
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acetylene-linked conjugated polymers for sacrificial photocatalytic hydrogen evolution from water†
Lunjie
Liu
a,
Michał A.
Kochman
b,
Yongjie
Xu
a,
Martijn A.
Zwijnenburg
 *b,
Andrew I.
Cooper
*a and
Reiner Sebastian
Sprick
*ac
*b,
Andrew I.
Cooper
*a and
Reiner Sebastian
Sprick
*ac
aDepartment of Chemistry and Materials Innovation Factory, University of Liverpool, 51 Oxford Street, Liverpool L7 3NY, UK. E-mail: aicooper@liverpool.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: m.zwijnenburg@ucl.ac.uk
cDepartment of Pure and Applied Chemistry, University of Strathclyde, Thomas Graham Building, 295 Cathedral Street, Glasgow G1 1XL, UK. E-mail: sebastian.sprick@strath.ac.uk
First published on 16th July 2021
Abstract
Conjugated organic polymers have shown potential as photocatalysts for hydrogen production by water splitting. Taking advantage of a high throughput screening workflow, two series of acetylene-linked co-polymers were prepared and studied for their potential as photocatalysts for sacrificial hydrogen production from water. It was found that a triethynylbenzene-based polymer with a dibenzo[b,d]thiophene sulfone linker (TE11) had the highest performance in terms of hydrogen evolution rate under visible illumination in the presence of a sacrificial hole-scavenger. Synthetically elaborating the triethynylbenzene linker in TE11 by changing the core and by introducing nitrogen, the resulting hydrogen evolution rate was further increased by a factor of nearly two.
Introduction
As a result of rapid human development and rising energy demand, the search for new sustainable energy supplies has become an important global challenge for society. Hydrogen has advantages compared to traditional fossil-fuels, owing to its higher energy density and clean combustion. Over the past three decades, photocatalytic and photoelectrochemical (PEC) hydrogen production from water under sunlight irradiation have been studied in an attempt to solve the environmental problems associated with fossil fuels.1–4Significant efforts have been made in this area since 1972 with the experiments reported by Fujishima and Honda using TiO2 as the anode material in a PEC cell for water splitting.5 So far, the search for photocatalytic water splitting materials has mainly focused around using inorganic semiconductors. While semiconducting organic materials were studied as early as three decades ago,6–8 they received little attention until 2009, when graphitic carbon nitride was reported as a photocatalyst for photocatalytic hydrogen production in the presence of a hole-scavenger.9 After this, many organic photocatalysts have shown potential as photocatalysts for hydrogen production from water, such as linear conjugated polymers,6,10–13 conjugated microporous polymers (CMPs),12–15 covalent organic frameworks (COFs),16–20 hydrogen-bonded organic frameworks (HOFs)21 and covalent triazine-based frameworks (CTFs).22–26 Recently, organic materials have also been reported to perform water oxidation24,27 and even overall water splitting.28–30 Due to the diversity of monomers and different polymerization routes, many different polymer photocatalysts have been synthesized,31 but their performance as photocatalysts for hydrogen evolution from water is still lower than for the best inorganic semiconductors.32
In previous work, a large series of conjugated polymers was synthesized by Suzuki–Miyaura polycondensation and tested as hydrogen production photocatalysts using a high-throughput workflow, showing that the screening allows for the accelerated discovery of high performance photocatalysts.11–13,17 Acetylene-linked polymer photocatalysts and photoelectrode materials have been used for water splitting or pollutants degradation under visible light illumination with good results.33–37 For example, acetylene-linked thiophene polymers on Cu foam supports exhibited high cathodic photocurrents, demonstrating the potential of acetylene-bridged materials.33 Similarly, 1,4-diethynylbenzene-based linear conjugated polymers were found to be more photocatalytically active compared with 1,4-benzene-based linear conjugated polymers, and introducing the ethynyl group into their backbones can enhance visible light absorption, photocurrent intensity, and hydrogen production activity.34
Motivated by these studies, we synthesized a new library of acetylene-linked conjugated polymers based on 1,3,5-triethynylbenzene (TE, see Fig. 1) and 1,4-diethynylbenzene (DE) via a palladium-catalysed Sonogashira cross-coupling reaction. The photocatalytic hydrogen evolution activities of these polymers in the presence of triethylamine (TEA) as a hole scavenger and platinum as a co-catalyst were tested using our high-throughput workflow. Among this library of polymers, TE11 was the best-performing polymer photocatalyst with the hydrogen evolution rate of 1080 μmol g−1 h−1 under visible illumination (λ > 420 nm). Based on this, we further optimized the structure by changing the linker length and using a triazine containing core to give TEBN11. The hydrogen evolution rate of this material was improved to 1894 μmol g−1 h−1 with an EQE of 2.7% at 420 nm.
