DOI:
10.1039/D1TA04174F
(Paper)
J. Mater. Chem. A, 2021,
9, 24528-24538
Analysis of H2O-induced surface degradation in SrCoO3-derivatives and its impact on redox kinetics†
Received
17th May 2021
, Accepted 30th September 2021
First published on 7th October 2021
Abstract
Substituted SrCoO3 perovskites have been proposed as promising mixed ionic electronic conductors for a range of applications including intermediate temperature solid oxide fuel cells (IT-SOFCs), electrolysers and thermochemical water splitting reactors for H2 production. In this work we investigate the effect of sample exposure to water in substituted SrCoO3 powders and thin films and correlate it with the degradation of oxygen mobility and kinetics. SrCo0.95Sb0.05O3−δ (SCS) thin films have been deposited on different single crystal substrates by pulsed laser deposition (PLD). After water cleaning and post annealing at 300 °C, the sample surface presented an increase of the SrO-surface species as observed by ex situ X-ray Photoemission Spectroscopy (XPS) analysis. This increase in SrO at the sample surface has also been confirmed by the Low Energy Ion Scattering (LEIS) technique on both SCS thin film and powder. Thermochemical water splitting experiments on SCS and SrCo0.95Mo0.05O3−δ (SCM) powder revealed a phase degradation under water oxidising conditions at high temperature with the formation of the trigonal phase Sr6Co5O15. Transmission Electron Microscopy (TEM) analysis of SCS powder treated with water suggests that this phase degradation could already superficially start at Room Temperature (RT). By isotope exchange depth profile experiments on SCS thin films, we were able to quantify the oxygen diffusivity in this SCS surface decomposed layer (D* = 5.1 × 10−17 cm2 s−1 at 400 °C). In the specific case of bulk powder, the effect of water superficial decomposition translates into a lower oxidation and reduction kinetics as demonstrated by comparative thermogravimetric analysis (TGA) studies.
1 Introduction
At intermediate-temperature-solid-oxide-fuel-cell working temperature and ambient pressure, unsubstituted SrCoOx (SCO) stabilises into a 2H hexagonal polytype.1–6 This low temperature phase is slightly Co-deficient and described by Harrison as the trigonal phase with stoichiometry Sr6Co5O15.5 This compound has both lower electronic conductivity and oxygen permeability, and consequently is expected to have lower catalytic activity towards the oxygen reduction reaction (ORR) compared with the high temperature cubic SCO.7–11 The SrCoO3−δ stabilization in a 3C arrangement occurs when the B-site is partially substituted with a high valence cation preventing the face-sharing arrangement of the hexagonal phase. It can also be achieved by decreasing the tolerance factor through partially substituting the A-site cation for one of smaller ionic radius or promoting the reduction of the Co oxidation state. For B-site substituent concentration generally lower or equal to 0.2, the SrCoO3−δ stabilized in the tetragonal superstructure at room temperature (P4/mmm).11 Over this concentration the cubic phase is the stable one at RT. At a relatively low temperature the tetragonal SrCoO3−δ presents a high peak electronic conductivity value, significantly greater than the unsubstituted high-temperature cubic phase (280 S cm−1 at 400 °C and 150 S cm−1 at 925 °C respectively).7,11 Stabilising the tetragonal or cubic SrCoO3−δ phase has been experimentally demonstrated through substituting either the B-site12–23 or the A-site24–29 of the SCO perovskite respectively. The high catalytic activity suggests that the substituted SCO phases are ideal cathodes for IT-SOFCs. From many studies reporting on SCO cathodes, Zhu et al., for example, measured an impressive peak power density of 1016 mW cm−2 and an electrode-area-specific resistance of 0.052 Ω cm2 at 500 °C for a full cell.23 These electrochemical performances were measured for a Sr0.95Nb0.1Co0.9O3−δ cathode on a Sm0.2Ce0.8O1.9–Ni (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v) anode-supported Sm0.2Ce0.8O1.9 (10 μm) electrolyte single cell. Although many studies have proposed substituted SCO as an ideal candidate for IT-SOFCs, only a few have considered the degradation of this cathode surface over time.23,24,30 Zhu et al. indicates SrCO3 formation as responsible for the loss of surface activity of the cathode surface.23 The increase of CO2 species in the gas flow during electrochemical impedance spectroscopy (EIS) measurements was responsible for the increase of area specific resistance (ASR) of the analysed symmetric cell. This detrimental surface degradation was described as partially reversible once CO2 was removed from the gas source. The reversible role of CO2 in the performance degradation of substituted SCO was also reported by Yang et al.24 For Sr substituted A-site perovskites, the issue of Sr atoms segregating at the cathode surface, with consequent deactivation of the surface catalytic activity, has been extensively explored. The thermodynamic driving forces of cation segregation in perovskites have been broadly reviewed by Koo et al.1 One of the causes for strontium segregation is related to the elastic energy produced by the substitution of the A-site cation for one of a larger size, as in the case of strontium atoms partially substituting the smaller lanthanum species:31–35 the larger the size difference between the dopant and host atom the larger the elastic energy driving force for Sr segregation will be. The same type of elastic energy on the A-site cation can also be generated when the B-site is substituted, as reported by Koo et al.35 studying the Sr segregation in SrTi0.5Fe0.5O3−δ epitaxial thin films. The second important driving force is an electrostatic force. Surfaces in solids are generally charged due to the loss of symmetry at the solid surface. In La0.8Sr0.2CoO3 (LSC), for example, Tsvetkov et al. investigated the relationship between oxygen vacancy
