring (500 rpm) at room temperature. Na2CO3 (70 mmol) was dissolved in 200 mL of deionized water under stirring (500 rpm) at 80 °C. Thereafter, the mixed metallic nitrate salt aqueous solution was added to a sodium carbonate aqueous solution using a peristaltic pump (ATTO, SJ-1211 II-H) at a rate of 1–2 mL min−1 under vigorous stirring (700 rpm). Because of the precipitation reaction, the clear solution gradually turned into light brown (Mn), dark brown (Fe), or navy (Co) suspensions. The suspensions were stirred for 30 min without heating them, followed by filtration under vacuum. The filtered precipitates (precursors) were rinsed with deionized water at 80 °C to remove the residual Na-containing by-products. Thereafter, the pH of the filtrate, which was measured using pH test paper, was adjusted to ∼7, followed by drying the precursors in air at 80 °C for 24 h. Subsequently, the agglomerated precursors were pulverized via ball milling at 300 rpm for 8 × 15 min in 5 min intervals, followed by calcination at 500 °C for 12 h at a heating rate of 10 °C min−1.Electrochemical tests
The electrochemical performance of the MgM2O4 (M = Mn, Fe, and Co) spinel oxides was evaluated using a three-electrode cell. We used a composite electrode, a Mg ribbon (99.9%) polished with sandpaper (#150), and a Ag/Ag+ electrode as the WE, CE, and RE, respectively. The Ag/Ag+ electrode was fabricated by immersing a Ag wire into a glass tube containing a porous vycor glass and 450 μL of triethyleneglycol dimethyl ether (G3) solution containing 0.01 M AgNO3 and 0.10 M Mg(TFSA)2. The composite electrode was prepared from a slurry comprising a mixture of the calcined nanocrystalline MgM2O4 powder (active material), SP (conductive agent), and PVdF (binder) with a mass ratio of 8:1:1 dissolved in N-methyl-2-pyrrolidone (NMP). After the addition of 600 μL of NMP, the slurry was mixed using a rotation–revolution mixer (Thinky, ARE-310) at 2000 rpm for 20 min, spread on an Al plate (current collector), and dried under vacuum at 80 °C overnight.
The [Mg(G4)][TFSA]2/[Pyr1,3][TFSA] electrolyte was prepared according to the method described in a previous paper.25 Mg[TFSA]2 (5 mmol) and G4 (5 mmol) were added to a 10 mL volumetric flask, and the flask was filled to the mark with [Pyr1,3][TFSA]. The mixture was vigorously stirred at 100 °C overnight. The G3, G4, and [Pyr1,3][TFSA] solutions were dried over molecular sieves (3 Å), heated at 250 °C for 3 h and then again heated at 300 °C for 4.5 h under vacuum before use. The three-electrode cell was fabricated in an Ar-filled glove box with a dew point lower than −80 °C.
All the electrochemical measurements were performed at 100 °C using a three-electrode cell set in an Al block placed on a hot plate to maintain a constant temperature. The cyclic voltammograms of MgM2O4 were obtained using a potentiostat (Biologic, VSP-300) at a scan rate of 25 μV s−1. The lowest cut-off potential was set at 0.8 V vs. Mg/Mg2+ because the reductive decomposition of the TFSA-based electrolyte occurred at lower potentials.
