DOI:
10.1039/D1TA05130J
(Paper)
J. Mater. Chem. A, 2021,
9, 23095-23105
Tuning the electrochemical properties by anionic substitution of Li-rich antiperovskite (Li2Fe)S1−xSexO cathodes for Li-ion batteries†
Received
17th June 2021
, Accepted 20th September 2021
First published on 6th October 2021
Abstract
The development of electrode materials with multielectron redox functionality is imperative for next-generation Li-ion batteries with a high gravimetric capacity. Within this context, a Li-rich (Li2Fe)SO antiperovskite cathode is a promising candidate exhibiting such multielectron cationic and anionic redox features, resulting in a reversible extraction/insertion of about 1.2 Li+ per formula unit. However, it suffers from poor structural and cycling stabilities which hinder its practical application. Herein, we systematically investigate the effect of anionic substitution of S with Se on the structural, thermal and electrochemical properties of the (Li2Fe)SO cathode. With increasing the Se content, higher thermal stability and lower sensitivity to moist air were obtained. Multi-stage cationic and anionic redox processes characterized the electrochemical activity of all the prepared (Li2Fe)S1−xSexO solid solutions. The cationic redox process was shifted to higher potentials while the anionic redox process was shifted to lower potentials upon the increase of the Se content. Among the various synthesized compositions, (Li2Fe)S0.7Se0.3O exhibited the best electrochemical performance with a high discharge capacity of ∼245 mA h g−1 and an outstanding cycling stability at 0.1C current rate as well as nearly 100% capacity recovery after rate capability tests of 50 cycles. To deeply characterize (Li2Fe)S0.7Se0.3O, various ex situ and in situ techniques were applied. In contrast to (Li2Fe)SO, the substituted (Li2Fe)S0.7Se0.3O material remains crystalline without the evolution of secondary phases or superstructures after the first charge/discharge cycle highlighting its enhanced structural stability. Similar to (Li2Fe)SO, both the cation (Fe) and anions (S/Se) from (Li2Fe)S0.7Se0.3O participate in the redox process.
1. Introduction
Rechargeable Li-ion batteries (LIBs) are widely used in portable electronics, electric tools and hybrid/full electric vehicles due to their high energy and power densities.1 Current commercial cathodes such as layered LiCoO2 (LCO), layered LiNi1/3Mn1/3Co1/3O2 (NMC), olivine LiFePO4 (LFP) and spinel LiMn2O4 (LMO) have some disadvantages despite their optimization.2 To be more specific, all these commercial cathodes have a limitation of one electron transfer in the redox processes, which restricts their gravimetric capacities, and hence, their energy densities.3 Therefore, multielectron redox battery chemistries with the capability of multielectron charge storage can be an attractive option.4 The multielectron redox approach can be realized either by (i) successive redox of transition metal cations such as the case of Li2Mn0.67Nb0.33O2F (Mn2+/4+)5 or (ii) redox including both transition metal cations and anions such as Li1+xCo1−x−y−zNiyMnzO2.6 Recently, a novel class of Li-rich antiperovskite cathodes with a general formula (Li2TM)ChO (TM = Fe, Mn, Co; Ch = S, Se) was discovered and its electrochemical performance was evaluated.7–10 These antiperovskite cathodes display a multielectron storage merit with a reversible transfer of 1.2 Li+ per formula unit, resulting in an outstanding specific capacity. Besides, they offer other advantages such as reasonable working voltage (∼1.2–3 V) and 3-dimensional (3D) Li+ diffusion with a low diffusion barrier (∼0.32 eV).11 Despite that the moderate working voltage limits the energy density to some extent, it enables the usage of various alternative electrolytes which cannot be used at high voltages due to their instabilities. Among the antiperovskite cathodes, the (Li2Fe)SO composition showed one of the best reported electrochemical performances with an outstanding average discharge capacity ∼285 mA h g−1 at 0.1C and high rate capability (∼200 mA h g−1 at 1C) when cycled from 1.2 to 3 V.12 However, this compound suffered from poor cycling stability at current rates below 1C and low capacity recovery at 0.1C after only 50 cycles (∼80%).12 Therefore, further fundamental studies are required to improve the electrochemical performance of (Li2Fe)SO in a targeted manner for its potential application in the future. Herein, we systematically investigated the influence of anionic substitution of S with Se in the (Li2Fe)SO cathode on its structural and electrochemical performance. Our findings verified the tunable structure–electrochemical property relationships by gradual modification of the stoichiometry. Among all the prepared solid-solutions, (Li2Fe)S0.7Se0.3O showed the best electrochemical performance in terms of capacity retention/recovery. Therefore, various in situ and ex situ techniques were used to explore the origin of its superior performance. Operando XRD measurements indicated a higher structural stability during cycling for (Li2Fe)S0.7Se0.3O compared to unsubstituted (Li2Fe)SO. Furthermore, X-ray absorption spectroscopy (XAS) confirmed that the multielectron capability originates from the contribution of the cation (Fe) and anions (S/Se) in the redox processes.
2. Experimental details
2.1. Synthesis
The synthesis of Li-rich antiperovskite cathodes was performed via a solid-state reaction method as follows. Compositions of (Li2Fe)S1−xSexO (where x = 0.1, 0.3, 0.5, 0.7, and 0.9) were prepared by weighing powder quantities (purchased from Alfa Aesar) according to the desired stoichiometry from Li2O, Fe, S and Se in an Ar-filled glovebox (MBraun, Germany) with O2 and H2O concentrations below 1 ppm. The precursors were mixed in an agate mortar for homogenization and the final product was loaded in a corundum crucible (Aliaxis, Frialit-Degussit, AL23). The crucible was placed into a silica tube (QSILAG; Quarzschmelze, Ilmenau) and temporarily closed with a rubber stopper. Outside the glovebox, the tube was evacuated to a pressure of less than 10−3 mbar, refilled with Ar to adjust the internal pressure to 0.2 bar, and sealed with a gas burner. The closed ampoule was placed into a furnace and heated to 850 °C with a heating rate of 50 °C h−1. This temperature was maintained for 3 h before quenching the ampoule in water. Quenching in water after the main reaction was found to be an essential synthesis step to prevent the partial decomposition of antiperovskites to other secondary phases such as transition metal oxides and sulfides.7
2.2. Characterization
2.2.1. X-ray diffraction (XRD).
The phase purity of each powder was checked by XRD using a STOE STADI P diffractometer (Stoe, Germany) in Debye–Scherrer geometry with a Mo Kα1 radiation source (λ = 0.70926 Å) and a Mythen 1K detector (Dectris). To protect the samples from any reactions with air, the samples were loaded into glass capillaries (Mark tubes, Glass No. 14, Hilgenberg) inside the glovebox and subsequently melt-sealed outside.
2.2.2. Elemental analysis.
Inductively coupled plasma-optical emission spectroscopy (ICP-OES) (iCAP 6500 Duo View, Fa. Thermo Fisher Scientific GmbH) was employed to estimate the average composition of the samples.
2.2.3. Morphology investigations.
Scanning electron microscopy (Nova-NanoSEM 200) together with energy dispersive spectroscopy (EDS) mapping were used to evaluate the morphology and the elemental distribution homogeneity, respectively.
2.2.4. Thermal analysis.
Differential thermal analysis (DTA) up to 1200 °C was carried out using a Setaram DTA92-2400 (alumina container) under a helium atmosphere with a heating rate of 10 °C min−1.
2.3. Electrochemical measurements
2.3.1. Galvanostatic cycling.
Galvanostatic cycling with potential limitation (GCPL) was carried out in Swagelok cells using a VMP3 potentiostat (Biologic Instruments) with metallic lithium (99.9%, G-Materials) as the anode and antiperovskite composition as the cathode. The measurements were performed at room temperature using 1 M lithium bis-(trifluoromethanesulfonyl)-imide (LiTFSI, 99.95%, Sigma Aldrich) in a mixture of dimethoxyethane (DME) and 1,3-dioxolane (DOL) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume, BASF) as an electrolyte. LiTFSI was used instead of the common LP30 electrolyte containing lithium hexafluorophosphate (LiPF6), since it has a lower S solubility, thus reducing S leaching from the cathode.12 A glass fiber cloth (Whatman, GF/D) served as a separator. The cathode was fabricated through pressing a mixture of the active material, CNT as a conductive additive and polytetrafluoroethylene binder (PTFE, Aldrich) (80:10:10 by weight ratio, respectively) onto a stainless-steel mech current collector. The rate capability tests were carried out in the potential range between 1.2 and 3 V vs. Li+/Li at different current rates between 0.1C and 1C. Noteworthily, 1C corresponds to the charge/discharge current needed for the removal/insertion of 1 Li+ from/in the antiperovskite cathode during 1 hour.
