
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of hydration on ion transport in Li2Sn2S5·xH2O†
Markus
Joos
 a,
Christian
Schneider
a,
Andreas
Münchinger
a,
Igor
Moudrakovski
a,
Robert
Usiskin
*a,
Joachim
Maier
a and
Bettina V.
Lotsch
ab
a,
Christian
Schneider
a,
Andreas
Münchinger
a,
Igor
Moudrakovski
a,
Robert
Usiskin
*a,
Joachim
Maier
a and
Bettina V.
Lotsch
ab
aMax Planck Institute for Solid State Research, Heisenbergstr. 1, 70569 Stuttgart, Germany. E-mail: r.usiskin@fkf.mpg.de
bDepartment of Chemistry, Ludwig-Maximilians-Universität München, Butenandtstr. 5-13, 81377 München, Germany
First published on 23rd July 2021
Abstract
This work investigates the structure and transport properties of the layered material Li2Sn2S5·xH2O. The anhydrous phase shows a room-temperature Li+ diffusivity below 10−9 cm2 s−1 and conductivity below 10−5 S cm−1. Upon exposure to humidity, water intercalates between the layers and increases the interlayer distance, inducing first-order transitions to a hydrated phase (x ≈ 2–4) and then to a second hydrated phase (x ≈ 8–10). The latter is soft and sticky but remains solid. Diffusion of both Li+ ions and H2O remains predominantly two-dimensional under all conditions. The Li+ diffusivity and conductivity both increase by three orders of magnitude upon hydration, reaching values of 5 × 10−7 cm2 s−1 and 10−2 S cm−1 in the second hydrate. These transport rates are extraordinary for a solid electrolyte and approach what is typically seen in aqueous solutions. The material Li2Sn2S5·xH2O thus bridges the gap between a hydrated solid electrolyte and a confined liquid electrolyte, which is scientifically interesting and potentially useful in battery applications. In the light of these findings, a previous work on Li2Sn2S5 from our groups is revisited.
Introduction
Hydration is relevant for both fundamental science and technological applications because it can strongly modify the charge transport properties of materials. In general, the reversible incorporation of water can proceed by a dissociative or a molecular mechanism. Dissociative incorporation is frequently encountered in oxides, hydroxides, and apatites,1 and it leads to the formation of OH species, which are often accommodated on oxygen sites. Molecular incorporation of H2O can arise in a broad range of structures, including oxides with roomy structures.2,3 Criteria as to whether water is incorporated in a dissociative or non-dissociative way are based on site and bond properties.4 Both mechanisms frequently lead to a substantial increase in H+ conductivity, as seen in a variety of materials including minerals5–13 and polymers.14,15 Some of these materials (Nafion, Y-doped BaZrO3) play a crucial role in state-of-the-art electrochemical devices.Hydration can also promote the transport of other cations besides protons. For example, perfluorinated ionomer membranes can be converted by ion exchange into single-ion conductors for various cations.16,17 Similarly, layered silicate compounds can be converted into Li+, Na+, Mg2+ or Ca2+ conductors. Water uptake can influence the cation mobility by solvation or by modifying the crystallographic geometry, usually by incorporating water into the SiO2–Al2O3 sheets and consequent coordination to the present cation, which increases the interlayer distance.5–13 The ability to undergo cation exchange is a general feature of layered structures and is observed in various chemical compositions.18–21 The two-dimensional (2D) arrangement must remain intact after hydration for the cations in the interlayer galleries to remain highly mobile.22 Another example is Li-exchanged Nafion, which shows high Li+ conductivity when hydrated.23 In the case of solid Li+ and Na+ conductors, hydration studies are rather scarce24–30 and tend to suggest that water uptake promotes Li+ and Na+ conduction;31 one recent paper reported a material in which the opposite is true.30
The compound Li2Sn2S5 is a good candidate for hydration studies. In an initial report, Li2Sn2S5 was shown to be a layered material consisting of covalently-bonded (and partially Li-substituted) tin sulfide sheets which are held together by ionic bonds to interlayer Li+ ions.32 Upon immersion in water, the ionic bonds are disrupted, and the material exfoliates into a suspension of monolayer sheets. A follow-up work explored reassembling the exfoliated sheets as part of a Bragg stack.33 The water content, interlayer spacing, and optical properties of the restacked sheets were shown to be sensitive to the surrounding water partial pressure, a property that can be used for humidity sensing. Another work characterizing the structure and ion transport properties of the restacked material is forthcoming.34 A fourth work confirmed by XRD refinement that 75% of the Li+ ions are on interlayer sites (Li+∥), while the remaining 25% substitute onto Sn4+ sites within the sheets (Li+⊥), such that the available interlayer Li+ sites are only 38% occupied.35 Accordingly, the general formula of this solid solution system can be written (Li∥)3z[(Li⊥)zSn1−zS2]. That work also reported that Li2Sn2S5 shows fast Li+ diffusion,35 which raises questions about the mechanism and about whether the transport properties can be further improved by hydration. One can be optimistic that the material will not decompose, since other lithium tin sulfides are reportedly stable under air and moisture36–42 (unlike the thiophosphates),40–56 while Li4SnS4 and other alkali tin sulfides can form stable hydrates.36,57
Here we show that the water content in Li2Sn2S5·xH2O can be varied from x = 0 to 10 by adjusting the humidity. The material undergoes first-order phase transitions to two hydrated phases, but the layered structure remains largely intact. Pulsed-field gradient nuclear magnetic resonance (PFG NMR) reveals that the Li+ transport remains predominantly 2D at all water contents. The transport in the anhydrous material is far slower than previously reported. NMR and impedance spectroscopy measurements reveal strong increases in the Li+ mobility, Li+ conductivity, and H2O mobility with increasing water content. The fully hydrated compound remains solid, but exhibits liquid-like Li+ conductivity and Li+ diffusivity values of 10−2 S cm−1 and 10−7 cm2 s−1 at room temperature. These transport rates are extraordinary in a solid and approach what is seen in concentrated aqueous electrolytes.58 The amount of water incorporated is sufficient to provide the interlayer Li+ ions with a full hydration shell. Connections between the behavior of hydrated layered solids and confined concentrated liquids are discussed, and the final two sections re-evaluate our previous work on Li2Sn2S5 and offer an outlook for applications.
Results
TGA and XRD: hydration study
A typical measurement of the water content x in Li2Sn2S5·xH2O is shown in Fig. 1a. The thermogravimetric analyzer (TGA) can resolve changes in x of about 0.01 or larger. After an increase in the water partial pressure pH2O, x stabilizes at a higher value on a typical timescale of several hours. Dehydration steps show far longer stabilization times, on the order of tens of hours. Due to the sluggish kinetics, for practical reasons the humidity was usually switched at the onset of a plateau in the mass–time trace, before the mass had truly stabilized. Follow-up experiments with longer dwell times suggest that the error in x introduced by this approach is below 0.1 (Fig. S4a†). A substantial contribution from surface adsorption is unlikely in light of the slow kinetics and the large particle size (∼50 μm). The variation of water content with humidity and temperature is summarized in Fig. 1b. Values of x in the range 0.4 to 1.6 do not appear, which suggests the presence of a miscibility gap, i.e., a first-order phase transition. The position of the phase boundary can be estimated by averaging the steepest and shallowest lines that fall between the brown and blue points in Fig. 1b. If the water content changes across the transition by y molecules of H2O per formula unit, the law of mass action can be written: | (1) |
 | ||
| Fig. 1 Average water content x in Li2Sn2S5·xH2O. (a) Typical gravimetric data (after buoyancy correction) measured under isothermal conditions (here 39 °C) while varying the absolute humidity pH2O. (b) Dependence of x on pH2O and temperature, as measured using a TGA (brown and light blue circles) or a balance and salt solutions (dark blue circles). Black line shows the estimated boundary between the anhydrous and hydrated phases. Typical ambient conditions are also shown, assuming a relative humidity of 50–70%. | ||
Samples that are cycled between the anhydrous and hydrated (x ≈ 2–3) states show a small but reproducible hysteresis. Specifically, the dehydration steps require a higher temperature by 20–30 °C than the rehydration steps (Fig. S1 and S2†). Similar hysteresis is observed in isothermal measurements (Fig. S3†). Hydrolysis can be ruled out, as discussed in the ESI.†
Fig. 2a shows XRD patterns measured from ten powder samples with different average water contents. The pattern obtained for x = 0 shows good agreement to that reported previously for anhydrous Li2Sn2S.35 A few small additional reflections are visible, which likely arise from stacking faults.60,61 This attribution is supported by XRD patterns acquired from a single anhydrous particle, which show multiple diffraction spots in close proximity, consistent with some degree of stacking faults, twinning, and/or mosaicism (Fig. S16a†). From the published structural determination, the reflection at 6.5° can be attributed to (001) planes. Using that reflection, the interlayer distance d is found from Bragg's law to be 6.24 Å, similar to the value of 6.17 Å obtained previously (Fig. 2b). The samples with an average water content of 1.0, 1.6, and 1.9 show similar patterns as the anhydrous sample, except a new reflection appears at 4.9°, and the intensity of the reflection at 6.5° is somewhat smeared to lower angles. With increasing x, the reflection intensity increases at 4.9° and decreases at 6.5°, until at x = 2.7 the reflection at 6.5° is entirely absent. This behavior is consistent with a first-order phase transition to a hydrated phase with an interlayer distance of 8.4 Å.
 | ||
| Fig. 2 (a) Powder X-ray diffraction patterns of Li2Sn2S5·xH2O samples with increasing degree of hydration. The patterns are vertically offset to facilitate comparisons. (b) Distance between (001) planes inferred from the patterns in (a). | ||
The patterns at x = 2.7–3.4 show no additional reflections, suggesting that in this range a single phase is present into which water is miscible. New reflections appear and increase in intensity when the average water content is increased to x = 5.9 or 7.4, and they are the only reflections visible at x = 10.5, consistent with a first-order transition to a second hydrated phase. Assuming the reflection at 3.7° corresponds to (001) planes, an interlayer distance of 11.1 Å is obtained. Thus, upon hydration, the interlayer distance jumps by 2.0 Å when transitioning to the first hydrate, and it jumps by another 2.7 Å when transitioning to the second hydrate. The latter jump matches the diameter of an oxygen ion in a water molecule (about 2.7 Å).62 The interlayer distance remains essentially constant within the single-phase ranges (x ≈ 2–4 for the hydrated phase, x ≈ 8–10 for the swollen phase), suggesting that in these ranges, incorporation is accommodated by denser packing of the water molecules rather than swelling of the material. Pattern refinements and a more detailed structural discussion will be given in a future work.