:50 v/v) anode-supported Sm0.2Ce0.8O1.9 (10 μm) electrolyte single cell. Although many studies have proposed substituted SCO as an ideal candidate for IT-SOFCs, only a few have considered the degradation of this cathode surface over time.23,24,30 Zhu et al. indicates SrCO3 formation as responsible for the loss of surface activity of the cathode surface.23 The increase of CO2 species in the gas flow during electrochemical impedance spectroscopy (EIS) measurements was responsible for the increase of area specific resistance (ASR) of the analysed symmetric cell. This detrimental surface degradation was described as partially reversible once CO2 was removed from the gas source. The reversible role of CO2 in the performance degradation of substituted SCO was also reported by Yang et al.24 For Sr substituted A-site perovskites, the issue of Sr atoms segregating at the cathode surface, with consequent deactivation of the surface catalytic activity, has been extensively explored. The thermodynamic driving forces of cation segregation in perovskites have been broadly reviewed by Koo et al.1 One of the causes for strontium segregation is related to the elastic energy produced by the substitution of the A-site cation for one of a larger size, as in the case of strontium atoms partially substituting the smaller lanthanum species:31–35 the larger the size difference between the dopant and host atom the larger the elastic energy driving force for Sr segregation will be. The same type of elastic energy on the A-site cation can also be generated when the B-site is substituted, as reported by Koo et al.35 studying the Sr segregation in SrTi0.5Fe0.5O3−δ epitaxial thin films. The second important driving force is an electrostatic force. Surfaces in solids are generally charged due to the loss of symmetry at the solid surface. In La0.8Sr0.2CoO3 (LSC), for example, Tsvetkov et al. investigated the relationship between oxygen vacancy  accumulation at the LSC surface and strontium segregation.36 The relatively positively charged oxygen vacancies electrostatically attract the negatively charged A-site dopants, described in the Kröger–Vink notation as
accumulation at the LSC surface and strontium segregation.36 The relatively positively charged oxygen vacancies electrostatically attract the negatively charged A-site dopants, described in the Kröger–Vink notation as  . Theoretical and experimental results proved that this could also be a cause for cation segregation. Due to the complex nature of the problem, both elastic and electrostatic driving forces concurrently affect cation segregation, and each contribution is not easily separable. The third and less explored aspect of the segregation is the reaction of the material with the gas atmosphere, in particular with H2O and CO2. These chemical reactions are ultimately responsible for the appearance of reactant species such as hydroxide, oxide or carbonates at the cathode surfaces.37–42 Niania et al. studied the effect of different gas atmospheres at high temperature on surface strontium segregation of a La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) bulk material and determined that a water enriched atmosphere had a much greater effect upon particle growth compared to air or oxygen atmospheres.42 In this study, we investigated the surface SrO-formation in the B-site substituted SCO perovskite (Sb, Mo). The reactivity of the B-site substituted SCO phase towards water has been explored both at room temperature and at high temperature by two different experimental approaches. The first method employed dense epitaxial SCS films grown by pulsed laser deposition (PLD) on different single crystals: SrTiO3 (001) (STO), (LaAlO3)0.3(Sr2TaAlO6)0.7 (001) (LSAT) and NdGaO3 (110) (NGO). The effect of water cleaning at room temperature before thermal treatment was investigated. The water reaction with the SCS surface was studied by comparative XPS and LEIS experiments. In addition, the quantification of the oxygen diffusion in the SCS surface phase after the water reaction was attempted using the isotope exchange depth profile technique (IEDP). In our complementary experiments we replicated the study with SrCo0.095Sb0.05O3−δ and SrCo0.095Mo0.05O3−δ (SCM) powder samples due to their commercial interest for water splitting application. Finally, transmission electron microscopy (TEM) and thermogravimetric analysis (TGA) were employed to explore both the material surface phase change and the redox kinetics behaviour after the materials' reaction with water.
. Theoretical and experimental results proved that this could also be a cause for cation segregation. Due to the complex nature of the problem, both elastic and electrostatic driving forces concurrently affect cation segregation, and each contribution is not easily separable. The third and less explored aspect of the segregation is the reaction of the material with the gas atmosphere, in particular with H2O and CO2. These chemical reactions are ultimately responsible for the appearance of reactant species such as hydroxide, oxide or carbonates at the cathode surfaces.37–42 Niania et al. studied the effect of different gas atmospheres at high temperature on surface strontium segregation of a La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) bulk material and determined that a water enriched atmosphere had a much greater effect upon particle growth compared to air or oxygen atmospheres.42 In this study, we investigated the surface SrO-formation in the B-site substituted SCO perovskite (Sb, Mo). The reactivity of the B-site substituted SCO phase towards water has been explored both at room temperature and at high temperature by two different experimental approaches. The first method employed dense epitaxial SCS films grown by pulsed laser deposition (PLD) on different single crystals: SrTiO3 (001) (STO), (LaAlO3)0.3(Sr2TaAlO6)0.7 (001) (LSAT) and NdGaO3 (110) (NGO). The effect of water cleaning at room temperature before thermal treatment was investigated. The water reaction with the SCS surface was studied by comparative XPS and LEIS experiments. In addition, the quantification of the oxygen diffusion in the SCS surface phase after the water reaction was attempted using the isotope exchange depth profile technique (IEDP). In our complementary experiments we replicated the study with SrCo0.095Sb0.05O3−δ and SrCo0.095Mo0.05O3−δ (SCM) powder samples due to their commercial interest for water splitting application. Finally, transmission electron microscopy (TEM) and thermogravimetric analysis (TGA) were employed to explore both the material surface phase change and the redox kinetics behaviour after the materials' reaction with water.
