
Achieving performance and longevity with butane-operated low-temperature solid oxide fuel cells using low-cost Cu and CeO2 catalysts†
Cam-Anh
Thieu
 ab,
Sungeun
Yang
*a,
Ho-Il
Ji
ab,
Hyoungchul
Kim
a,
Kyung Joong
Yoon
a,
Jong-Ho
Lee
ab and
Ji-Won
Son
*abc
ab,
Sungeun
Yang
*a,
Ho-Il
Ji
ab,
Hyoungchul
Kim
a,
Kyung Joong
Yoon
a,
Jong-Ho
Lee
ab and
Ji-Won
Son
*abc
aEnergy Materials Research Center, Korea Institute of Science and Technology (KIST), 14-5 Hwarang-ro, Seongbuk-gu, Seoul 02792, Republic of Korea. E-mail: syang@kist.re.kr; jwson@kist.re.kr
bDivision of Nano & Information Technology, KIST School, Korea University of Science and Technology (UST), Seoul 02792, Republic of Korea
cGraduate School of Energy and Environment, KU-KIST Green School, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
First published on 26th October 2021
Abstract
The use of thin-film solid oxide fuel cells (TF-SOFCs) can effectively lower the operating temperature of a typical solid oxide fuel cell (SOFC) below 600 °C, while maintaining high efficiency and using low-cost catalysts. However, the fuel flexibility in SOFCs becomes a significant challenge at lower operating temperatures, resulting in the need for expensive noble-metal catalysts. The effective implementation of low-cost catalysts, Cu and CeO2, in a TF-SOFC presents a solution to this problem. Cu is inserted directly near the electrolyte–anode interface via a combination of pulsed laser deposition and sputtering to assist the electrochemical reactions, and the anode support, which constitutes the main volume of the cell, is infiltrated with CeO2 to effectively facilitate thermochemical reforming reactions. A comprehensive study of catalyst-modified cells (Cu–Ce-cell, Ce-cell, and Cu-cell) and a Ni/YSZ reference cell (ref-cell) is performed over n-butane fuel in an operating temperature range of 500 to 600 °C. The cell incorporating Cu and CeO2 (Cu–Ce-cell) shows a record high performance for a hydrocarbon-fueled SOFC, with a peak power density of 1120 mW cm−2 at 600 °C. Cu and CeO2 improve the activity of the steam reforming reaction, and CeO2 expands the triple-phase boundary, increasing the electrochemical activity. Cu–Ce-cell also degrades at a much slower rate than ref-cell. Post-reaction analysis proves that the drastic improvement in longevity is achieved as a result of the enhanced carbon deposition resistance of Cu–Ce-cell.
1. Introduction
High-temperature fuel cells based on ceramic materials (solid oxide fuel cells, SOFCs) offer high electrical efficiency, low levels of contaminant emission, and fuel flexibility. This fuel flexibility is especially beneficial compared to other types of fuel cell technology, as hydrocarbon fuels can be used without complex pretreatment. SOFCs could play a crucial role in accelerating the transition from an economy based on fossil fuels to one based on renewable fuels.1–6 Despite their potential, the high operating temperatures (>800 °C) required pose a serious challenge, leading to issues such as fast cell degradation, the need for expensive construction materials, and slow startup–shutdown cycles. In particular, the latter impedes the application of SOFCs for small-scale and portable applications.SOFCs that operate below 600 °C (denoted as LT-SOFCs) have attracted increasing attention, as they can overcome the above-mentioned challenges associated with conventional SOFCs.7–9 LT-SOFCs will enable the application of SOFCs for small-scale and portable applications and support the adoption of low-cost materials. Challenges related to the development of LT-SOFCs include the limited mobilities of oxide ions, the slower kinetics of electrode reactions, and reduced reforming reaction rates caused by the lower operating temperature.10 Kilner has been a pioneer in the development of novel materials suitable for use in LT-SOFCs through pursuing a fundamental understanding of oxide materials that are involved in the diffusion and exchange of oxygen.11–16 Greatly benefiting from his work, LT-SOFCs have evolved over the last decade, with remarkable improvements achieved via applying suitable materials, implementing thin-film deposition techniques, and incorporating nanometer-scale tailored materials.17–19
The incorporation of nanostructures into LT-SOFCs via thin-film deposition decreases the thickness of the electrolyte and allows for the creation of newly developed functionalities that can significantly improve the performance of the LT-SOFC.20–25 Our group has been developing a thin-film-based SOFC that is built on a sintered, conventional anode–electrolyte composite (cermet) support. This is classified as a “multiscale-architecture thin-film SOFC” (denoted as TF-SOFC) because materials on a variety of different scales, from nanometer to centimeter, were integrated to construct the anode-supported thin-film-based SOFC.8 The performances of TF-SOFCs, operated below 600 °C, were comparable to or even better than those of high-temperature SOFCs operated above 800 °C.26
In addition to the operating temperature, the fuel selection and the accommodation of internal reforming are critical issues when aiming to minimize the system volume and weight for small-scale and portable SOFC applications. n-Butane (n-C4H10) has a high energy density (29.7 MJ L−1 and 49.5 MJ kg−1)27 and it can be easily stored and used in liquid form, unlike H2 and CH4. The vapor pressure of butane at ambient temperature, ∼2.43 bar at 25 °C, is low enough so as to not require a high-strength container but high enough to eliminate the need for a fuel pump.28 The reforming mode is also important for portable applications. In a typical Ni–YSZ cermet fuel anode, hydrocarbon fuel is reformed to H2 and CO before undergoing electrochemical oxidation. In internal reforming mode, the reforming reaction takes place inside the fuel cell anode where Ni serves as the reforming catalyst. Alternatively, an external catalytic reformer is added to the system in external reforming mode. An external reformer is not suitable for portable applications as it adds weight and volume to the system, and it may even lower the overall efficiency.29 Low-temperature operation, fuel selection, and the accommodation of internal reforming are key issues to be solved for small-scale and portable SOFCs.
Significant challenges arise when hydrocarbons are used in internal-reforming LT-SOFCs. The steam reforming reaction of hydrocarbons is highly endothermic, leading to poor reforming reaction rates at lower temperatures. Moreover, carbon deposition is thermodynamically favored at operating temperatures below 700 °C.30 Therefore, a high steam-to-carbon ratio (SCR), from 2 to 3, and a suitable catalyst are required to avoid carbon deposition and to increase the reforming reaction rate at lower temperatures. Ni is the most commonly used anode catalyst in SOFCs. Unfortunately, the high catalytic activity of Ni in hydrocarbon cracking can lead to severe carbon deposition on the Ni surface. Ni promotes the formation of carbon filaments from hydrocarbons under a range of operating conditions, and a high SCR is not sufficient in butane-fueled SOFCs to prevent carbon deposition.28,31,32 Therefore, it is vital to properly tailor the catalyst material in the fuel electrode. One of the approaches to overcome the limitations of Ni is to add a reforming catalyst, usually a noble metal such as Ru, Pt, and Rh,33–35 or a transition metal, such as Cu, to suppress carbon deposition.36–38 Generally, noble metals display better resistance to carbon deposition; however, their high cost and limited availability restrict their application. The costs of different catalyst materials are listed in Table S1.†
In our previous work, the effects of various catalyst materials (Ru, Pd, and Cu) on the performances of internal-steam-reforming butane-fueled TF-SOFCs (ISRB–TF-SOFCs) under low-temperature conditions were systematically investigated. The catalysts were incorporated near the electrolyte–anode interface, anode functional layer (AFL), of TF-SOFCs via the sandwich-like alternating deposition of sputtering and pulsed laser deposition (PLD). The metal catalyst was deposited via sputtering, and the NiO–YSZ composite was deposited via PLD.38,39 Among the catalysts tested, Ru showed the best performance; however, Cu also displayed promising results, especially at 500 °C. Therefore, in the current study, we tried to further improve the performance and durability of ISRB–SOFCs at lower operating temperatures using economical catalysts. To achieve this, Cu and CeO2 were chosen as the catalysts for use in ISRB–SOFCs. Cu, one of the most affordable transition metals, is extensively used as a catalyst in hydrocarbon oxidation reactions.36–38 Moreover, the use of Cu is an economical solution in processes involving hydrocarbon reforming and CO oxidation, including the water-gas-shift (WGS) reaction.40–42 CeO2 is a well-known oxidation catalyst and it has been commonly used in numerous catalytic reactions, including the WGS reaction, hydrocarbon oxidation, partial oxidation, and steam reforming.43–49 The superior catalytic capabilities of CeO2 are associated with the reversible redox pair Ce3+/Ce4+.49 The infiltration of the anode layer of a SOFC with CeO2 can assist the conversion of hydrocarbon fuels, thereby improving the performance of the fuel cell.4,50 In addition, CeO2 is also used in direct hydrocarbon fuel cells owing to its resistance to carbon deposition.51,52 The insertion of CeO2 into the anode layer has been effective in improving the electrocatalytic activity over both hydrogen and hydrocarbon fuels via expanding the triple phase boundary, leading to enhanced electrochemical reaction performance.53 However, most of these studies were performed under high-temperature conditions, from 750 to 900 °C, and with methane as the fuel.54–57
In this work, Cu and CeO2 were incorporated into two different regions of a TF-SOFC to achieve both high performance and durability of the ISRB-SOFC. Cu was sputter-deposited at the AFL via sandwich-like placement between the NiO–YSZ composite layers. This strategy enabled the homogeneous dispersion of the Ni–Cu alloy at the electrolyte–anode interface where electrochemical reactions occur. CeO2 nanoparticles were used to cover the entire anode region using urea-assisted infiltration. Urea acted as a complexing agent and in situ precipitation agent, ensuring the uniform distribution of the nanometer-sized CeO2.58 The catalytic properties of Cu- and CeO2-modified Ni–YSZ powder are first investigated at the powder level using a packed bed reactor. The reforming reaction rate and long-term stability were improved upon incorporating either Cu or CeO2 into Ni–YSZ powder. Then, TF-SOFCs with four different catalyst insertion configurations were tested as ISRB–SOFCs, and the electrochemical performances of these TF-SOFCs under different operating conditions were compared at temperatures below 600 °C. Enhancements in both the performance and longevity of TF-SOFCs upon the incorporation of Cu and CeO2 are clearly demonstrated.