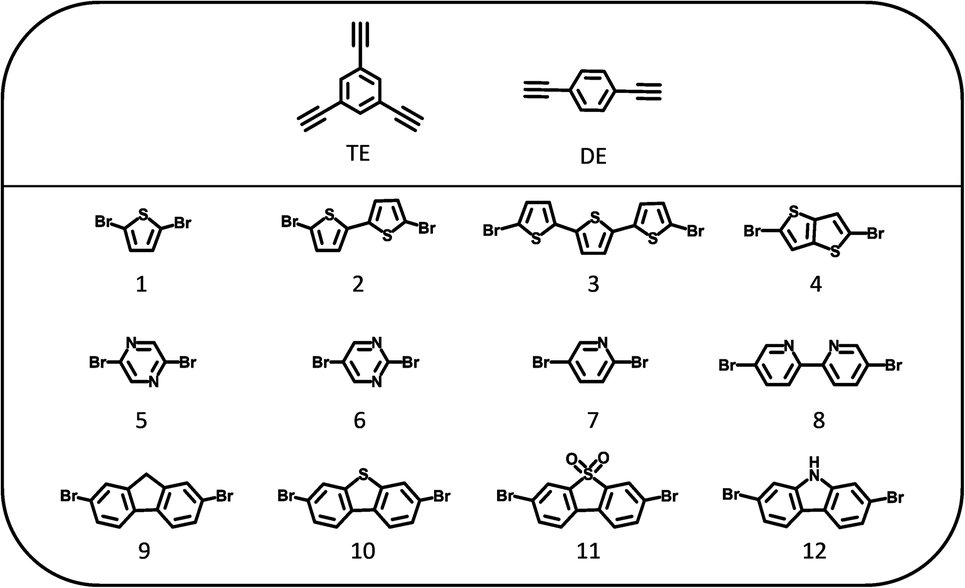 | ||
| Fig. 1 Structures of the 1,3,5-triethynylbenzene (TE), 1,4-diethynylbenzene (DE) and dibromo monomers (1–12) used to synthesize the polymer photocatalysts. | ||
Results and discussion
Synthesis and characterisation
All polymers were synthesized by Pd-catalyzed Sonogashira–Hagihara polycondensation of 1,3,5-triethynylbenzene or 1,4-diethynylbenzene with a range of dibromo-functionalized monomer linkers (1–12; Fig. 1) to give the corresponding TE-n or DE-n polymer photocatalysts (whereby n is the corresponding dibromo monomer linker). The polymers were classified into the three subsets: sulfur-heterocycles (1–4), nitrogen-heterocycles (5–8), and planar heterofluorenes (9–12).All materials were characterized by microanalysis and Fourier transform infrared (FTIR) spectroscopy (Fig. S1–S3†). All FTIR spectra show a peak around 2190 cm−1 that can be assigned to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretching modes of the acetylene linker present in all materials.38,39 In addition, we found that the crosslinked TE polymer networks were amorphous while the linear DE polymers were semi-crystalline, as evident by their powder X-ray diffraction patterns (Fig. S4–S6†). Scanning electron microscopy images (Fig. S7–S9†) showed variable morphologies for the polymers; most TE polymers tended to form spherical nano aggregates, while the linear DE polymers consisted mainly of flakes or blocks. Thermogravimetric measurements under N2 showed that all polymers were thermally stable to around 250 °C under N2 (Fig. S10–S12†). The apparent Brunauer–Emmett–Teller (SABET) surface areas were estimated using nitrogen adsorption isotherms at 77 K and up to 1 bar (Fig. S13–S15 and Table S1†). The BET surface areas of all linear DE polymers fell below 60 m2 g−1, while the TE polymers showed variable surface areas: 5 polymers had surface areas ranging from 150–600 m2 g−1 that denoted a degree of porosity, with other samples being essentially non-porous (SABET < 30 m2 g−1).
C stretching modes of the acetylene linker present in all materials.38,39 In addition, we found that the crosslinked TE polymer networks were amorphous while the linear DE polymers were semi-crystalline, as evident by their powder X-ray diffraction patterns (Fig. S4–S6†). Scanning electron microscopy images (Fig. S7–S9†) showed variable morphologies for the polymers; most TE polymers tended to form spherical nano aggregates, while the linear DE polymers consisted mainly of flakes or blocks. Thermogravimetric measurements under N2 showed that all polymers were thermally stable to around 250 °C under N2 (Fig. S10–S12†). The apparent Brunauer–Emmett–Teller (SABET) surface areas were estimated using nitrogen adsorption isotherms at 77 K and up to 1 bar (Fig. S13–S15 and Table S1†). The BET surface areas of all linear DE polymers fell below 60 m2 g−1, while the TE polymers showed variable surface areas: 5 polymers had surface areas ranging from 150–600 m2 g−1 that denoted a degree of porosity, with other samples being essentially non-porous (SABET < 30 m2 g−1).
UV-visible absorption spectra were measured in the solid state to study optical properties of the materials (Fig. S16–S18†). All polymers had absorption onsets in the visible part of the solar spectrum and most TE polymers had small red shift in their onset compared to their corresponding DE polymers. The optical gaps of the polymers (Table S2†) range from 1.85 eV for TE4 to 2.52 eV for TE8. Time-resolved single photon counting experiments were performed to study the lifetime of the excited state (Fig. S19–S42 and Table S3†). The weighted average fluorescence lifetimes of TE and DE polymers varied from 0.3 ns to 2.6 ns, with most materials having lifetimes shorter than 1 ns.
Contact angle measurements with water were performed to study the wetting behaviour of the polymers (Fig. S43–S44 and Table S4†). Polymers TE4, TE6, TE7 and TE11 absorbed water and swelled, while the TE8 polymer showed the highest water contact angle (115.7°). TE5 had the lowest contact angle of 59.5°; the contact angles of the DE polymers ranged from 75° to 95°.
The dispersibility of all polymers in TEA/methanol (MeOH)/water mixtures was determined by measuring the transmittance of the resulting photocatalyst dispersions (Table S5†), where lower transmission means better dispersibility (that is, less particle settling). The dispersibility of TE5 particles was the highest with a transmittance value of just 0.5%, while the transmittance of TE12 and DE10 were approximately 80%, indicating poor dispersibility in the reaction mixture, which could also be observed by eye. Particle size distributions of polymers were measured by static light scattering experiments (Fig. S45–S47 and Table S6†). The DE polymers were found to have smaller average particle sizes (8.9–18.5 μm) compared to the respective TE polymers (20.6–86.1 μm).
In addition, their electron affinity (EA) and ionization potential (IP) were predicted based on density functional theory (Fig. 2 and Table S7†). EA governs the driving force for proton reduction, while IP governs the driving force for water oxidation (or TEA oxidation in this case), which are often approximated in the literature (though not here) by the energy of the LUMO (lowest unoccupied molecular orbital) and HOMO (highest occupied molecular orbital), respectively.13,25,40 The EA values of all polymers are more negative than the proton reduction potential at pH 12.3 (the experimentally measured pH of the TEA/MeOH/water mixture), which means they all should be able to reduce protons when illuminated. All polymers should also be able drive the overall oxidation of TEA to diethylamine and acetaldehyde. However, for a number of the polymers, e.g., TE3/4 and DE3/4, the one-hole oxidation of TEA, the first step in the overall oxidation, is predicted to be endergonic, suggesting that this step might give rise to a thermodynamic barrier.