2 Experimental
2.1 SCS and SCM powder preparation by the citrate route
B-site substituted SrCoO3 (B = Sb, Mo) with the composition SrCo0.95B0.05O3−δ was synthesised using the citrate-modified method11 whereby stoichiometric amounts of precursor powders (Sr(NO3)2 (>99.0%, Sigma Aldrich), Co(NO3)2·6H2O (98.0–102.0%, Alfa Aesar), (NH4)6Mo7O24·4H2O (99.0%, Alfa Aesar), and Sb2O3 (99.0% Alfa Aesar)) were weighed and added to a stirring solution of 0.1 M citric acid (aq. 1.0% (v/v) 68.0% nitric acid). The precursors and the solution were heated to a temperature between 60 and 80 °C resulting in their complete dissolution. Water was subsequently evaporated by heating to 350 °C promoting the gelation of the solution. The dry gel was decomposed in an extraction furnace at 600 °C for 12 hours before further calcination steps in a muffle furnace at 900, 1000 and 1200 °C, for 12, 24 and 24 hours respectively with intermediate grinding stages using an agate mortar. To produce the SCS-PLD target, 7 g of powder prepared with this procedure was pressed first in a uniaxial press at 5 tons (pellet ϕ = 20 mm) and subsequently in an isostatic press at 300 MPa. A sintering step in a tubular furnace at 1200 °C for 12 h was necessary to obtain a pellet density of around 93% (measured by geometry).
In the comparative study of the substituted SCO phase decomposition, the as-sintered powder was compared with the water treated one. In this case the nomenclature “no water” and “water” was employed, respectively.
2.2 PLD thin film preparation
The PLD films were grown using a Compex Pro 201 KrF excimer laser (248 nm) at 1 Hz pulse repetition with a laser fluence of around 2 J cm−2 (80 mJ over 4 mm2 spot size). Ablation of the SCS dense target was performed in a 32 mbar atmosphere of pure O2 (99.999%). Substrates were commercial single crystals of SrTiO3 (001) (STO), (LaAlO3)0.3–(Sr2AlTaO6)0.7 (001) (LSAT) and NdGaO3 (110) (NGO) from Crystal, GmbH. Substrate temperatures were set to 800 °C. Target to substrate distance was fixed at about 50 mm. After the SCS thin film deposition, and before the film was cooled to room temperature, the PLD chamber was filled with 800 mbar of oxygen to compensate oxygen defects in the films which form due to the reducing conditions of the PLD deposition technique. In the study of the effect of water on the substituted SCO thin film surface decomposition, the effect of water and acetone was compared. Only for the thin film experiments, the nomenclature “water” and “acetone” was employed.
2.3 XPS analysis
The surface chemistry and the electronic structure of the cathode were characterized using high-resolution X-ray photoelectron spectroscopy (XPS). To reduce the adsorbed carbon on the sample surface, the samples were analysed just after an annealing in dry pure oxygen at 300 °C in a closed tube. The spectra were recorded on a Thermo Scientific K-Alpha+ X-ray photoelectron spectrometer operating at 2 × 10−9 mbar base pressure equipped with a monochromated, micro-focused Al Kα X-ray source (hν = 1486.6 eV) and a 180° double focusing hemispherical analyser with a 2D detector. The X-ray source was operated at 6 mA emission current and 12 kV anode bias providing an X-ray spot size of 400 μm2. Survey spectra were recorded at 200 eV pass energy, 20 eV pass energy for the core level. The binding energy was calibrated to the C 1s photoemission peak of adventitious hydrocarbons at 284.8 eV. The quantitative XPS analysis was performed using the Avantage software.
2.4 LEIS analysis
The outermost atomic surface and sub-surface chemical compositions were measured with a Qtac100 low-energy ion scattering spectroscopy (LEIS) instrument (IONTOF GmbH, Münster, Germany) operated with normal incidence He+ (3 keV) or Ne+ (5 keV) as primary beam sources. The instrument is coupled with a secondary ion beam sputtering source of 0.5–2 keV Ar+, incident at 45° to normal. This was employed to provide information about the chemical composition of the sub-surface region. The LEIS depth profile analysis was run by alternating the analysis Ne+ beam with the Ar+ sputtering beam with a dwell time of 0.5 s. The primary beam scanning area was set as 500 × 500 μm2 and the rastered area with the secondary ion beam was 1000 × 1000 μm2.
2.5 Isotope exchange depth profile (IEDP) experiments
SCS thin film surfaces were cleaned with acetone or water and dried with a cotton stick before the exchange experiment. The samples subjected to isotopic exchange were enclosed in a quartz tube and pumped down to 10−7 mbar. A first annealing in 200 mbar of pure oxygen N5 (99.999%) with 18O2 in the normal isotopic abundance was performed. This pre-annealing is employed to neutralize any oxygen material defects induced by the high reducing conditions in which the films were grown in the PLD chamber. After pre-annealing the tube was pumped down again and re-filled with an ∼90% 18O2 enriched gas (200 mbar). After annealing the sample was quenched to room temperature to avoid any oxygen back-exchange. The exchange temperature and time were around 400 °C and 10 min respectively. As reported by Chater et al. 10 min exchange is close to the lower time limit for an IEDP experiment. This will reduce the accuracy of the results.43 However, as these experiments were meant as a comparative study, and the samples treated with different solvents were exchanged at the same time, we believe that this limitation will not affect our conclusions. Further details about IEDP experiments are provided in Chater et al.43 Once exchanged, the 18O diffusion profile signal was recorded using a TOF-SIMS 5 instrument (ION-TOF GmbH, Munster, Germany). The negative ion analysis was performed using a liquid metal bismuth gun (LMIG). A 25 keV Bi+ primary ion beam was used to generate the secondary ion detected in the burst alignment mode. A secondary Cs+ ion beam (1 keV) was employed for the depth profile analysis. The analysis beam area was 50 μm × 50 μm while the sputtering area was 150 μm × 150 μm. The pause time between analysis and sputtering was 0.1 s.