2. Materials and methods
2.1. Evaluation of the catalytic properties at the powder level
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :44 wt%; NiO from Sumitomo Metal Mining, Japan; YSZ from Tosoh Corp., Japan) powder, which was pre-treated at 1200 °C for 1 h in air to homogenize the anode particle size; the samples were denoted as Ce–Ni–YSZ powder and Cu–Ni–YSZ powder, respectively. The same number of moles of Ce or Cu was deposited onto Ni–YSZ powder, at a molar ratio of 1:30 (Ce or Cu:Ni), and the weight fractions are 3.38 and 1.56 wt%, respectively. The catalyst-infiltrated powder was thermally treated in air for 2 h at 80 °C to decompose the urea. Subsequently, secondary thermal treatment was applied at 400 °C for 1 h to remove any remaining organic compounds. Then, the in situ crystallization of Ce–Ni–YSZ and Cu–Ni–YSZ powder was performed at 650 °C and 1200 °C, respectively, for 1 h. The thermal treatment temperatures for the two precursors were selected based on the fabrication processes for TF-SOFC cells containing a Cu-incorporated anode functional layer (post-annealed at 1200 °C) and a CeO2-infiltrated anode support (post-annealed at 650 °C).
:44 wt%; NiO from Sumitomo Metal Mining, Japan; YSZ from Tosoh Corp., Japan) powder, which was pre-treated at 1200 °C for 1 h in air to homogenize the anode particle size; the samples were denoted as Ce–Ni–YSZ powder and Cu–Ni–YSZ powder, respectively. The same number of moles of Ce or Cu was deposited onto Ni–YSZ powder, at a molar ratio of 1:30 (Ce or Cu:Ni), and the weight fractions are 3.38 and 1.56 wt%, respectively. The catalyst-infiltrated powder was thermally treated in air for 2 h at 80 °C to decompose the urea. Subsequently, secondary thermal treatment was applied at 400 °C for 1 h to remove any remaining organic compounds. Then, the in situ crystallization of Ce–Ni–YSZ and Cu–Ni–YSZ powder was performed at 650 °C and 1200 °C, respectively, for 1 h. The thermal treatment temperatures for the two precursors were selected based on the fabrication processes for TF-SOFC cells containing a Cu-incorporated anode functional layer (post-annealed at 1200 °C) and a CeO2-infiltrated anode support (post-annealed at 650 °C).
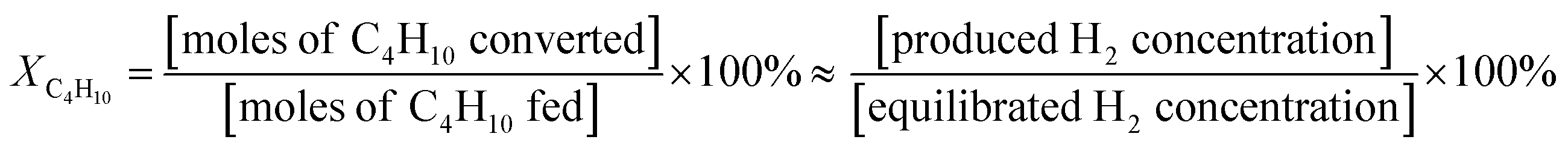 | (1) |
2.2. Catalytic properties investigated at the cell level
The standard reference cell consists of a Ni–YSZ anode support substrate (AS), a Ni–YSZ AFL, a YSZ and gadolinium-doped ceria (GDC) bilayer electrolyte, and a lanthanum strontium cobalt oxide (LSC) cathode. The anode substrate (1 mm thick) was fabricated via laminating 7 NiO–YSZ tape-cast layers (each 150 μm thick, containing poly(methyl methacrylate) as the pore former) and 1 NiO–YSZ layer (30 μm thick, no pore former, tape-AFL) at 75 °C under 15 MPa uniaxial pressure. The laminated cell was sintered at 1300 °C for 4 h prior to thin-film deposition.
PLD was used to fabricate the standard Ni/YSZ reference cell (ref-cell). The NiO–YSZ AFL (nanoAFL) with a thickness of 2 μm was deposited on the 2 cm × 2 cm AS at a substrate temperature (Ts) of 700 °C and a deposition oxygen ambient pressure (Pamb) of 50 mTorr, followed by post-annealing for 1 h at 1200 °C under air. Then, the YSZ/GDC bilayer electrolyte was deposited at a Ts value of 700 °C and a Pamb value of 50 mTorr (the thicknesses of the layers were 1 μm and 200 nm, respectively). A 1 cm × 1 cm 3 μm-thick LSC layer was deposited on the surface of GDC at 25 °C and a Pamb value of 100 mTorr, followed by post-annealing at 650 °C for 1 h under air for cathode crystallization.26,59–66
To fabricate Ce-cell, CeO2 precursor solution was infiltrated into the AS following the method described above. The infiltration and thermal treatments were repeated five times. For Cu–Ce-cell, Cu was incorporated in the AFL, followed by CeO2 infiltration into the AS. Energy-dispersive X-ray spectroscopy (EDS) was used to determine the distributions of the catalysts at the anode, and transmission electron microscopy (TEM, Titan) and FE-SEM were used to observe the cross-sectional microstructures of the catalyst-incorporated cells. A detailed method for preparing the Cu–Ni functional layer is described in our previous work.38
3. Results and discussion
3.1. Steam reforming of n-butane
The steam reforming of hydrocarbons mainly consists of two reactions: hydrocarbon splitting with steam (eqn (2)) and the water-gas-shift (WGS) reaction (eqn (3)). The overall reforming reaction is highly endothermic and requires high temperatures to achieve a sufficient reaction rate and conversion.| CnHm + nH2O ↔ nCO + (n + m/2)H2 | (2) |
| CO + H2O ↔ CO2 + H2 | (3) |
During SOFC operation, the electrochemical oxidation of H2 (eqn (4)) and CO (eqn (5)) also occurs. The removal of H2 and CO via electrochemical reactions further promotes hydrocarbon splitting with steam (eqn (2)).
| H2 + O2− ↔ H2O + 2e− | (4) |
| CO + O2− ↔ CO2 + 2e− | (5) |
Carbon deposition is a major obstacle in hydrocarbon reforming. During the operation of an internal reforming SOFC, carbon deposits can block electrochemical and catalytic active sites, eventually causing cell failure. Carbon is deposited on the catalyst surface via CO or CH4 dissociation (eqn (6) and (7)) and via the cracking of alkanes to alkenes, followed by carbon deposition (eqn (8)). Higher molecular weight hydrocarbons have a greater tendency towards carbon deposition, and thus greater care is required.
| 2CO ↔ C + CO2 | (6) |
| CH4 ↔ C + 2H2 | (7) |
| CnH2n+2 ↔ CnH2n + H2 ↔ Cn + (n + 1)H2 | (8) |
Carbon deposition can be conveniently predicted using a C–H–O diagram obtained from thermochemical calculations (Fig. S1†).68,69 The carbon deposition region expands significantly upon a decrease in the temperature from 800 to 500 °C. Therefore, severe carbon deposition is expected when operating an internal reforming SOFC at a lower temperature. The addition of an appropriate amount of steam can prevent carbon deposition via increasing the O and H fractions. For butane fuel, an SCR of 2 lies near the edge of the carbon deposition region, and thus an SCR higher than 2 is generally required. However, thermochemically calculated SCRs are often imprecise, as will be discussed below.70
3.2. Catalytic properties of Cu- and CeO2-modified Ni–YSZ anode powder
The thermal catalytic effects of Cu and CeO2 addition to Ni–YSZ powder for the steam reforming of butane were evaluated before incorporating the catalysts into TF-SOFCs. Cu and CeO2 were incorporated into Ni–YSZ powder via infiltration, and the samples were denoted as Cu–Ni–YSZ powder and Ce–Ni–YSZ powder, respectively. The powder samples were tested in a packed bed reactor and reduced before the reforming reaction. The morphologies and BET surface areas were almost identical for Ni–YSZ, Cu–Ni–YSZ, and Ce–Ni–YSZ powder (Fig. S2, and Table S3†), demonstrating that the catalytic properties are only governed by the insertion of the secondary catalyst. The XRD patterns given in Fig. S3† show phase information for Cu–Ni–YSZ and Ce–Ni–YSZ powder before and after reduction. Before reduction, the XRD patterns of both powder samples primarily show peaks from NiO and YSZ, while the XRD pattern of the Ce-infiltrated sample shows small peaks at ∼46.8° and 56.5°, which correspond to CeO2. These CeO2 peaks are also present in the XRD pattern after reduction. From the binary alloy phase diagram of Cu–Ni (Fig. S4†), Cu–Ni exists as a homogeneous solid solution, which was also confirmed in our previously reported results.38,71The steam reforming of butane was tested in a packed bed reactor in a temperature range of 500 to 600 °C with an SCR of 3. The experimentally observed butane conversion levels are shown in Fig. 1a, and the reaction products are shown in more detail in Fig. S5.† The reaction produced 20–30% H2, 5–7% CO2, and small amounts of CO and CH4. The butane conversion is calculated via dividing the experimentally produced H2 by the equilibrium concentration of H2 under the given conditions. The butane conversion levels of all the catalysts increased with temperature due to the endothermic nature of the reforming reaction. The addition of CeO2 and Cu enhanced the steam reforming of butane, with the CeO2-modified powder, Ce–Ni–YSZ, providing the best results. This enhancement was more prominent at lower temperatures.
 | ||
| Fig. 1 The steam reforming of butane using powder catalysts. (a) Butane conversion levels from 500 to 600 °C using various Ni–YSZ powder samples at an SCR of 3. (b) Long-term testing of the steam reforming of butane at 600 °C with an SCR of 3 for 20 h. (c) Raman spectra and (d) SEM images of various Ni–YSZ powder samples after 20 h of the steam reforming of butane. | ||
The promotional effects of CeO2 on hydrocarbon reforming are often explained as being due to its excellent redox capabilities for shuttling between Ce(III) and Ce(IV), which are related to its oxygen storage capacity. Surface oxygen vacancies created via the reduction of Ce(IV) to Ce(III)72 and metal–support interactions at the Ni–CeO2 interface73 provide additional pathways to activate and dissociate steam, facilitating steam reforming reactions. It has been also reported that the introduction of CeO2 or modified CeO2 to Ni catalysts can reduce carbon deposition to a great extent,74,75 and the amount of deposited carbon is strongly correlated with the oxygen storage capacity.76 The addition of Cu to Ni reduces carbon deposition as well. The Ni–Cu alloy shows increased resistance to carbon formation via reducing the adsorption energy of C while retaining the activity of Ni.77 Many reports state that hydrocarbons form carbon deposits at the step sites of Ni. In Ni–Cu alloys, Cu prefers to segregate at the surface of Ni, preferentially blocking step sites to reduce the total surface energy.78,79 Therefore, it is expected that the higher activity of Cu–Ni–YSZ powder compared with Ni–YSZ powder could be related to carbon deposition resistance.