In terms of mechanical properties, the material remains solid up to x = 10.5 but becomes noticeably softer and stickier with increasing water content. At high water contents (x ≥ 7.4), it seemed possible to squeeze some water out of the material by the application of pressure. At x > 11 the material was deliquescent and became a solid–liquid composite; this situation was observed at the saturation pressure of water (40–120 mbar) at 30–50 °C.
NMR: Li+ and H+ diffusion
Li2Sn2S5·xH2O has a layered structure, so anisotropic properties are expected. Electron density analysis of anhydrous Li2Sn2S5 predicted a far higher Li+ diffusivity in-plane than out-of-plane.35 To test this suggestion, the normalized echo signal attenuation data obtained by PFG NMR can be fit by a standard isotropic 3D model with a single diffusivity,63 as well as an anisotropic 2D model with different in-plane and out-of-plane diffusivities.64 For all water contents, the 2D model yields visually satisfactory fits (Fig. 3), acceptable values of the goodness of fit, and reasonable confidence intervals for the fit parameters. In contrast, the 3D model leads to substantially worse fits (Fig. S6†). Additional models were also attempted and discarded. By averaging over all possible crystal orientations, the effective Li+ diffusivity of a polycrystalline sample is obtained. More details are given in the ESI.†
of a polycrystalline sample is obtained. More details are given in the ESI.†
 | ||
| Fig. 3 Normalized echo signal versus the squared gyromagnetic ratio times the integral I(G), as measured by 7Li PFG NMR at 180 °C (green), 140 °C (orange) and 60 °C (red) from Li2Sn2S5·xH2O samples with the indicated water contents. Points are raw data; curves are fits by the 2D model. See ESI† for details. | ||
The dependence of  on temperature and average water content is shown in Fig. 4a. The overall trend is clear: the Li+ diffusivity increases monotonically with increasing x. For x ≈ 8.0, the diffusivity reaches 5 × 10−7 cm2 s−1 at room temperature, which is three orders of magnitude higher than the corresponding (extrapolated) value for the anhydrous material. Similar results are obtained when the diffusion time in the PFG measurement is increased from 10 to 100 ms (see also Fig. S5†). The activation energy determined from the linear fits in Fig. 4a is 0.28, 0.29, 0.28, and 0.31 eV for the x ≈ 0, 2.3, 4.0, and 8.0 samples. The fact that the activation energy is nearly constant suggests that the faster diffusivity is due to an increased exponential pre-factor, and specifically an increased attempt frequency, since the entropy of migration is expected to be small.
on temperature and average water content is shown in Fig. 4a. The overall trend is clear: the Li+ diffusivity increases monotonically with increasing x. For x ≈ 8.0, the diffusivity reaches 5 × 10−7 cm2 s−1 at room temperature, which is three orders of magnitude higher than the corresponding (extrapolated) value for the anhydrous material. Similar results are obtained when the diffusion time in the PFG measurement is increased from 10 to 100 ms (see also Fig. S5†). The activation energy determined from the linear fits in Fig. 4a is 0.28, 0.29, 0.28, and 0.31 eV for the x ≈ 0, 2.3, 4.0, and 8.0 samples. The fact that the activation energy is nearly constant suggests that the faster diffusivity is due to an increased exponential pre-factor, and specifically an increased attempt frequency, since the entropy of migration is expected to be small.
 | ||
| Fig. 4 Effective diffusivity of (a) Li+ and (b) H+ in Li2Sn2S5·xH2O samples, as determined from 7Li and 1H PFG NMR measurements using a diffusion time of 10 or 100 ms. In (a), solid lines are linear fits to all points at a given water content, except the data for x ≈ 4.0 below 80 °C show significant scatter and were excluded. For a few points in (b), the goodness of fit in the PFG data was noticeably worse; these are marked with an asterisk. | ||
At lower water contents and lower temperatures, the degree of attenuation was below the PFG NMR detection limit, so diffusivity values could not be reliably extracted. The 7Li data for x ≈ 4.0 show somewhat anomalous behavior near room temperature, which is tentatively attributed to small variations in water content due to condensation in the NMR capillary. The condensation issue was mitigated before acquiring the x ≈ 8.0 data, as described in the Experimental details.
Fig. 4b shows the results from 1H PFG NMR measurements. The effective proton diffusivity 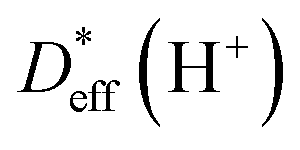 is comparable to the effective Li+ diffusivity in samples with x ≈ 4 and 8. This proton motion is attributed to the diffusion of water molecules, since there is no evidence for the presence of protonic defects, as discussed below. Assuming this attribution is correct, the data indicate that Li+ ions and water molecules in the interlayer galleries have a comparable mobility. The diffusivity of water decreases by a somewhat larger factor (3–10×) than the Li+ diffusivity when the diffusion time is increased from 10 to 100 ms, which hints that grain boundaries are more blocking for water molecules than for Li+ ions. The data show more scatter compared to Li+; the underlying reason may again be small variations in water content due to a tiny amount of condensation inside the sealed NMR capillary. Measurements by 1H PFG NMR for x ≈ 2.3 were also attempted, however, the T2 relaxation time was too short to apply feasible gradient durations.
is comparable to the effective Li+ diffusivity in samples with x ≈ 4 and 8. This proton motion is attributed to the diffusion of water molecules, since there is no evidence for the presence of protonic defects, as discussed below. Assuming this attribution is correct, the data indicate that Li+ ions and water molecules in the interlayer galleries have a comparable mobility. The diffusivity of water decreases by a somewhat larger factor (3–10×) than the Li+ diffusivity when the diffusion time is increased from 10 to 100 ms, which hints that grain boundaries are more blocking for water molecules than for Li+ ions. The data show more scatter compared to Li+; the underlying reason may again be small variations in water content due to a tiny amount of condensation inside the sealed NMR capillary. Measurements by 1H PFG NMR for x ≈ 2.3 were also attempted, however, the T2 relaxation time was too short to apply feasible gradient durations.
Magic angle spinning (MAS) 6Li NMR spectra acquired at various water contents and temperatures are shown in Fig. 5a. To facilitate comparisons, the spectra are normalized to have the same maximum intensity. The 6Li spectrum for x = 0 at room temperature can be fit by 5 signals. The strong signals at 2.2 and 1.2 ppm exhibit an intensity ratio of 0.8 and are nearly identical to those seen in previous work.35 The weak signals at 1.7 and −0.1 ppm can be attributed to the presence of ∼4% crystalline Li2SnS3.35 The signal at 0.8 ppm comprises ∼30% of the Li present and was not observed in the previous work. These observations are corroborated by a 119Sn MAS spectrum (Fig. S8b†), which shows three strong signals consistent with those previously observed for Li2Sn2S5; a weak (∼4%) signal consistent with Li2SnS3; and two broader signals which were not observed in the previous work and which comprise ∼40% of the Sn present.35 We assign these signals to a disordered Li–Sn–S side phase, reflecting the possible phase width and stacking disorder in this system. The presence of side phases adds some uncertainty to the interpretation of changes in overall water content; in principle, it is possible that the apparent phase widths at x = 2–4 and 8–10 are due to hydration of the side phases rather than Li2Sn2S5·xH2O. Otherwise the side phases are unlikely to affect the conclusions about Li2Sn2S5·xH2O in this work. One reason is that the PFG NMR data show the fast-conducting phase is a layered material; if the disordered side phase is not layered, then it is unlikely to be the fast conductor. As a note, the spectra acquired for x = 0 below room temperature required long measurements times and are less reliable due to possible drift.