Material characterization
The valence state of transition metal ions in spinel oxides before and after chronocoulometry under potentiostatic control of 4 V vs. Mg/Mg2+ was investigated by X-ray absorption spectroscopy (XAS) at the BL14B2 beamline of Spring-8. The samples were prepared by mixing active materials with boron nitride (diluent) and pressing them into pellets.The spinel-type MgM2O4 (M = Mn, Fe, and Co) compounds were packed in a Lindeman glass capillary with an outer diameter of 0.5 mm and a glass thickness of 0.01 mm and were subjected to synchrotron X-ray diffraction (SXRD) analysis at the BL02B2 beamline of SPring-8. The X-ray wavelength was 0.4200 Å for MgM2O4 powders and 0.5000 Å for the composite electrode as prepared and after chronocoulometry, which were calibrated using a CeO2 standard. The crystal structures were drawn using the VESTA software,47 and Rietveld refinement was performed using the SXRD data and the RIETAN-FP software (Table S2,and Fig. S5†).48
Computational details
First-principles calculations based on the DFT data were performed to optimize the crystal structures of the bulk and surface models of spinel-type MgM2O4 (M = Mn, Fe, and Co). We used the Vienna ab initio simulation package (VASP)49 with the modified Perdew–Burke–Ernzerhof GGA for solids (PBEsol-GGA)50,51 and the projector-augmented wave (PAW) method.52 On-site Coulomb correction (DFT + U) was performed for the localized electronic states, and the U values for the Mn, Fe, and Co 3d states were set to 3.9, 5.3, and 3.32 eV, respectively, using data reported in the literature.53 First, structural calculations were performed for the bulk structures of the MgM2O4 (M = Mn, Fe, and Co) spinel oxides using the DFT + U approach under the assumption that the Mg and M ions occupied tetrahedral and octahedral sites, respectively. This assumption was based on the symmetry requirements for surface electronic structure calculations, although we reported that Mg and M exchange occurred in the MgM2O4 compounds.46 A conventional Mg8M16O32 (Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) face-centered unit cell model and 3 × 3 × 3 k-point grids were used. After determining the cubic lattice parameters of the spinel oxides, the surface structures were computed using the slab technique, wherein a set of infinite layers separated by vacuum layers were repeated periodically along the surface normal. The low-index facets of the (001), (011), and (111) surfaces consisting of charge-balanced stoichiometric slabs were modeled in this study using data from the literature.42 The slab thicknesses for the (001), (011), and (111) surfaces were ∼16, ∼12, and ∼11 Å, respectively, and that of the vacuum layers was set to ∼30 Å. In addition, the slabs were constructed such that their sides were symmetrically equivalent and could be mapped into each other via an inversion or mirror type of symmetry operation in the middle of the slabs. The k-point grids for the surface models were set to be 3 × 3 × 1.
m) face-centered unit cell model and 3 × 3 × 3 k-point grids were used. After determining the cubic lattice parameters of the spinel oxides, the surface structures were computed using the slab technique, wherein a set of infinite layers separated by vacuum layers were repeated periodically along the surface normal. The low-index facets of the (001), (011), and (111) surfaces consisting of charge-balanced stoichiometric slabs were modeled in this study using data from the literature.42 The slab thicknesses for the (001), (011), and (111) surfaces were ∼16, ∼12, and ∼11 Å, respectively, and that of the vacuum layers was set to ∼30 Å. In addition, the slabs were constructed such that their sides were symmetrically equivalent and could be mapped into each other via an inversion or mirror type of symmetry operation in the middle of the slabs. The k-point grids for the surface models were set to be 3 × 3 × 1.
Conclusions
In summary, we demonstrated the oxidative decomposition mechanism of a TFSA-based electrolyte. The charge transfer from the electrolyte to the electrode was slow and was promoted or catalyzed by the temporary oxidation of spinel oxides. The Eox values of the electrolyte depended on the VBMs of spinel oxides; the lower the VBM, the higher the Eox value of the electrolyte. DFT calculations demonstrated that the VBM of MgFe2O4 was the lowest among all the spinel oxides, which was in agreement with the highest Eox value of the electrolyte on MgFe2O4 (i.e., the largest overpotential for oxidative electrolyte decomposition). Therefore, the addition of ions with lower VBMs, such as Fe ions, can suppress oxidative electrolyte decomposition, improving the stability of MRBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Professor Kiyoshi Kanamura (Tokyo Metropolitan University) and Dr Kohei Uosaki (National Institute for Materials Science) for their valuable advice. This study was supported by the Advanced Low Carbon Technology Research and Development Program, Specially Promoted Research for Innovative Next Generation Batteries (ALCA-SPRING: JPMJAL1301) of the Japan Science and Technology Agency (JST) and the Japan Society for the Promotion of Science (JSPS: 18H05249, 20H05180). This study was also financially supported by a Grant-in-Aid from The University of Tokyo Excellent Young Researcher and the GIMRT Program of the Institute for Materials Research, Tohoku University (Proposal No. 20K0086). XAS and SXRD measurements were conducted at the BL14B2 beamline (Proposal No. 2021B1726) and the BL02B2 beamline (Proposal No. 2020A0583 and 2021B1327) of SPring-8, respectively.Notes and references
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265 RSC.
- P. Canepa, G. Sai Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Chem. Rev., 2017, 117, 4287–4341 CrossRef CAS PubMed.
- I. Shterenberg, M. Salama, Y. Gofer, E. Levi and D. Aurbach, MRS Bull., 2014, 39, 453–460 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton 2003 Search PubMed.