The stability of the catalyst powder samples was studied at 600 °C with an SCR of 3 for 20 h. The unmodified Ni–YSZ powder showed the fastest deactivation (Fig. 1b), with a drop in the butane conversion from 68.6% to 30.5% after 20 h. During the same period, the Cu–Ni–YSZ and Ce–Ni–YSZ powder samples demonstrated much better stability, with butane conversion drops of only ∼13.7% and ∼5.6%, respectively. The extent of carbon deposition over the catalyst powder after 20 h of steam reforming was investigated using Raman spectroscopy (Fig. 1c) and SEM (Fig. 1d) studies. SEM analysis was performed at several positions on the powder samples and representative microstructures are illustrated in Fig. 1d. Severe carbon nanofiber formation is observed in the SEM image of the tested Ni–YSZ powder, while much fewer carbon nanofibers are observed in the SEM image of the tested Cu–Ni–YSZ powder. Carbon nanofibers were hard to detect in the tested Ce–Ni–YSZ powder via SEM. However, since SEM analysis only covers a small area with limited resolution, we cannot rule out the possibility of carbon deposition on Ce–Ni–YSZ. It is presumed that Ni–YSZ powder rapidly loses its activity due to carbon deposition on the Ni surface, which blocks the active sites. The improved stability of the CeO2- and Cu-incorporating powder samples indicates that CeO2 and Cu play crucial roles in suppressing carbon deposition. Although the C–H–O diagram (Fig. S1†) predicts no carbon deposition at an SCR of 3, significant carbon deposition was observed, especially on the unmodified Ni–YSZ catalyst. The endothermic nature of the reaction can induce local cold spots where carbon deposition is favored. In addition, differences in the diffusivities and adsorption energies of different species may cause local areas of inhomogeneity in the C fraction, resulting in carbon deposition.
To characterize the deposited carbon, Raman spectroscopy was performed (Fig. 1c). Two intense bands, which correspond to deposited carbon, appear in the spectra. The D band at ∼1350 cm−1 is associated with in-plane imperfections or the disordered structure of the carbon, and the G band at ∼1530 cm−1 is associated with graphitic carbon layers and the tangential lattice vibration mode. Amorphous carbon, which is D-band related, can be easily removed when oxidants are present (e.g., H2O and O2−), whereas graphitic carbon, which is G-band related, is much more difficult to remove.80,81 Unlike in SEM images, where carbon nanofibers were hardly identifiable in the Cu–Ni–YSZ and Ce–Ni–YSZ samples, carbon related peaks were present for all three samples but with different ratios of the two bands. The intensity ratio of the two bands, R = ID/IG, reflects the degree of graphitization. These values were 1.12, 1.33, and 1.38 for Ni–YSZ, Cu–Ni–YSZ, and Ce–Ni–YSZ powder, respectively. Although all samples show signs of carbon deposition, the higher R values of the modified samples imply that these materials could be more resistant to carbon deposition.80–82 The R values of the powder samples containing secondary catalysts are higher than that of Ni–YSZ powder, which is consistent with the catalytic activity and durability trends. The catalytic properties and post-reaction analysis of the three catalysts clearly demonstrated that the addition of Cu and CeO2 suppressed carbon deposition and enhanced the reforming activity and durability upon the steam reforming of butane. These characteristics are highly desirable for internal steam reforming anodes.
3.3. TF-SOFC performance using internally steam reformed butane fuel
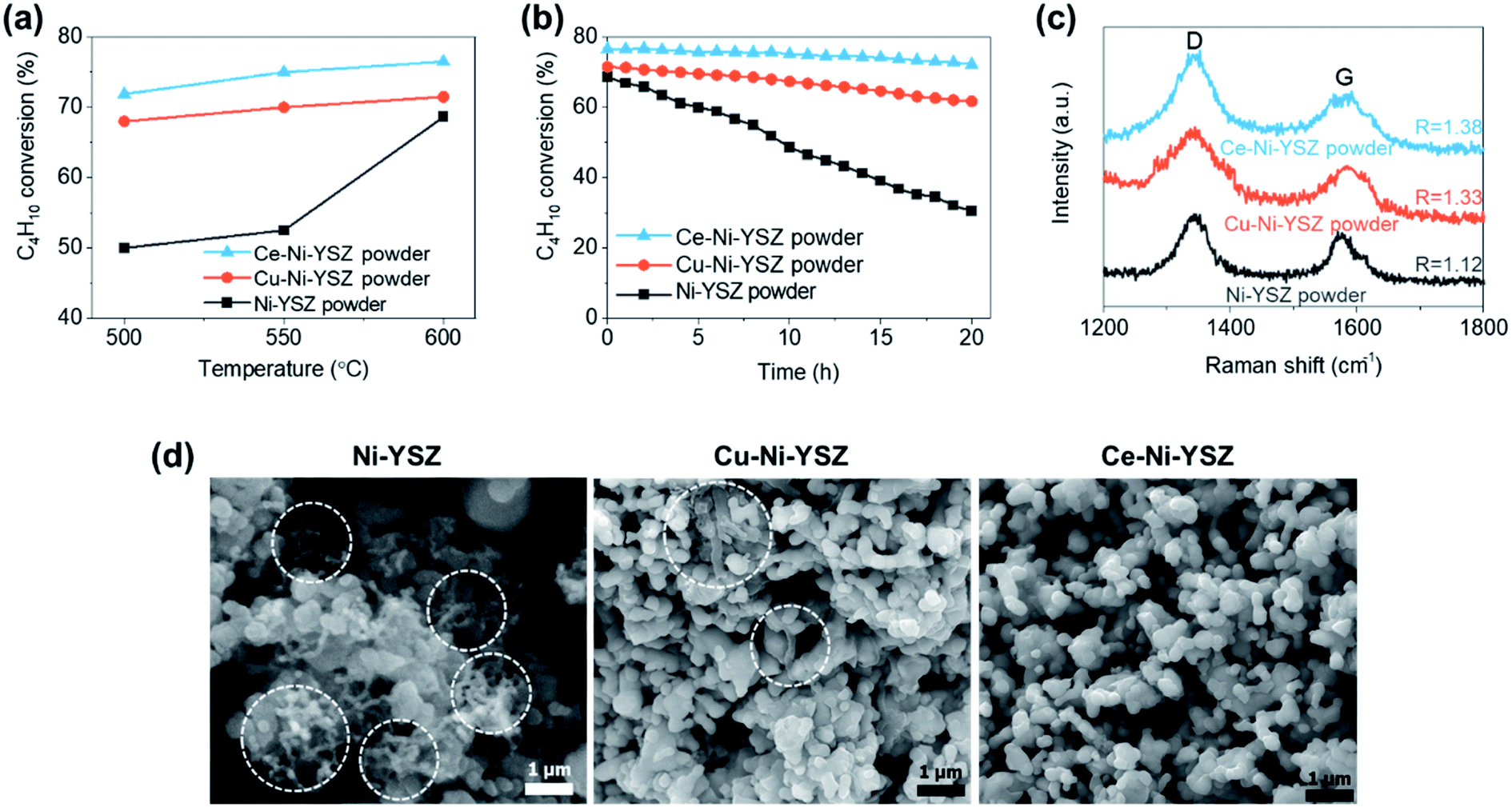
We incorporated Cu and CeO2 into two different regions of TF-SOFCs to improve the performance and carbon deposition resistance of ISRB–SOFCs. First, the electrochemically active region, the AFL, requires significant improvement in terms of preventing carbon deposition from blocking electrochemically active sites and fatally impacting the SOFC performance. To achieve this, Cu was sputter-deposited at the AFL with a sandwich-like configuration between the NiO–YSZ PLD layers (Fig. 2a). Upon reduction treatment, the sandwiched Cu layer diffused into the Ni–YSZ layer, forming a homogeneous Ni–Cu alloy at the anode functional layer. The amount of Cu incorporated in the cell was 0.5 mg cm−2. This content corresponds to a 30 nm-thick Cu layer at the AFL. This optimal configuration was found to prevent significant structural defects, such as delamination at the AFL, in our recent publications.21,38 CeO2, another low-cost catalyst, was used to cover the entire anode layer and improve the reforming activity and resistance to carbon deposition. Urea-assisted infiltration was employed to ensure the uniform distribution of small-sized CeO2 nanoparticles (Fig. 2a). The urea in the infiltration solution acts as a complexing agent for the metal cations and as a precipitating agent upon mild thermal treatment at 80 °C. Mild thermal treatment separates the precipitation and drying processes, resulting in the formation of uniformly distributed CeO2 nanoparticles.58 Three different CeO2 loading levels were tested to optimize the CeO2 loading. As shown in Fig. S6,† the optimum deposition amount of 2 wt% CeO2 results in finely distributed small nanoparticles. A loading amount of 1 wt% CeO2 deposition resulted in uncovered regions in the anode support, and 5 wt% CeO2 deposition resulted in agglomerated CeO2 particles.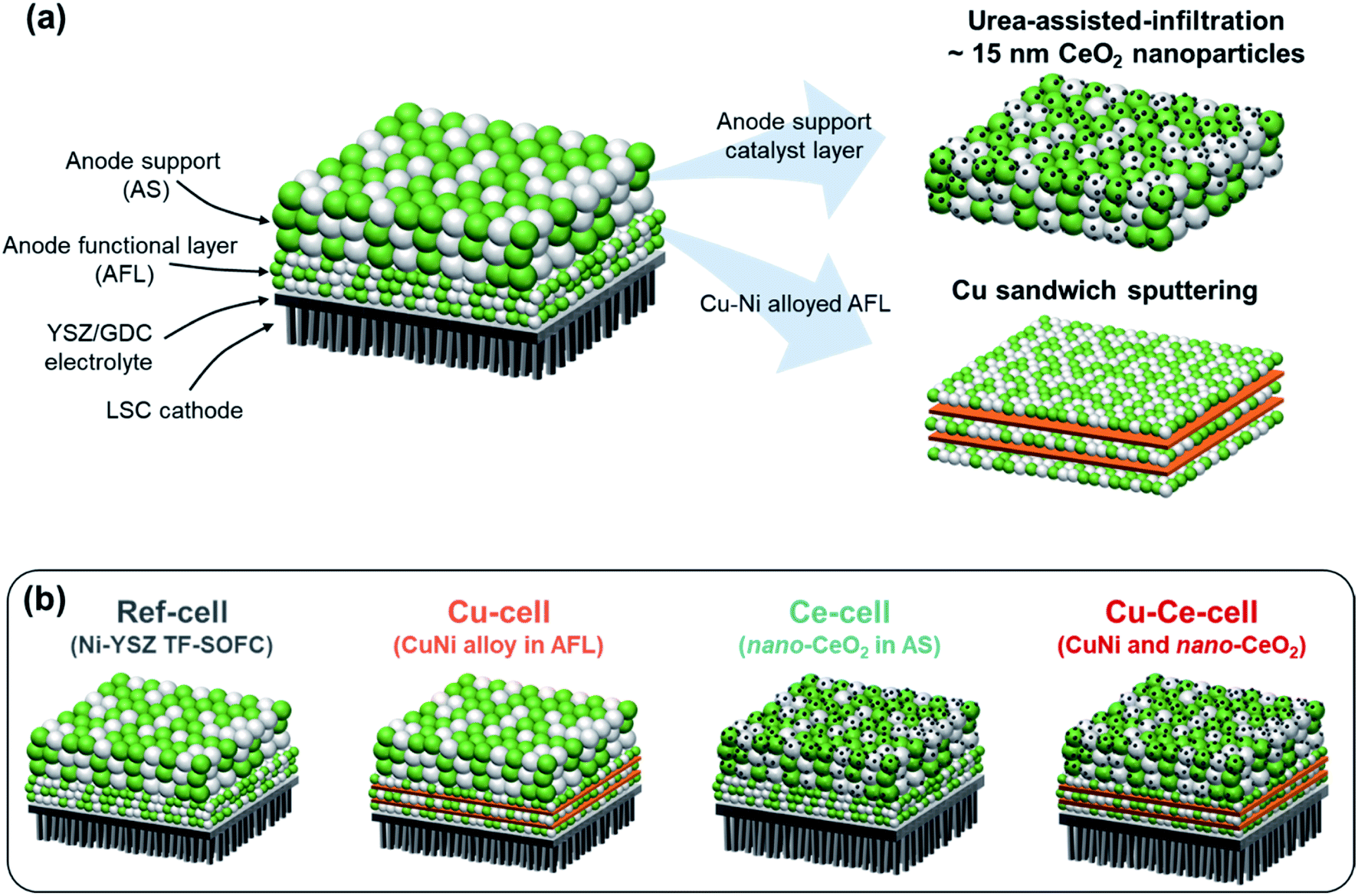 | ||
| Fig. 2 Schematic diagrams of TF-SOFCs. (a) The configuration of the TF-SOFC and catalyst incorporation strategies. (b) Schematic diagrams and naming of the tested TF-SOFCs. | ||
Four different TF-SOFCs were prepared and tested for ISRB–SOFC operation using temperatures between 500 and 600 °C: a conventional Ni–YSZ TF-SOFC (ref-cell); a sample in which Cu was added at the AFL of the TF-SOFC (Cu-cell); a CeO2-infiltrated TF-SOFC (Ce-cell); and a cell which both Cu and CeO2 were incorporated (Cu–Ce-cell). Cu-cell has been previously reported and is replotted together with the other cells for detailed analysis.38
