
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pt current collectors artificially boosting praseodymium doped ceria oxygen surface exchange coefficients†
Yuxi
Ma
 ab,
Theodore E.
Burye
ac and
Jason D.
Nicholas
*a
ab,
Theodore E.
Burye
ac and
Jason D.
Nicholas
*a
aMichigan State University, Chemical Engineering and Materials Science Dept., East Lansing, MI 48824, USA. E-mail: jdn@msu.edu
bContemporary Amperex Technology USA Inc, Rochester Hills, MI 48309, USA
cU.S. Army Combat Capabilities Development Command Ground Vehicle Systems Center, Warren, MI 48092, USA
First published on 26th October 2021
Abstract
The chemical oxygen surface exchange coefficient (kchem) values used to quantify and rank oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) catalyst performance for high-temperature, oxygen-exchange-enabled devices (such as Solid Oxide Fuel Cells, Solid Oxide Electrolysis Cells, oxygen sensors, etc.) are often determined electrically, with the aid of precious metal current collectors. However, the curvature relaxation (κR) and Time-of-Flight Secondary Ion Mass Spectroscopy (ToF-SIMS) analyses performed here on Pulsed Laser Deposited thin films of the oxygen exchange catalyst Pr0.1Ce0.9O2−x (PCO) show that unpolarized platinum current collectors dramatically improve the kchem of Si-contaminated PCO by reducing the Si concentration at the PCO surface and/or diffusing into the PCO; even for PCO thin films only exposed to mild temperatures of 500 °C. This suggests that precious metal current collectors are likely responsible for some of the large kchem variation reported in the literature for “identical” materials tested under “identical” conditions.
1. Introduction
Solid Oxide Fuel Cells (SOFCs) are solid state chemical to electrical energy conversion devices that exhibit high power density, high energy density, high energy conversion efficiency, and fuel flexibility.1–4 In addition, SOFCs can be used in reverse as Solid Oxide Electrolysis Cells (SOECs) to store energy, produce chemicals, and/or generate fuels.4–7 Unfortunately, high system costs, high operating temperatures, and high degradation rates have complicated the commercial deployment of these Solid Oxide Cells (SOCs).4 With time however, it seems likely that improved materials will help lower these commercialization barriers. Since today's “State-of-the-Art” SOCs are typically limited by poor oxygen ion transport (1) through the bulk of the solid electrolyte, (2) across the solid electrolyte-solid electrode interface, and/or (3) across the solid electrode-gas phase interface,8 it seems likely that future material sets will feature increased bulk oxygen ion conductivities, reduced interfacial resistivities, and/or increased chemical oxygen surface exchange coefficients.The chemical oxygen surface exchange coefficient, kchem, is commonly defined as:
| J = kchem(Co − Cs) | (1) |
 | (2) |
| ko = kchem/γo | (3) |
 | (4) |
A material's oxygen surface exchange properties (i.e. a material's kchem, ko, kq, Rsetc.) can also be combined with SOC structure–property-performance models16–18 to predict SOC electrode polarization resistances19–21 and/or identify optimal SOC electrode microstructures.22 Other devices (such as solar thermochemical cells,23 oxide memristors,24 oxygen separation membranes,25 catalytic converters,26 oxygen sensors,27etc.) can also have their performance and/or efficiency impacted by the oxygen surface exchange properties of the materials within them.
However, before the scientific community can effectively engineer oxygen surface exchange coefficients for improved device performance, it must first figure out how to reliably measure them. Specifically, Fig. 1 shows that, even for the most common MIEC materials, huge kchem, ko and Rs variations exists in literature for “identical” materials under “identical” conditions. For instance, in Fig. 1 there is a >1000 times variation in the extrapolated 700 °C Pr0.1Ce0.9O2−x (PCO) kchem values, a >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times variation in the measured 650 °C La0.6Sr0.4FeO3−x (LSF) kchem values, and a >100000 times variation in the measured 500 °C Ba0.6Sr0.4Co0.8Fe0.2O3−x (BSCF) kchem values.
000 times variation in the measured 650 °C La0.6Sr0.4FeO3−x (LSF) kchem values, and a >100000 times variation in the measured 500 °C Ba0.6Sr0.4Co0.8Fe0.2O3−x (BSCF) kchem values.
 | ||
| Fig. 1 Literature-reported oxygen surface exchange properties of La0.6Sr0.4FeO3−x (LSF, squares), La0.6Sr0.4Fe0.8Co0.2O3−x (LSFC, diamonds), La0.6Sr0.4CoO3−x (LSC, up-pointing triangles), Sm0.5Sr0.5CoO3−x (SSC, left-pointing triangles), Ba0.5Sr0.5Co0.8Fe0.2O3−x (BSCF, right-pointing triangles) and Pr0.1Ce0.9O1.95−x (PCO, pentagons) in air. Color denotes directly-measured values. Dark gray denotes values converted viaeqn (2)–(4) with data from the same study as the directly-measured value. Light gray denotes values converted viaeqn (2)–(4) with Co and γo values obtained from other studies (i.e. those shown in Fig. S1 of the ESI†). Here, ko was assumed to be ≈kq, as commonly done in the literature10. The ESI† contains a description of the minor equations and the actual spreadsheets used to perform the oxygen surface exchange conversions. A = Mosleh et al.,57B = Søgaard et al.,53 C = Søgaard et al.,113D = Yang et al.,79 E = ten Elshof et al.,15F = Armstrong et al.,114 G = Ishigaki et al.,115H = Baumann et al.,116I = Tripkovic et al.,36J = Plonczak et al.,60 K = Bouwmeester et al.,117 L = Cox-Galhorta and McIntosh,35 M = Dalslet et al.,13 N = Huang et al.,118 O = Lane et al.,119 P = Li et al.,120 Q = Ried et al.,121 R = Wang et al.,122 S = Chater et al.,123T = Steele and Bae,124U = Simrick et al.,62V = Moreno et al.,125 W = Berenov et al.,126 X = Egger et al.,127 Y = Søgaard et al.,128Z = Januschewsky et al.,28AA = Siebenhofer et al.,40AB = Adler,129AC = Hayd et al.,10 AD = Preis et al.,43 AE = Yeh et al.,130 AF = Fullarton et al.,131 AG = Fu et al.,132AH = Burriel et al.,56AI = Wang et al.,133 AJ = Bucher et al.,134 AK = Bucher et al.,135 AL = Chen and Shao,136 AM = Girdauskaite et al.,137AN = Baumann et al.,138AO = Wang et al.,139, AP = Falkenstein et al.,44 AQ = Lohne et al.,140 AR = Nicollet et al.,141 AS = Nicollet et al.,51AT = Chen et al.,102AU = Ma and Nicholas,75AV = Chen et al.,90AW = Simons et al.,61AX = Zhao et al.,105AY = Chen et al.102Bold studies were performed on thin films, italic studies were performed on loose or partially sintered powders, and unformatted studies were performed on bulk pellets with relative densities >92%. | ||
The reasons for these oxygen surface exchange property variations are manifold. For instance, previous studies have shown that systematic differences in film crystallinity,28,29 grain size,30 surface orientation,31–34 surface chemistry,35–39 and/or surface gas phase adsorbates34,40,41 can all impact a material's measured oxygen surface exchange properties. Experimental complications such as poor gas phase mixing,42 non-instantaneous atmospheric switching times,43 and/or large amounts of sample oxygen release that can undesirably alter the atmospheric pO2 (ref. 44) can also produce kchem variations.
However, despite the fact that precious metal surface additions are well known to improve the performance of the MIEC materials used as SOC electrodes, oxygen sensors, vehicular catalytic converters etc.,45–48 an examination of how current collectors (especially the precious metal current collectors commonly used in the existing oxygen surface exchange literature36,40,49–62), affect oxygen surface exchange properties is less clear. This uncertainty is perpetuated by the fact that (1) today's most-common kchem, ko, kq, and Rs measurement techniques (i.e. Electrical Conductivity Relaxation (ECR) and Electrochemical Impedance Spectroscopy (EIS)) require the electrical polarization of a current collector, and (2) many ECR and EIS studies do not include descriptions of the current collector microstructure, materials, thickness, pattern geometry, and/or electrical-polarization used to perform the oxygen surface exchange measurements. This is problematic because electrically-polarized current collectors could alter oxygen exchange through (1) the enhanced oxygen exchange properties some precious metals have been shown to catalyze at metal-MIEC–air interfaces,47,63–65 (2) the chemical gettering of surface-segregated MIEC ions/impurities that some current collecting materials are known to possess,66 (3) current collector coefficient of thermal expansion (CTE) mismatch stress induced alterations in the point defect and/or surface chemistry that some mechano-chemically-active MIECs are known to exhibit,67,68 (4) electric-field induced alterations in the point defect, surface chemistry, and/or metal-MIEC–air triple-phase-boundary widths that are known to occur in some MIECs,63,68–70etc.