2.6 Thermochemical water splitting of SCS and SCM powders
Thermochemical water splitting was conducted using an experimental rig as illustrated in Fig. S1.† Approximately 300 mg of oxide powder was loaded onto a quartz wool base in a quartz tube before being secured in a vertically orientated cylindrical flow furnace. The powder was heated to the reducing temperature, Tred at a rate of 20 °C min−1 under a continuous 60 ml min−1 nitrogen (≥99%) flow. A 20 minute isothermal hold at Tred was included to equilibrate the non-stoichiometry within the powder, before rapid cooling to the chosen oxidation temperature, Tox. An 80% water vapour to gas ratio atmosphere was subsequently introduced by controlled evaporation and mixing at 80 °C. Hydrogen, nitrogen, and oxygen output gas pressures were simultaneously measured using a Thermo Fisher ProLab Benchtop Mass Spectrometer. Isothermal water splitting experiments where Tred = Tox were also performed.
2.7 TEM analysis
Imaging of the samples at the nanoscale was carried out by transmission electron microscopy (TEM) using an FEI Titan operating at a 300 kV voltage equipped with a Cs aberration image corrector. TEM specimen preparation was carried out by drop-casting a suspension of the powder samples, previously sonicated in high purity isopropanol, on holey carbon film copper grids (3.05 mm diam. 300 mesh, TAAB). Lattice spacing measurement was performed by the analysis of FFT patterns generated from the regions of interest in the raw micrographs.
2.8 TGA analysis
The oxygen storage capability of the substituted SCO powders was investigated using a Netzsch F5 thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) system in tandem. Approximately 75 mg of powder was loaded into a platinum crucible and heated to the target temperature at a rate of 10 °C min−1 under a nitrogen flow of 60 ml min−1. The target temperature was held for 20 minutes to reach the equilibrium. Redox experiments were run isothermally changing the analysis gas atmosphere between N2 and synthetic air and vice versa.
3 Results and discussion
3.1 Low energy ion scattering surface analysis
One gram of SrCo0.95Sb0.05O3−δ powder, prepared by the citrate route, was immersed in water for 5 min and dried in air at 100 °C. Another gram was immersed only in acetone. The as-synthesised SCS and SCM powder purity was confirmed by Le Bail refinement of the XRD powder diffraction data (see Fig. S2†). After drying the powder at 100 °C, the two samples were analysed by LEIS. This technique allows the chemical composition of the first atomic layer of the sample to be determined. When employing the low energy argon secondary ion beam, it is possible to perform depth profile analysis and obtain the chemical information of the first 1–30 nm of the sample surface. Details about the LEIS technique can be found elsewhere.44Fig. 1 shows the normalised intensity of the LEIS spectra of Sr, Sb and Co, respectively. The sample that was treated in water presents less Co and Sb at the surface. Otherwise, the Sr concentration increased. LEIS provides accurate chemical information of the first monoatomic layer with the highest surface sensitivity of all the spectroscopic surface analysis techniques (i.e. SIMS, XPS). However, it is impossible to obtain information about the nature of the Sr-species enriched at the sample surface. For this reason, XPS was also employed. The SCS powder treated in water and dried at 100 °C was analysed by XRD as well. The powder diffraction was inconclusive and the presence of crystalline SrO-species was not clear (XRD pattern is presented in Fig. S3†). This could indicate that the reaction at this low temperature is constrained at the material surface.