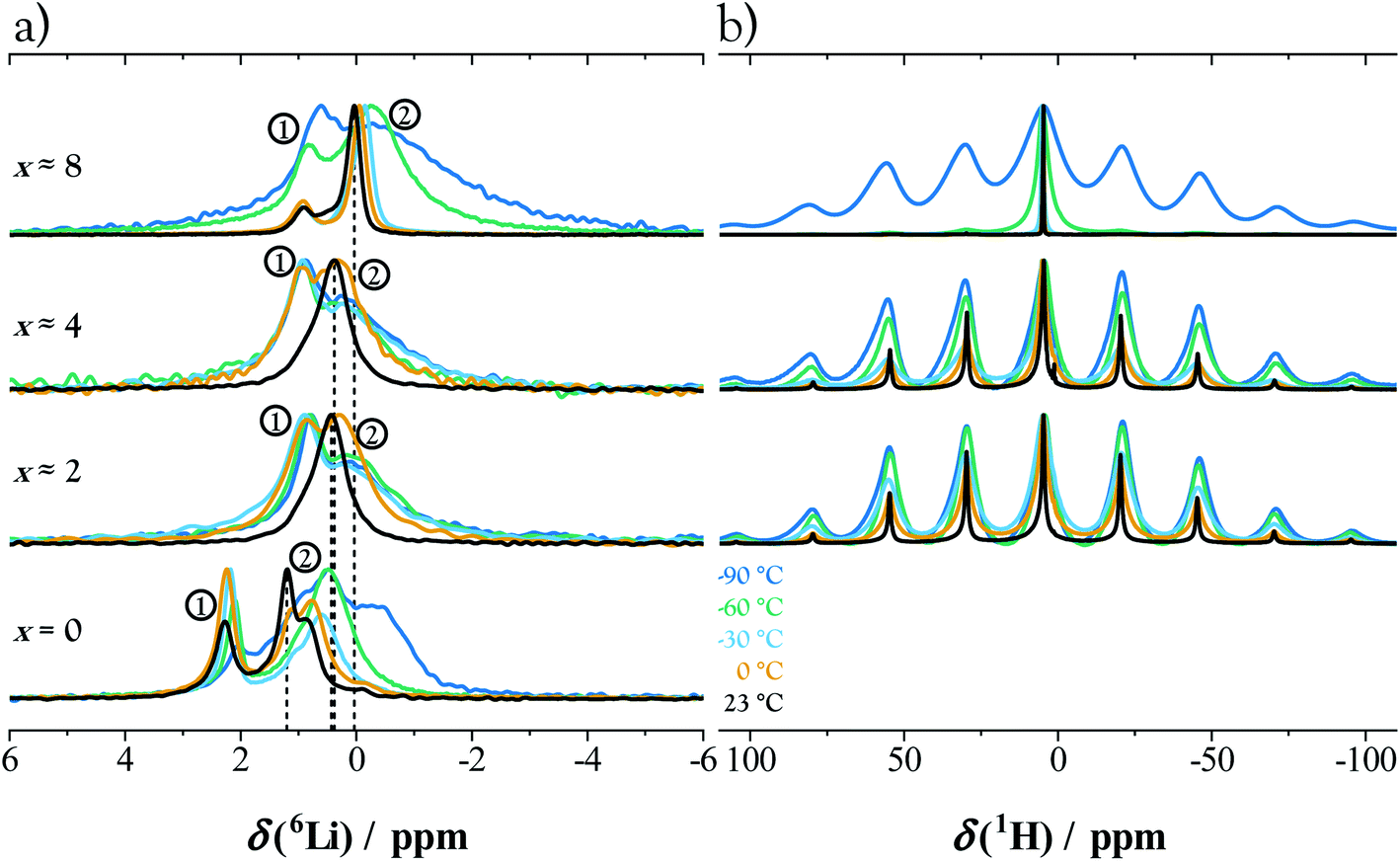 | ||
| Fig. 5 (a) 6Li and (b) 1H NMR measurements of Li2Sn2S5·xH2O for various water contents and temperatures. | ||
The spectra at x ≈ 2, x ≈ 3 (not shown), and x ≈ 4 are nearly identical, and they are clearly distinct from the spectra at x = 0 and x ≈ 8, consistent with the phase transitions seen by XRD between x = 0, 2–4, and 8. For simplicity, we fit all the 6Li spectra from the hydrated samples by two signals. The signals are labeled 1 and 2 in Fig. 5; example fits are shown in Fig. S8a;† and the full width at half maximum (FWHM) and chemical shift data are plotted in Fig. S9.† The FWHM of signal 1 remains approximately constant throughout, while signal 2 narrows as the temperature increases from −90 to 23 °C. The latter is a signature of motional narrowing.65 A likely explanation is that the Li+ ions on tin sites (Li⊥+) correspond to signal 1 and remain immobile, while the Li+ ions in the interlayer galleries (Li∥+) give rise to signal 2 and are increasingly mobile with increasing temperature or water content. Upon hydration from x ≈ 4 to 8, the chemical shift of signal 1 remains unchanged, while signal 2 shifts to ∼0 ppm, consistent with interlayer Li+ ions becoming solvated by the incorporated water. For comparison, fully solvated Li+ in liquid water shows a similar chemical shift, slightly below 0 ppm.66,67 Although we cannot exclude that the two signals are a convolution of multiple signals, the simple fitting approach used here leads to a reasonable physical interpretation.
The signals in the 1H spectra also narrow with increasing temperature, consistent with the presence of mobile protons (Fig. 5b). Indeed, the concurrent broadening of the 6Li and 1H signals suggests coupled short-range motion of Li+ and H2O under all hydrated conditions. Also, the 1H spectra at x ≈ 2 and 4 show spinning sidebands, consistent with a low diffusivity of water, while the sidebands disappear above −60 °C for x ≈ 8, suggesting a higher (more liquid-like) diffusivity of water.
To review, the MAS NMR results suggest: (i) for all x values there are Li+⊥ and Li+∥ present, but only the latter are mobile; (ii) the behavior in the range x ≈ 2–4 is similar; (iii) Li+ and H2O exhibit coupled short-range motion, with higher mobilities at x ≈ 8 than at x ≈ 4.
EIS: ion transport
The conductivity and dielectric properties of Li2Sn2S5·xH2O were characterized by impedance spectroscopy.68Fig. 6 shows representative spectra for three degrees of hydration. At all x, a depressed high frequency semicircle and a low-frequency blocking arc were observed. The latter is typical for blocking of ions at a metal electrode. For samples with x ≳ 4.0, cooling below 25 °C was required to observe the semicircle. The semicircle was fit by the simple equivalent circuit shown in Fig. 6. The resistance scales linearly with sample thickness (Fig. S10†), as expected for bulk conduction. The dielectric constant εr and conductivity σ were extracted using standard relations (see Experimental details). The εr values are unusually high, falling in the range 80–300 depending on temperature and humidity (Fig. S15†). The reason is unclear. For comparison, the dielectric constant of pure water in the kHz range is 80 at room temperature.69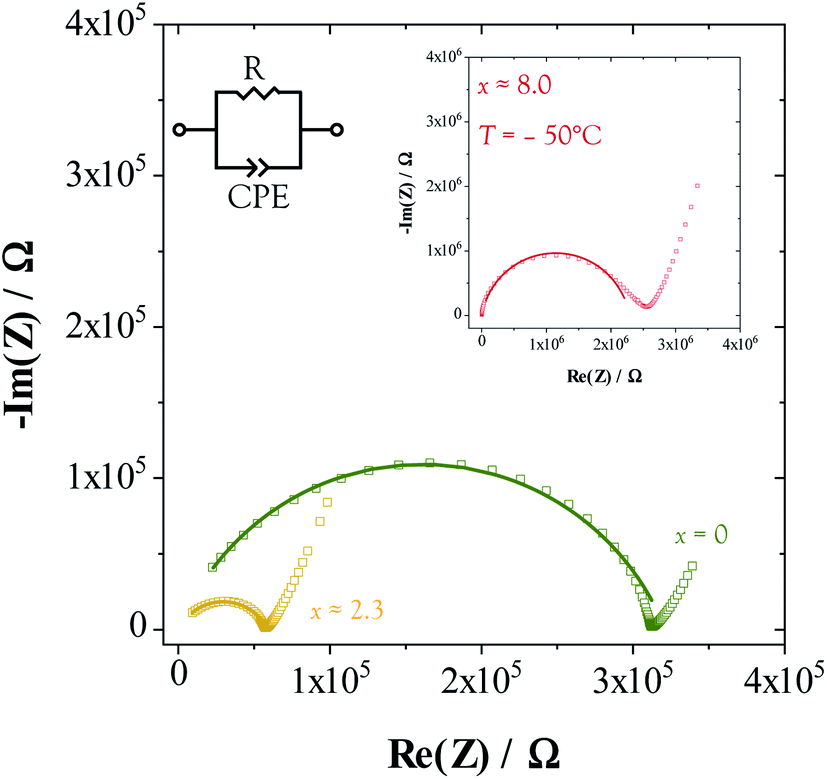 | ||
| Fig. 6 Typical impedance spectra measured from Li2Sn2S5·xH2O compacts at 30 °C (x = 0 and x ≈ 2.3) and −50 °C (x ≈ 8.0, inset). | ||
An apparent high dielectric constant seems to be a common feature in hydrates (cf. Li3InCl6·1.5H2O),30 which suggests that the presence of H2O molecules induces polarization effects. It can be ruled out that the capacitance is due to blocking grain boundaries; strong blocking behavior would show two distinct (non-overlapping) semicircles, but here only one is observed, even after cooling as low as −130 °C. The red-orange color of the material hardly changes upon hydration, which suggests that the bandgaps of the hydrated phases are near that measured previously32 for the anhydrous phase (1.9 eV).