:1 by volume, BASF) as an electrolyte. LiTFSI was used instead of the common LP30 electrolyte containing lithium hexafluorophosphate (LiPF6), since it has a lower S solubility, thus reducing S leaching from the cathode.12 A glass fiber cloth (Whatman, GF/D) served as a separator. The cathode was fabricated through pressing a mixture of the active material, CNT as a conductive additive and polytetrafluoroethylene binder (PTFE, Aldrich) (80:10:10 by weight ratio, respectively) onto a stainless-steel mech current collector. The rate capability tests were carried out in the potential range between 1.2 and 3 V vs. Li+/Li at different current rates between 0.1C and 1C. Noteworthily, 1C corresponds to the charge/discharge current needed for the removal/insertion of 1 Li+ from/in the antiperovskite cathode during 1 hour.
2.3.2. Cyclic voltammetry (CV).
A slow rate cyclic voltammetry (SRCV) technique was used to obtain information about the redox mechanism for the prepared cathodes. All measurements were carried out at room temperature and the potential range was selected between 1 and 3 V with a scan rate of 0.05 mV s−1. The cell configuration was the same as for GCPL measurements.
2.3.3. Galvanostatic intermittent titration technique (GITT).
GITT measurements at room temperature were performed to examine the kinetics of the extraction/insertion process of Li+ from/into the (Li2Fe)S0.7Se0.3O cathode. For this purpose, a three-electrode Swagelok cell was used with metallic Li as counter and reference electrodes and a 1 M solution of LiTFSI in a mixture of DME and DOL (1:1 by volume) as the electrolyte. The duration of the current pulse at 0.1C was 10 min, while the relaxation time was 8 h. The calculation of the chemical diffusion coefficient was accomplished as described in the literature.13
2.3.4.
Operando synchrotron X-ray diffraction (XRD).
The operando XRD technique at the P02.1 beamline (DESY, Hamburg) was employed for monitoring the structural changes of the (Li2Fe)S0.7Se0.3O cathode during charge and discharge of the battery.14 An 8-fold coin cell holder connected to a Biologic potentiostat was used, as described in the literature.15 A wavelength of 0.20696 Å was confirmed using a LaB6 standard material. The first diffraction pattern of the pristine compound was measured before starting the electrochemical testing. Afterwards, the cell was charged and discharged at a constant current rate of 0.1C while recoding the corresponding diffraction patterns.
2.3.5.
Operando X-ray absorption spectroscopy (XAS).
Operando XAS analysis was performed for the (Li2Fe)S0.7Se0.3O cathode to elaborate the redox activity of Fe and Se by recording Fe and Se K-edge spectra during Li+ insertion/extraction into/from the cathode. The measurements were done at the P65 beamline at PETRA III extension (DESY, Hamburg, Germany) in transmission and fluorescence modes.16 The cell setup and electrochemical measurements were the same as illustrated in the Operando XRD session. FeO and Fe2O3 compounds were used as reference materials for Fe2+ and Fe3+ valence states. To analyze the obtained results, the Demeter program was used.17
2.4. Magnetic measurements
Magnetic measurements of the (Li2Fe)S0.7Se0.3O composition were performed by using a SQUID magnetometer (MPMS, Quantum Design). The variation of magnetization with temperature was recorded in zero-field-cooled (ZFC) and field-cooled (FC) modes in the range from 2 K to 300 K.
3. Results and discussion
3.1. Structural analysis and morphology
Fig. 1a shows the XRD patterns for the series (Li2Fe)S1−xSexO where x = 0.1, 0.3, 0.5, 0.7, and 0.9. The data showed that all compositions crystallize in a cubic antiperovskite structure with the Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group without any additional impurities. A shift in all peak positions to lower 2θ angles was observed with the increasing Se content, which is highlighted in Fig. 1b. This confirms the increase of the cubic lattice parameter (a) (see Table S1 and Fig. S1 in the ESI†) due to the higher ionic radius of Se2− compared to S2− according to Vegard's law.18 Noteworthily, the ideal lattice parameter was estimated using the formula
m space group without any additional impurities. A shift in all peak positions to lower 2θ angles was observed with the increasing Se content, which is highlighted in Fig. 1b. This confirms the increase of the cubic lattice parameter (a) (see Table S1 and Fig. S1 in the ESI†) due to the higher ionic radius of Se2− compared to S2− according to Vegard's law.18 Noteworthily, the ideal lattice parameter was estimated using the formula  , where rCh and r(Li2Fe) represent the average ionic radii of (S1−xSex) and (Li2Fe) atomic sites (see Table S1 in the ESI†). The high spin ionic radius of Fe2+ was considered in the calculations based on previous Mössbauer spectroscopy.12 The experimentally obtained lattice parameters are higher than the ideal ones suggesting the existence of larger voids for Li+ diffusion. This finding is in a good agreement with cell metrics reported for (Li2Fe)SeO and (Li2Fe)SO cathodes.7 In order to get an indication about the structural stability upon S substitution by Se, the Goldschmidt tolerance factor (t) was calculated using the formula
, where rCh and r(Li2Fe) represent the average ionic radii of (S1−xSex) and (Li2Fe) atomic sites (see Table S1 in the ESI†). The high spin ionic radius of Fe2+ was considered in the calculations based on previous Mössbauer spectroscopy.12 The experimentally obtained lattice parameters are higher than the ideal ones suggesting the existence of larger voids for Li+ diffusion. This finding is in a good agreement with cell metrics reported for (Li2Fe)SeO and (Li2Fe)SO cathodes.7 In order to get an indication about the structural stability upon S substitution by Se, the Goldschmidt tolerance factor (t) was calculated using the formula  , where rCh and r(Li2Fe) are as mentioned before and rO refers to the ionic radius of O (see Fig. S2 in the ESI†).19 This factor can be regarded as a predictor of the structural stability of antiperovskites based on the degree of mismatch between the constituent ions. In general, a value of t close to 1 corresponds to an ideal undistorted cubic structure.20 It is evident that with the replacement of S by Se in the structure, t increases, suggesting that a higher structural stability is provided. This increase is attributed to the larger ionic size of Se (1.84 Å) compared to S (1.7 Å). In order to study the change in the internal structure, the XRD average crystallite size (DXRD) and the lattice strain (ε) were calculated for (Li2Fe)S1−xSexO series (Table S1 in the ESI†). The DXRD was calculated from the Scherrer's equation DXRD = 0.9λ/βcosθ where λ is the wavelength of the X-ray source, β is the full width at half maximum (FWHM) of the most intense peaks and θ is the Bragg angle, while ε was estimated using the formula ε = βcosθ/4.21,22 It was found that DXRD and ε change slightly with increasing Se content. These local structural parameters (DXRD and ε) may affect the electrochemical performance as reported for other antiperovskite compounds such as Li3OCl.23,24 However, the observed minor change in these parameters for (Li2Fe)S1−xSexO is not expected to have any effect on the overall electrochemical properties.
, where rCh and r(Li2Fe) are as mentioned before and rO refers to the ionic radius of O (see Fig. S2 in the ESI†).19 This factor can be regarded as a predictor of the structural stability of antiperovskites based on the degree of mismatch between the constituent ions. In general, a value of t close to 1 corresponds to an ideal undistorted cubic structure.20 It is evident that with the replacement of S by Se in the structure, t increases, suggesting that a higher structural stability is provided. This increase is attributed to the larger ionic size of Se (1.84 Å) compared to S (1.7 Å). In order to study the change in the internal structure, the XRD average crystallite size (DXRD) and the lattice strain (ε) were calculated for (Li2Fe)S1−xSexO series (Table S1 in the ESI†). The DXRD was calculated from the Scherrer's equation DXRD = 0.9λ/βcosθ where λ is the wavelength of the X-ray source, β is the full width at half maximum (FWHM) of the most intense peaks and θ is the Bragg angle, while ε was estimated using the formula ε = βcosθ/4.21,22 It was found that DXRD and ε change slightly with increasing Se content. These local structural parameters (DXRD and ε) may affect the electrochemical performance as reported for other antiperovskite compounds such as Li3OCl.23,24 However, the observed minor change in these parameters for (Li2Fe)S1−xSexO is not expected to have any effect on the overall electrochemical properties.