Although a few studies to the contrary exist,71 many more studies have found that precious metal surface decoration can significantly alter a material's measured oxygen surface exchange properties. For instance, ECR measurements by Egger and Sitte72 demonstrated that a thin layer of silver increased the 600 °C kchem of La2NiO4 by an order of magnitude, and Zhang et al.32 observed similar ECR kchem improvements for lanthanum strontium manganate decorated with platinum or palladium nanoparticles. EIS and oxygen isotope exchange depth profiling experiments by Sahibzada et al.73 showed that palladium surface decoration decreased the 700 °C ko of La0.6Sr0.4Fe0.8Co0.2O3−x (LSFC) by an order of magnitude but improved the electrochemical performance of LSFC at lower temperatures. Similarly, microelectrode EIS measurements performed by Riedl et al.74 showed that platinum surface decoration either increased or decreased the Rs of La0.6Sr0.4FeO3−x (LSF) by up to ∼100 times depending on the pO2. Further, Ma and Nicholas75 found that the 600 °C PCO kchem values obtained from current-collector-free curvature relaxation (κR) and current-collector-free optical transmission relaxation (OTR) experiments were identical, but more than 10–100 times lower than those obtained from kchem techniques utilizing precious metals. Similarly, simultaneous ∼300–600 °C measurements by Perry and coworkers59,76 on strontium titanium ferrite thin films showed that roughly identical kchem values were obtained from OTR and in-plane ECR measurements utilizing current collectors that only covered a small portion of the MIEC surface, but that ∼10 times higher kchem values were obtained via through-sample EIS measurements utilizing current collectors covering a large portion of the MIEC surface.
Despite these reports, precious metal current collectors are still being actively used to measure the oxygen surface exchange properties of MIEC materials.36,49–60 Hence, the first objective of this work was to dramatically demonstrate the role that even minute amounts of unpolarized precious metals can have on the kchem of MIEC materials. The second objective of this work was to go beyond the aforementioned past studies and identify any local chemistry changes likely responsible for the observed kchem alteration. These objectives were achieved by pairing (1) the remarkable ability of the curvature relaxation technique to obtain direct kchem measurements on PCO thin films that were either uncovered, partially Pt-covered, or completely Pt-covered, with (2) Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS) analyses of the sample chemistry before and after curvature relaxation.
2. Experimental methods
Here, PCO thin films were produced by Pulsed Laser Deposition (PLD), optionally coated with one of the platinum current collector geometries shown in Fig. 2, and tested via curvature relaxation (κR). | ||
| Fig. 2 A completely Pt-covered PCO|YSZ sample (left), an uncovered PCO|YSZ sample (middle), and a partially Pt-covered PCO|YSZ sample (right). Note, each sample was 25.4 mm in diameter. | ||
2.1 Sample fabrication
Prior to thin film deposition, 25.4 mm diameter, one-side-polished, 200-micron-thick, (100)-oriented (Y2O3)0.095(ZrO2)0.905 (YSZ) single crystal substrates (Crystec GmbH, Berlin, Germany) were air-annealed at 1450 °C for 20 h utilizing 5 °C min−1 nominal heating and cooling rates to remove any residual stress within them. This wafer annealing schedule was chosen because it had previously been shown capable of reducing the curvature changes observed upon heating these YSZ wafers from 25 to 700 °C to less than 0.005 m−1 (so that residual stress changes would not obscure the ∼0.002 m−1 curvature changes caused by film reduction or oxidation at a single, constant temperature).75 In addition, ∼94% dense Pr0.1Ce0.9O2−δ PLD targets were produced from PCO powder fabricated via the glycine nitrate combustion (GNC) method.77 This GNC process was conducted by first dissolving 99.9% pure cerium nitrate (Strem Chemicals, Newburyport MA), 99.9% pure praseodymium nitrate (Strem Chemicals, Newburyport MA), and 99% pure glycine (Millipore Sigma, Burlington, MA) in 18.2 MΩ water (Millipore Sigma, Burlington, MA) at a 1:1 glycine:nitrate molar ratio using a Teflon coated stir bar (Fischer Scientific, Pittsburgh, PA) in a Pyrex beaker (Fischer Scientific, Pittsburgh, PA). The resulting solutions were then ignited over a hotplate in a stainless-steel reaction vessel (Polar Ware, Kiel, WI). To remove unreacted glycine, the resulting powder was calcined in a 99.8% pure alumina crucible (CoorsTek, Golden, CO) at 1000 °C in air for 1 hour using 5 °C min−1 nominal heating and cooling rates. The calcined powder was then uniaxially compacted to ∼63 MPa in a 38 mm stainless-steel die (MTI, Richmond, CA) and sintered at 1450 °C for 20 h in air using nominal 3 °C min−1 heating and 10 °C min−1 cooling rates. The resulting sintered target was then ground down to 25 mm diameter and 2.5 mm thick using 240 grit SiC sandpaper and bonded with silicone to a ∼2.5 mm thick, 25 mm diameter copper backing.
PCO PLD thin films were produced from the aforementioned target by depositing ∼265 nm of PCO onto 700 °C-preheated YSZ wafers within 30 mTorr of oxygen using the PLD/MBE 2300 system (PVD Products, Inc) at the Northwestern University PLD User Facility. PLD was conducted using a 100 mm target-to-substrate distance and 15000, 10 Hz, 200 mJ, 248 nm ΚrF excimer laser pulses. All the PCO|YSZ samples reported here were (1) cooled to room temperature at 10 °C min−1 upon the completion of PCO deposition, (2) produced consecutively during a single PLD session, and (3) stored within individual polypropylene wafer containers when not undergoing subsequent processing or characterization. After thin film deposition, each PCO|YSZ sample had its oxygen vacancy defect concentration re-equilibrated with air via an hour-long 1100 °C hold utilizing nominal 3 °C min−1 heating and cooling rates.
To produce patterned platinum current collectors covering ∼2.7% of the PCO geometric surface area (such as those for the “partially Pt-covered PCO|YSZ samples shown on the right-hand side of Fig. 2) some of the 1100 °C re-equilibrated PCO|YSZ samples were subjected to an acetone rinse, a methanol rinse, a deionized water rinse, nitrogen gas drying, a 5–10 minute excursion to 115 °C, 10 seconds of 700 rpm rotation in a spin coater, and finally 30 seconds of 3000 rpm rotation in a spin coater. Next, the exposed PCO surfaces of these samples were covered with Photoresist S1813 (Microchem Corp, Westborough, MA) by slowing the PCO|YSZ sample rotation to 700 rpm, dripping ∼50 μL of photoresist solution onto the PCO|YSZ sample, removing the PCO|YSZ sample from the spin coater, and baking the spin-coated sample for 1 min at 115 °C. Current collector patterns were then introduced into the photoresist via UV curing in a Karl Suss MJB3 mask aligner (SUSS MicroTec SE, Garching, Germany) using a positive mask (Photoscience Inc, Lexington, KY) and 274 watts of 250 nm light for 90 seconds. The UV-exposed portion of the photoresist was then removed by exposing it to photoresist developer MF319 (Microchem Corp, Westborough, MA) for 45 seconds, rinsing it with deionized water, and then baking it in air at 115 °C for 5 min. The samples were then placed in an Axiss PVD System (Kurt J. Lesker, Jefferson Hills, PA), and the total chamber pressure was pumped down to 10−6 torr. After backfilling with Ar gas until the total chamber pressure was 20 mTorr, and after initiating 30 rpm of sample rotation, platinum (from a 99.95% pure Pt target) was sputtered onto the sample surface for 1 minute using 200 W of power. After removal from the sputtering chamber, these samples were dipped in acetone and then rinsed in isopropanol to remove the photoresist.