- M. Matsui, J. Power Sources, 2011, 196, 7048–7055 CrossRef CAS.
- C. You, X. Wu, X. Yuan, Y. Chen, L. Liu, Y. Zhu, L. Fu, Y. Wu, Y.-G. Guo and T. van Ree, J. Mater. Chem. A, 2020, 8, 25601–25625 RSC.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- K. Taniguchi, Y. Gu, Y. Katsura, T. Yoshino and H. Takagi, Appl. Phys. Express, 2016, 9, 011801 CrossRef.
- M. Wang, J. Han, W. Liu, M. Kamiko and S. Yagi, J. Alloys Compd., 2021, 883, 160822 CrossRef CAS.
- Y. Orikasa, T. Masese, Y. Koyama, T. Mori, M. Hattori, K. Yamamoto, T. Okado, Z.-D. Huang, T. Minato, C. Tassel, J. Kim, Y. Kobayashi, T. Abe, H. Kageyama and Y. Uchimoto, Sci. Rep., 2015, 4, 5622 CrossRef PubMed.
- Y. Nuli, Y. Zheng, Y. Wang, J. Yang and J. Wang, J. Mater. Chem., 2011, 21, 12437–12443 RSC.
- T. Hatakeyama, H. Li, N. L. Okamoto, K. Shimokawa, T. Kawaguchi, H. Tanimura, S. Imashuku, M. Fichtner and T. Ichitsubo, Chem. Mater., 2021, 33, 6983–6996 CrossRef CAS.
- T. Hatakeyama, N. L. Okamoto, K. Shimokawa, H. Li, A. Nakao, Y. Uchimoto, H. Tanimura, T. Kawaguchi and T. Ichitsubo, Phys. Chem. Chem. Phys., 2019, 21, 23749–23757 RSC.
- S. Okamoto, T. Ichitsubo, T. Kawaguchi, Y. Kumagai, F. Oba, S. Yagi, K. Shimokawa, N. Goto, T. Doi and E. Matsubara, Adv. Sci., 2015, 2, 1500072 CrossRef PubMed.
- K. Shimokawa, T. Atsumi, M. Harada, R. E. Ward, M. Nakayama, Y. Kumagai, F. Oba, N. L. Okamoto, K. Kanamura and T. Ichitsubo, J. Mater. Chem. A, 2019, 7, 12225–12235 RSC.
- X. Sun, P. Bonnick, V. Duffort, M. Liu, Z. Rong, K. A. Persson, G. Ceder and L. F. Nazar, Energy Environ. Sci., 2016, 9, 2273–2277 RSC.
- S. K. Kolli and A. Van der Ven, Chem. Mater., 2021, 33, 6421–6432 CrossRef CAS.
- T. Chen, G. Sai Gautam and P. Canepa, Chem. Mater., 2019, 31, 8087–8099 CrossRef CAS.
- Z. Rong, R. Malik, P. Canepa, G. Sai Gautam, M. Liu, A. Jain, K. Persson and G. Ceder, Chem. Mater., 2015, 27, 6016–6021 CrossRef CAS.
- K. Ishii, S. Doi, R. Ise, T. Mandai, Y. Oaki, S. Yagi and H. Imai, J. Alloys Compd., 2020, 816, 152556 CrossRef CAS.
- K. Shimokawa, T. Atsumi, N. L. Okamoto, T. Kawaguchi, S. Imashuku, K. Wagatsuma, M. Nakayama, K. Kanamura and T. Ichitsubo, Adv. Mater., 2021, 33, 2007539 CrossRef CAS PubMed.
- Y. Idemoto, N. Kawakami, N. Ishida and N. Kitamura, J. Power Sources, 2020, 455, 227962 CrossRef CAS.
- Y. Idemoto, M. Ichiyama, N. Ishida and N. Kitamura, J. Power Sources, 2021, 482, 228920 CrossRef CAS.
- S. Yagi, A. Tanaka, Y. Ichikawa, T. Ichitsubo and E. Matsubara, J. Electrochem. Soc., 2013, 160, C83–C88 CrossRef CAS.
- T. Mandai, K. Tatesaka, K. Soh, H. Masu, A. Choudhary, Y. Tateyama, R. Ise, H. Imai, T. Takeguchi and K. Kanamura, Phys. Chem. Chem. Phys., 2019, 21, 12100–12111 RSC.