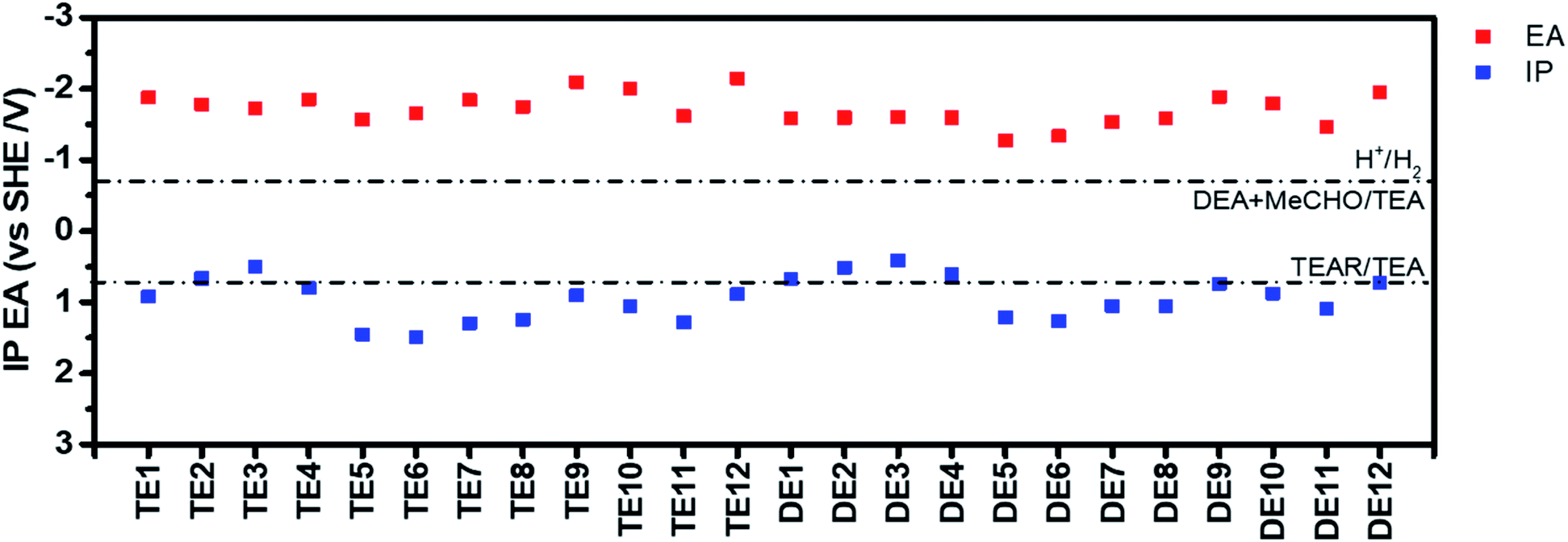 | ||
| Fig. 2 Predicted IP and EA potentials of the different polymers. Solution half reactions are shown for pH 12.3, the measured pH of the TEA/MeOH/water mixture (MeCHO-acetaldehyde; TEAR-deprotonated TEA radical). | ||
To rapidly screen the photocatalytic activity of all polymers, we used the same high-throughput workflow that we reported previously (Table S8†).13,25 TEA was used as a hole scavenger and platinum was added by photodeposition from H2PtCl6 (3 wt% based on polymer) as the cocatalyst for hydrogen evolution. After 2 h illumination under simulated solar light (1 sun), the TE11 photocatalyst showed the highest hydrogen production rate (Table S8†) by some margin of 1811.0 μmol g−1 h−1. The correlation of the hydrogen evolution rates and factors were plotted to study the relationship between the performance of the polymers and their properties (Fig. S49–S56†), it seems once more that there is no single property that dominates the photocatalytic activity, as found in previous studies.13,25
Structural modification of TE11
In an attempt to further improve the photocatalytic activity of the best-performing polymer, TE11, we modified the linker by using TEB instead of TE, which bears additional phenylenes and therefore extends the conjugation length of the linker. We also explored TEBN, which is the triazine analogue of TEB (Fig. 3a).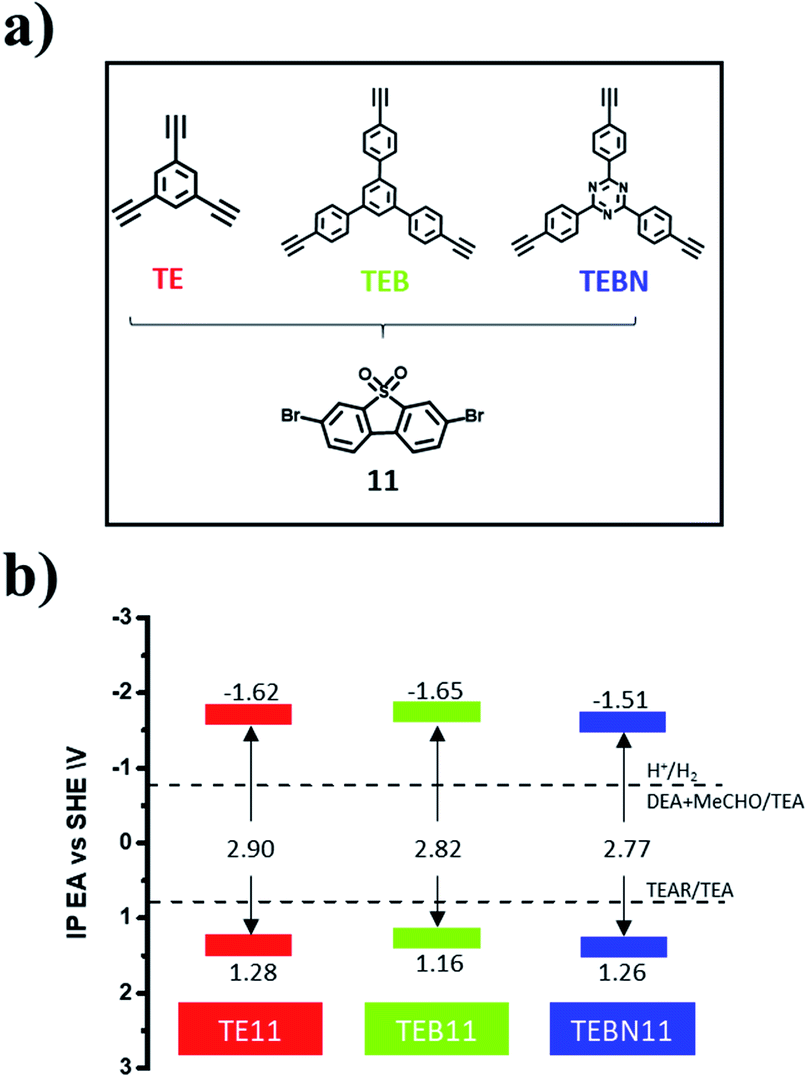 | ||
| Fig. 3 (a) Chemical structures of expanded TE analogues; (b) predicted EA and IP positions for the resulting copolymers. Solution half reactions are shown for pH 12.3, the measured pH of the TEA/MeOH/water mixture (MeCHO-acetaldehyde; TEAR-deprotonated TEA radical). | ||
TEB11 and TEBN11 were characterised by FT-IR (Fig. S57†), and characteristic bands at 1160 cm−1 and 2190 cm−1 were attributed to the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration and CC stretching modes, respectively.39 The presence of alkyne bonds was furthermore confirmed by solid-state 13C cross-polarization magic angle spinning (CP-MAS) NMR spectra (Fig. S58†) showing a characteristic peak at approximately 90 ppm while Raman spectra showed a feature at 2210 cm−1 indicative of carbon–carbon triple bonds (Fig. S59†).38 PXRD patterns of all three materials showed that these materials were amorphous (Fig. S60†). Their morphologies varied: TE11 comprised porous flake-like structures while TEB11 was composed of non-uniform nanospheres; TEBN11 consisted mainly of large, fused aggregates (Fig. S61†). The absorption onset of TEB11 was found to be similar to TE11, while TEBN11 showed a slight red-shift (Fig. 4a). In line with this observation, TEBN11 was also predicted to have a narrower fundamental gap than TE11 (Fig. 