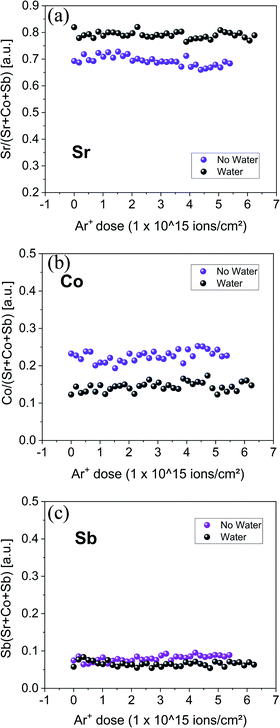 |
| | Fig. 1 Ne+ 5 keV depth profile LEIS analysis of SCS powders. For each spectrum, the Sr, Co and Sb species energy peaks were integrated, and the intensities plotted as a function of the total Ar+ sputtering dose. The as-synthesized powder (purple dots) and the same batch of powder but analyzed after being dipped in water and dried at 100 °C (black dots) are compared. | |
3.2 XPS analysis on substituted SCO
A thin film sample of SCS (∼15 nm), grown on NGO (001) single crystal by PLD, was cut into two parts with a diamond saw. One part was immersed in acetone and the other half in Milli-Q purified water for 5 min. The two samples were dried with a cotton stick and heated in a controlled atmosphere (200 mbar of pure oxygen N5) at 300 °C for 1 h. This procedure was optimized to eliminate as much adsorbed carbon as possible, which could otherwise complicate the XPS analysis. After heating in a closed tube, the two samples were directly loaded into the XPS chamber to reduce air exposure. In Fig. 2, XPS analysis of Sr 3d core level is presented. Generally, the fitting of this core level reported in the literature is obtained with two features: one narrow contribution at lower binding energy, assigned to lattice Sr–O, and a wider contribution at higher binding energy, assigned to Sr at the outermost surface such as Sr–CO3 and surface Sr–O or Sr–(OH)2.45–48 Similar to Van der Heide,49 this model did not fit our data well and a three-contribution model of similar full width half maximum (FWHM) was chosen instead, with contributions from lower to higher binding energy of lattice Sr–O, surface Sr–O or Sr–(OH)2 and Sr–CO3, respectively. Details about the fitting are shown in Table S1.† The sample treated in water shows an increase of the surface Sr–O or Sr–(OH)2 – species and a reduction of the lattice Sr–O (corresponding to the Sr in the perovskite structure). This was proved reproducible for different samples grown on different substrates (see Fig. S4†). The increasing Sr-concentration at the material surface agrees with the LEIS measurement results. Even though the contact with water was relatively short, this proved to be sufficiently long to trigger the strontium reaction and formation of a SrO-enriched layer at room temperature. Tunney et al. reported an XPS study for the SrFeyCo1−yOx phases (y between 0 and 0.9).50 Their study was performed on 250–350 nm thin films deposited by PLD on a (1−102) single-crystal sapphire substrate. For y = 0, the film adopted the SrCoO2.52 hexagonal structure.50 XPS analysis showed two main features for the O 1s spectra: binding energy at 529.5 eV and between 531 and 532 eV respectively, similar to our observations for the O 1s spectra of the SCS sample dipped in water, Fig. 3. In this sample the O-lattice binding energy doublet reduces dramatically in comparison with the sample cleaned in acetone. A more detailed fitting of the O 1s spectra can be found in Fig. S5.† Some similarities can also be observed for the Sr 3d doublet, where, for the sample treated in water, there is a reduction of the lattice Sr–O contribution and an increase of the surface Sr–O contribution. This could be suggesting a partial decomposition of the film surface and the possible formation of the Sr6Co5O15 trigonal phase. To separate the effect of the water cleaning from the heating at 300 °C, a 15 nm SCS thin film sample deposited on NGO (110) single crystal was cut and subjected to the following treatment: sample-1 (S1) cleaned in acetone, dried with a cotton stick and heated in pure oxygen at 300 °C, sample-2 (S2) cleaned in water and dried at room temperature, sample-3 (S3) cleaned in water, dried with a cotton stick and heated in pure oxygen at 300 °C. Fig. 4 shows the comparative XPS results for the three samples. The results were normalised to the peak of the Co 2p3/2. The C 1s spectra reveal the effect of heating–cleaning at 300 °C in oxygen: as a matter of fact, sample 2 (S2), only dried at RT, is the one with the higher adventitious carbon at a binding energy of 284.8 eV. This adsorbed carbon is removed during the heat treatment in oxygen. Looking then at Sb 3d3/2, a difference between the samples cleaned in water and acetone is also apparent. When the sample was washed with water, the Sb 3d3/2 originally located at 538 eV and assigned to the lattice SCS moves towards higher binding energy (539 eV) corresponding to the Sb2O3 species.51 This is in agreement with an SCS film surface decomposition mechanism into SrO, Sb2O3 and SrCoOx species. From these comparison experiments we can conclude that the degradation of the surface in the presence of water is already starting at room temperature. XPS analysis typically samples between 7 and 10 nm of the sample surface. Since the Sr2+-bulk and the O-lattice signals are still present in the Sr 3d and O 1s spectra respectively, we can estimate a decomposition volume that does not overcome the first 10 nm of the SCS surface. Rupp et al. reported the effect of water in removing the SrO forming at the La0.6Sr0.4CoO3−δ thin film surface.45 From the surface of the La0.6Sr0.4CoO3−δ cathode analysed in Rupp's study, the insulating SrO-species formed at the surface during the PLD deposition was partially water-dissolved at room temperature. For substituted SCO material, instead, water seems to trigger a surface material decomposition process related to the thermodynamic instability of the phase as we will discuss later. This phase instability was also demonstrated for both the Mo-substituted SCO (XPS results in Fig. S6†) and for A-site deficient Sr0.95Co0.95Sb0.05O3−δ material (XPS results in Fig. S7†) both analysed in the powder state. The change in the Co 2p spectra for the powder sample treated with water showed a clear decrease of Co3+/4+ and an increase of Co2+, suggesting a decrease of the total formal oxidation state of the cobalt, in agreement with the formation of Sr6Co5O15.