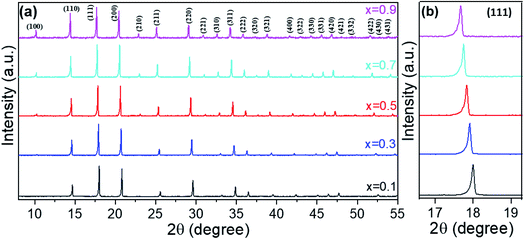 |
| | Fig. 1 XRD patterns (a) and reflection position shift (b) for the series (Li2Fe)S1−xSexO, where x = 0.1, 0.3, 0.5, 0.7, and 0.9. | |
The chemical composition of the materials was verified by ICP-OES and the obtained molar ratios are in good agreement with the expected stoichiometries (see Table S2 in the ESI†). The morphology of all samples can be described as irregularly shaped particles with a large size distribution (Fig. S3 in the ESI†). Overall, no significant difference in the particle size was observed between (Li2Fe)S1−xSexO series. According to the EDS-mapping results, the elements distribute homogeneously throughout all compositions suggesting a monophasic synthesis process. It should be noted that Li cannot be detected by EDS due to its low energy X-ray emission.
3.2. Thermodynamic investigations
It was reported that (Li2Fe)SO and (Li2Fe)SeO end members of the (Li2Fe)S1−xSexO solid solutions undergo a congruent melting (see Fig. S4 in the ESI†).7 Therefore, similar thermal behavior is expected for their intermediate compositions. Fig. 2a shows the DTA results of (Li2Fe)S1−xSexO series. The thermograms reveal that all solid solutions melt congruently without any intermediate phase transition/decomposition. The congruent melting is supported by the appearance of only one peak for the melting/crystallization process.25 Furthermore, no significant difference in the melting/crystallization temperatures was realized when carrying out DTA for two cycles for Li2FeS0.5Se0.5O, as an example, which confirms our assumption (see Fig. S5 in the ESI†). A negligible 0.1% mass loss was detected for (Li2Fe)S0.5Se0.5O after two cycles of measurements which might be attributed to the desorption of moisture. These features reflect the better thermal stability of this class of antiperovskites compared to other cathodes such as LiNi0.33Co0.33Mn0.33O2.26Fig. 2b shows the endothermic thermograms in order to monitor the variation of the melting temperature with the composition. Obviously, the melting temperature gradually increases with increasing the Se content indicating that the thermal stability is increased. The highest melting point of about 1025 °C was obtained for the (Li2Fe)S0.1Se0.9O composition. It is worth mentioning that various factors can affect the magnitude of the melting temperature such as the particle size, crystallite size and structural defects.27 A higher melting point is expected for the sample with larger crystallite size and reduced defects. However, the slight change in these parameters observed from XRD and SEM for the (Li2Fe)S1−xSexO series suggests a minor contribution to the melting temperature. One possible reason for such an increase in the melting point is the difference between electronegativities for S (2.58) and Se (2.55) which may affect the ionic character and hence the lattice energy. Furthermore, the degree of local distortion of the cation octahedra (indicated by t value) in these antiperovskites such as rotation or tilting might have an effect on the melting point.
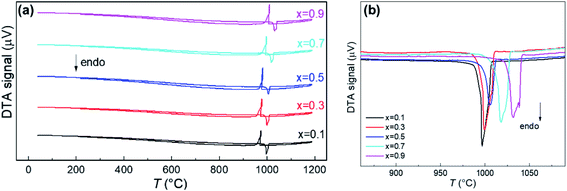 |
| | Fig. 2 Full DTA spectra (a) and the variation of the melting temperature (b) for the series (Li2Fe)S1−xSexO (where x = 0.1, 0.3, 0.5, 0.7, and 0.9). | |
3.3. Stability to air exposure
Based on previous DFT calculations,11 it was found that (Li2Fe)SO is a metastable compound at 0 K and it tends to decompose to more stable phases such as FeS, Li2S and Li2FeO2. The existence of (Li2Fe)SO at room temperature was attributed to the entropy stabilization and the slow decomposition kinetics.11 Exposure to air can activate the decomposition process through the side reactions occurring at the surface between water and oxygen with the main compound. Indeed, Lai et al.7 reported the phase deterioration of (Li2Fe)ChO (where Ch = S or Se) cathodes up on exposure to air. One possible mechanism is that water in combination with oxygen can overcome the energy barrier for leaching Li ions from the structure, and hence, initiates the phase decomposition. We tested the air stability and the dependance of the materials deterioration rate on the substitution degree. Fig. S6 (in the ESI†) compares the XRD patterns of the prepared samples without and after 24 h exposure to air. All samples suffered from amorphization to some degree during exposure to air. However, the chemical stability seems to enhance with increasing the Se content, and the phase deterioration rate becomes slower. This can be realized by observing the broadening and the shift of the peak positions. Obviously, the sample with the highest Se content exhibited the lowest broadening and shift. Such higher stability with increasing the Se content coincides well with the results reported by Lai et al.7 To explore to which extent the amorphization occurs, long-range in situ XRD measurements while exposing (Li2Fe)S0.3Se0.7O to air for 90 h were carried out, as seen in Fig. S7 (in the ESI†). Even after 90 h exposure to air, the (Li2Fe)S0.3Se0.7O phase exists, however, with significant loss of its crystallization combined with the weak appearance of the Li(OH)·H2O phase. Interestingly, Kim et al.20 reported that the increase of distortions in the crystal structure (decrease of t) is accompanied by lower energy barriers for Li+ migration as a result of the suppression of potential energy surface corrugations and destabilization of the equilibrium configurations. Therefore, this higher stability with increasing the Se content might be attributed to the increase of the energy barrier caused by the decrease in the lattice distortions as suggested by the tolerance factor calculations.
3.4. Electrochemical investigations
Fig. 3 shows the cyclic voltammograms of the series (Li2Fe)S1−xSexO scanned in the potential range from 1.2 to 3 V. The data illustrated that the deintercalation process (cell charge) of Li+ is accomplished through several stages. During intercalation (cell discharge), a similar behavior with much broader and overlapped peaks was noticed. A possible explanation for the multi-stage behavior in CV involves a multi-phase redox process. The multi-stage oxidation behavior is composed of cationic (Fe2+/Fe3+) and anionic (S2−/0 and Se2−/0) processes. The initial oxidation of Fe2+ facilitates such anion (S2−) oxidation as was reported for Li2FeS2−ySey series.28 Particularly, the accompanied change in the electronic structure during Fe2+ oxidation leads to a higher Fe–S covalency, pushing the S 2p states closer to the Fermi level and facilitating anion oxidation.28 Based on previously reported XPS measurements for (Li2Fe)SO,12 the oxidation peak at ∼3 V is attributed to a partial oxidation of sulfide (S2−) to elemental sulfur (S0). Similarly, the oxidation peak in this work at ∼2.9 V for (Li2Fe)S0.9Se0.1O can correspond to partial S2− to S0 oxidation while the other two peaks at 2.1 and 2.5 V are expected to be a two-stage oxidation of Fe2+ to Fe3+. Interestingly, the first oxidation peak at around 2.1 V for the composition with x(Se) = 0.1 is shifted to higher potentials with the increased Se content while the second oxidation peak at 2.55 V is nearly independent of the Se content. In contrast, as the Se content increases, the intensity of the anionic oxidation peak grows while its position shifts to a lower potential. This means that either (i) Se facilitates the oxidation of S2− or (ii) Se2− is also oxidized at lower potentials, and the peaks are partially overlapping in the voltammogram. However, the second scenario seems to be more correct based on previous studies. For example, it was found that the Se substitution at the S site in Li2FeS2−ySey solid solutions increases the energy of frontier states, and therefore, reduces the anion oxidation potential.28 This reduction of the anion oxidation potential was attributed to the participation of the Se 4p orbital to the density of states at the expense of S 3p.28 Separated peaks due to S2− and Se2− oxidation were not observed in our CV data, suggesting that the complete mixing of the anionic (S2− and Se2−) states occurs instead of oxidizing independently. This finding highlights the role of anionic substitution in controlling the anionic redox and agrees well with the results reported for Li-rich chalcogenide Li2FeS2−ySey series.28 Rate capability tests were carried out for the compositions with different S/Se ratios. Fig. S8a–e† shows the charge/discharge capacities combined with their relative coulombic efficiency at different current rates for the series under study. As a matter of comparison facilitation, only discharge capacities at different current rates (0.1C–1C) versus the cycle number is shown in Fig. 4. Clearly, the Se incorporation inside the (Li2Fe)SO lattice plays a critical role in tailoring the electrochemical performance and the cycling stability. Table 1 compares some electrochemical parameters between this work and the reported (Li2Fe)SO phase.12 These parameters can be regarded as a direct indicator of the cycling stability and performance. Noteworthily, the calculated theoretical capacities in Table 1 correspond to the extraction/insertion of 2 Li+ from the structure while the obtained practical capacities correspond to the extraction/insertion of ∼1.2 Li+ from the compound. Extracting/inserting more than 1.2 Li+ from the structure is first principally expected to damage the antiperovskite structure.11 Although the specific capacity should decrease with the increasing Se content due to its higher molar weight, the positive effects of Se in terms of the enhanced capacity retention can compensate the overall cycling performance.29 Generally, the enhanced capacity recovery was realized with increasing Se ratio, as seen in Table 1. However, the capacity value and capacity retention upon cycling depend on the relative atomic percentages between S and Se. Fig. 4 shows that the replacement of a small amount of S by Se (10 at%) in (Li2Fe)SO reduces the specific discharge capacity while it is not enough to stabilize the performance in terms of capacity retention/recovery in comparison to (Li2Fe)SO.12 Further increase of Se resulted in the improved capacity accompanied by promising cycling stability, particularly at low current rates (<1C), as seen in Fig. 4 for (Li2Fe)S0.7Se0.3O. This composition exhibited an average discharge capacity of 245 mA h g−1 at 0.1C close to that of (Li2Fe)SO.12 However, it shows improved cycling stability in terms of capacity retention at low current rates and a capacity recovery of ∼100% after rate capability tests of 50 cycles.