3b). Compared with TE11, the photoluminescence emission spectra of TEB11 and TEBN11 showed a red shift (Fig. S62†), which was consistent with the absorption spectrum. X-ray photoelectron spectra showed a shift in the high-resolution peaks of C 1s, O 1s and S 2p after modification of TE11 (Fig. S63†), which could be attributed to the changes of electronic properties induced by extending the linker length and introduction of nitrogen into the benzene-ring.41 TEB11 and TEBN11 also showed increased fluorescence lifetime (Fig. 4b and Table S9†) and improved thermal stability compared with TE11 (Fig. S64†); by contrast, the materials were found to be more hydrophobic against water (Fig. S65 and Table S10†), but in mixtures of TEA/MeOH/water, thanks to the wettability of organic solvents, the dispersibility was improved (Table S10†). Finally, average particle sizes were found to be similar for TE11, TEB11 and TEBN11 (Fig. S66 and Table S12†).
O stretching vibration and CC stretching modes, respectively.39 The presence of alkyne bonds was furthermore confirmed by solid-state 13C cross-polarization magic angle spinning (CP-MAS) NMR spectra (Fig. S58†) showing a characteristic peak at approximately 90 ppm while Raman spectra showed a feature at 2210 cm−1 indicative of carbon–carbon triple bonds (Fig. S59†).38 PXRD patterns of all three materials showed that these materials were amorphous (Fig. S60†). Their morphologies varied: TE11 comprised porous flake-like structures while TEB11 was composed of non-uniform nanospheres; TEBN11 consisted mainly of large, fused aggregates (Fig. S61†). The absorption onset of TEB11 was found to be similar to TE11, while TEBN11 showed a slight red-shift (Fig. 4a). In line with this observation, TEBN11 was also predicted to have a narrower fundamental gap than TE11 (Fig. 3b). Compared with TE11, the photoluminescence emission spectra of TEB11 and TEBN11 showed a red shift (Fig. S62†), which was consistent with the absorption spectrum. X-ray photoelectron spectra showed a shift in the high-resolution peaks of C 1s, O 1s and S 2p after modification of TE11 (Fig. S63†), which could be attributed to the changes of electronic properties induced by extending the linker length and introduction of nitrogen into the benzene-ring.41 TEB11 and TEBN11 also showed increased fluorescence lifetime (Fig. 4b and Table S9†) and improved thermal stability compared with TE11 (Fig. S64†); by contrast, the materials were found to be more hydrophobic against water (Fig. S65 and Table S10†), but in mixtures of TEA/MeOH/water, thanks to the wettability of organic solvents, the dispersibility was improved (Table S10†). Finally, average particle sizes were found to be similar for TE11, TEB11 and TEBN11 (Fig. S66 and Table S12†).