The distributions of Cu and Ce in the cell anode layer were analyzed via SEM-EDS as a function of depth from the anode–electrolyte interface (Fig. 3a) after reduction treatment. Cu was initially sputter-deposited within a 2 μm-thick nano-AFL region at the anode–electrolyte interface. After annealing and reduction treatment, Cu was distributed to a depth of over 60 μm (Fig. 3a) from the anode–electrolyte interface. EDX mapping analysis after reduction treatment shows a homogeneous distribution of Cu at the Ni grains of the AFL region, confirming the formation of a homogeneous Ni–Cu alloy at the AFL (Fig. 3b). A small amount of Cu is also located at the YSZ grains. CeO2 was introduced from the top surface of the anode support (AS) substrate using a urea-assisted infiltration technique. Therefore, the concentration of Ce is higher at the top of the AS, and Ce is distributed throughout the entire anode region down to the anode–electrolyte interface. CeO2 nanoparticles, with a size of ∼15 nm, were uniformly distributed at the AS layer, as shown in Fig. 3c. The nanometer-sized structure of CeO2 will facilitate the effects of CeO2via providing a greater interfacial area between CeO2 and Ni. The particle size of CeO2 is also related to its oxygen storage capacity, where small nanoparticles have a larger oxygen storage capacity, higher reforming activity, and better resistance to carbon deposition.83,84
 | ||
| Fig. 3 Characterization of Cu–Ce-cell. (a) Distributions of the catalytic promoters (Cu and Ce) as a function of distance from the anode functional layer after reduction, obtained via SEM-EDS. The Cu distribution is adapted from our previous work.38 (b) A HAADF-STEM image and energy dispersive X-ray spectroscopy analysis showing the Cu, Zr, and Ni distributions after reduction at the anode functional layer. The white dashed lines indicate Ni grains. (c) An SEM image of the anode support region showing the uniform distribution of the CeO2 nanoparticles. | ||
Before the TF-SOFCs were tested with butane fuel, the integrity of the cells and the differences in cell performances were investigated using wet H2 (97% H2/3% H2O) as a fuel. The results are shown in Fig. S7–10, Tables S4 and 5.† The open-circuit voltage (OCV) of the fabricated TF-SOFCs was approximately 1.1 V, which is a suitable OCV value for TF-SOFCs, revealing that the thin-film electrolyte was deposited with microstructural integrity. The current–voltage–power (I–V–P) results indicate that the fuel cell performances of ref-cell and Cu-cell38 using humidified H2 are similar, whereas Ce-cell and Cu–Ce-cell appeared to exhibit slightly better performances. The polarization area specific resistance (RP) of the TF-SOFCs noticeably drops upon the infiltration of CeO2. As shown in Fig. 3a, infiltrated CeO2 nanoparticles are also present at the AFL, where electrochemical reactions occur. These CeO2 nanoparticles are likely to promote electrochemical reactions via expanding the triple phase boundary.85,86 Detailed analysis of the H2-fueled performance is discussed in the ESI.†
The performances of the TF-SOFCs were tested with butane fuel at various SCRs at 600 °C, as shown in Fig. 4a. The incorporation of Cu and CeO2 improved the performances of the ISRB–TF-SOFCs at all SCRs. Compared with the power improvement using wet H2 fuel, as presented in the ESI† (30% for Cu–Ce-cell compared to ref-cell), the enhancement is much more pronounced in ISRB mode. The peak power densities (PPDs) of Cu–Ce-cell were improved by 43%, 51%, and 60% compared with ref-cell at SCRs of 3, 2.5, and 2, respectively. Therefore, the performance enhancement is not only electrochemical but is also due to the reforming capabilities of the inserted catalysts.
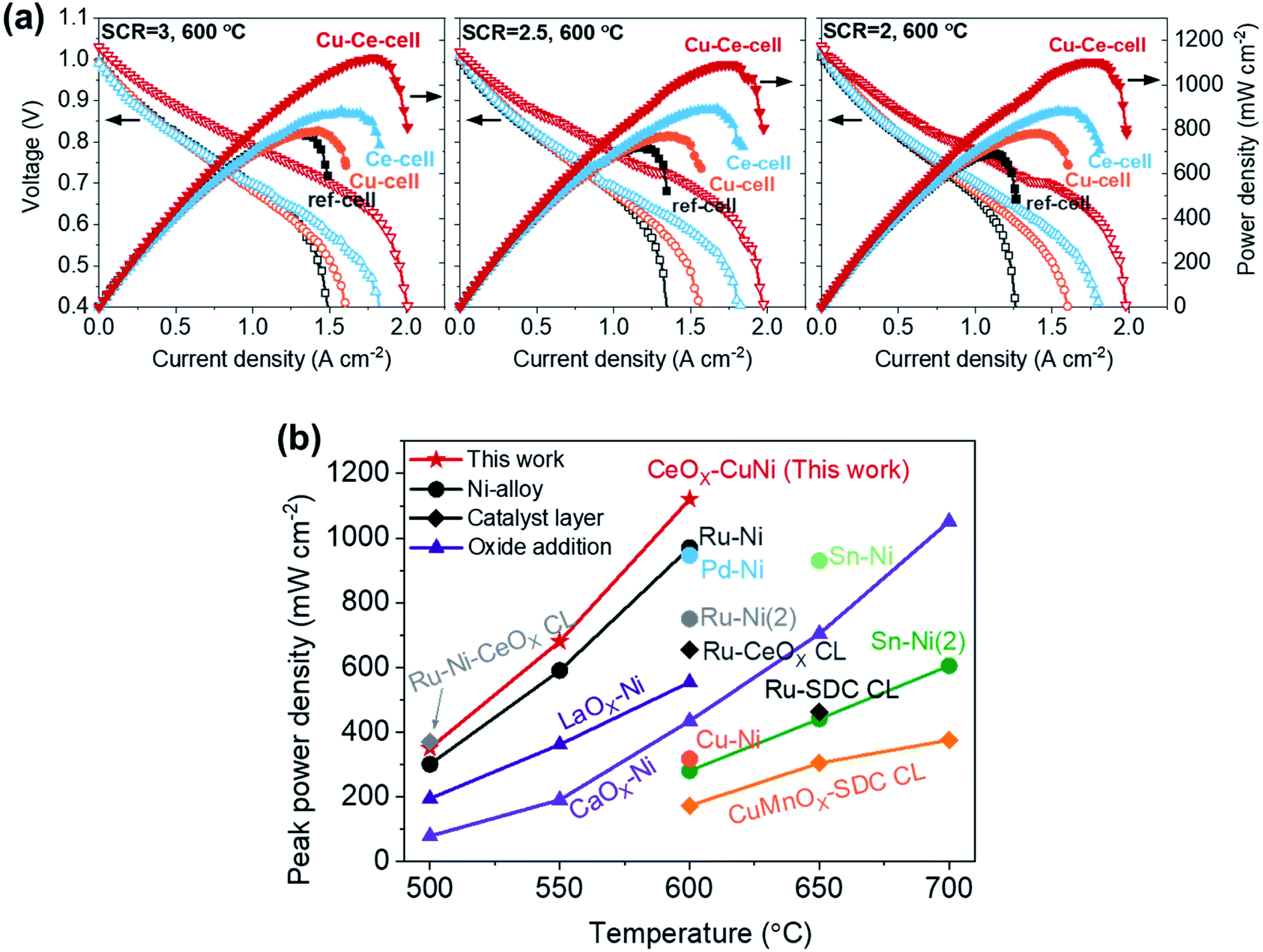 | ||
| Fig. 4 Cell performances of ISRB–TF-SOFCs. (a) I–V–P curves of TF-SOFCs operated via the internal steam reforming of butane at 600 °C at different SCRs. The I–V–P data for Cu-cell were previously reported38 and are plotted together with the data obtained in this study. (b) A comparison of the peak power densities of internal hydrocarbon reforming LT-SOFCs. The data were adapted from the following references: Ru–Ni from ref. 38, Pd–Ni from ref. 39, Ru–Ni(2) from ref. 87, Ru–Ni–CeOX CL from ref. 48, Ru-CeOX CL from ref. 91, Sn–Ni from ref. 88, Sn–Ni(2) from ref. 89, LaOX–Ni from ref. 94, CaOX–Ni from ref. 95, Cu–Ni from ref. 90, Ru-SDC CL from ref. 92, and CuMnOX–SDC CL from ref. 93. A detailed description of the data included in the plot is presented in Table S6.† | ||
The significant improvements shown by Ce-cell and Cu–Ce-cell, as shown in Fig. 4a, demonstrate that the uniformly distributed CeO2 nanoparticles act as an excellent reforming catalyst. These reforming capabilities are also observable from the mass transport limited currents. The mass transport limited current reflects the availability of the reactants, H2 and CO, produced by the steam reforming reaction. While the mass transport limited current density of ref-cell at an SCR of 3 was approximately 1.5 A cm−2, the insertion of secondary catalysts (Cu–Ce-cell) increased the mass transport limited current density to 2.0 A cm−2. The PPDs of the catalyst-incorporating cells, especially the Ce-incorporating cell, did not significantly change with the SCR; the PPD changed from 1120 to 1092 mW cm−2 for Cu–Ce-cell, from 891 to 882 mW cm−2 for Ce-cell, and from 794 to 770 for Cu-cell. However, ref-cell showed a significant performance drop from 783 to 689 mW cm−2. The mass transport limited current also decreased significantly only for ref-cell with a decrease in the SCR. The impact of the inserted catalysts in ISRB–SOFCs is more significant upon decreasing the SCR, when more demanding reforming conditions are present.