 |
| | Fig. 2 Sr 3d core level XPS analysis of 15 nm SCS thin film grown by PLD on an NGO single crystal. The sample was cut into two, half treated in acetone (left) and half in water (right) for 5 min. After drying, both samples were treated at 300 °C in pure oxygen before the XPS analysis. | |
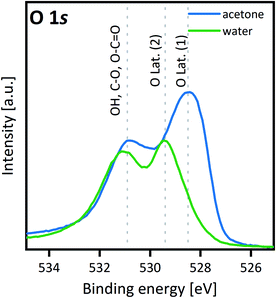 |
| | Fig. 3 O 1s core level comparison XPS analysis of 15 nm SCS thin film. For the film previously treated in water a binding energy peak at 529.4 eV appears and the M–O peak intensity decreases at the same time. The O 1s binding energy spectra are comparable to the one previously reported by Tunney measuring a hexagonal SCO thin film.50 | |
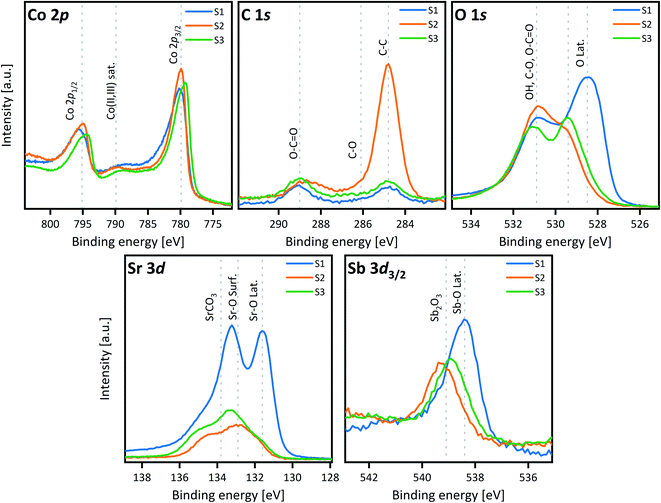 |
| | Fig. 4 XPS study on the effect of temperature and cleaning solvent in SCS thin film surface chemical change: sample-1 (S1) cleaned in acetone, dried with a cotton stick and heated in pure oxygen at 300 °C, sample-2 (S2) cleaned in water and dried at room temperature, sample-3 (S3) cleaned in water, dried with a cotton stick and heated in pure oxygen at 300 °C. | |
3.3 Isotope exchange depth profile experiments
The effect of SCS surface decomposition was also studied by IEDP experiments. The same sample surface water treatment was applied prior to the IEDP analysis experiments. A 15 nm SCS thin film grown on LSAT (100) sample was cut into two. One piece was cleaned in water, the other in acetone for 5 min. Afterwards both were dried using a cotton stick. To reduce the experimental uncertainty, both pieces were exchanged at the same time in the same exchange tube. After pre-annealing in pure 16O2 atmosphere, the samples were heated at 400 °C for 10 min in 200 mbar of 18O2 enriched gas atmosphere (∼90% enriched). By Time-Of-Flight-Secondary-Ion-Mass Spectrometry (TOF-SIMS) the 18O isotope-diffusion concentration profile was measured. Fig. 5 shows the oxygen concentration profile for the sample cleaned in water (orange dots). Under these exchange conditions, since the substrate will act as an oxygen barrier, this very thin film (15 nm) should fully saturate with 18O, however a diffusion profile was measured instead. From the 18O diffusion curve fitting, D* and k* of 5 × 10−17 cm2 s−1 and 2 × 10−11 cm s−1 were measured respectively for the exchange at 400 °C. The fact that an 18O diffusion profile was observed confirms the hypothesis that between the decomposition products there must be a phase capable, even if slowly, of conducting oxygen such as the trigonal unsubstituted Sr6Co5O15 phase. In contrast, the sample cleaned only in acetone (green dots) presents an 18O saturation plateau at higher 18O concentration compared with the piece cleaned in water, indicating faster oxygen diffusion. It is worth commenting on the apparent uphill oxygen diffusion at the surface of the sample. This unusual behaviour is consistent with a partial surface reaction with atmospheric water. Samples were kept under atmospheric conditions after PLD deposition. A partially decomposed top-surface, not covering the full sample surface, would appear in the TOF-SIMS depth profile as a lower 18O concentration. This concentration would increase with depth as soon as the decomposed layer would be sputtered, reaching an 18O saturation level that primarily depends on the annealing temperature (see the schematic in Fig. S8†). Since acetone is hygroscopic and can absorb till an 8% of water, the initial uphill 18O increase can otherwise be related to the partial surface reaction with the water contained in the acetone. In the experiments on thin films, no measurement of the water content in the acetone was performed. However, similar XPS results obtained on the SSC powders (Fig. S7†), where the as-sintered powder (no acetone treatment) is compared with the water-treated one, seem to indicate a low/null effect of the acetone absorbed water on the thin film IEDP results. From these experiments, it can be concluded that the surface degradation reaction occurs already at RT. The change in oxygen diffusion behaviour depending on the cleaning solvent (water or acetone) was confirmed for the films grown on NGO and STO single crystal substrates (Fig. S9†).