To produce “completely” Pt-covered PCO|YSZ samples such as those shown in the left side of Fig. 2 (i.e. those where 100% of the geometric PCO surface area was covered with 89% dense78 Pt films), some PCO|YSZ samples were placed in the aforementioned Axiss PVD system and subjected to the same platinum sputtering conditions used for the partially Pt-covered PCO|YSZ samples. However, these completely Pt-covered samples were not subject to the solvent cleaning, photoresist application, UV curing, or photoresist removal processes needed for the partially Pt-covered samples.
To make sure the κR signal of the completely Pt-covered PCO|YSZ samples came from oxygen exchange into/out of the PCO (instead of the Pt), a bare YSZ wafer was completely covered with platinum using the same procedures as those used for the completely Pt-covered PCO|YSZ samples.
2.2 Oxygen surface exchange measurements
Here, thin film PCO oxygen surface exchange coefficient measurements were performed using the κR method described, and derived, previously in the literature.75,79–82 In brief, the curvature relaxation techniques determines kchem from the PCO volume changes accompanying small, isothermal, pO2-induced changes in the oxygen nonstoichiometry (Δδ) that occur in PCO above ∼400 °C in air via the reaction:75,83–85 | (5) |
Assuming that as this mechano-chemically-active defect reaction proceeds (1) the oxygen-exchange-active thin film remains well bonded to an oxygen-exchange-inactive substrate, (2) the film and substrate only deform in a linear elastic manner, (3) the film is more than ∼500 times thinner than the substrate but is thick enough to ensure that its volume changes bend the wafer in a detectable manner, (4) the film thickness is at least ∼500 times lower than the film materials characteristic thickness (LC = Dchem/kchem where Dchem is the chemical diffusion coefficient) to ensure that the sample response is only controlled by surface exchange and not partially or fully controlled by oxygen diffusion in the bulk of the film, and (5) the pO2 step size is small enough to ensure that (a) the oxygen surface exchange process remains linear, and (b) the resulting change in oxygen nonstoichiometry (Δδ) is small enough to ensure that the film strain (εC) can be described by:
 | (6) |
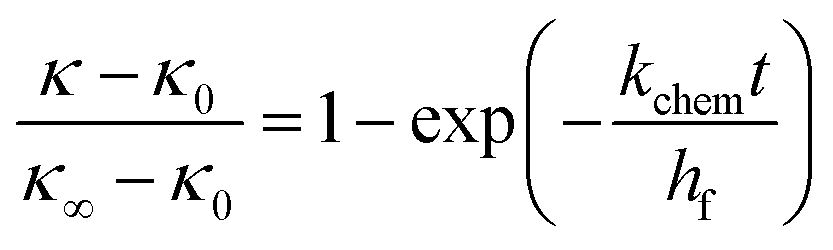 | (7) |
To determine if multiple processes with different time constants (or different rates of the same process in different parts of the sample) were active, all curvature data was replotted to search for slope changes in  vs. time plots during each relaxation, as has been done previously.31,79,86–88
vs. time plots during each relaxation, as has been done previously.31,79,86–88
Fig. 3 shows a schematic of the controlled atmosphere multi-beam optical stress sensor setup used to perform the κR measurements reported here (see Nicholas81 for the exact dimensions). In preparation for κR experiments, each PCO|YSZ sample was placed atop the central quartz support tube shown in Fig. 3, covered with the controlled-atmosphere gas-preheating manifold shown in Fig. 3, heated to 500 °C at 5 °C min−1, and held at 500 °C for 1 hour in air to equilibrate and homogenize the PCO oxygen vacancy concentration. Then, the uncovered, partially Pt-covered, and completely Pt-covered PCO|YSZ samples were measured in 25 °C increments from 675 to 725 °C, 500 to 725 °C, and 500 to 425 °C, respectively, after holding for at least 30 minutes at each temperature to promote thermal equilibrium. Curvature relaxations were triggered by switching the pO2 around each sample from 100 sscm of 0.21 (21% O2-79% Ar, i.e. synthetic air) to 100 sccm 0.021 (2.1% O2–97.9% Ar, i.e. 10 times diluted synthetic air) using a four-way valve. Since the same synthetic gas mixtures were used to test all the samples, atmospheric impurity content differences should not have to contributed to the observed differences in sample behavior. As shown in Fig. S2–S4 of the ESI,† each sample was allowed to re-equilibrate at each pO2 for at least 5 times the characteristic relaxation time (to allow for reliable κR fitting, as explained in den Otter et al.).89 Only kchem values that were not reactor flush time limited (i.e. those where switching between two 100 sccm gas flows or between two 300 sccm gas flows yielded the same kchem values) were reported here.
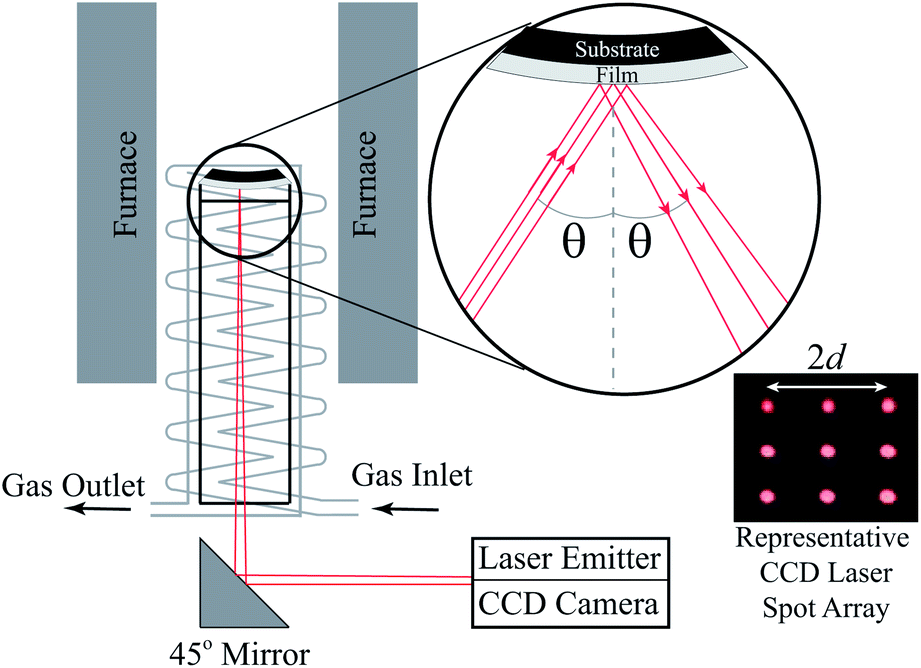 | ||
| Fig. 3 Schematic of the Multi-beam Optical Stress (MOS) sensor setup used to perform the curvature relaxation oxygen surface exchange coefficient measurements reported here. Reprinted from Nicholas,81 with permission from Elsevier. | ||
2.3 Additional sample characterization
To evaluate PCO thin film phase purity, X-Ray Diffraction (XRD) was performed in room temperature air from 20 to 80° with a 0.01° step size and a 1 s per step dwell time using unfiltered Cu kα radiation from a SmartLab diffractometer (Rigaku, The Woodlands, TX) operated at 44 kV and 40 mA with 10 mm-wide detector and source slits.To evaluate PCO thin film surface composition, X-Ray Photoelectron Spectroscopy (XPS) was performed at room temperature and 10−9 torr on uncovered PCO|YSZ samples after (1) post-deposition re-equilibration in air and (2) after curvature relaxation. XPS was performed using the Al Kα X-ray radiation inside a Phi 5600 XPS (PerkinElmer, Waltham, MA). XPS survey scans were collected with a step size of 0.4 eV and a pass energy of 187.5 eV. XPS peak positions were calibrated with the carbon 1s peak at 284.8 eV. Due to concerns that the vacuum encountered during XPS analysis might alter the PCO oxygen vacancy concentration, the samples used for XPS analyses were not used for κR experiments.
To evaluate film thickness, Scanning Electron Microscopy (SEM) was performed on fractured PCO|YSZ samples coated with ∼5 nm of Ti and imaged using a TESCAN MIRA3 Field Emission SEM (Tescan, Brno, Czechia).