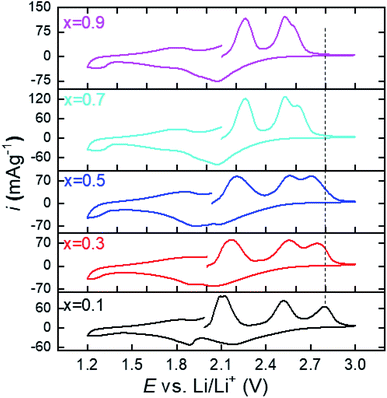 |
| | Fig. 3 Cyclic voltammograms of the series (Li2Fe)S1−xSexO where x = 0.1, 0.3, 0.5, 0.7, and 0.9. | |
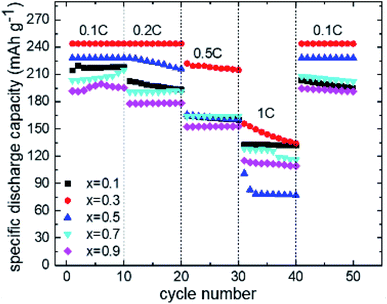 |
| | Fig. 4 Discharge capacity retention profile at different current rates as a function of cycle number for the series (Li2Fe)S1−xSexO where x = 0.1, 0.3, 0.5, 0.7, and 0.9, respectively. | |
Table 1 Some electrochemical parameters for the series (Li2Fe)S1−xSexO (x = 0.1–0.9) compared to the reported (Li2Fe)SO cathode
| Composition |
Theoretical capacitya (mA h g−1) |
Average discharge capacity at 0.1C (mA h g−1) |
Discharge capacity retention after 20 cycles (%) |
Capacity recovery at 0.1C after 50 cycles (%) |
|
The calculated theoretical capacity corresponds to the extraction/insertion of 2 Li from/in the antiperovskite composition.
|
| (Li2Fe)SO12 |
455 |
285 |
85 |
80 |
| (Li2Fe)S0.9Se0.1O |
438 |
215 |
90 |
91 |
|
(Li
2
Fe)S
0.7
Se
0.3
O
|
407
|
245
|
100
|
∼100 |
| (Li2Fe)S0.5Se0.5O |
379 |
228 |
95 |
∼100 |
| (Li2Fe)S0.3Se0.7O |
356 |
207 |
95 |
∼100 |
| (Li2Fe)S0.1Se0.9O |
335 |
195 |
93 |
∼100 |
Samples with Se contents higher than 30 at% displayed promising capacity recovery, however, with a lower average capacity at all studied current densities compared to (Li2Fe)S0.7Se0.3O. Due to the superior performance of the (Li2Fe)S0.7Se0.3O phase, we chose this composition for further detailed studies to closely figure out its properties, as will be shown in the following sections. Fig. 5 shows the potential dependence on the amount of extracted/inserted Li+ from/in (Li2Fe)S0.7Se0.3O during 5 cycles at 0.1C. As seen, a reversible extraction/insertion of 1.2 Li+ from/in (Li2Fe)S0.7Se0.3O without significant change for the 5 cycles is realized indicating its stable multielectron cycling behavior. Table S3† compares the electrochemical properties of antiperovskite (Li2Fe)S0.7Se0.3O with the most common cathode materials, indicating the competitiveness of antiperovskites in terms of the specific capacity. An additional protective coating with carbon or metal oxides such as Al2O3, TiO2, ZrO2 may be a possible solution to overcome the air sensitivity and the modest cycling stability at high current rates for these antiperovskite cathodes.30 For example, carbon coating could enhance the stability in air and cycling performance by providing a protective layer, that prevents the direct contact of LiFePO4 particles with air and the electrolyte.31
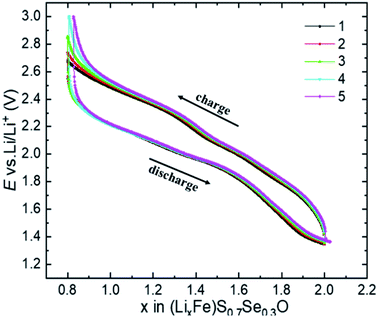 |
| | Fig. 5 Potential dependence on the amount of inserted/extracted Li+, during 5 cycles at 0.1C for the (Li2Fe)S0.7Se0.3O cathode. | |
3.5. Comprehensive investigations of (Li2Fe)S0.7Se0.3O
3.5.1. Diffusion coefficient (DLi+) measurements.
In order to examine the chemical diffusion coefficient of Li ions (DLi+) in (Li2Fe)S0.7Se0.3O during cycling, GITT was used, as seen in Fig. 6.13 The DLi+ was recorded during charge and discharge of the cell in a potential range between 2.7 and 1.4 V. A minimum in the DLi+ plot is commonly related to the structural re-arrangement due to oxidation or reduction.32 As seen, the variation of DLi+ is not linear and resembles the multi-phase behavior of the CV redox process. Three minima in the DLi+ plot during cell charge were observed at ∼2, 2.5 and 2.7 V which coincide well with the oxidation peaks observed in CV (Fig. 3) for (Li2Fe)S0.7Se0.3O. A similar behavior can be noted during discharge. The vacancy and dumbbell diffusion mechanisms of Li+ inside the antiperovskite compounds were previously reported.20 The first mechanism involves the hopping of Li+ through the edges of the octahedra while the second represents the hopping of Li+ interstitial dumbbell.20 In both mechanisms, the ionic migration involves a much weaker lattice strain upon ionic hopping and, therefore, exhibits a much lower activation energy barrier.33 Lu et al. reported a vacancy diffusion mechanism for (Li2Fe)SO with a low activation energy barrier of ∼0.32 eV comparable with the value obtained for the Li3ClO superionic conductor (∼0.35 eV).11 Such a low energy barrier is beneficial for battery applications and allows charging/discharging the cell at high current densities. Besides, vacancy/dumbbell diffusion mechanisms usually result in a small lattice change during cycling which is an essential requirement for any cathode used in LIBs. At the beginning of charge, DLi+ decreased due to the lack of vacancies until the oxidation process occured at ∼2.1 V (first minimum in the DLi+ plot). Afterwards, it increased dramatically owing to the increase of the available vacancies reaching a maximum value of 10−12 cm2 s−1 at 2.14 V. Further increase of the cell voltage resulted in a decrease in DLi+ until the occurrence of the second oxidation process (second minimum at ∼2.5 V). This decrease might be associated with the mobility limitation due to the accompanied structural changes. The same explanation can be given for the third minimum at ∼2.7 V. Obviously, large hysteresis in the ionic diffusion between charge and discharge processes can be realized from Fig. 6, particularly in the range between 2 and 2.5 V. This gap was already reported for (Li2Fe)SO and might be due to a lattice shrinking or an unknown reaction at the grain boundaries which prohibits subsequent lithiation to some extent.12 Fig. S9† compares the DLi+ for (Li2Fe)S0.7Se0.3O and (Li2Fe)SO to investigate the role of Se incorporation in the kinetic behavior of Li+. Although the behavior is similar during charge, a much higher DLi+ for (Li2Fe)S0.7Se0.3O was observed during discharge in the entire potential range. This reflects that more irreversible structural/chemical changes occur in (Li2Fe)SO, which impede the subsequent Li+ insertion.