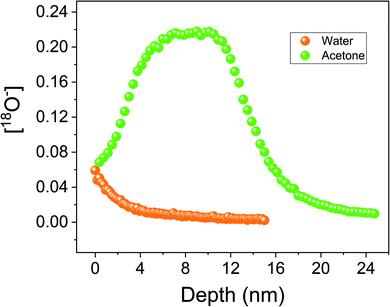 |
| | Fig. 5 IEDP experiment was performed on 15 nm SCS thin film deposited on LSAT single crystal cut into two parts. TOF-SIMS depth profile analysis is presented here. Before the isotopic exchange, half a sample was cleaned in acetone (green dots) and the other half in water (orange dots). The oxygen-18 concentration is plotted as a function of the sputtering time. Exchange temperature and time were around 400 °C and 10 minutes, respectively. | |
3.4 Substituted SrCoO3−δ thermochemical water splitting
To prove the surface degradation effect for practical applications, we performed an experiment to evaluate the activity of this material in thermochemical water splitting (TCWS) for hydrogen production applications. Typically, a two-step cyclical process is employed: in the first step both changes in temperature and oxygen partial pressure are used to reduce the material; in the second step the material is re-oxidized in a water-enriched atmosphere with consequent hydrogen production.52 In recent years, perovskites have gained more interest for TCWS application due to their large oxygen storage capabilities and lower operating temperatures compared to the current state of the art material, ceria. Even though at these lower water splitting temperatures (<800 °C) the total free energy change of the two step process is generally endergonic, by employing a significant H2O excess, it is possible to push the reaction towards the water splitting direction as reviewed by Kubicek et al.53 SCS and SCM were considered promising materials for TCWS due to their known large oxygen storage content and fast surface reaction kinetics.54,55 Powders were produced by a citrate route and their purity checked by XRD. The preliminary TGA analyses of SCS and SCM powder samples in a nitrogen atmosphere have shown maximum O2 exchange capacities of 36 and 37 mmol per mol of SCS and SCM at 600 °C respectively (the SCM TGA analysis, as an example, is shown in Fig. S10†). Both phases, SCS and SCM, were tested in a furnace system connected to a mass spectrometer, a schematic of the system is presented in Fig. S1.† Mass spectrometry was employed to monitor both oxygen evolution during the material reduction, and hydrogen evolution during water oxidation. The gas atmosphere was regulated by mass flow controllers, changing from synthetic air to a low pO2 nitrogen atmosphere. Water partial pressure was instead controlled using a water mass flow controller (Bronkhorst). The system was initially tested by employing ceria powder to verify that the appropriate oxidation/reduction processes were observed. In Fig. 6 the mass spectra for the SCM powder sample reduced at 1000 °C and water-oxidized at 800 °C are presented. While the oxygen evolution peak is clearly observable between 500 °C and 1000 °C, there was no sign of hydrogen evolution when water (80%) was added to the system gas flow. The experiment was repeated several times for both SCS and SCM and it consistently showed no or very low hydrogen production compared to the theoretically expected values from the oxygen evolution. XRD analysis before and after the TCWS experiment, Fig. 7, revealed that the tetragonal phase partially decomposes towards the trigonal Sr6Co5O15. For completeness, an isothermal TCWS at 600 °C, where a very low hydrogen evolution was measured, and the XRD analysis of an SSC powder after TCWS are also presented in Fig. S11.† LEIS analysis of the powder before and after the water splitting experiment (1000–800 °C experiment) confirmed an increase of the Sr surface species (Fig. S12†). An increase of Sr concentration similar to that observed for the PLD SCS thin film samples after water cleaning at room temperature was identified. The formation of a phase with very low oxygen permeability such as the Sr6Co5O15 trigonal phase, agrees with the measured low oxygen yield under reduction conditions. Sr6Co5O15 oxygen permeability was previously investigated by Kruidhof employing the oxygen permeability technique.8
 |
| | Fig. 6 Mass spectra of a thermochemical water splitting experiment performed on the SCM powder. The O2 evolution signal during the powder reduction in N2 is shown in blue. Two oxygen evolution peaks are present. This is probably related to the phase transition from tetragonal to the cubic phase. When water is introduced in the system, blue area, no hydrogen evolution was detected. | |
 |
| | Fig. 7 XRD analysis of SCM powder as synthesized (green) and after the TCWS experiment (red). Part of the material has decomposed during the thermochemical water splitting experiment, forming the trigonal phase. | |
3.5 Mechanism, thermodynamics and kinetics considerations on the water induced substituted SCO phase decomposition