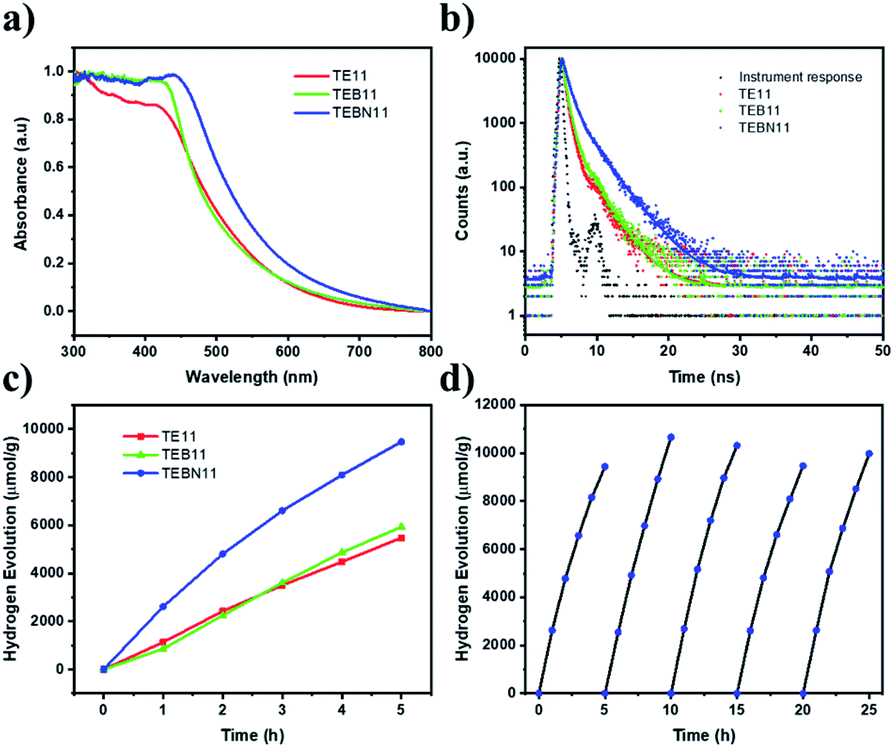 | ||
| Fig. 4 (a) Solid state UV-vis spectra for copolymers; (b) fluorescence lifetime of TE11, TEB11, and TEBN11 measured in ethanol suspensions; (c) photocatalytic HER performance of TE11, TEB11 and TEBN11; (d) repeat photocatalysis runs for TEBN11 under visible light (300 W Xe light source with λ > 420 nm filter) in TEA/MeOH/water mixture with 3 wt% Pt, the TEBN11 was recovered and the reaction solution was replaced between runs. | ||
To further investigate the photocatalytic performance, kinetic hydrogen evolution experiments were carried out under visible light irradiation (300 W Xe light source, λ > 420 nm filter). A sacrificial hydrogen production rate of 1894 μmol g−1 h−1 was determined for TEBN11 in TEA/MeOH/water mixtures loaded with platinum, which was nearly twice as high as for TE11 (1080 μmol g−1 h−1), while the HER rate of TEB11 (1186 μmol g−1 h−1) was only slightly increased compare with TE11 (Fig. 4c and Table S10†). The materials were found to contain similar amounts of platinum of approximately 1 wt% (Table S11†). All three materials are also active without additional platinum, in this case residual palladium from the synthesis facilitates the proton reduction reaction (Table S10†).12,42
The higher photocatalytic performance of TEBN11 compared to TEB11 and TE11 seems to originate from its deeper IP and red-shifted absorption onset allowing for enhanced light absorption. This seems to be much more important than the accessible surface area in this case, which is different to what was observed in a previous study.12
Photocurrent and impedance measurements (Fig. S67†) also showed that TEBN11 had a larger photo-response current and smaller resistance. In addition, the photocatalytic activity had no obvious decrease over 25 hours (Fig. 4d) and the morphology also did not change (Fig. S68†). PXRD and FT-IR results (Fig. S69 and S70†) before and after the photocatalytic experiments were nearly the same, which indicating that TEBN11 has good photocatalytic stability. The external quantum efficiency (EQE) was measured at different wavelength to evaluate the HER performance, the EQE value of TEBN11 was 2.7% at 420 nm, which is higher than TEB11 (1.6% at 420 nm) and TE11 (1.6% at 420 nm), and also relatively high compared to some organic polymer catalysts reported, such as covalent triazine-based framework (CTF-2, 1.6% at 420 nm),43 cyano-substituted carbon-linked framework (OB-POP-3, 2.0% at 420 nm),44 and diacetylene functionalized COF (TP-BDDA, 1.3% at 420 nm),36 although lower than other dibenzo[b,d]thiophene sulfone co-polymers (P7, 7.2% at 420 nm,45 P10, 11.6% at 420 nm (ref. 46) and P64, 20.7% at 420 nm (ref. 13)) (Table S13†). The EQEs determined at different wavelengths of TEBN11 broadly follow the absorption profile (Fig. S71†), as expected from a photocatalytic process.12
Conclusions
In summary, 24 acetylene-linked organic polymers were synthesized via Sonogashira poly-condensation and used as photocatalysts for sacrificial hydrogen production from water under visible illumination. Using a high-throughput workflow, photocatalyst TE11, a co-polymer of 1,3,5-triethynylbenzene and dibenzo[b,d]thiophene sulfone, was identified as the best-performing material with a sacrificial hydrogen evolution rate of 1811.0 μmol g−1 h−1. We further optimized the structure of polymer of TE11 by increasing the size of the core and adding a triazine ring. This resulted in a further increase in hydrogen production rate under visible light illumination from 1080 μmol g−1 h−1 for TE11 to 1894 μmol g−1 h−1 for TEBN11 with an EQE of 2.7% at 420 nm. The material was found to be stable under irradiation for 25 hours, which demonstrating the potential as robust organic photocatalysts for hydrogen production.Experimental section
Materials
All reagents were obtained from Sigma-Aldrich, Fluorochem, TCI, Alfa Aesar and used as received. 1,3,5-Tris-(4-ethynyl-phenyl)-benzene,47 2,4,6-tris[(4-ethynyl)phenyl]-1,3,5-triazine,48 were synthesized using reported literature procedures.Characterizations
The absorption spectra of the polymers were recorded on a Cary 5000 UV-visible-NIR spectrophotometer as powders. Transmission FT-IR spectra were obtained with an attenuated total reflectance (ATR) method on a Bruker Tensor-27 spectrometer at room temperature. Photoluminescence spectra were performed on a Shimadzu RF-5301PC fluorescence spectrometer. Time-correlated single photon counting (TCSPC) experiments were measured on an Edinburgh Instruments LS980-D2S2-STM spectrometer (EPL-375 diode, λ = 371 nm). The instrument response of the TCSPC spectrometer was measured with colloidal silica (LUDOX HS-40, Sigma-Aldrich) at the excitation wavelength. Decay times were fitted in the FAST software using three decay exponents. Thermogravimetric analysis was performed by