From these analyses, including catalytic performance tests using a packed bed reactor, we conclude that the introduction of small amounts of secondary catalysts promotes the reforming of butane to H2 and CO, improving the fuel cell performance. The impact of CeO2 is more significant than that of Cu. As seen in Fig. 3a, the amount of incorporated Ce is much larger than that of Cu. In addition, CeO2 exists as nanoparticles, which provide a higher surface area than alloyed Ni–Cu.
To estimate the conversion during the butane reforming reaction, PPD values from ISRB operations are compared with respect to PPD values from H2-fueled operations. If 5% butane, the inlet composition for ISRB operations, is completely converted at an SCR of 3 and 600 °C, 51% H2 would be produced.38 First, the TF-SOFCs were operated at 50 vol% H2 fuel content (N2 balance) at 600 °C (Fig. S11†) to acquire reference PPD values. When considering the ratio of PPD(ISRB)/PPD(50% wet H2), the estimated butane conversions of ref-, Cu-, Ce-, and Cu–Ce-cell were approximately 70%, 70%, 80%, and 90%, respectively. The butane conversion capabilities of the TF-SOFCs are in the order: Cu–Ce- > Ce-, > Cu- ≈ ref-cell.
The performance of the Cu–Ce-cell, with a PPD of 1120 mW cm−2 at 600 °C, is a significant improvement on previously reported internal hydrocarbon reforming fuel cells operated below 700 °C. The PPDs of state-of-the-art SOFCs are plotted together with the Cu–Ce-cell in Fig. 4b, with data given in Table S6.† Previously reported data can be categorized based on three different strategies: Ni-alloy anodes,38,39,87–90 the addition of a catalyst layer on top of the anode,48,91–93 and basic oxide addition to the anode.94,95 Our strategy involves coupling a Ni–Cu alloy at the electrochemical interface and using an AS layer as the catalyst layer via incorporating CeO2 nanoparticles. This synergistic approach is responsible for the high performance. We expect that our approach could provide an economic framework for high-performance hydrocarbon-fueled SOFCs, especially under low-temperature operating conditions. Although our Cu–Ce cell shows remarkable performance, a direct comparison of its performance to those of reported systems can be misleading because the reported systems operate under different reforming conditions involving different fuels, SCRs, and C/O ratios.
Electrochemical impedance spectroscopy (EIS) and distribution relaxation time (DRT) analysis was used to investigate the electrochemical characteristics of the four different TF-SOFCs (Fig. 5) under ISRB–SOFC conditions at 600 °C and an SCR of 3 at a current density of 0.2 A cm−2. From the Nyquist plots, all TF-SOFCs demonstrated almost identical ohmic area specific resistance (ASR) values, mainly originating from the YSZ electrolyte and independent of the secondary catalysts. The polarization ASR (RP) noticeably decreases upon catalyst insertion, and the relative order of the TF-SOFC RP values is Cu–Ce-cell < Ce-cell < Cu-cell < ref-cell. The Bode plots in Fig. 5b indicate that the improved performance can be primarily correlated with a reduction in the impedance in the mid-frequency range, which is dependent on the inserted catalysts. This order demonstrates the effectiveness of the incorporated catalysts in enhancing the internal steam reforming reaction.
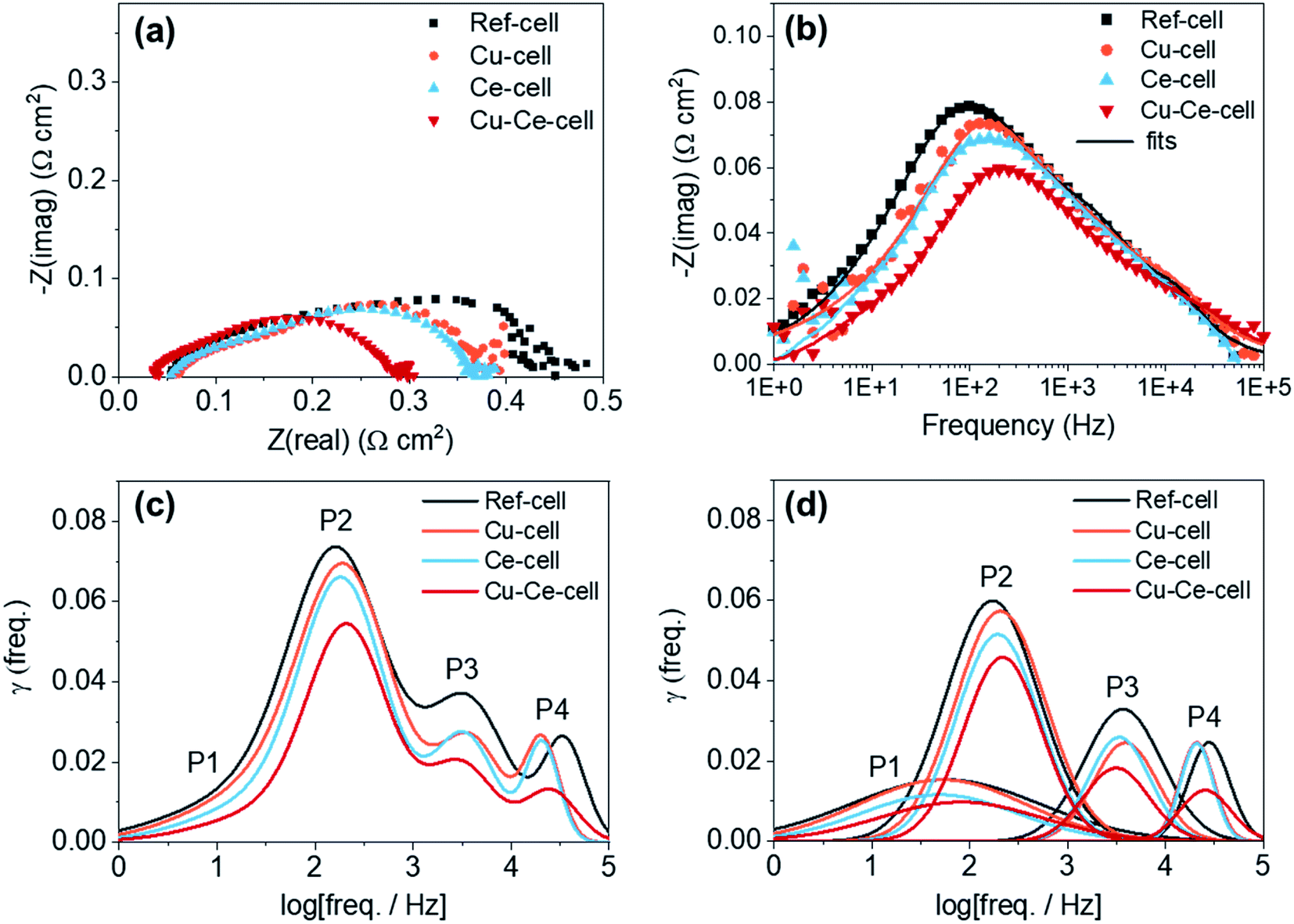 | ||
| Fig. 5 Electrochemical impedance analysis of ISRB–TF-SOFCs. EIS data from the four TF-SOFCs in ISRB mode at 600 °C with an SCR of 3 at 0.2 A cm−2: (a) Nyquist plots; (b) Bode plots; (c) DRT results; and (d) DRT fitting plots demonstrating distinct peaks for each TF-SOFC (note that the P4 peak of Cu-cell overlaps with the P4 peak of Ce-cell in (d)). | ||
DRT analysis is used to distinguish different electrochemical mechanisms that often overlap in EIS spectra. The fitted EIS data in Fig. 5b were used for DRT calculations. The DRT results, showing four distinctive peaks, are compared in Fig. 5c, and DRT plots of the deconvoluted peaks for each TF-SOFC are shown in Fig. 5d. DRT analysis indicates the following 4 distinguishable mechanisms: (i) P1 (101–102 Hz) is attributed to gas diffusion at the anode96,97 and is related to the reactant (H2) concentration. The P1 peak reduces with an increase in the hydrogen concentration at the anode98 and, therefore, an increased supply of reformed fuel will reduce the P1 peak; (ii) P2 (102–103 Hz) is attributed to the electrode/gas interfacial reaction; (iii) P3 (103–104 Hz) is correlated to the charge transfer reaction; and (iv) P4 (104–105 Hz) is associated with the ohmic resistance of the fuel electrode99 and the charge transfer reaction at the triple phase boundary.100 Based on our prior work, the incorporation of Cu into Cu-cell and Cu–Ce-cell mainly affects P2 through improving the surface-related electrode reaction component.38 CeO2 infiltration reduces the responses at all frequency ranges in both Ce-cell and Cu–Ce-cell. P1, which is related to the reactant concentration, is reduced owing to the improved supply of reformed fuel due to the high reforming capabilities of the CeO2 catalyst. The CeO2 catalyst slightly reduces P3. This effect is understandable considering the deposition behavior of CeO2; the infiltrated CeO2 is deposited on the surfaces of the Ni and YSZ grains, as shown in Fig. 3c. The charge transfer reaction occurs on the electrode surface; thus, infiltrated CeO2 modifies the electrode structure, thereby improving the charge transfer kinetics at the surface.97 The decrease in P2 in the cases of Ce-cell and Cu–Ce-cell originates from the additional active sites on the anode surface and at the anode interface provided by the CeO2 nanoparticles. CeO2 is a mixed ionic–electronic conductor and a decent redox catalyst; therefore, more active sites are available for H2 and CO electro-oxidation to occur, improving the electrode reaction process. The high-frequency P4 peak reduces slightly upon the infiltration of CeO2. While this peak is related to the ohmic resistance of the fuel electrode99 or the charge transfer reaction at the triple phase boundary100 in the literature, the origin of this phenomenon requires further investigation since it was temperature independent under H2 fuel operation (see the ESI†).