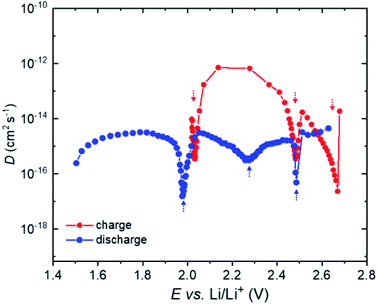 |
| | Fig. 6 Diffusion coefficient (DLi+) during charge and discharge for the (Li2Fe)S0.7Se0.3O composition. | |
3.5.2.
Operando XRD studies.
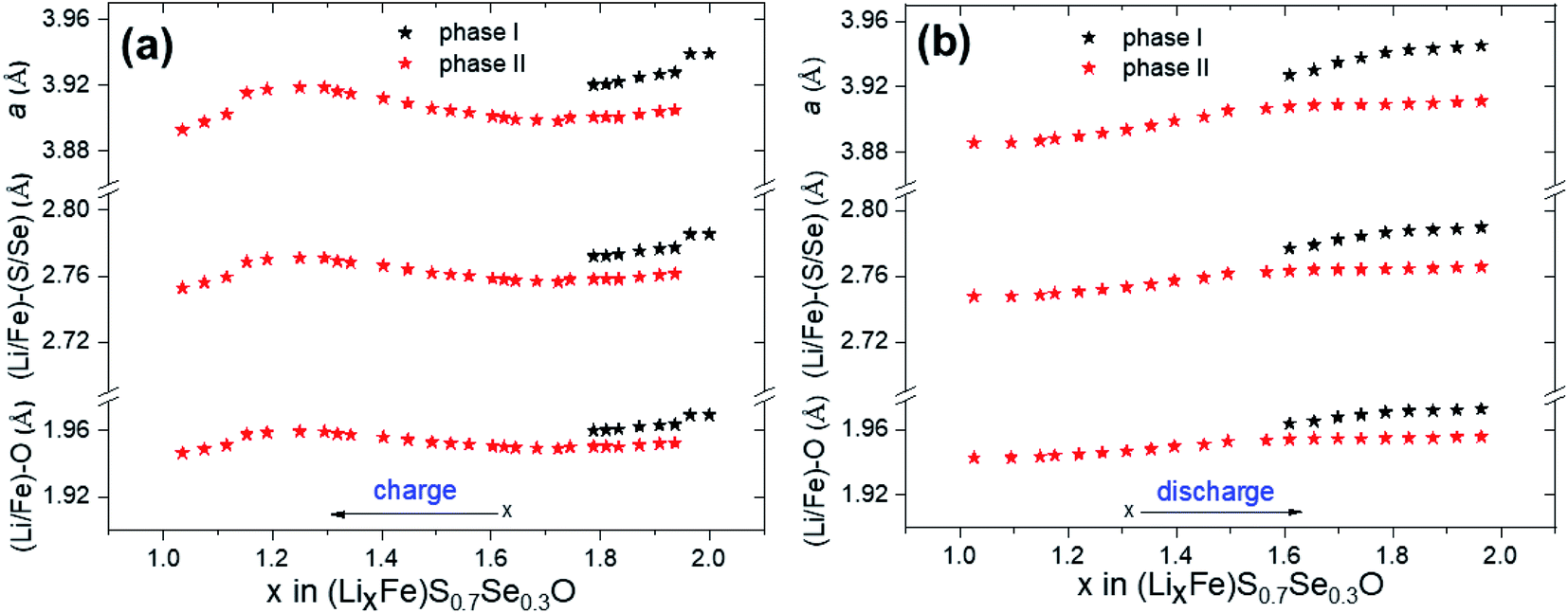
Fig. 7a shows the structural evolution during cycling of the (Li2Fe)S0.7Se0.3O cathode using the operando XRD technique. The long-range atomic order partially diminished after the first cycle. Bragg reflections corresponding to the main compound significantly widened and forfeited their initial intensity as a result of the lack of crystallinity and atomic disorder increase. Furthermore, a significant shift in the peak reflections associated with the main compound was detected, as highlighted in Fig. 7b during cycling (as illustrated in the corresponding voltage profile in Fig. 7c) compared to other inactive components of the in situ cell such as Al, Li and PTFE. In contrast to the reported (Li2Fe)SO cathode (see Fig. S10 in the ESI†),12 no significant reflections for additional secondary phases or superstructures were noticed after the first cycle for (Li2Fe)S0.7Se0.3O, highlighting its improved structural stability. This observation is in agreement with the Goldschmidt tolerance factor closer to 1 in the case of the Se-substituted materials. Moreover, a stabilizing effect of the S2−/Se2− ionic mixing at a high state of charge for local coordination was also reported by Martinolich et al. for Li-rich chalcogenide Li2FeS2−ySey solid solutions.28 In addition, this observation agrees well with the higher Li+ diffusion coefficient during discharge obtained for (Li2Fe)S0.7Se0.3O compared to (Li2Fe)SO. After complete Li insertion at the end of discharge, the original diffraction pattern was not fully recovered in terms of peak intensities or peak positions referring to a slight irreversible structural change. This irreversible structural change can be attributed to various reasons such as (i) a local mechanical stress and strain, (ii) a local random off-centering of ions in the antiperovskite structure instated of localization at high-symmetrical positions after cycling and (iii) Li+ concentration gradient in the particles, which leads to the broadening of Bragg reflections observed in the operando XRD experiments.12Fig. 8a and b show the variation of the lattice parameter (a) and (Li/Fe)–O and (Li/Fe)–(S/Se) distances versus the extracted/inserted amount of Li+ from/in (LixFe)S0.7Se0.3O during charge/discharge obtained from operando XRD results, respectively. The average (Li/Fe)–O and (Li/Fe)–(S/Se) distances were computed using the formulas a/2 and  , respectively. Generally, for most of the intercalation cathodes, the contraction of the unit cell volume during charge (Li+ extraction) is common which is mainly due to the shrinking of the transition metal ionic radius through the oxidation process, and the reverse occurs during discharge (Li+ insertion).34 However, there are several reported exceptions in this trend which were attributed to different mechanisms. For instance, the unit cell volume of LiVO2 (ref. 35) almost did not change, while for LiNbO2 (ref. 36) and Li2MoO3 (ref. 34) it increased during the charge process (Li+ extraction). Before charging (x = 2 in (LixFe)S0.7Se0.3O), one phase was detected with a bigger lattice parameter which would be denoted as phase I, as seen in Fig. 8a. Subsequent extraction of Li up to x ∼ 1.8 in (LixFe)S0.7Se0.3O resulted in the coexistence of phase I with the simultaneous appearance of another phase with a lower lattice parameter (denoted as phase II). In this range, both lattice parameters aphase I and aphase II decreased with the increased amount of extracted Li in good coincidence with the expectation. This phase coexistence might be due to the impeded kinetics of Li+ which can lead to a significant inhomogeneous distribution of Li+ between the surface and bulk of the particles. Afterwards, phase I totally disappeared while the lattice parameter aphase II unexpectedly increased with the increase of Li+ extraction up to x = 1.25 in (LixFe)S0.7Se0.3O.
, respectively. Generally, for most of the intercalation cathodes, the contraction of the unit cell volume during charge (Li+ extraction) is common which is mainly due to the shrinking of the transition metal ionic radius through the oxidation process, and the reverse occurs during discharge (Li+ insertion).34 However, there are several reported exceptions in this trend which were attributed to different mechanisms. For instance, the unit cell volume of LiVO2 (ref. 35) almost did not change, while for LiNbO2 (ref. 36) and Li2MoO3 (ref. 34) it increased during the charge process (Li+ extraction). Before charging (x = 2 in (LixFe)S0.7Se0.3O), one phase was detected with a bigger lattice parameter which would be denoted as phase I, as seen in Fig. 8a. Subsequent extraction of Li up to x ∼ 1.8 in (LixFe)S0.7Se0.3O resulted in the coexistence of phase I with the simultaneous appearance of another phase with a lower lattice parameter (denoted as phase II). In this range, both lattice parameters aphase I and aphase II decreased with the increased amount of extracted Li in good coincidence with the expectation. This phase coexistence might be due to the impeded kinetics of Li+ which can lead to a significant inhomogeneous distribution of Li+ between the surface and bulk of the particles. Afterwards, phase I totally disappeared while the lattice parameter aphase II unexpectedly increased with the increase of Li+ extraction up to x = 1.25 in (LixFe)S0.7Se0.3O.