To evaluate PCO thin film bulk composition, PCO|YSZ samples were analyzed via ToF-SIMS depth profiling conducted by EAG Labs (East Windsor, NJ, USA). The absolute concentrations of Zr, Y, and O in the films were calculated from the relative concentrations produced by SIMS by setting the SIMS-detected concentrations in the bulk of the YSZ substrate equal to those found in (Y2O3)0.095(ZrO2)0.905. Likewise, the absolute concentrations of Pr and Ce were calculated from the measured SIMS profiles by offsetting each Pr and Ce SIMS profile until the Pr and Ce concentrations in the interior of the film equaled those found in Pr0.1Ce0.9O2. Likewise again, the Pt concentration profiles were offset until those in the Pt current collector equaled those found in dense Pt metal. In contrast, a single Ta2O5 standard with a known amount of Si was used to estimate the Si concentrations in all the PCO films, assuming that the SIMS matrix effects in Ta2O5 and PCO were similar. Even if this assumption was incorrect and as a result introduced errors in the absolute Si concentration values, the fact that the same Si calibration, SIMS collection settings, and SIMS equipment were used for all the samples suggests that any relative Si concentration differences between the samples, and within each sample, should be valid.
3. Results and discussion
Fig. 4 shows representative XRD scans indicating that only crystalline, fluorite-structured PCO and crystalline, fluorite-structured YSZ peaks were present in the PCO|YSZ samples. Consistent with previous literature reports,75,90–95 all the PCO films here exhibited (100) preferred orientation, consistent with the (100) oriented YSZ substrates they were grown upon. | ||
| Fig. 4 XRD spectrum of an uncovered PCO|YSZ sample immediately before κR testing. Identical results (not shown) were obtained on uncoated samples analyzed after κR testing. | ||
Fig. 5 shows representative XPS scans indicating that the sample fabrication procedures used here produced “clean” PCO surfaces containing only Ce, Pr and O. No Si, Zr, Y or any other element was found in any of the XPS scans, even for uncovered PCO|YSZ samples κR tested to 725 °C in a Si-containing κR test rig known to vapor deposit Si on samples held for extended times at and above 600 °C.96 This suggests that any surface impurities in the PCO|YSZ samples here were present in amounts below the ∼1 atomic percent XPS detection limit.97
 | ||
| Fig. 5 XPS analysis of an uncovered PCO|YSZ sample immediately before κR testing. Note, the Mo peaks here are from the Mo XPS sample retaining mesh. Identical results (not shown) were obtained on uncoated PCO|YSZ samples analyzed after κR testing. | ||
Fig. S2–S4 of the ESI† show representative curvature relaxation data for the uncovered, partially Pt-covered, and completely Pt-covered PCO|YSZ samples. These plots show that all the PCO|YSZ samples here exhibited reproducible equilibrium stress levels and oxygen surface exchange constants with pO2 cycling. Further, as indicated by the linear nature of the  data during the portion of time each sample was equilibrating to a new pO2, only a single oxygen exchange process was active for all the measured samples. The small deformations involved, the mechano-chemical inactivity of YSZ under the conditions used here,98 the chemical compatibility of YSZ and ceria under the conditions used here,99 a hf/hS < 0.002, a hf < 0.0001 × LC (based on the reported 450–725 °C PCO LC values,90Dchem activation energies100,101 and kchem activation energies51,75,90,102), and the reproducible curvature behavior with pO2 switching for all the PCO|YSZ samples tested here and shown in Fig. S5 of the ESI,† ensured that all the assumptions needed to ensure the validity of eqn (7) were met.
data during the portion of time each sample was equilibrating to a new pO2, only a single oxygen exchange process was active for all the measured samples. The small deformations involved, the mechano-chemical inactivity of YSZ under the conditions used here,98 the chemical compatibility of YSZ and ceria under the conditions used here,99 a hf/hS < 0.002, a hf < 0.0001 × LC (based on the reported 450–725 °C PCO LC values,90Dchem activation energies100,101 and kchem activation energies51,75,90,102), and the reproducible curvature behavior with pO2 switching for all the PCO|YSZ samples tested here and shown in Fig. S5 of the ESI,† ensured that all the assumptions needed to ensure the validity of eqn (7) were met.
Fig. 6 shows that covering either ∼2.7% or 100% of the PCO surface with unpolarized Pt current collectors significantly boosted the PCO oxygen surface exchange coefficient. Further, uncovered PCO|YSZ samples measured at the beginning (green circles) and end (red triangles) of the kchem measurement series yielded identical kchem values, attesting to the high reproducibility of the sample fabrication and measurement procedures used here. Interestingly, compared to uncovered PCO|YSZ samples from Ma and Nicholas75 that were prepared in a different PLD chamber, grown at a lower temperature, and (unlike the present samples) alkaline etched to remove surface-segregated impurities, the uncovered PCO|YSZ samples here had lower kchem values. However, the ∼0.6 eV activation energies observed for the present study's uncovered PCO|YSZ samples were similar to those measured above 500 °C in the curvature relaxation measurements on uncovered PCO PLD thin films from Ma and Nicholas,75 the optical transmission relaxation measurements on uncovered PCO PLD thin films from Chen et al.,102 the electrochemical impedance spectroscopy measurements on Ag-paste-covered PCO PLD thin films from Chen et al.,90 and the optical transmission relaxation measurements on uncovered PCO PLD thin films from Nicollet et al.51 This suggests that the same rate-determining step for oxygen incorporation into PCO was active above ∼500 °C for all the PCO samples here and in the literature, even its rate was faster in some samples versus others.
 | ||
| Fig. 6 Oxygen surface exchange coefficients for the uncovered PCO (green circles and red triangles), partially Pt-covered PCO (orange squares), and completely Pt-covered PCO (blue diamonds) thin films measured here, compared to the PCO literature.51,61,75,90,102,105 “-R” denotes kchem values obtained upon reduction. “-O” denotes kchem values obtained upon oxidation. Note, unlike the uncovered PCO κR samples from Ma and Nicholas,75 none of the samples here were alkaline etched to remove PCO surface impurities prior to kchem measurement. | ||
In contrast, Fig. 6 shows that at temperatures less than ∼500 °C, the completely Pt-covered PCO|YSZ samples (blue diamonds) had activation energies essentially double those of the uncoated PCO|YSZ samples measured above ∼500 °C. Similarly, although not mentioned by the authors, high sub-500 °C PCO activation energies of ∼1.3 eV can also be seen in the optical transmission spectroscopy data of Chen et al.102 on uncovered (i.e. Pt-free) PCO PLD thin films that transitioned to kchem activation energies of ∼0.5 eV above ∼500 °C. (Unfortunately, the exceeding-fast, low-temperature oxygen exchange kinetics of the completely Pt-covered PCO|YSZ samples meant that reactor flush time limitations prevented them from being measured above 500 °C, when their kchem > 1 × 10−6 cm s−1). The fact that this previously unrecognized kchem activation energy transition was seen in Chen et al.'s102 samples without platinum current collectors indicated that it is a general characteristic of PCO, and is not related to the absence or presence of precious metal current collectors. The observation of a higher activation energy at lower temperatures is the opposite of (1) what is commonly observed in other doped ceria compositions,103 and (2) what might be expected based on the idea that lower temperatures would de-activate higher activation energy barrier pathways for oxygen exchange into/out of a static structure. Hence, it is likely that the ∼500 °C transition to a higher kchem activation energy upon cooling below 500 °C is related to a change in the rate-determining step for oxygen exchange brought about by the increased difficulty in exchanging oxygen as the thin film PCO Pr3+/Pr4+ ratio and oxygen nonstocichometry rapidly trend toward zero below ∼500 °C.75
Regardless of the exact reason for this newly-recognized 500 °C activation energy change, Fig. 6 makes it clear that completely covering the PCO surface with sputtered Pt dramatically boosts the measured kchem. The fact that sputtered Pt did not reduce the measured kchem is consistent with the thin nature of the sputtered Pt current collectors, the low grain boundary and/or surface-path resistivities observed previously for oxygen transport in sputtered Pt films,64,104 and the likely-percolated ∼11% porosity of the sputtered Pt films78 facilitating gaseous oxygen transport to the PCO surface.
Fig. 6 also shows that the partially Pt-covered PCO|YSZ samples exhibited kchem values between those of the uncovered and completely Pt-covered PCO|YSZ samples. With only ∼2.7% of the PCO surface covered by Pt, the observed kchem enhancements were modest and hence the partially Pt-covered PCO|YSZ samples could only be measured above 500 °C, where they displayed activation energies similar to those measured previously.