heating samples under N2 in open platinum pans from 25 to 800 °C at 10 °C min−1 on an EXSTAR6000 instrument. PXRD measurements were performed on a Panalytical Empyrean diffractometer with a Cu X-ray source. Static light scattering measurements were performed on a Malvern Mastersizer 3000 Particle Sizer at a laser obscuration of 5–10%. Particle sizes were fitted according to the Mie theory with the Malvern ‘General Purpose’ analysis model. Transmittance of high-throughput samples was measured on a Formulaction S.A.S. Turbiscan AGS system with an 880 nm NIR diode and a detector at 180° (relative to the light source) in a cylindrical glass cell. Surface areas were tested on Micromeritics 2420 instrument at a temperature of 77 K. Before analysis, polymer samples were degassed offline at 110 °C for 15 hours under dynamic vacuum (10−5 bar). CHNS-O Analyzer was measured using standard microanalytical procedures. Palladium, platinum and copper contents were determined using ICP-OES Agilent 5110 equipped with a collision/reaction cell after a microwave digest of the materials in nitric acid (67–69%, trace metal analysis grade) in a microwave. Water contact angles were measured using pressed pellets and a drop-shape analysis apparatus (Krüss DSA100) with the Young–Laplace fitting method. The morphology of the polymer particles was studied using a Hitachi S4800 SEM. X-Ray photoelectron spectroscopy (XPS) measurement were performed on a Thermo Fisher ESCALAB 250Xi instrument. The Raman measurements were performed on an inVia Reflex Qontor Confocal Raman microscope upon excitation of 785 nm laser. 13C Magic-angle spinning measurements were carried out at 100.63 MHz using a Bruker Avance III HD spectrometer and 4 mm (rotor o.d.) probe. Spectra were acquired at a spin rate of 10 kHz. Cross-polarisation (CP) spectra were recorded with TOSS spinning sideband suppression, 4 ms contact time and with a recycle delay of 4 s. Carbon spectral referencing is relative to neat tetramethylsilane, carried out by setting the high frequency signal from an external sample of adamantane to 38.5 ppm.Polymer synthesis procedure
General procedure for the Sonogashira polycondensation: in a glovebox, a glass vial was charged with the monomers, Pd(PPh3)2Cl2, CuI, triphenylphosphine, anhydrous N,N-dimethylformamide and triethylamine, and then sealed with a silicone septum. The sealed vials were taken out of the glovebox, inserted in a sand bath preheated to 100 °C and kept at this temperature for 2 days. After cooling to room temperature, the mixture was quenched by addition of methanol, and the solids were filtered off. Further purification was carried out by Soxhlet extraction with methanol and chloroform. The final product was dried in the vacuum oven at 80 °C overnight. See the ESI† for detail results of all polymers.High-throughput photocatalytic hydrogen evolution experiments
High-throughput screening experiments were carried out using an automatic platform. For this, a sample vial (10 mL) was charged with 5 mg of polymer powders and H2PtCl6 solution (0.01 mL, 3 wt% in water), then all vials along with degassed jars of triethylamine, methanol, and water were transferred to the Chemspeed Accelerator SWING and purged with nitrogen for 6 hours. After that, the liquid transfer head was used to transfer liquids (trimethylamine/methanol/water = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1; total volume = 5 mL) form the jars to the vials automatically under inert conditions, then the vials were capped with the capper/crimper tool. The sealed vials were subsequently taken out and transferred to an ultrasonic bath to finely disperse polymers by ultrasonication for 10 minutes. Photocatalytic hydrogen evolution experiments were performed on an Oriel Solar Simulator 94123A with an output of 1.0 Sun (Class AAA, 1440 W Xe lamp, 12 × 12 in.). After 2 hours, the amount of hydrogen evolved was determined on an Agilent HS-GC (7890B). Control experiments of mixtures of trimethylamine/methanol/water or trimethylamine/methanol/water/H2PtCl6 showed that no hydrogen was produced in the absence of a photocatalyst.
:1:1; total volume = 5 mL) form the jars to the vials automatically under inert conditions, then the vials were capped with the capper/crimper tool. The sealed vials were subsequently taken out and transferred to an ultrasonic bath to finely disperse polymers by ultrasonication for 10 minutes. Photocatalytic hydrogen evolution experiments were performed on an Oriel Solar Simulator 94123A with an output of 1.0 Sun (Class AAA, 1440 W Xe lamp, 12 × 12 in.). After 2 hours, the amount of hydrogen evolved was determined on an Agilent HS-GC (7890B). Control experiments of mixtures of trimethylamine/methanol/water or trimethylamine/methanol/water/H2PtCl6 showed that no hydrogen was produced in the absence of a photocatalyst.