Fig. 7 shows that the conductivity increases strongly and monotonically with increasing water content. At room temperature the increase is by more than three orders of magnitude, from 7 × 10−6 to 2 × 10−2 S cm−1 as x increases from 0 to 10.0. The activation energy (determined from the slope of σT) is 0.40, 0.57, 0.39, 0.34, and 0.24 eV for x ≈ 0, 2.3, 4.0, 8.0, and 10.0. The influence of porosity is judged to be minor due to adequate densification and the soft nature of the hydrated materials. Additional conductivity data showing reproducibility as well as heating and cooling steps are given in Fig. S11.†
 | ||
| Fig. 7 Conductivity values measured from Li2Sn2S5·xH2O by electrochemical impedance spectroscopy (EIS, open squares). Solid lines are linear fits. Also shown are conductivity values estimated from the 7Li PFG NMR results in Fig. 4 using the Nernst–Einstein equation (triangles), as well as values measured previously for a 1 M aqueous solution of LiCl (dashed line).58 | ||
For comparison, one can estimate the conductivity from the effective diffusivity obtained by PFG NMR using the Nernst–Einstein equation:
 | (2) |
A few final experiments are noteworthy. Below room temperature, the conductivity of Li2Sn2S5·8H2O was found to drop far more quickly than that of Li2Sn2S5, such that the conductivity trends for those phases cross at −60 °C (Fig. S12†). Thus, below −60 °C the presence of H2O is detrimental to ion transport. In addition, conductivity and electromotive force (EMF) experiments performed using Li and LiAl electrodes indicate that Li2Sn2S5 reacts in contact with Li to yield a composite material with a high overall electronic conductivity (Fig. S13†). Simultaneously, the apparent dielectric constant εr increases by a factor of ∼50 (Fig. S14†), suggesting that the electronic conductivity is dominated by pathways within the grain boundary network.
Discussion
(a) Overall interpretation
Reasonable quantitative agreement between the Li+ diffusivity and the total conductivity is obtained if all Li+ ions in the interlayer galleries are assumed to be mobile. The agreement suggests that the improved conductivity upon hydration is due almost entirely to increased Li+ mobility, while the concentration of mobile Li+ carriers remains roughly constant (decreasing slightly with increasing x due to volume expansion). Such behavior is reasonable, given the ability of water to promote ion motion. It is also chemically intuitive that the Li+ ions in the interlayer galleries would be hydrated by adjacent water molecules. The coupling offers a natural explanation for why Li+ and H+ exhibit comparable diffusivities in this system.Because the water diffusivity in Li2Sn2S5·xH2O strongly increases with increasing water content, water incorporation effectively accelerates during hydration and decelerates during dehydration. This phenomenon explains why dehydration is far more sluggish than hydration, and it suggests that the hydration process is diffusion-limited, at least for the ∼50 μm particle size used in this work.
The protonic conductivity is probably quite low, due to a lack of protonic charge carriers. Significant dissociation of the incorporated water is not observed in the MAS NMR data. An internal ion exchange process (whereby H+ ions exchange positions with Li+ in the layers, leaving behind excess OH− carriers in the interlayer galleries) is also unsupported by the data. By comparison, pure liquid H2O also shows a high water diffusivity and a low proton conductivity.58,70–72
In unconfined aqueous solutions, Li+ ions are hydrated by 4–5 H2O molecules.73 To provide the same coordination for the 1.5 interlayer Li+ ions per formula unit of Li2Sn2S5·xH2O, approximately 6–8 water molecules per formula unit are required. One might therefore predict that the Li+ transport rates in Li2Sn2S5·xH2O will approach “liquid-like” values when x > 6. The data are broadly consistent with this simple picture. For example, a 1 M aqueous solution of LiCl shows a total conductivity of 2.3 × 10−2 S cm−1 at room temperature (Fig. 7),58 which—since the cation transference number in liquids is usually around 1/3—implies a Li+ conductivity of 8 × 10−3 S cm−1. The second hydrate, Li2Sn2S5·8H2O, exhibits a virtually identical value of 9 × 10−3 S cm−1 at 25 °C. However, since the negatively-charged tin sulfide layers are immobile, the Li+ transference number in Li2Sn2S5·xH2O is probably unity. Thus, Li2Sn2S5·8H2O achieves a liquid-like conductivity with a solid-like Li+ transference number. Both of these features are beneficial for electrolyte applications.
Further hydration up to x ≈ 10 provides almost no additional conductivity benefit, which is consistent with the idea that the electrostatic influence of the tin sulfide sheets on the interlayer Li+ ions is already fully screened out at x ≈ 8. This point suggests more broadly that the transport rates in concentrated aqueous solutions represent an upper bound to what is achievable in hydrated layered solids such as Li2Sn2S5·xH2O. In this work the highest measured conductivity at 27 °C was 2 × 10−2 S cm−1 in Li2Sn2S5·10.2H2O, surpassing prominent oxide Li+ electrolytes such as Li7La3Zr2O12 (3 × 10−4 S cm−1)74 and on par with state-of-the-art sulfide electrolytes such as Li9.54Si1.74P1.44S11.7Cl0.3 (2.5 × 10−2 S cm−1).75
To recapitulate, the simple model discussed so far involves a nearly constant mobile Li+ concentration, a Li+ mobility which is promoted by increasing water content up to x ≈ 8, and H2O molecules which coordinate to the Li+ ions and show similar mobility. Further hydration above x ≈ 8 yields little or no additional benefit in conductivity. The Li+ migration enthalpy remains in the range 0.3–0.6 eV. Fig. 8 summarizes the basic picture: At x = 0 conductivity is poor because of electrostatic interactions (trapping as extreme case) with the tin sulfide layers (A). The interaction is weakened by increasing temperature or introducing water. A moderate water content (x ≈ 4) leads to efficient screening (B), while a high water content (x ≈ 8) enables liquid-like nano-ionic transport (C).
 | ||
| Fig. 8 Conductivity at 30 °C as a function of the average water content in Li2Sn2S5·xH2O. Dashed line is a guide for the eye. Schematics show the interlayer water (red/white) and Li∥+ ions (gray). | ||
This model explains the major trends at 25 °C and above. Below room temperature, some refinement may be needed. Aqueous solutions freeze slightly below 0 °C, and the formation of a rigid ice lattice causes the conductivity of dissolved ions to drop sharply by several orders of magnitude.76 In contrast, Li2Sn2S5·8H2O is solid at room temperature, and the ionic conductivity shows no first-order transition during cooling to −70 °C (Fig. S12†). Evidently the formation of an ice lattice is hindered by the water molecules being confined between the tin sulfide layers.77,78 Instead, during cooling the activation energy increases steadily for x ≈ 8. It is unclear if this change is due to a concentration or a mobility effect. One can speculate that “ice-like” clusters of water molecules gradually form upon cooling and associate with Li+ ions (reducing the mobile Li+ concentration) and/or change the potential landscape (causing the Li+ mobility to decrease more rapidly with decreasing temperature).
(b) Previous work revisited
The previous work from our groups35 on anhydrous Li2Sn2S5 must be reinterpreted in two respects:First, in the current work we consistently find that anhydrous Li2Sn2S5 exhibits a Li+ diffusivity below 10−9 cm2 s−1 at 25 °C. The higher value of 10−7 cm2 s−1 reported previously is obtained here only at hydration levels of x ≥ 8. The simplest and most likely explanation is that the PFG NMR measurements published previously were inadvertently conducted on hydrated samples. An alternative possibility is that the different synthesis protocols in the previous work led to structural differences which facilitate ion transport, such as fewer stacking faults or reduced amorphous content. Our numerous attempts to confirm this alternative were unsuccessful, but it cannot be entirely ruled out.
Second, anhydrous Li2Sn2S5 exhibits an ionic conductivity slightly below 10−5 S cm−1 at 25 °C. The value of 10−4 S cm−1 reported previously using LiAl electrodes appears to correspond not to ion conduction, but rather to electron conduction that arises when Li2Sn2S5 is reduced by LiAl and electronic pathways form in the microstructure (Fig. S13 and S14†).
(c) Outlook
An interesting avenue for future work is to incorporate other solvents besides water into Li2Sn2S5, to promote Li+ conductivity while making the material compatible with non-aqueous conditions. Another promising direction is to investigate hydrated layered compounds based on higher-valent cations such as Mg2+ or Zn2+. The anhydrous forms of such materials will likely show quite poor cation conductivity due to strong coulombic interactions with the immobile lattice. However, the hydrated forms may achieve a liquid-like ionic conductivity while remaining solid, as in the present work, due to the ability of water to promote ion motion. A few examples of this approach have already been demonstrated.9,79,80Although not attempted in this work, it should be possible to use Li2Sn2S5·xH2O as a battery electrolyte. At x ≈ 8 the Li+ conductivity is competitive with that of liquid electrolytes, and the material is soft enough that simple pressing may be sufficient to assemble composite electrodes with good interfacial contact at the electrode–electrolyte interfaces. As with any solid-state battery, the key challenge is to maintain good interfacial contact during cycling despite interfacial reactivity and volume changes. The softness of Li2Sn2S5·xH2O at high water contents could plausibly help in this regard. The practical voltage window for battery cycling may extend beyond the limits associated with splitting of liquid water, since Li2Sn2S5·8H2O is conceptually similar to water-in-salt electrolytes,81 and the geometric confinement of H2O might even widen the window further. The fact that the Li+ transference number is probably near unity is also beneficial for avoiding concentration polarization effects at high current densities. However, some concerns are worth noting. Despite the possible suppression of water splitting, the electrochemical stability window of Li2Sn2S5·xH2O is narrow, as seen for other sulfides.82 In particular, contact with low-voltage anodes may be impractical due to Sn4+ reduction and the formation of a mixed-conducting passivation layer, similar to the impact of Ti4+ reduction in well-known electrolyte materials such as LiLaTiO3 or Li1+xAlxTi2−x(PO4)3.83,84 The addition of water could mitigate this issue, because the typical hydrolysis products of lithium (LiOH, Li2O, LiH) are electronically blocking,85 but it seems risky to rely on this point.86,87 Another concern is that the application of pressure—a standard method for restoring interfacial contact in solid-state batteries—may squeeze water out of Li2Sn2S5·8H2O and thereby reduce the Li+ conductivity. The surrounding water partial pressure may also need to be controlled to avoid deliquescence. Finally, high current densities may deplete water on one side of the electrolyte due to the coupled motion of Li+ and H2O; it seems reasonable to expect that this phenomenon is a general feature of hydrated layered materials.