3.4. Durability of the TF-SOFCs and the post-test microstructures
Catalyst infiltration into the Ni–YSZ powder reduced carbon deposition, thereby improving the operational stability during butane reforming (see above) in the packed bed reactor. Therefore, catalyst incorporation in TF-SOFCs is expected to improve the longevity of the cell during ISRB mode operation. Herein, the long-term performances of ref-cell and Cu–Ce-cell are compared under a constant current load of 0.15 A cm−2 using butane fuel with an SCR of 3 at 600 °C, as shown in Fig. 6. The voltage of ref-cell decreases drastically, by ∼14%, after 60 h of operation. Excessive carbon deposition and the deactivation of Ni are thought to be the main reasons for the voltage drop. The initial PPD of 785 mW cm−2 decreased significantly to ∼510 mW cm−2 after only 2 h of operation, and eventually it decreased to ∼235 mW cm−2 after 60 h of operation (Fig. 6b). EIS analysis showed a huge increase in Rp, resulting from a decreased electrochemical interface due to carbon deposition. An increase in ROhm was also visible in the EIS analysis, showing significant degradation due to carbon deposition. In contrast, Cu–Ce-cell yielded a stable voltage output for 250 h with only slight voltage degradation of ∼2.76% over 250 h. Fig. 6d shows I–P curves that were intermittently recorded during long-term testing, and Fig. 6e displays Nyquist plots from EIS measurements collected every 50 h. Even after the 250 h durability test, Cu–Ce-cell exhibited a PPD of 1020 mW cm−2 at an operation temperature of 600 °C, which is an 8.9% drop from the initial value of 1120 mW cm−2. The EIS changes shown in Fig. 6e indicate that the ASR of Cu–Ce-cell did not change significantly as long-term testing progressed. This demonstrates that the electrode did not degrade during 250 h of operation. Considering the demanding operating conditions of ISRB, this degradation rate is truly remarkable. To the best of our knowledge, the durability of Cu–Ce-cell is the best among low-temperature internal reforming SOFCs reported in the literature, as it has the longest operating time and the lowest degradation rate (Fig. 7).38,39,88–90,93–95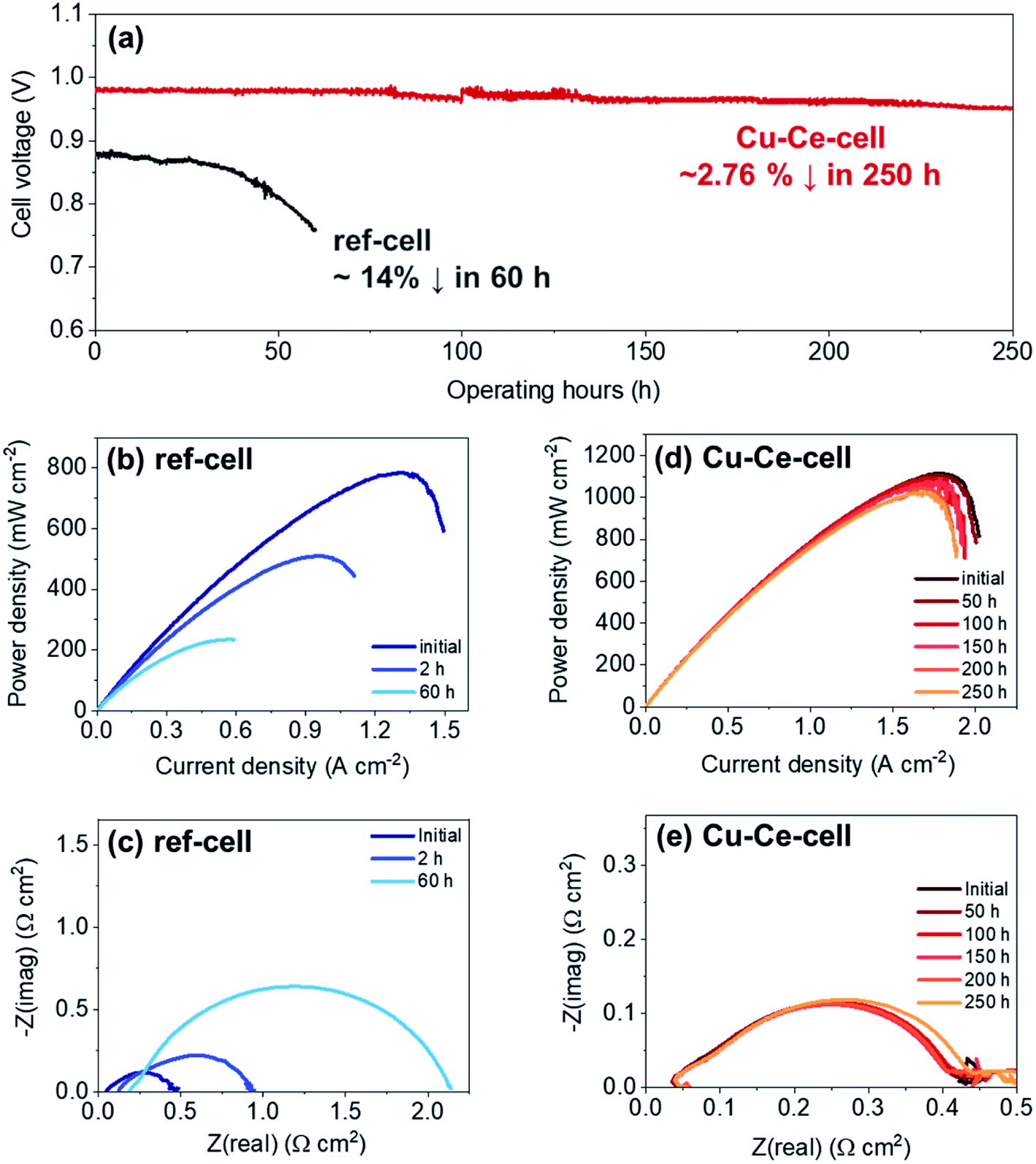 | ||
| Fig. 6 Durability testing of ISRB–TF-SOFCs. (a) Durability testing of Cu–Ce-cell and ref-cell at 0.15 A cm−2 during the internal steam reforming of butane at an SCR of 3 at 600 °C. I–P curves of (b) ref-cell and (d) Cu–Ce-cell and Nyquist plots of (c) ref-cell and (e) Cu–Ce-cell measured intermittently during long-term operation. | ||
 | ||
| Fig. 7 Durability comparison. A comparison of the long-term stabilities of previously reported internal hydrocarbon reforming LT-SOFCs. The reported operating hours are plotted along with the degradation rate in %/h. The data were adapted from the following sources: ref. 38 for Ru–Ni, ref. 39 for Pd–Ni, ref. 88 for Sn–Ni, ref. 89 for Sn–Ni(2), ref. 90 for Cu–Ni, ref. 93 for CuMnOX–SDC CL, ref. 94 for LaOX, and ref. 95 for CaOX. A detailed description of the data included in the plot is presented in Table S6.† | ||
Carbon deposition is considered to be the main cause of cell performance degradation. The primary reason for Cu–Ce-cell outlasting ref-cell is its ability to suppress carbon deposition, as suggested by the powder catalyst tests (see above). After long-term SOFC operation tests, SEM images at different positions of the anodes of both cells were examined, and the representative microstructures are shown in Fig. 8. The microstructures of as-prepared cells are displayed along with cells subjected to long-term testing. Fig. 8a and d, illustrating as-prepared cells, show almost identical microstructures. Ref-cell suffered severe carbon nanofiber formation during long-term operation (Fig. 8b and c). Carbon deposition on the surface of Ni led to the deactivation of the Ni catalyst. This is considered to be the reason for the cell voltage drop of ref-cell during the 60 h test. On the contrary, no clear carbon deposition was detected on the anode of Cu–Ce-cell after 250 h of operation (Fig. 8e and f). Negligible carbon formation on Cu–Ce-cell in ISRB operation mode is considered to enhance the longevity of the cell. The CeO2 nanoparticles were still well dispersed and covered the entire anode region, even after long-term testing (Fig. 8f). However, the size of the CeO2 nanoparticles increased to ∼30 nm after long-term operation compared to an initial size of ∼15 nm.