- S.-Y. Ha, Y.-W. Lee, S. W. Woo, B. Koo, J.-S. Kim, J. Cho, K. T. Lee and N.-S. Choi, ACS Appl. Mater. Interfaces, 2014, 6, 4063–4073 CrossRef CAS PubMed.
- H. Kuwata, M. Matsui and N. Imanishi, J. Electrochem. Soc., 2017, 164, A3229–A3236 CrossRef CAS.
- S. J. Kang, H. Kim, S. Hwang, M. Jo, M. Jang, C. Park, S. T. Hong and H. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 517–524 CrossRef CAS PubMed.
- J. Han, S. Yagi and T. Ichitsubo, J. Power Sources, 2019, 435, 226822 CrossRef CAS.
- S. Yagi, A. Tanaka, T. Ichitsubo and E. Matsubara, ECS Electrochem. Lett., 2012, 1, D11–D14 CrossRef CAS.
- S. Yagi, A. Tanaka, Y. Ichikawa, T. Ichitsubo and E. Matsubara, Res. Chem. Intermed., 2014, 40, 3–9 CrossRef CAS.
- O. Mizrahi, N. Amir, E. Pollak, O. Chusid, V. Marks, H. Gottlieb, L. Larush, E. Zinigrad and D. Aurbach, J. Electrochem. Soc., 2008, 155, A103 CrossRef CAS.
- H. S. Kim, T. S. Arthur, G. D. Allred, J. Zajicek, J. G. Newman, A. E. Rodnyansky, A. G. Oliver, W. C. Boggess and J. Muldoon, Nat. Commun., 2011, 2, 427 CrossRef PubMed.
- Y. Guo, F. Zhang, J. Yang, F. Wang, Y. NuLi and S. Hirano, Energy Environ. Sci., 2012, 5, 9100 RSC.
- R. E. Doe, R. Han, J. Hwang, A. J. Gmitter, I. Shterenberg, H. D. Yoo, N. Pour and D. Aurbach, Chem. Commun., 2014, 50, 243–245 RSC.
- O. Tutusaus, R. Mohtadi, T. S. Arthur, F. Mizuno, E. G. Nelson and Y. V. Sevryugina, Angew. Chem., Int. Ed., 2015, 54, 7900–7904 CrossRef CAS PubMed.
- H. Dong, O. Tutusaus, Y. Liang, Y. Zhang, Z. Lebens-Higgins, W. Yang, R. Mohtadi and Y. Yao, Nat. Energy, 2020, 5, 1043–1050 CrossRef CAS.
- T. Mandai, Y. Akita, S. Yagi, M. Egashira, H. Munakata and K. Kanamura, J. Mater. Chem. A, 2017, 5, 3152–3156 RSC.
- T. Mandai, K. Yoshida, S. Tsuzuki, R. Nozawa, H. Masu, K. Ueno, K. Dokko and M. Watanabe, J. Phys. Chem. B, 2015, 119, 1523–1534 CrossRef CAS PubMed.
- S. Terada, T. Mandai, S. Suzuki, S. Tsuzuki, K. Watanabe, Y. Kamei, K. Ueno, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2016, 120, 1353–1365 CrossRef CAS.
- A. Karim, S. Fosse and K. A. Persson, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 075322 CrossRef.
- M. Nakayama, H. Taki, T. Nakamura, S. Tokuda, R. Jalem and T. Kasuga, J. Phys. Chem. C, 2014, 118, 27245–27251 CrossRef CAS.
- W. B. Park, S. U. Hong, S. P. Singh, M. Pyo and K.-S. Sohn, ACS Omega, 2016, 1, 483–490 CrossRef CAS PubMed.
- J.-H. Yuan, Q. Chen, L. R. C. Fonseca, M. Xu, K.-H. Xue and X.-S. Miao, J. Phys. Commun., 2018, 2, 105005 CrossRef.
- M. Nakayama, K. Ishida, K. Watanabe, N. Tanibata, H. Takeda, H. Maeda and T. Kasuga, ACS Omega, 2020, 5, 4083–4089 CrossRef CAS PubMed.
- S. Yagi, Y. Ichikawa, I. Yamada, T. Doi, T. Ichitsubo and E. Matsubara, Jpn. J. Appl. Phys., 2013, 52, 025501 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- F. Izumi and K. Momma, Solid State Phenom., 2007, 130, 15–20 CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08115b |
| This journal is © The Royal Society of Chemistry 2021 |