Fig. 7 provides a graphical summary of just how much faster oxygen exchange was in the completely Pt-covered PCO|YSZ samples than in either the uncovered or partially Pt-covered PCO|YSZ samples. Specifically, even though the temperature for the completely Pt-covered PCO|YSZ curvature relaxation is 250 °C lower than the uncovered or partially Pt-covered PCO|YSZ samples in Fig. 6, the time required to complete the oxygen exchange process is ∼2 times less.
 | ||
| Fig. 7 κR behavior of a completely Pt-covered PCO|YSZ sample at 475 °C compared to a partially Pt-covered PCO|YSZ sample at 725 °C and an uncovered PCO|YSZ sample at 725 °C. The dashed lines are eqn (7) fits to the data. | ||
Fig. 8 suggests that the platinum current collectors used here improved the PCO kchem by removing Si from the PCO surface and/or diffusing into the PCO. Specifically Fig. 8a and b show that the uncovered PCO films had approximately 1 Si atom for every 100 Pr atoms in their bulk and had enriched amounts of Si at their air-PCO and PCO-YSZ interfaces. This Si contamination probably resulted from the silica-based glassware used to produce the PCO powder utilized in PLD target fabrication (this contamination could likely have been avoided through the use of polyethylene beakers but was retained here to illustrate the interaction common processing and environmental contaminants can have with PCO platinum current collectors). Si enrichment at the air-PCO interface may help explain the low kchem values observed for the uncovered PCO|YSZ samples, since surface-segregated Si impurities are well-known for inhibiting oxygen surface exchange and bulk oxygen transport in PCO and a variety of other ceria-based compositions.96,105–109Fig. 8a and b also show a slight enhancement of Zr and Ce at the PCO surface, but since those elements occurred at concentrations 10 to 100 times less than the Si concentration, Si was assumed to be responsible for the low kchem values observed in the uncovered PCO|YSZ samples. Fig. 8a and b also show interdiffusion of Pr, Ce, Zr, and Y between the film and substrate, and diffusion of Si from the film into the substrate, the majority of which presumably happened during the 1 hour, 1100 °C re-equilibration in air. Interestingly, a comparison of Fig. 8a and b shows that although the bulk Si level in the film and near the exposed surface roughly doubled (presumably due to silica vaporization from the silica κR test rig known to occur at and above ∼600 °C)96,105 and some of the bulk Si migrated to the PCO|YSZ interface, no other significant changes in the sample chemistry, or chemical distribution, occurred during κR testing up to 725 °C.
 | ||
| Fig. 8 SIMS analysis of (a) an uncovered PCO\YSZ sample after PLD and 1100 °C re-equilibration in air, (b) an uncovered PCO|YSZ sample after PLD, 1100 °C re-equilibration in air, and κR testing, and (c) a completely Pt-covered PCO|YSZ sample after PLD, 1100 °C re-equilibration in air, Pt current collector (C.C) deposition, and κR testing. The position of the PCO–YSZ interface was determined from the intersection of the film and substrate majority cation concentrations (i.e. the Ce and the Zr concentrations). This position also corresponded to the midpoint in oxygen concentration between the film and substrate. The position of the current collector (C.C) – PCO interface in part c) was determined by the transition from a flat Pt concentration in the C.C. to a diffusion Pt concentration profile in the film. This C.C. – PCO interface location also produced identical near-surface reductions in the Ce and Pr film concentrations as those observed in the uncovered PCO films in parts (a) and (b). | ||
Fig. 8c suggests that the completely Pt-covered PCO|YSZ current collectors applied here cleaned the air-PCO interface by serving as a chemical getter for the surface-segregated Si, Zr and Ce present on uncoated films. The ability of Pt current collectors to act as Si getters to improve the PCO kchem is not completely surprising since (1) Ma and Nicholas96 recently quantified the relationship between the PCO Si surface content and kchem, and (2) Zhao et al.105 found that La or Sm PCO surface additions improved kchem by gettering surface Si. However, it is also possible that the near-surface dips in the Si, Zr, and Ce profiles could be artifacts caused by the SIMS front transitioning from the metal to the oxide.
In addition to suggesting that Pt cleared Si from the PCO surface, Fig. 8c shows that significant quantities of Pt diffused into the PCO film (with the Pt concentration exceeding the Pr doping level for more than 50 nm into the PCO film). This is somewhat surprising because the completely Pt-covered PCO|YSZ samples only saw temperatures up to 500 °C during κR testing. However, this behavior is consistent with prior reports that platinum can diffuse into ceria at room temperature,110 especially when ceria experiences reducing conditions,111 as likely occurred during the Pt current collector sputtering process.
Whether from the removal of Si from the PCO surface, Pt diffusion into the ceria, or some of the other mechanisms proposed in the literature,63,111,112 the results here make it clear that completely covering a significant portion of a material's surface with Pt current-collectors can led to kchem values significantly different from those the material would display on its own. As such, the small kchem enhancements observed here for the partially Pt-covered samples in Fig. 6 were assumed to be due to the portion of the PCO close to the Pt current collectors behaving like the completely Pt-covered PCO|YSZ samples, while those portions of the PCO further away from the Pt current collectors behaved more like the uncoated PCO|YSZ samples.
Fig. 9 compares the curvature response of a completely Pt-covered PCO|YSZ sample to a completely Pt-covered YSZ wafer. The fact that essentially no curvature response was observed in the completely Pt-covered YSZ wafer with pO2 switching confirms that the observed kchem enhancements were not the result of oxygen exchange into/out of the bulk of the Pt, but instead were related to Pt current collectors influencing oxygen exchange into/out of the PCO.
 | ||
| Fig. 9 Curvature changes upon 500 °C pO2 switching for (a) completely Pt-covered PCO|YSZ sample and (b) a completely Pt-covered YSZ wafer. | ||
4. Conclusions
Despite many studies suggesting that precious metal current collectors and/or impurities can impact the performance of oxygen-exchange-enabled devices,45–48 precious metal current collectors are still routinely used to transport electronic species into/out of oxygen exchange materials in Electrical Conductivity Relaxation,36,49–58 and Electrical Impedance Spectroscopy59,60 oxygen exchange experiments. The results here demonstrate that precious metal current collectors are not necessarily inert (i.e. they can diffuse into and/or chemically clean the surface of the oxygen exchange material of interest), can unexpectedly alter the measured kchem values, and, hence, should be treated with caution when used for oxygen surface exchange measurements. This is especially true when performing oxygen exchange measurements on either bulk or thin film samples where a significant fraction of the oxygen-exchange-active surface is in close proximity to a precious metal current collector (such as ECR kchem measurements on materials with low electronic resistivities that require the close placement of interdigitated electrodes to get measurable electronic conductivities, experiments performed with fine-grained colloidal precious metal pastes and/or precious metal thin films applied over most of the oxygen-exchanging surface, etc.). Further, if precious metal current collectors dramatically improve the oxygen exchange kinetics of the neighboring/underlying MIEC, artificially high kchem values could also be recorded for electrical (ECR, EIS, etc.) kchem measurements performed using small precious metal current collectors placed a significant distance apart (since the precious-metal-activated portions of the MIEC surface would act as “major leaks” for oxygen incorporation into the bulk of the material and hence would “drown-out” the response from slower oxygen incorporation along the bare MIEC surfaces). For all these reasons, it is likely that precious metal current collectors are responsible for a significant portion of the kchem variation reported in the literature for “identical” materials tested under “identical” conditions.Author contributions
Y. Ma performed the thin film deposition and characterization, T. E. Burye performed the kchem literature review, and J. D. Nicholas conceptualized and led the write-up of this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. Ma acknowledges support from U.S. Department of Energy Office of Fossil Energy Award Number DE-FE0031672. T. E. Burye and J.D. Nicholas acknowledge support from U.S. National Science Foundation CAREER Award Number CBET-1254453. The PLD thin films were grown at the Materials Research Center at Northwestern University, which is supported by the National Science Foundation MRSEC program (DMR-1720139) and the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205). Scanning electron microscopy was conducted at the Michigan State University Composite Materials and Structures Center, which is supported by the NSF Major Research Instrumentation Program and Michigan State University.References
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935–939 CrossRef CAS PubMed.
- S. McIntosh and R. J. Gorte, Chem. Rev., 2004, 104, 4845–4865 CrossRef CAS PubMed.
- S. H. Chan, C. F. Low and O. L. Ding, J. Power Sources, 2002, 103, 188–200 CrossRef CAS.