Conclusions
Upon exposure to humidity, water intercalation into the layered structure of Li2Sn2S5 is facile. The resulting material Li2Sn2S5·xH2O shows a water content x that varies from 0 to about 10 depending on the humidity and temperature. Both Li+ and H2O are transported within the material predominantly by 2D diffusion in the interlayer galleries. The transport rates increase strongly with increasing water content up to about x ≈ 8, which corresponds approximately to a full hydration shell of 4–5 water molecules for each interlayer Li+ ion. At x ≈ 8 the material remains a soft solid, but the Li+ diffusivity and conductivity reach liquid-like values of 5 × 10−7 cm2 s−1 and 10−2 S cm−1. Anhydrous Li2Sn2S5 consistently showed far lower transport rates than those reported in a previous work by our groups. The discrepancy is most likely due to inadvertent hydration of the material in the previous work, but an alternative explanation based on structural differences cannot be ruled out. In any case, Li2Sn2S5·xH2O offers a case study of the transition between a hydrated solid electrolyte and a confined liquid electrolyte, which is both scientifically interesting and potentially useful in applications.Experimental
General
Anhydrous samples were prepared, handled, and measured under dry inert gas (Ar or N2 with <1 ppm H2O) or vacuum. Hydrated samples were sealed in containers with small dead volumes to minimize water loss. The water content, x, was assessed gravimetrically; for a two-phase mixture, the reported value of x is an average over both phases. Some transfer steps required exposing hydrated samples to dry inert gas for a short time, on the order of minutes. The water loss during these transfer steps was estimated by weighing representative samples before and after exposure. The cumulative decrease in x from these transfers was estimated to be 0.5 or less and was neglected. Although Li2Sn2S5·xH2O appears to be stable in ambient air, the water content is sensitive to water partial pressure (pH2O), so air exposure was avoided except where indicated. See the ESI† for more details.Sample preparation
Anhydrous Li2Sn2S5 was synthesized by solid state reaction of Li2S (99.9%, Alfa Aesar) and SnS2 at 650 °C. Hydrated Li2Sn2S5·xH2O was prepared using either a TGA or saturated salt solutions. Both materials were compacted by uniaxial pressing for impedance measurements; anhydrous Li2Sn2S5 with 5–10 kbar and various pressure values for Li2Sn2S5·xH2O depending on x. Various heat treatments were attempted to increase the density of anhydrous Li2Sn2S5 further. See ESI† for more details.Thermogravimetric analysis (TGA)
The equilibrium water content in Li2Sn2S5·xH2O was measured by installing anhydrous powder samples (70–600 mg) inside a quartz crucible in a TGA system. The sample was exposed to ambient air for approx. 30 s during transfers. The TGA was a Netzsch STA 449 instrument with a protective N2 gas flow (<10 ppm H2O including trace leaks, 60 ml min−1). To remove water absorbed during sample installation, a pre-anneal was typically conducted under flowing N2. The sample mass was measured after a typical dwell time of 1–2 h under humid N2 at various temperatures in the range 27–100 °C and pH2O in the range 1.4–17.1 mbar (0.4–48.2% relative humidity). The humidity was set by mixing dry and humidified N2 in different ratios (0–60 ml min−1 dry or wet N2). The N2 gas was humidified by bubbling dry N2 gas through deionized water in a double-walled glass container, which was temperature controlled using a thermostat in the range 5–18 °C. The flow rates were sufficiently low that saturation could be assumed, such that the humidity is given by the saturation pressure of water at the thermostat set point. The exhaust gas of the TGA was analyzed by a Balzers Prisma quadrupole mass spectrometer. A buoyancy correction was performed by subtraction of mass changes at measurement temperatures from data obtained of an empty crucible in a separate measurement. The recorded mass changes of the empty crucible were fit with a polynomial and the extrapolation line was used for the buoyancy correction.X-ray diffraction (XRD)
Powder XRD measurements were acquired using a STOE StadiP diffractometer (curved Ge (111) monochromator, DECTRIS Mythen2R 1K detector, Debye–Scherrer geometry, Mo-Kα1, wavelength 0.7093 Å). Powder samples with lower x values (0–4.0) were packed in borosilicate capillaries (Hilgenberg, 0.3 mm diameter) and flame sealed under argon. Due to their soft and sticky consistency, samples at higher x values (4.0–10.0) were prepared in Kapton tubes of 0.8 mm diameter. These data were analyzed using WinXPow (STOE). Phase analysis checks were performed on a second setup, as described in the ESI.†Pulsed-field-gradient nuclear magnetic resonance (PFG NMR)
Li2Sn2S5·xH2O powder was packed at one end of an NMR glass capillary (Deutero, D400) with ground joints, and a solid glass rod that matched the capillary inner radius was inserted to reduce the dead volume. The capillary was then sealed under slightly reduced pressure (∼0.5 bar) of inert gas. Water loss during the sealing was negligible. Capillaries prepared in this way were installed in the NMR instrument (Bruker Avance III 400) equipped with either a diff60 gradient probe (max. 2900 G cm−1, specialized RF inserts for 1H and 7Li) or a double resonance broad band probe (max. 1750 G cm−1, BBO 400 MHz W1/S2 5 mm with Z-gradient) with tuneable frequency for different nuclei. The sample end of the capillary was heated stepwise between 25–180 °C. After a ∼0.5 h effective dwell time at each temperature to allow for equilibration, NMR measurements were acquired. At all temperatures the intent was to keep the water content in the powder constant. However, some amount of condensation was visible at the protruding cold (non-sample) end of the capillary, suggesting the samples underwent a small (and reversible) amount of water loss during heating. For the x ≈ 8.0 sample, the capillary sealing was modified to essentially eliminate the dead volume of gas, and no condensation was observed. Data were acquired using a stimulated echo sequence with spoiler gradients. Various diffusion times in the range 10–100 ms were used, with effective gradient durations of 1–3 ms. Remagnetization times were chosen to be 4 to 5 times higher than the spin–lattice relaxation time, T1. The measured echo-signal attenuation peaks were phase corrected, and the integrated areas were used to extract the diffusivities. The main analysis approach assumed anisotropic diffusion with negligible out-of-plane diffusivity, so the procedure given in ref. 64 was used to calculate the diffusivity.Solid state nuclear magnetic resonance (ssNMR)
Powder samples were filled in a Pyrex magic angle spinning (MAS) rotor insert (Rototec) under argon. The insert was custom fitted with a ground glass joint for transfers. The sample bottom part of the insert was immersed in liquid nitrogen, and under slight static vacuum (∼0.5 bar) the sample was flame sealed. Spectra were acquired on a Bruker Avance III 400 MHz instrument (magnetic field of 9.4 T) at Larmor frequencies of 400 MHz (1H), 155.5 MHz (7Li), 149.15 MHz (119Sn) and 58.8 MHz (6Li), using a Bruker BL4 double resonance MAS probe and 4 mm OD ZrO2 spinners (10 kHz if not stated otherwise). The MAS spectra for all nuclei were acquired with a simple Bloch Decay experiment (i.e., pulse-acquisition), and acquisition delays sufficiently long for a complete magnetization recovery. The 7Li experiments were performed using central transition selective 90° pulses (the non-selective liquid 90° pulses were scaled by a factor of (I + 0.5)−1 = 0.5).88,897Li and 6Li variable temperature spin–lattice T1 relaxation times measurements were performed both on spinning and non-spinning samples using saturation-recovery method. Experiments with static samples were performed in a Bruker static double resonance PE400 probe with a horizontal 5 mm coil. Between 8 and 64 scans were commonly averaged for a good signal-to-noise ratio. The temperature in the probes was regulated with a Bruker BVT3000 temperature controller. Actual temperature of the sample was calibrated on 207Pb signal of powdered Pb(NO3)2.90 Chemical shifts were referenced externally relative to tetramethylsilane (1H, δiso = 0.0 ppm), to tetramethyltin (119Sn, δiso = 0.0 ppm), using SnO2 as a secondary standard (−603 ppm), and to 1 M LiCl (6,7Li, δiso = 0.0 ppm).91Electrochemical impedance spectroscopy (EIS)
Depending on x, samples were contacted either by sputtering ruthenium electrodes or with stainless steel rods. For x = 0, measurements were conducted under inert gas flow, and for x > 0, a closed set-up was used. In either case the impedance was measured with a Novocontrol Alpha-A analyzer (2-wire measurement, 106 to 10−1 Hz, 0.1 V amplitude).When necessary, the stray impedance of the measurement setup was measured separately (by short-circuiting the electrodes at the respective temperatures) and pointwise subtracted out. Impedance spectra were fit using either Zview (Scribner, Version 3.5.c) or RelaxIS3 (rhd instruments) in the high-frequency range by an equivalent circuit consisting of a resistor and constant phase element (CPE) in parallel. The obtained values for resistance and capacitance when including the low-frequency blocking feature by an additional CPE and having the dielectric capacitance in parallel were comparable. The conductivity σ and dielectric constant εr were extracted using the standard relations σ = L/(RA) and εr = CeffL/(ε0A), where R is resistance, L is sample thickness, A is cross-sectional area, ε0 is the electrical permittivity of free space, and Ceff is the effective capacitance calculated from Ceff = Qn−1R(n−1−1), where Q and n are the magnitude and exponent of the CPE.68 See ESI† for more details.