 | ||
| Fig. 8 SEM images of as-prepared and tested cells. SEM images of as-prepared cells: (a) ref-cell and (d) Cu–Ce-cell. SEM images of (b and c) ref-cell and (e and f) Cu–Ce-cell after the long-term testing procedure demonstrated in Fig. 6. | ||
4. Conclusions
The output performance and durability of ISRB–LT-SOFCs were improved upon incorporating economical and low-cost Cu and CeO2 catalysts at the anode. The effectiveness of catalyst incorporation into the Ni–YSZ anode for internal butane reforming was evaluated both at the powder level and at the cell level. The catalytic performances of the fabricated catalyst powder samples and the SOFC performances of fabricated TF-SOFCs operating in ISRB mode demonstrated the beneficial effects of the CeO2 and Cu catalysts at low operating temperatures. CeO2 incorporation dramatically improved the performance under all operating conditions. The PPD of the Cu- and CeO2-incorporating cell, Cu–Ce-cell, at an SCR of 3 and a temperature of 600 °C was 1120 mW cm−2, which is the highest value ever reported for an internal hydrocarbon reforming LT-SOFC. A comparison between the stabilities of ref-cell and Cu–Ce-cell using butane fuel during exposure to a constant current density of 0.15 A cm−2 at 600 °C revealed a significant enhancement in the durability upon the incorporation of the catalysts. This study demonstrates that the efficient utilization of economical catalysts, Cu and CeO2, can offer an affordable solution to issues associated with ISRB–LT-SOFCs. Such a strategy increases the fuel flexibility of LT-SOFCs and widens their application potential, including making small-scale and portable applications possible. We believe that our strategy can be expanded to similar internal reforming reactions of alternative energy carriers, utilizing renewable energy sources like biofuels101 and ammonia.102Author contributions
C.-A. T. contributed to conceptualization, investigation, writing – original draft; S. Y. contributed to visualization, supervision, writing – original draft, writing – review and editing; H.-I. J., and K. J. Y. contributed to formal analysis and the investigation of electrochemical data; H. K. contributed to the investigation of microstructures; J.-H. L. contributed to conceptualization and funding acquisition; and J.-W. S. contributed to conceptualization, supervision, writing – review and editing, project administration, and funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Abbreviations
| LT-SOFC | Low-temperature solid oxide fuel cells |
| TF-SOFC | Thin-film solid oxide fuel cells |
| ISRB | Internal steam reformed butane |
Acknowledgements
This work was financially supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) (no. 20213030030040) and by the Institutional Research Program (no. 2E30994) of the Korea Institute of Science and Technology (KIST). This work was also partially supported by the National Research Foundation of Korea (NRF) (NRF-2017M1A2A2044982).References
- S. D. Vora, G. Jesionowski and M. C. Williams, ECS Trans., 2019, 91, 27–39 CrossRef CAS.
- A. Pappacena, L. Bardini, M. Boaro, in Advances in Medium and High Temperature Solid Oxide Fuel Cell Technology, ed. M. Boaro and A. S. Aricò, Springer International Publishing, 1st edn, 2017, pp. 155–222, DOI:10.1007/978-3-319-46146-5.
- S. C. Singhal, Solid State Ionics, 2000, 135, 305–313 CrossRef CAS.
- Z. Lyu, Y. Wang, Y. Zhang and M. Han, Chem. Eng. J., 2020, 393, 124755 CrossRef CAS.
- H.-W. Lee, H.-I. Ji, J.-H. Lee, B.-K. Kim, K. J. Yoon and J.-W. Son, J. Korean Ceram. Soc., 2019, 56, 130–145 CrossRef CAS.
- K. J. Lee, J. H. Chung, M. J. Lee and H. J. Hwang, J. Korean Ceram. Soc., 2019, 56, 160–166 CrossRef CAS.
- Z. Gao, L. V. Mogni, E. C. Miller, J. G. Railsback and S. A. Barnett, Energy Environ. Sci., 2016, 9, 1602–1644 RSC.
- J. W. Son, in Metal Oxide-Based Thin Film Structures, ed. N. Pryds and V. Esposito, Elsevier, Amsterdam, 2018, pp. 331–359 Search PubMed.
- J. T. S. Irvine, G. P. G. Corre and X. Xu, in Energy Storage, ed. G. M. Crawley, World Scientific, Singapore, 2017 Search PubMed.
- H. Su and Y. H. Hu, Chem. Eng. J., 2020, 402, 126235 CrossRef CAS.
- M. Li, M. J. Pietrowski, R. A. De Souza, H. Zhang, I. M. Reaney, S. N. Cook, J. A. Kilner and D. C. Sinclair, Nat. Mater., 2014, 13, 31–35 CrossRef CAS PubMed.
- J. A. Kilner and M. Burriel, Annu. Rev. Mater. Res., 2014, 44, 365–393 CrossRef CAS.
- T. Akbay, A. Staykov, J. Druce, H. Téllez, T. Ishihara and J. A. Kilner, J. Mater. Chem. A, 2016, 4, 13113–13124 RSC.
- J. Druce, H. Téllez, M. Burriel, M. D. Sharp, L. J. Fawcett, S. N. Cook, D. S. McPhail, T. Ishihara, H. H. Brongersma and J. A. Kilner, Energy Environ. Sci., 2014, 7, 3593–3599 RSC.
- A. Tarancón, M. Burriel, J. Santiso, S. J. Skinner and J. A. Kilner, J. Mater. Chem., 2010, 20, 3799–3813 RSC.
- A. Tarancón, S. J. Skinner, R. J. Chater, F. Hernández-Ramírez and J. A. Kilner, J. Mater. Chem., 2007, 17, 3175–3181 RSC.
- Y. H. Lee, I. Chang, G. Y. Cho, J. Park, W. Yu, W. H. Tanveer and S. W. Cha, Int. J. Precis. Eng. Manuf. - Green Technol., 2018, 5, 441–453 CrossRef.
- Y. Lyu, F. Wang, D. Wang and Z. Jin, Mater. Technol., 2020, 35, 212–227 CrossRef.
- L. Pelegrini, J. B. Rodrigues Neto and D. Hotza, Rev. Adv. Mater. Sci., 2016, 46, 6–21 CAS.
- K. Bae, D. Y. Jang, H. J. Choi, D. Kim, J. Hong, B.-K. Kim, J.-H. Lee, J.-W. Son and J. H. Shim, Nat. Commun., 2017, 8, 14553 CrossRef CAS.
- C.-A. Thieu, J. Hong, H. Kim, K. J. Yoon, J.-H. Lee, B.-K. Kim and J.-W. Son, J. Mater. Chem. A, 2017, 5, 7433–7444 RSC.
- C.-C. Chao, C.-M. Hsu, Y. Cui and F. B. Prinz, ACS Nano, 2011, 5, 5692–5696 CrossRef CAS PubMed.
- Y. B. Kim, T. P. Holme, T. M. Gür and F. B. Prinz, Adv. Funct. Mater., 2011, 21, 4684–4690 CrossRef CAS.
- A. Karimaghaloo, J. Koo, H.-S. Kang, S. A. Song, J. H. Shim and M. H. Lee, Int. J. Precis. Eng. Manuf. - Green Technol., 2019, 6, 611–628 CrossRef.
- D. H. Myung, J. Hwang, J. Hong, H. W. Lee, B. K. Kim, J. H. Lee and J. W. Son, J. Electrochem. Soc., 2011, 158, B1000–B1006 CrossRef CAS.
- H.-S. Noh, K. J. Yoon, B.-K. Kim, H.-J. Je, H.-W. Lee, J.-H. Lee and J.-W. Son, J. Power Sources, 2014, 247, 105–111 CrossRef CAS.
- Handbook of Hydrogen Energy, CRC Press, Boca Raton, 1st edn, 2014 Search PubMed.
- A. Atkinson, S. Barnett, R. J. Gorte, J. T. S. Irvine, A. J. McEvoy, M. Mogensen, S. C. Singhal and J. Vohs, Nat. Mater., 2004, 3, 17 CrossRef CAS PubMed.
- E. A. Liese and R. S. Gemmen, J. Eng. Gas Turbines Power, 2005, 127, 86–90 CrossRef CAS.
- K. Sasaki and Y. Teraoka, J. Electrochem. Soc., 2003, 150, A885 CrossRef CAS.
- H. Sumi, Y.-H. Lee, H. Muroyama, T. Matsui, M. Kamijo, S. Mimuro, M. Yamanaka, Y. Nakajima and K. Eguchi, J. Power Sources, 2011, 196, 4451–4457 CrossRef CAS.
- S. McIntosh and R. J. Gorte, Chem. Rev., 2004, 104, 4845–4866 CrossRef CAS PubMed.
- G. Valderrama, M. R. Goldwasser, C. U. d. Navarro, J. M. Tatibouët, J. Barrault, C. Batiot-Dupeyrat and F. Martínez, Catal. Today, 2005, 107–108, 785–791 CrossRef CAS.
- Y. Im, J. H. Lee, B. S. Kwak, J. Y. Do and M. Kang, Catal. Today, 2018, 303, 168–176 CrossRef CAS.
- Y. Li, X. Wang and C. Song, Catal. Today, 2016, 263, 22–34 CrossRef CAS.
- W. K. Lewis, E. R. Gilliland and W. A. Reed, Ind. Eng. Chem., 1949, 41, 1227–1237 CrossRef CAS.
- B. Bochentyn, M. Chlipała, M. Gazda, S. F. Wang and P. Jasiński, Solid State Ionics, 2019, 330, 47–53 CrossRef CAS.
- C.-A. Thieu, S. Yang, H.-I. Ji, H. Kim, K. J. Yoon, J.-H. Lee and J.-W. Son, Appl. Catal., B, 2020, 263, 118349 CrossRef CAS.
- C. A. Thieu, S. Park, H. Kim, H. I. Ji, J. H. Lee, K. J. Yoon, S. Yang and J. W. Son, Int. J. Energy Res., 2020, 44, 9995–10007 CrossRef CAS.
- D. Gamarra, C. Belver, M. Fernández-García and A. Martínez-Arias, J. Am. Chem. Soc., 2007, 129, 12064–12065 CrossRef CAS.
- X. Wang, J. A. Rodriguez, J. C. Hanson, D. Gamarra, A. Martínez-Arias and M. Fernández-García, J. Phys. Chem. B, 2006, 110, 428–434 CrossRef CAS.
- X. Garcia, L. Soler, N. J. Divins, X. Vendrell, I. Serrano, I. Lucentini, J. Prat, E. Solano, M. Tallarida, C. Escudero and J. Llorca, Catalysts, 2020, 10 CrossRef CAS.
- S. Patel, P. F. Jawlik, L. Wang, G. S. Jackson and A. Almansoori, J. Fuel Cell Sci. Technol., 2012, 9, 041002–041007 CrossRef.