- J. D. Nicholas, ECS Interface, 2013, 22, 49–54 CAS.
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS.
- M. B. Mogensen, M. Chen, H. L. Frandsen, C. Graves, J. B. Hansen, K. V. Hansen, A. Hauch, T. Jacobsen, S. H. Jensen, T. L. Skafte and X. Sun, Clean Energy, 2019, 3, 175–201 CrossRef.
- M. Hecht, J. Hoffman, D. Rapp, J. McClean, J. SooHoo, R. Schaefer, A. Aboobaker, J. Mellstrom, J. Hartvigsen, F. Meyen, E. Hinterman, G. Voecks, A. Liu, M. Nasr, J. Lewis, J. Johnson, C. Guernsey, J. Swoboda, C. Eckert, C. Alcalde, M. Poirier, P. Khopkar, S. Elangovan, M. Madsen, P. Smith, C. Graves, G. Sanders, K. Araghi, M. D. Juarez, D. Larsen, J. Agui, A. Burns, K. Lackner, R. Nielsen, T. Pike, B. Tata, K. Wilson, T. Brown, T. Disarro, R. Morris, R. Steinkraus, R. Surampudi, T. Werne and A. Ponce, Space Sci. Rev., 2021, 217, 76 CrossRef.
- Y. Zhang and J. D. Nicholas, J. Electrochem. Soc., 2021, 168, 034513 CrossRef CAS.
- J. Crank, The Mathematics of Diffusion, Oxford University Press, London, 2nd edn, 1975 Search PubMed.
- J. Hayd, H. Yokokawa and E. Ivers-Tiffee, J. Electrochem. Soc., 2013, 160, F351–F359 CrossRef CAS.
- J. Maier, Physical Chemistry of Ionic Materials: Ions and Electrons in Solids, John Wiley & Sons, West Sussex, England, 2004 Search PubMed.
- F. S. Baumann, J. Fleig, H.-U. Habermeier and J. Maier, Solid State Ionics, 2006, 177, 1071–1081 CrossRef CAS.
- B. T. Dalslet, M. Søgaard and P. V. Hendriksen, J. Electrochem. Soc., 2007, 154, B1276–B1287 CrossRef CAS.
- J. Maier, Solid State Ionics, 1998, 112, 197–228 CrossRef CAS.
- J. E. tenElshof, M. H. R. Lankhorst and H. J. M. Bouwmeester, J. Electrochem. Soc., 1997, 144, 1060–1067 CrossRef.
- S. B. Adler, J. A. Lane and B. C. H. Steele, J. Electrochem. Soc., 1996, 143, 3554–3564 CrossRef CAS.
- C. W. Tanner, K.-Z. Fung and A. V. Virkar, J. Electrochem. Soc., 1997, 144, 21–30 CrossRef CAS.
- M. Shah, J. D. Nicholas and S. A. Barnett, Electrochem. Commun., 2009, 11, 2–5 CrossRef CAS.
- J. D. Nicholas, L. Wang, A. V. Call and S. A. Barnett, Phys. Chem. Chem. Phys., 2012, 14, 15379–15392 RSC.
- J. D. Nicholas and S. A. Barnett, J. Electrochem. Soc., 2010, 157, B536–B541 CrossRef CAS.
- J. D. Nicholas and S. A. Barnett, J. Electrochem. Soc., 2009, 156, B458–B464 CrossRef CAS.
- X. Song, A. Diaz, A. Benard and J. Nicholas, Struct. Multidiscip. Optim., 2013, 47, 453–464 CrossRef.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797–1801 CrossRef CAS PubMed.
- T. Heisig, C. Baeumer, U. N. Gries, M. P. Mueller, C. La Torre, M. Luebben, N. Raab, H. Du, S. Menzel, D. N. Mueller, C.-L. Jia, J. Mayer, R. Waser, I. Valov, R. A. De Souza and R. Dittmann, Adv. Mater., 2018, 30, 1800957 CrossRef PubMed.
- H. J. M. Bouwmeester, Catal. Today, 2003, 82, 141–150 CrossRef CAS.
- A. Trovarelli, Catal. Rev.: Sci. Eng., 1996, 38, 439–509 CrossRef CAS.
- M. W. Ahn, K. S. Park, J. H. Heo, J. G. Park, D. W. Kim, K. J. Choi, J. H. Lee and S. H. Hong, Appl. Phys. Lett., 2008, 93, 263103 CrossRef.
- J. Januschewsky, M. Ahrens, A. Opitz, F. Kubel and J. Fleig, Adv. Funct. Mater., 2009, 19, 3151–3156 CrossRef CAS.
- D. Tripkovic, R. Kungas, M. B. Mogensen and P. V. Hendriksen, J. Mater. Chem. A, 2019, 7, 11782–11791 RSC.
- E. Navickas, T. M. Huber, Y. Chen, W. Hetaba, G. Holzlechner, G. Rupp, M. Stoeger-Pollach, G. Friedbacher, H. Hutter, B. Yildiz and J. Fleig, Phys. Chem. Chem. Phys., 2015, 17, 7659–7669 RSC.
- K. Kerman, C. Ko and S. Ramanathan, Phys. Chem. Chem. Phys., 2012, 14, 11953–11960 RSC.
- L. Zhang, S. Wang, H. Huang, Y. Li, Y. Lu and C. Xia, J. Electrochem. Soc., 2017, 164, F610–F615 CrossRef CAS.
- M. Burriel, H. Téllez, R. J. Chater, R. Castaing, P. Veber, M. Zaghrioui, T. Ishihara, J. A. Kilner and J.-M. Bassat, J. Phys. Chem. C, 2016, 120, 17927–17938 CrossRef CAS.
- S. P. Waldow, B. J. Statham, H. F. Wardenga, T. E. Weirich, A. Klein and R. A. De Souza, ACS Appl. Mater. Interfaces, 2020, 12, 36768–36777 CrossRef CAS PubMed.
- R. A. Cox-Galhotra and S. McIntosh, Solid State Ionics, 2010, 181, 1429–1436 CrossRef CAS.
- D. Tripkovic, R. Kungas, M. B. Mogensen and P. V. Hendriksen, Phys. Chem. Chem. Phys., 2020, 22, 15418–15426 RSC.
- Y. Cheng, A. S. Raman, J. Paige, L. Zhang, D. Y. Sun, M. U. Chen, A. Vojvodic, R. J. Gorte and J. M. Vohs, J. Phys. Chem. Lett., 2019, 10, 4082–4088 CrossRef CAS PubMed.
- C. Argirusis, S. Wagner, W. Menesklou, C. Warnke, T. Damjanovic, G. Borchardt and E. Ivers-Tiffée, Phys. Chem. Chem. Phys., 2005, 7, 3523–3525 RSC.
- Y. Cao, M. J. Gadre, A. T. Ngo, S. B. Adler and D. D. Morgan, Nat. Commun., 2019, 10, 1346 CrossRef PubMed.
- M. Siebenhofer, T. M. Huber, G. Friedbacher, W. Artner, J. Fleig and M. Kubicek, J. Mater. Chem. A, 2020, 8, 7968–7979 RSC.
- E. Bucher, W. Sitte, F. Klauser and E. Bertel, Solid State Ionics, 2012, 208, 43–51 CrossRef CAS.
- Ø. F. Lohne, M. Søgaard and K. Wiik, J. Electrochem. Soc., 2013, 160, F1282–F1292 CrossRef.
- W. Preis, E. Bucher and W. Sitte, J. Power Sources, 2002, 106, 116–121 CrossRef CAS.
- A. Falkenstein, D. N. Mueller, R. A. De Souza and M. Martin, Solid State Ionics, 2015, 280, 66–73 CrossRef CAS.
- J. Zheng, X. Wang, J. Yu and N. Tian, Mater. Res. Express, 2021, 8, 035502 CrossRef CAS.
- H. Uchida, M. Yoshida and M. Watanabe, J. Electrochem. Soc., 1999, 146, 1–7 CrossRef CAS.
- M. Cargnello, V. V. T. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Science, 2013, 341, 771–773 CrossRef CAS PubMed.
- H. G. Seo, Y. Choi and W. Jung, Adv. Energy Mater., 2018, 8, 1703647 CrossRef.