Kinetic photocatalytic hydrogen evolution measurements
A quartz reaction flask was charge with 25 mg polymer and 25 mL mixtures of water/triethylamine/methanol = 1:1:1 and 0.02 mL H2PtCl6 solution (8 wt% in water), the flask was sealed with a rubber stopper. After ultrasonication for 10 minutes, the well-dispersed suspension was degassed by bubbling with N2 for 30 minutes before illuminating with a 300 W Newport Xe light source with a filter (λ > 420 nm). The concentration of hydrogen was determined by a Bruker 450 GC gas chromatograph equipped with TCD detector. For longer-term stability experiments, after illumination for 5 hours, the polymer was recovered through filtration, and after drying, 25 mL mixtures of water/triethylamine/methanol = 1:1:1 were added again for the next run. For external quantum efficiency (EQE) measurements, experiments were performed under a light-emitting diode irradiation (λ = 420, 490, 700 nm), and the EQE was estimated using the following formula: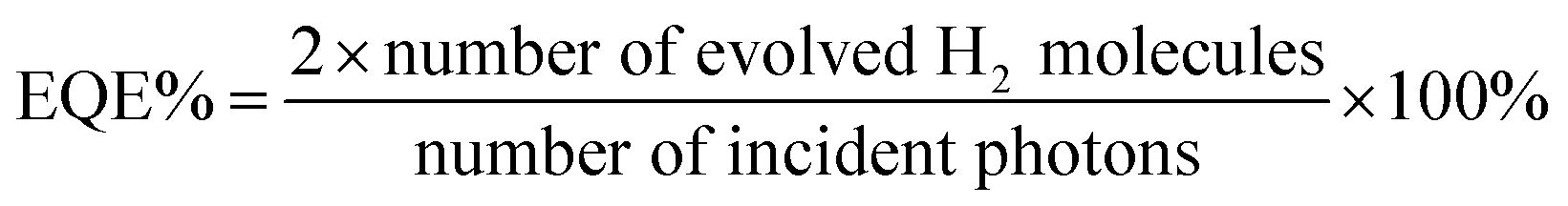
Electrochemical measurements
Electrochemical experiments were performed on the BioLogic SP200 workstation. The transient photocurrent responses (I–t) and electrochemical impedance spectra (EIS) of samples were investigated in a three-electrode system (FTO, Pt plate, and Ag/AgCl as working, counter, and reference electrode, respectively) using a 300 W Xe light source (Newport). The electrolyte was 0.5 M Na2SO4 aqueous solution and was degassed for 30 minutes by N2 at room temperature before the measurement. The working electrode was prepared as follows: 2 mg of the photocatalyst was ultrasonicated with 10 μL Nafion (5 wt%) aqueous solution and 100 μL ethanol giving a slurry. The slurry was then coated onto FTO glass electrodes with an active area of 1 cm2. The applied bias for intermittent photocurrent intensity measurement was 0.6 V vs. Ag/AgCl. The EIS spectra were determined by applying a 10 mV AC signal over the frequency range of 100 kHz to 10 mHz at a DC bias of 0.6 V vs. Ag/AgCl.Density functional theory (DFT) calculations
DFT calculations are helpful for computationally screening of candidate polymer photocatalysts and to gain a deep understanding of the structure and property relationships. For the DFT calculations, the TE and DE polymers were represented by finite (i.e., nonperiodic) oligomeric models; linear oligomers of composition (L(DE))6 in the case of DE polymers and a TE centered (TE)(L(TE))3 oligomers in the case of the TE polymers (Fig. S48†). The starting structures of the oligomers were constructed with the program STK.49 Having identified the representative conformer of each oligomer, we proceeded to optimize its geometry using density functional theory (DFT). To calculate the adiabatic ionization potential (IP) and adiabatic electron affinity (EA) of the oligomer, the geometries of the radical cation and radical anion forms of the representative conformer were likewise optimized using DFT. The excitation spectrum of the charge-neutral form of the representative conformer was calculated using time-dependent DFT (TD-DFT).The DFT and TDDFT calculations were performed with the program Turbomole, version 6.5. The B3LYP exchange–correlation functional50,51 was used in combination with the DZP basis set.52,53 The effects of aqueous solvation were included via the conductor-like screening model (COSMO).54,55 At all times, the energies and gradients were corrected for dispersion effects with the D3 semiempirical dispersion correction scheme of Grimme and coworkers.56
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC) for financial support under Grant EP/N004884/1. L. L. thanks the China Scholarship Council for a PhD studentship. R. S. S. thanks the University of Strathclyde for financial support through The Strathclyde Chancellor's Fellowship Scheme.Notes and references
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- Y. Miseki and K. Sayama, Adv. Energy Mater., 2019, 9, 1801294 CrossRef.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 8, 474–475 RSC.
- T. Shibata, A. Kabumoto, T. Shiragami, O. Ishitani, C. Pac and S. Yanagida, J. Phys. Chem., 1990, 94, 2068–2076 CrossRef CAS.
- T. Zhang, G. Xing, W. Chen and L. Chen, Mater. Chem. Front., 2020, 4, 332–353 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 559–565 CrossRef CAS PubMed.
- D. J. Woods, S. A. J. Hillman, D. Pearce, L. Wilbraham, L. Q. Flagg, W. Duffy, I. McCulloch, J. R. Durrant, A. A. Y. Guilbert, M. A. Zwijnenburg, R. S. Sprick, J. Nelson and A. I. Cooper, Energy Environ. Sci., 2020, 13, 1843–1855 RSC.
- R. S. Sprick, Y. Bai, A. A. Y. Guilbert, M. Zbiri, C. M. Aitchison, L. Wilbraham, Y. Yan, D. J. Woods, M. A. Zwijnenburg and A. I. Cooper, Chem. Mater., 2019, 31, 305–313 CrossRef CAS.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef CAS PubMed.
- L. Wang, X. Zheng, L. Chen, Y. Xiong and H. Xu, Angew. Chem., Int. Ed., 2018, 57, 3454–3458 CrossRef CAS PubMed.
- C. Zhao, Z. Chen, R. Shi, X. Yang and T. Zhang, Adv. Mater., 2020, 32, 1907296 CrossRef CAS PubMed.
- T. Banerjee, K. Gottschling, G. Savasci, C. Ochsenfeld and B. V. Lotsch, ACS Energy Lett., 2018, 3, 400–409 CrossRef CAS PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- Y. Wan, L. Wang, H. Xu, X. Wu and J. Yang, J. Am. Chem. Soc., 2020, 142, 4508–4516 CrossRef CAS PubMed.
- W. Chen, L. Wang, D. Mo, F. He, Z. Wen, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 16902–16909 CrossRef CAS PubMed.