Author contributions
All authors contributed to discussing content and writing the manuscript. B. L. and J. M. conceived the work. R. U., B. L., and J. M. supervised the work. M. J., C. S., A. M., I. M., and R. U. performed experiments. M. J. and R. U. coordinated and edited the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful for support from Rotraut Merkle and Klaus-Dieter Kreuer (TGA and discussions); Günter Majer (NMR); Tanja Holzmann, Leslie Schoop, Mazhar Ali, and Robert Cava (initial measurements and discussions); Helga Hoier (XRD); Uwe Traub, Udo Klock, and Florian Kaiser (technical support); Jürgen Nuss (SXRD); and Bernhard Fenk (SEM). Financial support was provided by the Deutsche Forschungsgemeinschaft (DFG) via the Cluster of Excellence e-conversion (Grant No. EXC2089) and the German Federal Ministry of Research and Education (BMBF), project 03XP0177B (FestBatt). Open Access funding provided by the Max Planck Society.References
- K. D. Kreuer, Proton-Conducting Oxides, Annu. Rev. Mater. Res., 2003, 33(1), 333–359, DOI:10.1146/annurev.matsci.33.022802.091825.
- J. G. V. Bhat, A2Ln2Ti3O10 (A = K or Rb; Ln = La or Rare Earth): A New Series of Layered Perovskites Exhibiting Ion Exchanged, Inorg. Chem., 1987, 26(26), 4299–4301, DOI:10.1021/ic00273a001.
- V. Øygarden, H. Fjellvåg, M. H. Sørby and A. O. Sjåstad, Crystal Structure of LaSr3Fe3O8(OH)2·xH2O, Inorg. Chem., 2016, 55(15), 7630–7636, DOI:10.1021/acs.inorgchem.6b01085.
- M. Spaeth, K. D. Kreuer, J. Maier and C. Cramer, Giant Haven Ratio for Proton Transport in Sodium Hydroxide, J. Solid State Chem., 1999, 148(1), 169–177, DOI:10.1006/jssc.1999.8495.
- J. Hougardy, W. E. E. Stone and J. J. Fripiat, NMR Study of Adsorbed Water. I. Molecular Orientation and Protonic Motions in the Two-Layer Hydrate of a Na Vermiculite, J. Chem. Phys., 1976, 64(9), 3840–3851, DOI:10.1063/1.432702.
- M. Suzuki, N. Wada, D. R. Hines and M. S. Whittingham, Hydration States and Phase Transitions in Vermiculite Intercalation Compounds, Phys. Rev. B, 1987, 36(5), 2844–2851, DOI:10.1103/PhysRevB.36.2844.
- M. Stanley Whittingham, Transport Properties of the Mineral Vermiculite, Solid State Ionics, 1989, 32/33, 344–349, DOI:10.1016/0167-2738(89)90239-7.
- H. Maraqah, J. Li and M. S. Whittingham, The Ionic Conductivity of Hydrogen Vermiculite, J. Electrochem. Soc., 1991, 138(11), L61–L63, DOI:10.1149/1.2085449.
- M. S. Whittingham, Hydrogen Motion in Oxides: From Insulators to Bronzes, Solid State Ionics, 2004, 168(3–4), 255–263, DOI:10.1016/j.ssi.2003.08.056.
- S. H. Sheffield and A. T. Howe, High Proton Conductivity in Pressed Pellets of H-Montmorillonite, HAl-Montmorillonite and HAlFe-Montmorillonite Clays, Mater. Res. Bull., 1979, 14(7), 929–935, DOI:10.1016/0025-5408(79)90159-4.
- R. C. T. Slade, J. Barker, P. R. Hirst, T. K. Halstead and P. I. Reid, Conduction and Diffusion in Exchanged Montmorillonite Clays, Solid State Commun., 1987, 24, 289–295, DOI:10.1016/0167-2738(87)90135-4.
- T. Kawada, H. Yokokawa and M. Dokiya, Ionic Conductivity of Montmorillonite/Alkali Salt Mixtures, Solid State Ionics, 1988, 28–30, 210–213, DOI:10.1016/S0167-2738(88)80035-3.
- M. S. Whittingham, Sodium Ion Conduction in Single Crystal Vermiculite, Solid State Ionics, 1987, 25, 295–300, DOI:10.1016/0167-2738(87)90193-7.
- K. D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Transport in Proton Conductors for Fuel-Cell Applications: Simulations, Elementary Reactions, and Phenomenology, Chem. Rev., 2004, 104(10), 4637–4678, DOI:10.1021/cr020715f.
- A. Kusoglu and A. Z. Weber, New Insights into Perfluorinated Sulfonic-Acid Ionomers, Chem. Rev., 2017, 117(3), 987–1104, DOI:10.1021/acs.chemrev.6b00159.
- C. Heitner-Wirguin, Recent Advances in Perfluorinated Ionomer Membranes: Structure, Properties and Applications, J. Membr. Sci., 1996, 120, 1–33, DOI:10.1016/0376-7388(96)00155-X.
- K. D. Kreuer, A. Wohlfarth, C. C. De Araujo, A. Fuchs and J. Maier, Single Alkaline-Ion (Li+, Na+) Conductors by Ion Exchange of Proton-Conducting Ionomers and Polyelectrolytes, ChemPhysChem, 2011, 12(14), 2558–2560, DOI:10.1002/cphc.201100506.
- M. J. Manos and M. G. Kanatzidis, Metal Sulfide Ion Exchangers: Superior Sorbents for the Capture of Toxic and Nuclear Waste-Related Metal Ions, Chem. Sci., 2016, 7(8), 4804–4824, 10.1039/c6sc01039c.
- J. Zhao, S. M. Islam, O. Y. Kontsevoi, G. Tan, C. C. Stoumpos, H. Chen, R. K. Li and M. G. Kanatzidis, The Two-Dimensional AxCdxBi4−xQ6 (A = K, Rb, Cs; Q = S, Se): Direct Bandgap Semiconductors and Ion-Exchange Materials, J. Am. Chem. Soc., 2017, 139(20), 6978–6987, DOI:10.1021/jacs.7b02243.
- R. Wang, H. Chen, Y. Xiao, I. Hadar, K. Bu, X. Zhang, J. Pan, Y. Gu, Z. Guo, F. Huang and M. G. Kanatzidis, Kx[Bi4−xMnxS6], Design of a Highly Selective Ion Exchange Material and Direct Gap 2D Semiconductor, J. Am. Chem. Soc., 2019, 141(42), 16903–16914, DOI:10.1021/jacs.9b08674.
- A. R. Sotiles, L. M. Baika, M. T. Grassi and F. Wypych, Cation Exchange Reactions in Layered Double Hydroxides Intercalated with Sulfate and Alkaline Cations (A(H2O)6)[M2+6Al3(OH)18(SO4)2]·6H2O (M2+ = Mn, Mg, Zn; A+ = Li, Na, K), J. Am. Chem. Soc., 2019, 141(1), 531–540, DOI:10.1021/jacs.8b11389.
- N. Li, L. Zhang, Y. Chen, M. Fang, J. Zhang and H. Wang, Highly Efficient, Irreversible and Selective Ion Exchange Property of Layered Titanate Nanostructures, Adv. Funct. Mater., 2012, 22(4), 835–841, DOI:10.1002/adfm.201102272.
- M. Doyle, L. Wang, Z. Yang and S. K. Choi, Polymer Electrolytes Based on Ionomeric Copolymers of Ethylene with Fluorosulfonate Functionalized Monomers, J. Electrochem. Soc., 2003, 150(11), D185, DOI:10.1149/1.1610467.
- P. Hartwig and W. Weppner, Ionic Conductivities of Lithium-Halide-Based Quaternary Compounds, Solid State Ionics, 1981, 3/4, 249–254, DOI:10.1016/0167-2738(81)90092-8.
- F. W. Poulsen, Ionic Conductivity of Solid Lithium Iodide and Its Monohydrate, Solid State Ionics, 1981, 2(1), 53–57, DOI:10.1016/0167-2738(81)90020-5.
- F. W. Poulsen, Ionic Conductivity of Solid and Molten Lithiumthiocyanate and Its Hydrate, Acta Chem. Scand., Ser. A, 1985, 39(4), 290–292, DOI:10.3891/acta.chem.scand.39a-0290.
- G. Schwering, A. Hönnerscheid, L. Van Wüllen and M. Jansen, High Lithium Ionic Conductivity in the Lithium Halide Hydrates Li3−n(OHn)Cl (0.83 ≤ n ≤ 2) and Li3−n(OHn)Br (1 ≤ n ≤ 2) at Ambient Temperatures, ChemPhysChem, 2003, 4(4), 343–348, DOI:10.1002/cphc.200390060.
- A. Sagua, A. Rivera, C. León, J. Santamaría, J. Sanz and E. Morán, High Ionic Conductivity of Hydrated Li0.5FeOCl, Solid State Ionics, 2006, 177(11–12), 1099–1104, DOI:10.1016/j.ssi.2006.03.031.