- R. Gao, A. Fernandez, T. Chakraborty, A. L. Luo, D. Pesquera, S. Das, G. Velarde, V. Thoreton, J. Kilner, T. Ishihara, S. Nemsak, E. J. Crumlin, E. Ertekin and L. W. Martin, Adv. Mater., 2021, 33, 2100977 CrossRef CAS PubMed.
- G. Yang, S.-Y. Kim, C. Sohn, J. K. Keum and D. Lee, J. Appl. Sci., 2021, 11, 3778 CrossRef CAS.
- C. Nicollet, D. Kalaev and H. L. Tuller, Solid State Ionics, 2019, 331, 96–101 CrossRef CAS.
- R. A. Cox-Galhotra and S. McIntosh, Solid State Ionics, 2012, 228, 14–18 CrossRef CAS.
- M. Søgaard, A. Bieberle-Hutter, P. V. Hendriksen, M. Mogensen and H. L. Tuller, J. Electroceram., 2011, 27, 134–142 CrossRef.
- L. Yan, K. R. Balasubramaniam, S. Wang, H. Du and P. A. Salvador, Solid State Ionics, 2011, 194, 9–16 CrossRef CAS.
- A. Zomorrodian, H. Salamati, Z. G. Lu, X. Chen, N. J. Wu and A. Ignatiev, Int. J. Hydrogen Energy, 2010, 35, 12443–12448 CrossRef CAS.
- M. Burriel, C. Niedrig, W. Menesklou, S. F. Wagner, J. Santiso and E. Ivers-Tiffée, Solid State Ionics, 2010, 181, 602–608 CrossRef CAS.
- M. Mosleh, M. Søgaard and P. V. Hendriksen, J. Electrochem. Soc., 2009, 156, B441–B457 CrossRef CAS.
- X. Chen, S. Wang, Y. L. Yang, L. Smith, N. J. Wu, B. I. Kim, S. S. Perry, A. J. Jacobson and A. Ignatiev, Solid State Ionics, 2002, 146, 405–413 CrossRef CAS.
- N. H. Perry, J. J. Kim and H. L. Tuller, Sci. Technol. Adv. Mater., 2018, 19, 130–141 CrossRef CAS PubMed.
- P. Plonczak, M. Søgaard, A. Bieberle-Hütter, P. V. Hendriksen and L. J. Gauckler, J. Electrochem. Soc., 2012, 159, B471–B482 CrossRef CAS.
- P. Simons, H. I. Ji, T. C. Davenport and S. M. Haile, J. Am. Ceram. Soc., 2017, 100, 1161–1171 CrossRef CAS.
- N. J. Simrick, A. Bieberle-Hutter, T. M. Ryll, J. A. Kilner, A. Atkinson and J. L. M. Rupp, Solid State Ionics, 2012, 206, 7–16 CrossRef CAS.
- A. K. Opitz, A. Schintlmeister, H. Hutter and J. Fleig, Phys. Chem. Chem. Phys., 2010, 12, 12734–12745 RSC.
- A. K. Opitz, A. Lutz, M. Kubicek, F. Kubel, H. Hutter and J. Fleig, Electrochim. Acta, 2011, 56, 9727–9740 CrossRef CAS PubMed.
- A. Bruix, A. Migani, G. N. Vayssilov, K. M. Neyman, J. Libuda and F. Illas, Phys. Chem. Chem. Phys., 2011, 13, 11384–11392 RSC.
- H. Baker and H. Okamoto, ASM Handbook, Vol 03-Alloy Phase Diagrams, 1992 Search PubMed.
- Y. Zhang, Y. Wen, K. Huang and J. D. Nicholas, ACS Appl. Energy Mater., 2020, 3, 4057–4067 CrossRef CAS.
- S. P. Jiang and J. G. Love, Solid State Ionics, 2003, 158, 45–53 CrossRef CAS.
- M. Rohnke, K. Schaepe, A. K. Bachmann, M. Laenger and J. Janek, Appl. Surf. Sci., 2017, 422, 817–827 CrossRef CAS.
- M. Chanthanumataporn, T. Nagasawa and K. Hanamura, J. Electrochem. Soc., 2019, 166, F581–F586 CrossRef.
- V. A. C. Haanappel, D. Rutenbeck, A. Mai, S. Uhlenbruck, D. Sebold, H. Wesemeyer, B. Röwekamp, C. Tropartz and F. Tietz, J. Power Sources, 2004, 130, 119–128 CrossRef CAS.
- A. Egger and W. Sitte, Solid State Ionics, 2014, 258, 30–37 CrossRef CAS.
- M. Sahibzada, S. J. Benson, R. A. Rudkin and J. A. Kilner, Solid State Ionics, 1998, 113–115, 285–290 CrossRef CAS.
- C. Riedl, A. Schmid, A. Nenning, H. Summerer, S. Smetaczek, S. Schwarz, J. Bernardi, A. Optiz, A. Limbeck and J. Fleig, J. Electrochem. Soc., 2020, 167, 104514 CrossRef CAS.
- Y. Ma and J. D. Nicholas, Phys. Chem. Chem. Phys., 2018, 20, 27350–27360 RSC.
- E. J. Skiba, T. Chen and N. H. Perry, ACS Appl. Mater. Interfaces, 2020, 12, 48614–48630 CrossRef CAS PubMed.
- L. A. Chick, L. R. Pederson, G. D. Maupin, J. L. Bates, L. E. Thomas and G. J. Exarhos, Mater. Lett., 1990, 10, 6–12 CrossRef CAS.
- I. Chang, S. Woo, M. H. Lee, J. H. Shim, Y. Piao and S. W. Cha, Appl. Surf. Sci., 2013, 282, 463–466 CrossRef CAS.
- Q. Yang, T. E. Burye, R. R. Lunt and J. D. Nicholas, Solid State Ionics, 2013, 249–250, 123–128 CrossRef CAS.
- Q. Yang and J. D. Nicholas, J. Electrochem. Soc., 2014, 161, F3025–F3031 CrossRef CAS.
- J. D. Nicholas, Extreme Mechanics Letters, 2016, 9, 405–421 CrossRef.
- J. D. Nicholas, in Electro-Chemo-Mechanics of Solids, ed. S. Bishop, D. Marrocchelli, N. Perry and B. Sheldon, Springer, New York, 2017, DOI:10.1007/978-3-319-51407-9_5, ch. 5, pp. 103–136.
- D. Chen, S. R. Bishop and H. L. Tuller, Chem. Mater., 2014, 26, 6622–6627 CrossRef CAS.
- J. Sheth, D. Chen, J. J. Kim, W. J. Bowman, P. A. Crozier, H. L. Tuller, S. T. Misture, S. Zdzieszynski, B. W. Sheldon and S. R. Bishop, Nanoscale, 2016, 8, 16499–16510 RSC.
- S. R. Bishop, H. L. Tuller, Y. Kuru and B. Yildiz, J. Eur. Ceram. Soc., 2011, 31, 2351–2356 CrossRef CAS.
- G. Kim, S. Wang, A. J. Jacobson and C. L. Chen, Solid State Ionics, 2006, 177, 1461–1467 CrossRef CAS.
- C. H. Ko, A. Karthikeyan and S. Ramanathan, J. Chem. Phys., 2011, 134, 014704 CrossRef PubMed.
- G. Kim, S. Wang, A. J. Jacobson, Z. Yuan, W. Donner, C. L. Chen, L. Reimus, P. Brodersen and C. A. Mims, Appl. Phys. Lett., 2006, 88, 024103 CrossRef.
- M. W. den Otter, H. J. M. Bouwmeester, B. A. Boukamp and H. Verweij, J. Electrochem. Soc., 2001, 148, J1–J6 CrossRef CAS.
- D. Chen, S. R. Bishop and H. L. Tuller, J. Electroceram., 2012, 28, 62–69 CrossRef CAS.
- N. Savvides, A. Thorley, S. Gnanarajan and A. Katsaros, Thin Solid Films, 2001, 388, 177–182 CrossRef CAS.
- C.-M. Wang, S. Thevuthasan and C. H. F. Peden, J. Am. Ceram. Soc., 2003, 86, 363–365 CrossRef CAS.
- D. Q. Shi, M. Ionescu, J. McKinnon and S. X. Dou, Physica C, 2001, 356, 304–310 CrossRef CAS.