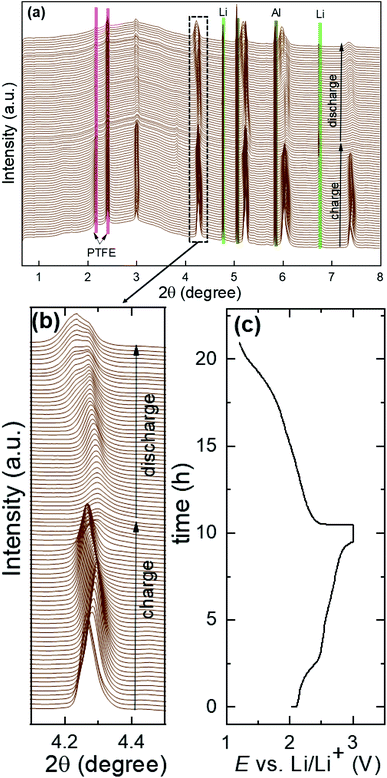 |
| | Fig. 7
Operando XRD pattern (a), peak position variation (b) and potential versus time profile (c) during cycling of the (Li2Fe)S0.7Se0.3O composition at 0.1C. | |
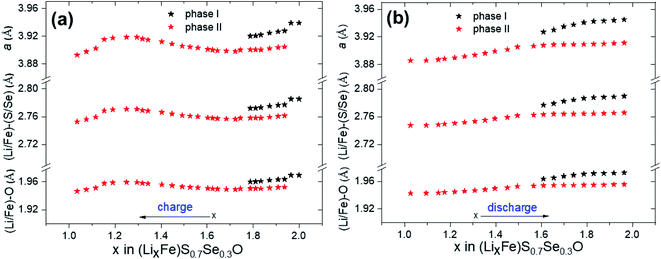 |
| | Fig. 8 Variation of the lattice parameter (a) and (Li/Fe)–O and (Li/Fe)–(S/Se) distances versus the extracted/inserted amount of Li+ from/in (LixFe)S0.7Se0.3O during charge (a) and discharge (b) obtained from operando XRD, respectively. | |
Accordingly, it seems that there are other factors that play a critical role in determining the trend of a. One possible factor is the screening effect. Extracting a large amount of Li+ from the cathode might lead to decreasing the screening effect between adjacent S/Se ions which increases the repulsion forces and, hence, increases the unit cell volume. This mechanism is called “unit cell breathing” and was reported for some layered cathodes.34 Therefore, the effect of screening might be higher than the effect of ionic shrinking in this range. Further extraction of Li+ with x < 1.25 in (LixFe)S0.7Se0.3O led to an abrupt decrease of aphase II which may result from the additional shrinking of the unit cell, originating from the oxidation of chalcogen atoms and the accompanied smaller ionic radii. During discharge, the change of aphase II followed back the charge process (Fig. 8b). Even a two-phase coexistence region (phase I and phase II) was observed as well when more than 1.6 Li+ was re-inserted into (LixFe)S0.7Se0.3O, similar to the charge process. The difference in the trend of the unit cell variation during charge and discharge processes might be attributed to the different kinetic behaviors where a slow diffusion coefficient (Fig. 6) with overlapping reduction peaks (Fig. 3) was observed during discharge.
3.5.3.
Operando XAS spectroscopy studies on (Li2Fe)S0.7Se0.3O.
In situ X-ray absorption spectroscopy is an element-specific powerful technique to characterize the valence state and the chemical environment during cycling.37 Fig. S11a† and 9a show the XAS results for the Fe K edge during various stages of charging combined with FeO and Fe2O3 spectra as standards corresponding to Fe2+ and Fe3+ valence states, respectively. The absorption edge (Eo) in the Fe K edge spectrum depends on the valence state, representing the electronic transition from a core state 1s to continuum 4p. It can be experimentally estimated as the energy value at 0.8 of the normalized post-edge intensity (xμ = 0.8),9 as shown in Fig. 9a. Generally, increasing the transition metal valence state increases the binding energy of the core electrons to the nucleus due to the increase in the effective nuclear charge Zeff, therefore, higher energy is required to eject the electron to a free state. In our case, the absorption edge (Eo) gradually shifts towards a higher energy with charging (Li+ extraction), confirming the change in the valence state from +2 to +3 for Fe. Besides, a remarkable drop in the intensity of the white line (highest intense absorption above the edge) was found during charging, indicating the increase in the structural disorder around the Fe atoms.38 The pre-edge feature (in the range from ∼7105 to 7112 eV) of the Fe K spectra originates from an electronic transition from the 1s to the 3d unoccupied bounded state. Although this transition is prohibited according to dipole selection rules, it appears due to the hybridization between 3d and 4p orbitals besides direct quadrupolar coupling.39 Similar to the main absorption edge, the pre-edge peak position shifts towards a higher energy. However, its intensity increases upon charging. The increase of the pre-edge peak upon Li extraction can be ascribed to the improvement in 3d–4p orbital intermixing as well as local distortion of highly symmetrical Fe surrounding. Fig. S11b† and 9b show the XAS spectra of the Se K edge at different potentials during charge. Compared to Fe, the analysis of the Se K edge is more complicated because the Se K edge is not only dependent on the valence state, but is also more sensitive to the bonding environment. For example, although ZnSe40 and Na2Se41 compounds have the same valence state for Se (−2), the edge energies for them are 12656.5 eV and 12656.17 eV, respectively. Herein, we recorded a spectrum of the uncycled (Li2Fe)S0.7Se0.3O compound at the open circuit voltage (OCV) as a reference for the −2 valence state of Se. For precise estimation of the Se K edge energy (Eo), the first derivative was computed and depicted, as seen in Fig. 10. Clearly, two main features can be noticed from the 1st derivative spectrum for all states of charge. The first group at lower energies corresponds to the pre-edge feature, originating mainly from the Se 1s to 4p dipole-allowed transition while the second group (at higher energies) is related to the edge transition and arises from the Se 1s to 5p dipole-allowed transition.
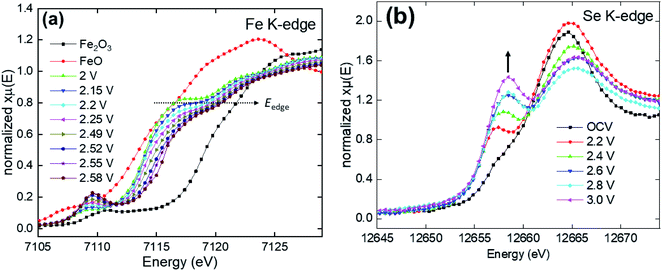 |
| | Fig. 9 XAS data of (Li2Fe)S0.7Se0.3O for Fe (a) and Se (b) K edges. | |
 |
| | Fig. 10 1st derivative Se K edge spectrum of (Li2Fe)S0.7Se0.3O during charge at different cut-off potentials. | |
Similar behavior of the Se K edge with the growing of the pre-edge feature was reported by Henthorn et al.,42 which was attributed to an increased interaction between Se and the neighboring atom. As the valence states for Fe increases from Fe2+ to Fe3+, the pre-edge feature is expected to be more intense due to the increase of overlapping between Fe 3d and Se 4p orbitals.42,43 Indeed, such an increase in the pre-edge feature was observed during charging, as indicated by arrow in Fig. 9b. At 2.2 V, the Eo has the same value compared to the OCV state, meaning that Se is electrochemically inactive until this state of charge. However, further increase in voltage (>2.4 V) resulted in a gradual shift in the Eo towards higher energies indicating a partial oxidation of Se2− to elemental Se0, as shown in Fig. 10.
3.5.4. Particle morphology of (Li2Fe)S0.7Se0.3O before and after cycling.
Fig. S12a and b† show the SEM image and the EDS spectrum of (Li2Fe)S0.7Se0.3O before cycling. The sample consists of predominantly large particles with the size higher than 10 μm. The EDS investigations confirmed the existence of the main elements Fe, S, Se and O with the expected atomic percentages without the emergence of any other impurities. After 50 cycles of (Li2Fe)S0.7Se0.3O at different current rates varied between 0.1C and 1C, the battery was disassembled and the material was tested again by SEM and EDS, as seen in Fig. S12c and d.† The investigations confirmed that the main elements Fe, S, Se, O are still present in amounts comparable with the pristine material. This observation supports the assumption that the redox process of the chalcogen atoms (S/Se) is not accompanied by their dissolution in significant amounts in the electrolyte solution.