- A. Takano, I. Oikawa, A. Kamegawa and H. Takamura, Enhancement of the Lithium-Ion Conductivity of LiBH4 by Hydration, Solid State Ionics, 2016, 285, 47–50, DOI:10.1016/j.ssi.2015.06.004.
- R. L. Sacci, T. H. Bennett, A. R. Drews, V. Anandan, M. J. Kirkham, L. L. Daemen and J. Nanda, Phase Evolution during Lithium-Indium Halide Superionic Conductor Dehydration, J. Mater. Chem. A, 2021, 9(2), 990–996, 10.1039/d0ta10012a.
- A. Adhikary, H. Yaghoobnejad Asl, P. Sandineni, S. Balijapelly, S. Mohapatra, S. Khatua, S. Konar, N. Gerasimchuk, A. V. Chernatynskiy and A. Choudhury, Unusual Atmospheric Water Trapping and Water Induced Reversible Restacking of 2D Gallium Sulfide Layers in NaGaS2 Formed by Supertetrahedral Building Unit, Chem. Mater., 2020, 32(13), 5589–5603, DOI:10.1021/acs.chemmater.0c00836.
- A. Kuhn, T. Holzmann, J. Nuss and B. V. Lotsch, A Facile Wet Chemistry Approach towards Unilamellar Tin Sulfide Nanosheets from Li4xSn1−xS2 Solid Solutions, J. Mater. Chem. A, 2014, 2(17), 6100–6106, 10.1039/C3TA14190J.
- K. Szendrei-Temesi, O. Sanchez-Sobrado, S. B. Betzler, K. M. Durner, T. Holzmann and B. V. Lotsch, Lithium Tin Sulfide—A High-Refractive-Index 2D Material for Humidity-Responsive Photonic Crystals, Adv. Funct. Mater., 2018, 28(14), 1–10, DOI:10.1002/adfm.201705740.
- A.-K. Hatz, I. Moudrakovski, S. Bette, M. Terban, M. Etter, M. Joos, N. Vargas-Barbosa, R. E. Dinnebier and B. V. Lotsch, Fast Water-Assisted Lithium Ion Conduction in Restacked Lithium Tin Sulfide Nanosheets, submitted.
- T. Holzmann, L. M. Schoop, M. N. Ali, I. Moudrakovski, G. Gregori, J. Maier, R. J. Cava and B. V. Lotsch, Li0.6[Li0.2Sn0.8S2] – a Layered Lithium Superionic Conductor, Energy Environ. Sci., 2016, 9(8), 2578–2585, 10.1039/C6EE00633G.
- T. Kaib, S. Haddadpour, M. Kapitein, P. Bron, C. Schröder, H. Eckert, B. Roling and S. Dehnen, New Lithium Chalcogenidotetrelates, LiChT: Synthesis and Characterization of the Li+-Conducting Tetralithium Ortho-Sulfidostannate Li4SnS4, Chem. Mater., 2012, 24(11), 2211–2219, DOI:10.1021/cm3011315.
- J. A. Brant, D. M. Massi, N. A. W. Holzwarth, J. H. Macneil, A. P. Douvalis, T. Bakas, S. W. Martin, M. D. Gross and J. A. Aitken, Fast Lithium Ion Conduction in Li2SnS3: Synthesis, Physicochemical Characterization, and Electronic Structure, Chem. Mater., 2015, 27(1), 189–196, DOI:10.1021/cm5037524.
- K. H. Park, D. Y. Oh, Y. E. Choi, Y. J. Nam, L. Han, J. Y. Kim, H. Xin, F. Lin, S. M. Oh and Y. S. Jung, Solution-Processable Glass LiI-Li4SnS4 Superionic Conductors for All-Solid-State Li-Ion Batteries, Adv. Mater., 2016, 28(9), 1874–1883, DOI:10.1002/adma.201505008.
- H. Kwak, K. H. Park, D. Han, K. W. Nam, H. Kim and Y. S. Jung, Li+ Conduction in Air-Stable Sb-Substituted Li4SnS4 for All-Solid-State Li-Ion Batteries, J. Power Sources, 2020, 446, 227338, DOI:10.1016/j.jpowsour.2019.227338.
- J. H. MacNeil, D. M. Massi, J.-H. Zhang, K. A. Rosmus, C. D. Brunetta, T. A. Gentile and J. A. Aitken, Synthesis, Structure, Physicochemical Characterization and Electronic Structure of Thio-Lithium Super Ionic Conductors, Li4GeS4 and Li4SnS4, J. Alloys Compd., 2014, 586, 736–744, DOI:10.1016/j.jallcom.2013.10.011.
- G. Sahu, Z. Lin, J. Li, Z. Liu, N. Dudney and C. Liang, Air-Stable, High-Conduction Solid Electrolytes of Arsenic-Substituted Li4SnS4, Energy Environ. Sci., 2014, 7(3), 1053–1058, 10.1039/c3ee43357a.
- Y. E. Choi, K. H. Park, D. H. Kim, D. Y. Oh, H. R. Kwak, Y. G. Lee and Y. S. Jung, Coatable Li4SnS4 Solid Electrolytes Prepared from Aqueous Solutions for All-Solid-State Lithium-Ion Batteries, ChemSusChem, 2017, 10(12), 2605–2611, DOI:10.1002/cssc.201700409.
- B. Radhakrishnan and S. P. Ong, Aqueous Stability of Alkali Superionic Conductors from First-Principles Calculations, Front. Energy Res., 2016, 4(16), 1–12, DOI:10.3389/fenrg.2016.00016.
- S. Wenzel, S. Randau, T. Leichtweiß, D. A. Weber, J. Sann, W. G. Zeier and J. Janek, Direct Observation of the Interfacial Instability of the Fast Ionic Conductor Li10GeP2S12 at the Lithium Metal Anode, Chem. Mater., 2016, 28(7), 2400–2407, DOI:10.1021/acs.chemmater.6b00610.
- Z. D. Hood, C. Kates, M. Kirkham, S. Adhikari, C. Liang and N. A. W. Holzwarth, Structural and Electrolyte Properties of Li4P2S6, Solid State Ionics, 2016, 284, 61–70, DOI:10.1016/j.ssi.2015.10.015.
- J. Lau, R. H. DeBlock, D. M. Butts, D. S. Ashby, C. S. Choi and B. S. Dunn, Sulfide Solid Electrolytes for Lithium Battery Applications, Adv. Energy Mater., 2018, 8(27), 1–24, DOI:10.1002/aenm.201800933.
- K. H. Park, Q. Bai, D. H. Kim, D. Y. Oh, Y. Zhu, Y. Mo and Y. S. Jung, Design Strategies, Practical Considerations, and New Solution Processes of Sulfide Solid Electrolytes for All-Solid-State Batteries, Adv. Energy Mater., 2018, 8(18), 1–24, DOI:10.1002/aenm.201800035.
- Ö. U. Kudu, T. Famprikis, B. Fleutot, M. D. Braida, T. Le Mercier, M. S. Islam and C. Masquelier, A Review of Structural Properties and Synthesis Methods of Solid Electrolyte Materials in the Li2S–P2S5 Binary System, J. Power Sources, 2018, 407, 31–43, DOI:10.1016/j.jpowsour.2018.10.037.
- S. Banerjee, X. Zhang and L. W. Wang, Motif-Based Design of an Oxysulfide Class of Lithium Superionic Conductors: Toward Improved Stability and Record-High Li-Ion Conductivity, Chem. Mater., 2019, 31(18), 7265–7276, DOI:10.1021/acs.chemmater.9b01639.
- H. Muramatsu, A. Hayashi, T. Ohtomo, S. Hama and M. Tatsumisago, Structural Change of Li2S-P2S5 Sulfide Solid Electrolytes in the Atmosphere, Solid State Ionics, 2011, 182(1), 116–119, DOI:10.1016/j.ssi.2010.10.013.
- S. P. Ong, Y. Mo, W. D. Richards, L. Miara, H. S. Lee and G. Ceder, Phase Stability, Electrochemical Stability and Ionic Conductivity of the Li10±1MP2X12 (M = Ge, Si, Sn, Al or P, and X = O, S or Se) Family of Superionic Conductors, Energy Environ. Sci., 2013, 6(1), 148–156, 10.1039/c2ee23355j.
- T. Ohtomo, A. Hayashi, M. Tatsumisago and K. Kawamoto, Suppression of H2S Gas Generation from the 75Li2S·25P2S5Glass Electrolyte by Additives, J. Mater. Sci., 2013, 48(11), 4137–4142, DOI:10.1007/s10853-013-7226-8.
- T. Ohtomo, A. Hayashi, M. Tatsumisago and K. Kawamoto, Characteristics of the Li2O–Li2S–P2S5 Glasses Synthesized by the Two-Step Mechanical Milling, J. Non-Cryst. Solids, 2013, 364(1), 57–61, DOI:10.1016/j.jnoncrysol.2012.12.044.
- D. Y. Oh, Y. J. Nam, K. H. Park, S. H. Jung, S. J. Cho, Y. K. Kim, Y. G. Lee, S. Y. Lee and Y. S. Jung, Excellent Compatibility of Solvate Ionic Liquids with Sulfide Solid Electrolytes: Toward Favorable Ionic Contacts in Bulk-Type All-Solid-State Lithium-Ion Batteries, Adv. Energy Mater., 2015, 5(22), 1–7, DOI:10.1002/aenm.201500865.