- N. L. Edleman, A. Wang, J. A. Belot, A. W. Metz, J. R. Babcock, A. M. Kawaoka, J. Ni, M. V. Metz, C. J. Flaschenriem, C. L. Stern, L. M. Liable-Sands, A. L. Rheingold, P. R. Markworth, R. P. H. Chang, M. P. Chudzik, C. R. Kannewurf and T. J. Marks, Inorg. Chem., 2002, 41, 5005–5023 CrossRef CAS PubMed.
- A. Cavallaro, F. Sandiumenge, J. Gazquez, T. Puig, X. Obradors, J. Arbiol and H. C. Freyhardt, Adv. Funct. Mater., 2006, 16, 1363–1372 CrossRef CAS.
- Y. Ma and J. D. Nicholas, J. Electrochem. Soc., 2021, 168, 104518 CrossRef.
- A. G. Shard, Surf. Interface Anal., 2014, 46, 175–185 CrossRef CAS.
- Y. Wang, K. Duncan, E. D. Wachsman and F. Ebrahimi, Solid State Ionics, 2007, 178, 53–58 CrossRef CAS.
- A. Martínez-Amesti, A. Larrañaga, L. M. Rodríguez-Martínez, M. L. Nó, J. L. Pizarro, A. Laresgoiti and M. I. Arriortua, J. Power Sources, 2009, 192, 151–157 CrossRef.
- P. P. Dholabhai, J. B. Adams, P. Crozier and R. Sharma, J. Chem. Phys., 2010, 132, 094104 CrossRef PubMed.
- H. L. Tuller and A. S. Nowick, J. Phys. Chem. Solids, 1977, 38, 859–867 CrossRef CAS.
- T. Chen, G. F. Harrington, J. Masood, K. Sasaki and N. H. Perry, ACS Appl. Mater. Interfaces, 2019, 11, 9102–9116 CrossRef CAS PubMed.
- P. S. Manning, J. D. Sirman and J. A. Kilner, Solid State Ionics, 1997, 93, 125–132 CrossRef.
- T. M. Huber, A. K. Opitz and J. Fleig, Solid State Ionics, 2015, 273, 8–12 CrossRef CAS PubMed.
- L. Zhao, N. H. Perry, T. Daio, K. Sasaki and S. R. Bishop, Chem. Mater., 2015, 27, 3065–3070 CrossRef CAS.
- D. Pérez-Coll, P. Núñez and J. R. Frade, J. Power Sources, 2011, 196, 8383–8390 CrossRef.
- T. S. Zhang, J. Ma, Y. J. Leng, S. H. Chan, P. Hing and J. A. Kilner, Solid State Ionics, 2004, 168, 187–195 CrossRef CAS.
- Y. H. Cho, P. S. Cho, G. Auchterlonie, D. K. Kim, J. H. Lee, D. Y. Kim, H. M. Park and J. Drennan, Acta Mater., 2007, 55, 4807–4815 CrossRef CAS.
- T. S. Zhang, J. Ma, S. H. Chan, P. Hing and J. A. Kilner, Solid State Sci., 2004, 6, 565–572 CrossRef CAS.
- Y. Zhou, M. Nakashima and J. M. White, J. Phys. Chem., 1988, 92, 812–818 CrossRef CAS.
- P. Luches, G. Gasperi, M. Sauerbrey, S. Valeri, J. Falta and J. I. Flege, Front. Chem., 2019, 7, 57 CrossRef CAS PubMed.
- W. Yang, Z. Wang, Z. Wang, Z. Yang, C. Xia, R. Peng, X. Wu and Y. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 21051–21059 CrossRef CAS PubMed.
- M. Søgaard, P. Vang Hendriksen and M. Mogensen, J. Solid State Chem., 2007, 180, 1489–1503 CrossRef.
- E. N. Armstrong, K. L. Duncan and E. D. Wachsman, Phys. Chem. Chem. Phys., 2013, 15, 2298–2308 RSC.
- T. Ishigaki, S. Yamauchi, K. Kishio, J. Mizusaki and K. Fueki, J. Solid State Chem., 1988, 73, 179–187 CrossRef CAS.
- F. S. Baumann, J. Fleig, G. Cristiani, B. Stuhlhofer, H. U. Habermeier and J. Maier, J. Electrochem. Soc., 2007, 154, B931–B941 CrossRef CAS.
- H. J. M. Bouwmeester, M. W. Den Otter and B. A. Boukamp, J. Solid State Electrochem., 2004, 8, 599–605 CrossRef CAS.
- Y. L. Huang, C. Pellegrinelli, K. T. Lee, A. Perel and E. D. Wachsman, J. Electrochem. Soc., 2015, 162, F965–F970 CrossRef CAS.
- J. A. Lane, S. J. Benson, D. Waller and J. A. Kilner, Solid State Ionics, 1999, 121, 201–208 CrossRef CAS.
- Y. H. Li, K. Gerdes, T. Horita and X. B. Liu, J. Electrochem. Soc., 2013, 160, F343–F350 CrossRef CAS.
- P. Ried, P. Holtappels, A. Wichser, A. Ulrich and T. Graule, J. Electrochem. Soc., 2008, 155, B1029–B1035 CrossRef CAS.
- S. Wang, P. A. W. van der Heide, C. Chavez, A. J. Jacobson and S. B. Adler, Solid State Ionics, 2003, 156, 201–208 CrossRef CAS.
- R. J. Chater, S. Carter, J. A. Kilner and B. C. H. Steele, Solid State Ionics, 1992, 53, 859–867 CrossRef.
- B. C. H. Steele and J. M. Bae, Solid State Ionics, 1998, 106, 255–261 CrossRef CAS.
- R. Moreno, J. Zapata, J. Roqueta, N. Bagués and J. Santiso, J. Electrochem. Soc., 2014, 161, F3046–F3051 CrossRef CAS.
- A. V. Berenov, A. Atkinson, J. A. Kilner, E. Bucher and W. Sitte, Solid State Ionics, 2010, 181, 819–826 CrossRef CAS.
- A. Egger, E. Bucher, M. Yang and W. Sitte, Solid State Ionics, 2012, 225, 55–60 CrossRef CAS.
- M. Søgaard, P. V. Hendriksen, M. Mogensen, F. W. Poulsen and E. Skou, Solid State Ionics, 2006, 177, 3285–3296 CrossRef.
- S. B. Adler, Solid State Ionics, 1998, 111, 125–134 CrossRef CAS.
- T. C. Yeh, J. L. Routbort and T. O. Mason, Solid State Ionics, 2013, 232, 138–143 CrossRef CAS.
- I. C. Fullarton, J. P. Jacobs, H. E. van Benthem, J. A. Kilner, H. H. Brongersma, P. J. Scanlon and B. C. H. Steele, Ionics, 1995, 1, 51–58 CrossRef CAS.
- Y.-P. Fu, J. Ouyang, C.-H. Li and S.-H. Hu, J. Power Sources, 2013, 240, 168–177 CrossRef CAS.
- L. Wang, R. Merkle, J. Maier, T. Acarturk and U. Starke, Appl. Phys. Lett., 2009, 94, 071908 CrossRef.
- E. Bucher, A. Egger, G. B. Caraman and W. Sitte, J. Electrochem. Soc., 2008, 155, B1218 CrossRef CAS.
- E. Bucher, A. Egger, P. Ried, W. Sitte and P. Holtappels, Solid State Ionics, 2008, 179, 1032–1035 CrossRef CAS.
- D. Chen and Z. Shao, Int. J. Hydrogen Energy, 2011, 36, 6948–6956 CrossRef CAS.
- E. Girdauskaite, H. Ullmann, V. V. Vashook, U. Guth, G. B. Caraman, E. Bucher and W. Sitte, Solid State Ionics, 2008, 179, 385–392 CrossRef CAS.
- F. S. Baumann, J. Fleig, H. U. Habermeier and J. Maier, Solid State Ionics, 2006, 177, 3187–3191 CrossRef CAS.
- L. Wang, R. Merkle and J. Maier, J. Electrochem. Soc., 2010, 157, B1802–B1808 CrossRef CAS.
- Ø. F. Lohne, M. Søgaard and K. Wiik, J. Electrochem. Soc., 2013, 160, F1282–F1292 CrossRef.
- C. Nicollet, C. Toparli, G. F. Harrington, T. Defferriere, B. Yildiz and H. L. Tuller, Nat. Catal., 2020, 3, 913–920 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta06237a |
| This journal is © The Royal Society of Chemistry 2021 |