- M. Liao, W. Wang, R. Ran and Z. Shao, J. Power Sources, 2011, 196, 6177–6185 CrossRef CAS.
- J. M. Klein, M. Hénault, C. Roux, Y. Bultel and S. Georges, J. Power Sources, 2009, 193, 331–337 CrossRef CAS.
- J. M. Klein, M. Hénault, P. Ǵlin, Y. Bultel and S. Georges, Electrochem. Solid-State Lett., 2008, 11, B144–B147 CrossRef CAS.
- Z. Zhan and S. A. Barnett, Science, 2005, 308, 844–847 CrossRef CAS.
- Y. Chen, B. deGlee, Y. Tang, Z. Wang, B. Zhao, Y. Wei, L. Zhang, S. Yoo, K. Pei, J. H. Kim, Y. Ding, P. Hu, F. F. Tao and M. Liu, Nat. Energy, 2018, 3, 1042–1050 CrossRef CAS.
- M. Boaro, S. Colussi and A. Trovarelli, Front. Chem., 2019, 7, 28 CrossRef CAS.
- J. Qiao, N. Zhang, Z. Wang, Y. Mao, K. Sun and Y. Yuan, Fuel Cells, 2009, 9, 729–739 CrossRef CAS.
- G. Kim, S. Lee, J. Y. Shin, G. Corre, J. T. S. Irvine, J. M. Vohs and R. J. Gorte, Electrochem. Solid-State Lett., 2009, 12, B48 CrossRef CAS.
- S. McIntosh and R. J. Gorte, Chem. Rev., 2004, 104, 4845–4865 CrossRef CAS PubMed.
- A. Trovarelli and P. Fornasiero, Ceria and Related Materials, Imperial College Press, 2002 Search PubMed.
- B. J. M. Sarruf, J.-E. Hong, R. Steinberger-Wilckens and P. E. V. de Miranda, Int. J. Hydrogen Energy, 2020, 45, 5297–5308 CrossRef CAS.
- B. Hołówko, P. Błaszczak, M. Chlipała, M. Gazda, S. F. Wang, P. Jasiński and B. Bochentyn, Int. J. Hydrogen Energy, 2020, 45, 12982–12996 CrossRef.
- J. M. Escudero, P. M. Yeste, Á. M. Cauqui and Á. M. Muñoz, Materials, 2020, 13 CrossRef.
- M. Shahid, C. He, S. Sankarasubramanian, V. Ramani and S. Basu, Int. J. Hydrogen Energy, 2020, 45, 11287–11296 CrossRef CAS.
- K. Joong Yoon, M. Biswas, H.-J. Kim, M. Park, J. Hong, H. Kim, J.-W. Son, J.-H. Lee, B.-K. Kim and H.-W. Lee, Nano Energy, 2017, 36, 9–20 CrossRef.
- H.-S. Noh, J. Hong, H. Kim, K. J. Yoon, B.-K. Kim, J.-H. Lee and J.-W. Son, ECS Trans., 2015, 68, 1915–1920 CrossRef CAS.
- H.-S. Noh, J. Hwang, K. Yoon, B.-K. Kim, H.-W. Lee, J.-H. Lee and J.-W. Son, J. Power Sources, 2013, 230, 109–114 CrossRef CAS.
- H.-S. Noh, H. Lee, B.-K. Kim, H.-W. Lee, J.-H. Lee and J.-W. Son, J. Power Sources, 2011, 196, 7169–7174 CrossRef CAS.
- H.-S. Noh, J.-S. Park, H. Lee, H.-W. Lee, J.-H. Lee and J.-W. Son, Electrochem. Solid-State Lett., 2011, 14, B26–B29 CrossRef CAS.
- H.-S. Noh, J.-S. Park, J.-W. Son, H. Lee, J.-H. Lee and H.-W. Lee, J. Am. Ceram. Soc., 2009, 92, 3059–3064 CrossRef CAS.
- H.-S. Noh, J.-W. Son, H. Lee, H.-I. Ji, J.-H. Lee and H.-W. Lee, J. Eur. Ceram. Soc., 2010, 30, 3415–3423 CrossRef CAS.
- H.-S. Noh, K. J. Yoon, B.-K. Kim, H.-J. Je, H.-W. Lee, J.-H. Lee and J.-W. Son, J. Power Sources, 2014, 249, 125–130 CrossRef CAS.
- J. H. Park, S. M. Han, K. J. Yoon, H. Kim, J. Hong, B.-K. Kim, J.-H. Lee and J.-W. Son, J. Power Sources, 2016, 315, 324–330 CrossRef CAS.
- C.-A. Thieu, H.-I. Ji, H. Kim, K. J. Yoon, J.-H. Lee and J.-W. Son, Appl. Energy, 2019, 243, 155–164 CrossRef CAS.
- Z. Jaworski, B. Zakrzewska and P. Pianko-Oprych, Rev. Chem. Eng., 2017, 33, 217–235 CAS.
- K. Sasaki and Y. Teraoka, J. Electrochem. Soc., 2003, 150, A885–A888 CrossRef CAS.
- K. Kochloefl, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger and J. Weitkamp, Wiley-VCH, Weinheim, 1997, pp. 1819-1831 Search PubMed.
- H. Okamoto, M. E. Schlesinger, E. M. Mueller, ASM Handbook Volume 3: Alloy Phase Diagrams, ASM International, Ohio, 2016 Search PubMed.
- W. Xu, Z. Liu, A. C. Johnston-Peck, S. D. Senanayake, G. Zhou, D. Stacchiola, E. A. Stach and J. A. Rodriguez, ACS Catal., 2013, 3, 975–984 CrossRef CAS.
- J. Carrasco, D. Lopez-Duran, Z. Liu, T. Duchon, J. Evans, S. D. Senanayake, E. J. Crumlin, V. Matolin, J. A. Rodriguez and M. V. Ganduglia-Pirovano, Angew. Chem., Int. Ed. Engl., 2015, 54, 3917–3921 CrossRef CAS.
- T. Takeguchi, S.-n. Furukawa and M. Inoue, J. Catal., 2001, 202, 14–24 CrossRef CAS.
- Y. Chen, F. Chen, W. Wang, D. Ding and J. Gao, J. Power Sources, 2011, 196, 4987–4991 CrossRef CAS.
- N. Laosiripojana, W. Sangtongkitcharoen and S. Assabumrungrat, Fuel, 2006, 85, 323–332 CrossRef CAS.
- W. An, D. Gatewood, B. Dunlap and C. H. Turner, J. Power Sources, 2011, 196, 4724–4728 CrossRef CAS.
- N. Galea, D. Knapp and T. Ziegler, J. Catal., 2007, 247, 20–33 CrossRef CAS.
- L. Zhu and A. E. DePristo, J. Catal., 1997, 167, 400–407 CrossRef CAS.
- B. Hua, M. Li, J. Pu, B. Chi and L. Jian, J. Mater. Chem. A, 2014, 2, 12576–12582 RSC.
- M. Li, B. Hua, J.-l. Luo, S. P. Jiang, J. Pu, B. Chi and L. Jian, J. Mater. Chem. A, 2015, 3, 21609–21617 RSC.
- B. Hua, N. Yan, M. Li, Y.-F. Sun, J. Chen, Y.-Q. Zhang, J. Li, T. Etsell, P. Sarkar and J.-L. Luo, J. Mater. Chem. A, 2016, 4, 9080–9087 RSC.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- W. Sutthisripok, S. Sattayanurak and L. Sikong, J. Porous Mater., 2007, 15, 519–525 CrossRef.
- S. P. Jiang, S. Zhang, Y. D. Zhen and A. P. Koh, Electrochem. Solid-State Lett., 2004, 7, A282 CrossRef CAS.
- B. Timurkutluk, C. Timurkutluk, M. D. Mat and Y. Kaplan, Int. J. Energy Res., 2011, 35, 1048–1055 CrossRef CAS.
- T. Hibino, A. Hashimoto, M. Yano, M. Suzuki and M. Sano, Electrochim. Acta, 2003, 48, 2531–2537 CrossRef CAS.
- J.-h. Myung, S.-D. Kim, T. H. Shin, D. Lee, J. T. S. Irvine, J. Moon and S.-H. Hyun, J. Mater. Chem. A, 2015, 3, 13801–13806 RSC.
- P. Li, Z. Wang, X. Yao, N. Hou, L. Fan, T. Gan, Y. Zhao, Y. Li and J. W. Schwank, Catal. Today, 2019, 330, 209–216 CrossRef CAS.
- Z. Wang, W. Weng, K. Cheng, P. Du, G. Shen and G. Han, J. Power Sources, 2008, 179, 541–546 CrossRef CAS.
- C. Sun, Z. Xie, C. Xia, H. Li and L. Chen, Electrochem. Commun., 2006, 8, 833–838 CrossRef CAS.
- K. Wang, R. Ran and Z. Shao, J. Power Sources, 2007, 170, 251–258 CrossRef CAS.
- C. Jin, C. Yang, H. Zheng and F. Chen, J. Power Sources, 2012, 201, 66–71 CrossRef CAS.
- A. Yan, M. Phongaksorn, D. Nativel and E. Croiset, J. Power Sources, 2012, 210, 374–380 CrossRef CAS.
- J. Qu, W. Wang, Y. Chen, X. Deng and Z. Shao, Appl. Energy, 2016, 164, 563–571 CrossRef CAS.
- G. Jeanmonod, S. Diethelm and J. Van Herle, J. Phys.: Energy, 2020, 2, 034002 CAS.
- P. J. Gasper, Y. Lu, A. Y. Nikiforov, S. N. Basu, S. Gopalan and U. B. Pal, J. Power Sources, 2020, 447, 227357 CrossRef CAS.
- H. Sumi, H. Shimada, Y. Yamaguchi, Y. Mizutani, Y. Okuyama and K. Amezawa, Sci. Rep., 2021, 11, 10622 CrossRef CAS PubMed.
- J. Hong, A. Bhardwaj, H. Bae, I.-h. Kim and S.-J. Song, J. Electrochem. Soc., 2020, 167 CrossRef CAS.
- A. Leonide, V. Sonn, A. Weber and E. Ivers-Tiffée, J. Electrochem. Soc., 2008, 155 CrossRef CAS.
- D. P. Ho, H. H. Ngo and W. Guo, Bioresour. Technol., 2014, 169, 742–749 CrossRef CAS PubMed.
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta06922e |
| This journal is © The Royal Society of Chemistry 2022 |