- H. M. Chen, C. Maohua and S. Adams, Stability and Ionic Mobility in Argyrodite-Related Lithium-Ion Solid Electrolytes, Phys. Chem. Chem. Phys., 2015, 17(25), 16494–16506, 10.1039/c5cp01841b.
- Y. S. Jung, D. Y. Oh, Y. J. Nam and K. H. Park, Issues and Challenges for Bulk-Type All-Solid-State Rechargeable Lithium Batteries Using Sulfide Solid Electrolytes, Isr. J. Chem., 2015, 55(5), 472–485, DOI:10.1002/ijch.201400112.
- T. Jiang and G. A. Ozin, New Directions in Tin Sulfide Materials Chemistry, J. Mater. Chem., 1998, 8(5), 1099–1108, 10.1039/a709054d.
- K. Tanaka and R. Tamamushi, A Physico-Chemical Study of Concentrated Aqueous Solutions of Lithium Chloride, Z. Naturforsch., A: Phys. Sci., 1991, 46, 141–147, DOI:10.1515/zna-1991-1-22.
- R. Zohourian, R. Merkle, G. Raimondi and J. Maier, Mixed-Conducting Perovskites as Cathode Materials for Protonic Ceramic Fuel Cells: Understanding the Trends in Proton Uptake, Adv. Funct. Mater., 2018, 28(35), 1–10, DOI:10.1002/adfm.201801241.
- S. Bette, R. E. Dinnebier and D. Freyer, Structure Solution and Refinement of Stacking-Faulted NiCl(OH), J. Appl. Crystallogr., 2015, 48(1936), 1706–1718, DOI:10.1107/S1600576715017719.
- L. Diehl, S. Bette, F. Pielnhofer, S. Betzler, I. Moudrakovski, G. A. Ozin, R. Dinnebier and B. V. Lotsch, Structure-Directing Lone Pairs: Synthesis and Structural Characterization of SnTiO3, Chem. Mater., 2018, 30(24), 8932–8938, DOI:10.1021/acs.chemmater.8b04261.
- Y. Zhang and Z. Xu, Atomic Radii of Noble Gas Elements in Condensed Phases, Am. Mineral., 1995, 80(7–8), 670–675, DOI:10.2138/am-1995-7-803.
- W. S. Price, Pulsed-Field Gradient Nuclear Magnetic Resonance as a Tool for Studying Translational Diffusion: Part 1. Basic Theory, Concepts Magn. Reson., 1997, 9(5), 299–336, DOI:10.1002/(sici)1099-0534(1997)9:5<299::aid-cmr2>3.3.co;2-2.
- F. Kimmerle, G. Majer, U. Kaess, A. J. Maeland, M. S. Conradi and A. F. McDowell, NMR Studies of Hydrogen Diffusion in ZrBe2H1.4, J. Alloys Compd., 1998, 264(1–2), 63–70, DOI:10.1016/S0925-8388(97)00273-9.
- A. Abragam, The Principles of Nuclear Magnetism, Oxford University Press, 1961 Search PubMed.
- C. Hall, G. L. Haller and R. E. Richards, N.M.R. Studies of Lithium Chloride and Lithium Bromide Solutions in Methanol-Water Mixtures, Mol. Phys., 1969, 16(4), 377–394, DOI:10.1080/00268976900100431.
- T. Umecky, T. Takamuku, T. Matsumoto, E. Kawai, M. Takagi and T. Funazukuri, Effects of Dissolved Water on Li+ Solvation in 1-Ethyl-3-Methylimidazolium Bis(Trifluoromethanesulfonyl)Amide Ionic Liquid Studied by NMR, J. Phys. Chem. B, 2013, 117(50), 16219–16226, DOI:10.1021/jp409324k.
- M. E. Orazem and B. Tribollet, Electrochemical Impedance Spectroscopy, 2008, vol. 48, DOI:10.1002/9780470381588.
- F. W. Sears, M. W. Zemansky and H. D. Young, University Physics, Addison-Wesley Pub. Co.: Reading, Mass, 1982 Search PubMed.
- R. Mills, Self-Diffusion in Normal and Heavy Water in the Range 1–45°, J. Phys. Chem., 1973, 77(5), 685–688, DOI:10.1021/j100624a025.
- Van London pHoenix Co., Education Series Conductivity Guide, 6103 Glenmont Dr. Houston, http://www.vl-pc.com Search PubMed.
- K. Tanaka and M. Nomura, Measurements of Tracer Diffusion Coefficients of Lithium Ions, Chloride Ions and Water in Aqueous Lithium Chloride Solutions, J. Chem. Soc., Faraday Trans. 1, 1987, 83(6), 1779–1782, 10.1039/F19878301779.
- P. E. Mason, S. Ansell, G. W. Neilson and S. B. Rempe, Neutron Scattering Studies of the Hydration Structure of Li+, J. Phys. Chem. A, 2015, 119(5), 2003–2009, DOI:10.1021/jp511508n.
- R. Murugan, V. Thangadurai and W. Weppner, Fast Lithium Ion Conduction in Garnet-Type Li7La3Zr2O12, Angew. Chem., Int. Ed., 2007, 46(41), 7778–7781, DOI:10.1002/anie.200701144.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, High-Power All-Solid-State Batteries Using Sulfide Superionic Conductors, Nat. Energy, 2016, 1(4), 16030, DOI:10.1038/nenergy.2016.30.
- R. A. Horne and R. A. Courant, The Electrical Conductivity of Aqueous Electrolytic Solutions near the Freezing Point, J. Phys. Chem., 1964, 68(5), 1258–1260, DOI:10.1021/j100787a518.
- C. Eyraud, J. F. Quinson and M. Brun, The Role of Thermoporometry in the Study of Porous Solids, in Characterization of Porous Solids, ed. K. K. Unger, J. Rouquerol, K. S. W. Sing and H. Kral, Studies in Surface Science and Catalysis, Elsevier, 1988, vol. 39, pp. 295–305, DOI:10.1016/S0167-2991(09)60753-9.
- C. Alba-Simionesco, B. Coasne, G. Dosseh, G. Dudziak, K. E. Gubbins, R. Radhakrishnan and M. Sliwinska-Bartkowiak, Effects of Confinement on Freezing and Melting, J. Phys.: Condens. Matter, 2006, 18, R15–R68, DOI:10.1088/0953-8984/18/6/R01.
- P. Novák, Electrochemical Insertion of Magnesium into Hydrated Vanadium Bronzes, J. Electrochem. Soc., 1995, 142(8), 2544, DOI:10.1149/1.2050051.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, A High-Capacity and Long-Life Aqueous Rechargeable Zinc Battery Using a Metal Oxide Intercalation Cathode, Nat. Energy, 2016, 1(10), 1–8, DOI:10.1038/nenergy.2016.119.
- R. Demir-Cakan, M. R. Palacin and L. Croguennec, Rechargeable Aqueous Electrolyte Batteries: From Univalent to Multivalent Cation Chemistry, J. Mater. Chem. A, 2019, 7(36), 20519–20539, 10.1039/c9ta04735b.
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Interface Stability in Solid-State Batteries, Chem. Mater., 2016, 28(1), 266–273, DOI:10.1021/acs.chemmater.5b04082.
- Y. Inaguma, L. Chen, M. Itoh and T. Nakamura, High Ionic Conductivity in Lithium Lanthanum Titanate, Solid State Commun., 1993, 86(10), 689–693 CrossRef CAS.
- P. Knauth, Inorganic Solid Li Ion Conductors: An Overview, Solid State Ionics, 2009, 180(14–16), 911–916, DOI:10.1016/j.ssi.2009.03.022.
- Y. Tian, Y. Sun, D. C. Hannah, Y. Xiao, H. Liu, K. W. Chapman, S. H. Bo and G. Ceder, Reactivity-Guided Interface Design in Na Metal Solid-State Batteries, Joule, 2019, 3(4), 1037–1050, DOI:10.1016/j.joule.2018.12.019.
- R. D. Armstrong and K. Landles, Electrochemical Behaviour of Lithium Iodide Monohydrate, J. Appl. Electrochem., 1981, 11(2), 247–251, DOI:10.1007/BF00610986.
- R. D. Armstrong and K. Landles, Lithium Ion Conducting Solids for Ambient Applications, J. Appl. Electrochem., 1982, 12(5), 533–535, DOI:10.1007/BF00614979.
- K. MacKenzie and M. E. Smith, Multinuclear Solid-State NMR of Inorganic Materials, Pergamon, 1st edn, 2002 Search PubMed.
- D. C. Apperley, R. K. Harris and P. Hodgkinson, Solid State NMR: Basic Principles and Practice, Momentum Press, New York, 2012 Search PubMed.
- A. Bielecki and D. P. Burum, Temperature Dependence of 207Pb MAS Spectra of Solid Lead Nitrate. An Accurate, Sensitive Thermometer for Variable-Temperature MAS, J. Magn. Reson., Ser. A, 1995, 116(2), 215–220, DOI:10.1006/jmra.1995.0010.
- R. K. Harris, E. D. Becker, S. M. C. De Menezes, R. Goodfellow and P. Granger, NMR Nomenclature: Nuclear Spin Properties and Conventions for Chemical Shifts - IUPAC Recommendations 2001, Solid State Nucl. Magn. Reson., 2002, 22(4), 458–483, DOI:10.1006/snmr.2002.0063.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, discussion, and data by TGA, PFG NMR, ssNMR, electrochemical measurements, ICP-OES, SEM and single crystal XRD. See DOI: 10.1039/d1ta04736a |
| This journal is © The Royal Society of Chemistry 2021 |