- C. M. Aitchison, C. M. Kane, D. P. McMahon, P. R. Spackman, A. Pulido, X. Wang, L. Wilbraham, L. Chen, R. Clowes, M. A. Zwijnenburg, R. S. Sprick, M. A. Little, G. M. Day and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 7158–7170 RSC.
- K. Schwinghammer, S. Hug, M. B. Mesch, J. Senker and B. V. Lotsch, Energy Environ. Sci., 2015, 8, 3345–3353 RSC.
- L. Guo, Y. Niu, H. Xu, Q. Li, S. Razzaque, Q. Huang, S. Jin and B. Tan, J. Mater. Chem. A, 2018, 6, 19775–19781 RSC.
- J. Xie, S. A. Shevlin, Q. Ruan, S. J. A. Moniz, Y. Liu, X. Liu, Y. Li, C. C. Lau, Z. X. Guo and J. Tang, Energy Environ. Sci., 2018, 11, 1617–1624 RSC.
- C. B. Meier, R. Clowes, E. Berardo, K. E. Jelfs, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Chem. Mater., 2019, 31, 8830–8838 CrossRef CAS PubMed.
- J. Yu, X. Sun, X. Xu, C. Zhang and X. He, Appl. Catal., B, 2019, 257, 117935 CrossRef CAS.
- R. S. Sprick, Z. Chen, A. J. Cowan, Y. Bai, C. M. Aitchison, Y. Fang, M. A. Zwijnenburg, A. I. Cooper and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 2–8 CrossRef PubMed.
- Y. Bai, K. Nakagawa, A. J. Cowan, C. M. Aitchison, Y. Yamaguchi, M. A. Zwijnenburg, A. Kudo, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 16283–16290 RSC.
- L. Lin, Z. Lin, J. Zhang, X. Cai, W. Lin, Z. Yu and X. Wang, Nat. Catal., 2020, 3, 649–655 CrossRef CAS.
- Z. Qin, Z. Huang, M. Wang, D. Liu, Y. Chen and L. Guo, Appl. Catal., B, 2020, 261, 118211 CrossRef CAS.
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- M. Rahman, H. Tian and T. Edvinsson, Angew. Chem., Int. Ed., 2020, 59, 2–18 CrossRef.
- H. J. Sun, I. H. Oner, T. Wang, T. Zhang, O. Selyshchev, C. Neumann, Y. B. Fu, Z. Q. Liao, S. Q. Xu, Y. Hou, A. Turchanin, D. R. T. Zahn, E. Zschech, I. M. Weidinger, J. Zhang and X. L. Feng, Angew. Chem., Int. Ed., 2019, 58, 10368–10374 CrossRef CAS PubMed.
- X.-H. Zhang, X.-P. Wang, J. Xiao, S.-Y. Wang, D.-K. Huang, X. Ding, Y.-G. Xiang and H. Chen, J. Catal., 2017, 350, 64–71 CrossRef CAS.
- T. Zhang, Y. Hou, V. Dzhagan, Z. Q. Liao, G. L. Chai, M. Loffler, D. Olianas, A. Milani, S. Q. Xu, M. Tommasini, D. R. T. Zahn, Z. K. Zheng, E. Zschech, R. Jordan and X. L. Feng, Nat. Commun., 2018, 9, 1140 CrossRef PubMed.
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomäcker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423–1427 CrossRef CAS PubMed.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed.
- L. Wang, Y. Wan, Y. Ding, S. Wu, Y. Zhang, X. Zhang, G. Zhang, Y. Xiong, X. Wu, J. Yang and H. Xu, Adv. Mater., 2017, 29, 1702428 CrossRef PubMed.
- X. Wang, B. Chen, W. Dong, X. Zhang, Z. Li, Y. Xiang and H. Chen, Macromol. Rapid Commun., 2019, 40, 1800494 CrossRef PubMed.
- P. Guiglion, C. Butchosa and M. A. Zwijnenburg, J. Mater. Chem. A, 2014, 2, 11996–12004 RSC.
- Z. Wu, J. Wang, K. Xia, W. Lei, X. Liu and D. Wang, J. Mater. Chem. A, 2018, 6, 616–622 RSC.
- M. Sachs, H. Cha, J. Kosco, C. M. Aitchison, L. Francàs, S. Corby, C.-L. Chiang, A. A. Wilson, R. Godin, A. Fahey-Williams, A. I. Cooper, R. S. Sprick, I. McCulloch and J. R. Durrant, J. Am. Chem. Soc., 2020, 142, 14574–14587 CrossRef CAS PubMed.
- C. B. Meier, R. S. Sprick, A. Monti, P. Guiglion, J.-S. M. Lee, M. A. Zwijnenburg and A. I. Cooper, Polymer, 2017, 126, 283–290 CrossRef CAS.
- S. Bi, Z.-A. Lan, S. Paasch, W. Zhang, Y. He, C. Zhang, F. Liu, D. Wu, X. Zhuang, E. Brunner, X. Wang and F. Zhang, Adv. Funct. Mater., 2017, 27, 1703146 CrossRef.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- X. Wang, S.-m. Lu, J. Li, Y. Liu and C. Li, Catal. Sci. Technol., 2015, 5, 2585–2589 RSC.
- D. Schwarz, Y. S. Kochergin, A. Acharjya, A. Ichangi, M. V. Opanasenko, J. Čejka, U. Lappan, P. Arki, J. He, J. Schmidt, P. Nachtigall, A. Thomas, J. Tarábek and M. J. Bojdys, Chem.–Eur. J., 2017, 23, 13023–13027 CrossRef CAS PubMed.
- L. Turcani, E. Berardo and K. E. Jelfs, J. Comput. Chem., 2018, 39, 1931–1942 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- J. Gauss, J. Chem. Phys., 1993, 99, 3629–3643 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 2, 799–805 RSC.
- A. Schäfer, A. Klamt, D. Sattel, J. C. W. Lohrenz and F. Eckert, Phys. Chem. Chem. Phys., 2000, 2, 2187–2193 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta04288b |
| This journal is © The Royal Society of Chemistry 2021 |