3.5.5. Magnetic properties of (Li2Fe)S0.7Se0.3O.
Fig. S13a† shows the results of magnetization measurements performed on the as-synthesized polycrystalline (Li2Fe)S0.7Se0.3O cathode material. The sample does not display a typical paramagnetic behavior and, therefore, does not obey the Curie–Weise equation. Instead, the low signal and odd temperature susceptibility (χ)-dependence may support strong spin–spin interactions in the material. Below the anomaly at about 50 K, the spin system seems to freeze. The obvious difference between field-cooled and zero field-cooled data suggests that the magnetic ground state has a degeneracy, i.e. forms domains and exhibits, probably, a glassy-like feature. The suggestion about spin-domains is reasonable, considering the atomic disorder of Fe on the shared (Li2Fe)-site. As estimated from magnetization measurements at room temperature, about 0.01% metallic Fe is present in (Li2Fe)S0.7Se0.3O, which was determined by a linear fitting (red line) the field-dependent isothermal magnetization curve (black line), as shown in Fig. S13b.†
4. Conclusion
(Li2Fe)S1−xSexO solid solutions (where x = 0.1, 0.3, 0.5, 0.7, and 0.9) were successfully synthesized by the solid state reaction method. Higher thermal stability combined with lower sensitivity to air were realized by increasing the Se content at the expense of S. Cyclic voltammetry measurements revealed a multi-stage cationic and anionic redox processes during Li+ extraction/insertion. Galvanostatic measurements showed that Se can play a significant role in enhancing the capacity recovery and the cycling stability, especially at low current rates. (Li2Fe)S0.7Se0.3 displayed the best electrochemical performance among all compositions with an average discharge capacity of about 245 mA h g−1 at 0.1C and about 100% capacity recovery in the rate performance investigations. A large gap in Li+ diffusion coefficients between charge and discharge processes was observed from the GITT measurements of (Li2Fe)S0.7Se0.3 highlighting the different kinetics. The operando XRD confirmed the non-linear variation of the lattice parameter during cycling of (Li2Fe)S0.7Se0.3 without the appearance of other phases or superstructures. This finding reflects the improved structural stability of (Li2Fe)S0.7Se0.3 during cycling compared to the unsubstituted (Li2Fe)SO. The XAS results of the Fe K edge confirmed the oxidation of Fe during charge through shifting the edge energy position to higher energies. Analyses of the Se K edge showed that Se2− is electrochemically active and oxidizes above 2.4 V vs. Li/Li+. SEM-EDS analyses after cycling of (Li2Fe)S0.7Se0.3 suggest the negligible dissolution of components because comparable atomic percentages before and after cycling were observed. This work paves the way for an anionic substitution approach for optimizing the functional properties of other antiperovskite cathodes such as (Li2Co)SO and (Li2Mn)SO.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the European Regional Development Fund through Sächsische Aufbaubank and project LUKSIAK (Project number 100350438). M. Valldor thanks DFG (German Science Foundation) for the financial support via project VA831/4-1. M. A. A. Mohamed and M. Gorbunov thank the IFW excellence program for the financial support. The authors thank Andrea Voss and Anne Voidel (IFW Dresden) for performing ICP-OES measurements. This research has benefitted from beamtime allocation at beamlines P02.01 and P65 at the PETRA III synchrotron (DESY, Hamburg, Germany).
References
- D. Deng, Energy Sci. Eng., 2015, 3, 385–418 CrossRef
 .
.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS .
- X. Shen, X.-Q. Zhang, F. Ding, J.-Q. Huang, R. Xu, X. Chen, C. Yan, F.-Y. Su, C.-M. Chen, X. Liu and Q. Zhang, Energy Material Advances, 2021, 2021, 1–15 CrossRef .
- C. J. Hansen, J. J. Zak, A. J. Martinolich, J. S. Ko, N. H. Bashian, F. Kaboudvand, A. V. der Ven, B. C. Melot, J. N. Weker and K. A. See, J. Am. Chem. Soc., 2020, 142, 6737–6749 CrossRef CAS PubMed .
- J. Lee, D. A. Kitchaev, D.-H. Kwon, C.-W. Lee, J. K. Papp, Y.-S. Liu, Z. Lun, R. J. clément, T. Shi, B. D. McCloskey, J. Guo, M. Balasubramanian and G. Ceder, Nature, 2018, 556, 185–190 CrossRef CAS PubMed .
- K. Luo, M. R. Roberts, R. Hao, N. Guerrini, D. M. Pickup, Y.-S. Liu, K. Edström, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, Nat. Chem., 2016, 8, 684–691 CrossRef CAS PubMed .
- K. T. Lai, I. Antonyshyn, Y. Prots and M. Valldor, J. Am. Chem. Soc., 2017, 139, 9645–9649 CrossRef CAS PubMed .
- K. T. Lai, I. Antonyshyn, Y. Prots and M. Valldor, Inorg. Chem., 2018, 57, 13296–13299 CrossRef CAS PubMed .
- M. V. Gorbunov, S. Carrocci, S. Maletti, M. Valldor, T. Doert, S. Hampel, I. G. G. Martinez, D. Mikhailova and N. Gräßler, Inorg. Chem., 2020, 59, 15626–15635 CrossRef CAS PubMed .
- M. V Gorbunov, S. Carocci, I. G. G. Martinez, V. Baran and D. Mikhailova, Front. Energy Res., 2021, 9, 1–10 Search PubMed .
- Z. Lu and F. Ciucci, J. Mater. Chem. A, 2018, 6, 5185–5192 RSC .
- D. Mikhailova, L. Giebeler, S. Maletti, S. Oswald, A. Sarapulova, S. Indris, Z. Hu, J. Bednarcik and M. Valldor, ACS Appl. Energy Mater., 2018, 1, 6593–6599 CrossRef .
- W. Weppner and R. A. Huggins, J. Electrochem. Soc., 1977, 124, 1569–1578 CrossRef CAS .
- F. Fauth, I. Peral, C. Popescu and M. Knapp, Powder Diffr., 2013, 28, S360–S370 CrossRef CAS .
- M. Herklotz, J. Weiß, E. Ahrens, M. Yavuz, L. Mereacre, N. Kiziltas-Yavuz, C. Dräger, H. Ehrenberg, J. Eckert, F. Fauth, L. Giebeler and M. Knapp, J. Appl. Crystallogr., 2016, 49, 340–345 CrossRef CAS .
- E. Welter, R. Chernikov, M. Herrmann and R. Nemausat, AIP Conf. Proc., 2019, 2054, 040002 CrossRef .
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed .
- C. P. Kempter, Phys. Status Solidi, 1966, 18, K117–K118 CrossRef CAS .
- Y. Sun, Y. Wang, X. Liang, Y. Xia, L. Peng, H. Jia, H. Li, L. Bai, J. Feng, H. Jiang and J. Xie, J. Am. Chem. Soc., 2019, 141, 5640–5644 CrossRef CAS PubMed .
- K. Kim and D. J. Siegel, J. Mater. Chem. A, 2019, 7, 3216–3227 RSC .
- M. Ashokkumar and S. Muthukumaran, J. Lumin., 2014, 145, 167–174 CrossRef CAS .
- A. M. Ahmed, M. Rabia and M. Shaban, RSC Adv., 2020, 10, 14458–14470 RSC .
- J. A. Dawson, P. Canepa, T. Famprikis, C. Masquelier and M. S. Islam, J. Am. Chem. Soc., 2018, 140, 362–368 CrossRef CAS PubMed .
- K. Shen, Y. Wang, J. Zhang, Y. Zong, G. Li, C. Zhao and H. Chen, Phys. Chem. Chem. Phys., 2020, 22, 3030–3036 RSC .
- E. B. Ferreira, M. L. Lima and E. D. Zanotto, J. Am. Ceram. Soc., 2010, 93, 3757–3763 CrossRef CAS .
- S. Muhammad, S. Lee, H. Kim, J. Yoon, D. Jang, J. Yoon, J.-H. Park and W.-S. Yoon, J. Power Sources, 2015, 285, 156–160 CrossRef CAS .
- T. Wejrzanowski, M. Lewandowska, K. Sikorski and K. J. Kurzydlowski, J. Appl. Phys., 2014, 116, 164302 CrossRef .
- A. J. Martinolich, J. J. Zak, D. N. Agyeman-Budu, S. S. Kim, N. H. Bashian, A. Irshad, S. R. Narayan, B. C. Melot, J. N. Weker and K. A. See, Chem. Mater., 2021, 33, 378–391 CrossRef CAS .
- A. Eftekhari, Sustainable Energy Fuels, 2017, 1, 14–29 RSC .
- P. Guan, L. Zhou, Z. Yu, Y. Sun, Y. Liu, F. Wu, Y. Jiang and D. Chu, J. Energy Chem., 2020, 43, 220–235 CrossRef .
- H. Li and H. Zhou, Chem. Commun., 2012, 48, 1201–1217 RSC .
- K. Fröhlich, I. Abrahams and M. Jahn, Sci. Technol. Adv. Mater., 2020, 21, 653–660 CrossRef PubMed .
- M. Wu, B. Xu and C. Ouyang, Chin. Phys. B, 2016, 25, 018206 CrossRef .
- Y.-N. Zhou, J. Ma, E. Hu, X. Yu, L. Gu, K.-W. Nam, L. Chen, Z. Wang and X.-Q. Yang, Nat. Commun., 2014, 5, 1–8 Search PubMed .
- L. A. de Picciotto and M. M. Thackeray, Mater. Res. Bull., 1984, 2, 1497–1506 CrossRef .
- N. Kumada, S. Muramatu, F. Muto and N. Kinomura, J. Solid State Chem., 1988, 73, 33–39 CrossRef CAS .
- W.-T. Chen, C.-W. Hsu, J.-F. Lee, C.-W. Pao and I.-J. Hsu, ACS Omega, 2020, 5, 4991–5000 CrossRef CAS PubMed .
- A. Muthiah, T. Baikie, S. Shukla, S. Ball, M. Copley, T. I. Hyde, Y. Du, G. Sankar, V. Aravindan and M. Srinivasan, J. Mater. Chem. A, 2017, 5, 19963–19971 RSC .
- M. L. Baker, M. W. Mara, J. J. Yan, K. O. Hodgson, B. Hedman and E. I. Solomon, Coord. Chem. Rev., 2017, 345, 182–208 CrossRef CAS PubMed .
- A. L. Ryser, D. G. Strawn, M. A. Marcus, J. L. Johnson-Maynard, M. E. Gunter and G. Möller, Geochem. Trans., 2005, 6, 1–11 CrossRef CAS .
- J. T. Olegario, N. Yee, M. Miller, J. Sczepaniak and B. Manning, J. Nanopart. Res., 2010, 12, 2057–2068 CrossRef CAS .
- J. T. Henthorn, R. J. Arias, S. Koroidov, T. Kroll, D. Sokaras, U. Bergmann, D. C. Rees and S. DeBeer, J. Am. Chem. Soc., 2019, 141, 13676–13688 CrossRef CAS PubMed .
- C. L. Chen, S. M. Rao, C. L. Dong, J. L. Chen, T. W. Huang, B. H. Mok, M. C. Ling, W. C. Wang, C. L. Chang, T. S. Chan, J. F. Lee, J.-H. Guo and M. K. Wu, EPL, 2011, 93, 47003 CrossRef .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta05130j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Mikhail V.
Gorbunov
*ab,
Mikhail V.
Gorbunov
 , where rCh and r(Li2Fe) represent the average ionic radii of (S1−xSex) and (Li2Fe) atomic sites (see Table S1 in the ESI†). The high spin ionic radius of Fe2+ was considered in the calculations based on previous Mössbauer spectroscopy.12 The experimentally obtained lattice parameters are higher than the ideal ones suggesting the existence of larger voids for Li+ diffusion. This finding is in a good agreement with cell metrics reported for (Li2Fe)SeO and (Li2Fe)SO cathodes.7 In order to get an indication about the structural stability upon S substitution by Se, the Goldschmidt tolerance factor (t) was calculated using the formula
, where rCh and r(Li2Fe) represent the average ionic radii of (S1−xSex) and (Li2Fe) atomic sites (see Table S1 in the ESI†). The high spin ionic radius of Fe2+ was considered in the calculations based on previous Mössbauer spectroscopy.12 The experimentally obtained lattice parameters are higher than the ideal ones suggesting the existence of larger voids for Li+ diffusion. This finding is in a good agreement with cell metrics reported for (Li2Fe)SeO and (Li2Fe)SO cathodes.7 In order to get an indication about the structural stability upon S substitution by Se, the Goldschmidt tolerance factor (t) was calculated using the formula  , where rCh and r(Li2Fe) are as mentioned before and rO refers to the ionic radius of O (see Fig. S2 in the ESI†).19 This factor can be regarded as a predictor of the structural stability of antiperovskites based on the degree of mismatch between the constituent ions. In general, a value of t close to 1 corresponds to an ideal undistorted cubic structure.20 It is evident that with the replacement of S by Se in the structure, t increases, suggesting that a higher structural stability is provided. This increase is attributed to the larger ionic size of Se (1.84 Å) compared to S (1.7 Å). In order to study the change in the internal structure, the XRD average crystallite size (DXRD) and the lattice strain (ε) were calculated for (Li2Fe)S1−xSexO series (Table S1 in the ESI†). The DXRD was calculated from the Scherrer's equation DXRD = 0.9λ/β
, where rCh and r(Li2Fe) are as mentioned before and rO refers to the ionic radius of O (see Fig. S2 in the ESI†).19 This factor can be regarded as a predictor of the structural stability of antiperovskites based on the degree of mismatch between the constituent ions. In general, a value of t close to 1 corresponds to an ideal undistorted cubic structure.20 It is evident that with the replacement of S by Se in the structure, t increases, suggesting that a higher structural stability is provided. This increase is attributed to the larger ionic size of Se (1.84 Å) compared to S (1.7 Å). In order to study the change in the internal structure, the XRD average crystallite size (DXRD) and the lattice strain (ε) were calculated for (Li2Fe)S1−xSexO series (Table S1 in the ESI†). The DXRD was calculated from the Scherrer's equation DXRD = 0.9λ/β





 , respectively. Generally, for most of the intercalation cathodes, the contraction of the unit cell volume during charge (Li+ extraction) is common which is mainly due to the shrinking of the transition metal ionic radius through the oxidation process, and the reverse occurs during discharge (Li+ insertion).34 However, there are several reported exceptions in this trend which were attributed to different mechanisms. For instance, the unit cell volume of LiVO2 (ref. 35) almost did not change, while for LiNbO2 (ref. 36) and Li2MoO3 (ref. 34) it increased during the charge process (Li+ extraction). Before charging (x = 2 in (LixFe)S0.7Se0.3O), one phase was detected with a bigger lattice parameter which would be denoted as phase I, as seen in Fig. 8a. Subsequent extraction of Li up to x ∼ 1.8 in (LixFe)S0.7Se0.3O resulted in the coexistence of phase I with the simultaneous appearance of another phase with a lower lattice parameter (denoted as phase II). In this range, both lattice parameters aphase I and aphase II decreased with the increased amount of extracted Li in good coincidence with the expectation. This phase coexistence might be due to the impeded kinetics of Li+ which can lead to a significant inhomogeneous distribution of Li+ between the surface and bulk of the particles. Afterwards, phase I totally disappeared while the lattice parameter aphase II unexpectedly increased with the increase of Li+ extraction up to x = 1.25 in (LixFe)S0.7Se0.3O.
, respectively. Generally, for most of the intercalation cathodes, the contraction of the unit cell volume during charge (Li+ extraction) is common which is mainly due to the shrinking of the transition metal ionic radius through the oxidation process, and the reverse occurs during discharge (Li+ insertion).34 However, there are several reported exceptions in this trend which were attributed to different mechanisms. For instance, the unit cell volume of LiVO2 (ref. 35) almost did not change, while for LiNbO2 (ref. 36) and Li2MoO3 (ref. 34) it increased during the charge process (Li+ extraction). Before charging (x = 2 in (LixFe)S0.7Se0.3O), one phase was detected with a bigger lattice parameter which would be denoted as phase I, as seen in Fig. 8a. Subsequent extraction of Li up to x ∼ 1.8 in (LixFe)S0.7Se0.3O resulted in the coexistence of phase I with the simultaneous appearance of another phase with a lower lattice parameter (denoted as phase II). In this range, both lattice parameters aphase I and aphase II decreased with the increased amount of extracted Li in good coincidence with the expectation. This phase coexistence might be due to the impeded kinetics of Li+ which can lead to a significant inhomogeneous distribution of Li+ between the surface and bulk of the particles. Afterwards, phase I totally disappeared while the lattice parameter aphase II unexpectedly increased with the increase of Li+ extraction up to x = 1.25 in (LixFe)S0.7Se0.3O.