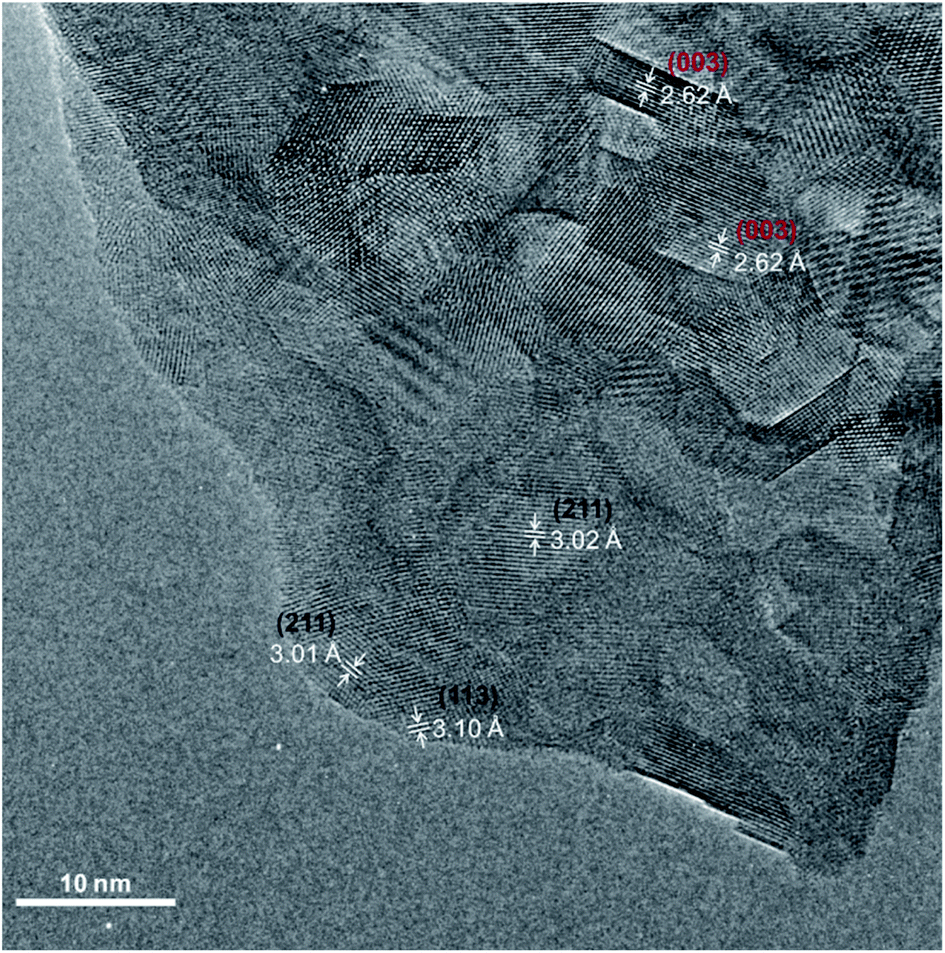
TEM analysis was performed on SCS powder treated in water and dried at 100 °C for 12 h. In Fig. 8, the High-Resolution Transmission Electron Microscopy (HRTEM) image of the powder is presented. Even though the untreated powder grain-size is roughly 3–5 μm, the powder surface, after water treatment, is covered with small grains in the size range of 5–20 nm. HR-TEM images of the powder after water treatment, showed a d-spacing of ∼3.11 Å and 3.01 Å corresponding unequivocally to the Sr6Co5O15 (113) and (211) planes (black labelling), respectively.56 The presence of the tetragonal SCS phase grains was also confirmed by d-spacings of 2.62 Å (003) and 2.59 Å (111) (red labelling) in Fig. 8. This finding is in good agreement with our hypothesis that the tetragonal SCS phase is not stable in contact with water, even at RT. The trigonal phase, also represented as SrCo0.86O2.5, has a deficiency in the B-site cation. This cobalt deficiency is supported by the XPS observation. When the SCS thin film surface decomposed after contact with water, the Sb 3d3/2 spectra present a higher binding energy with respect to the Sb in the unreacted SCS film, indicating the formation of Sb2O3 species. The same can be said for the Mo substituted SCO. Note that the presence of Co substituent (Sb or Mo in this study) stabilises a 3C-disposition of the octahedra, whereas the lack of it, stabilises a face sharing hexagonal disposition.11 Antimony or molybdenum being released from the perovskite structure would cause, inevitably, a deficiency in the cobalt content. The substituent in this composition occupies only 5% of the B-site position. It is therefore possible that more cobalt from the perovskite structure reacts, forming a different species. This was observed after the TCWS experiments by XRD analysis of the post-treated powder, Fig. 7, showing the formation of a CoO phase as well as the other decomposition oxide species. SrO species enrichment at the surface was observed by LEIS and XPS for the samples treated with water. In this case, the main mechanism driving force for the surface strontium segregation is clearly the reaction with water. However, it is difficult to confirm if this enrichment has a role in the phase degradation mechanism. Otherwise, the formation of Sb2O3 or MoO3 could be the triggering reaction for the decomposition of the substituted SCO in the presence of water.
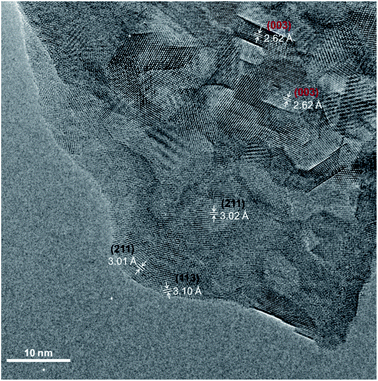 |
| | Fig. 8 High-resolution TEM micrograph of the SCS powder dipped in water and dried at 100 °C. Lattice spacings were obtained from analysis of FFTs generated on different ROIs of the image. Crystallographic planes correspond uniquely to the Sr6Co5O16 phase (labelled in black) and to the SCS tetragonal phase (labelled in red). | |
In the powder water treated at RT and analysed by HR-TEM the degradation mechanism is expected to be the same as in the case of the high temperature thermochemical water splitting experiment. No crystalline CoO or SrO species could nevertheless be found by HR-TEM analysis, indicating in this case that these species could be in an amorphous state. Actually, at the surface, in some areas of the water-treated powder, an amorphous phase (thickness < 2 nm) was also observed, Fig. S13.†
Zhang et al., employing a predictive model, calculated the Gibbs energy for all of the phases in the Sr–Co–O system. From the calculated phase diagram, the trigonal phase Sr6Co5O15 results as the more stable phase in a large Co composition range at temperatures below 1200 K.57 Usiskin et al. investigated the thermodynamic stability of SrCo0.9Nb0.1O3−δ (SCN) in relation with the oxygen stoichiometry variation at different temperatures and p(O2).58 They concluded that the SCN phase decomposition is thermodynamically favoured when the oxygen stoichiometry is greater than 2.58 (the Co formal oxidation state is greater than ∼3.0+). For SCN at atmospheric pO2 pressure the phase is stable only at a temperature above 850 °C. The prediction and this last experimental work agree with the phase instability issues of the substituted-SCO tetragonal phase observed in this work. It can be then argued that the effect of water is associated with an increase of the kinetics of the substituted SCO decomposition reaction, thermodynamically unstable under the employed temperature conditions.
3.6 Effect of the decomposed surface on the material redox kinetics
The effect of water on the SCS powder was also investigated by TGA analysis. In Fig. 9, the oxidation cycle from nitrogen to air atmosphere is compared for the water-treated and untreated powders. The weight gain curve for the as-prepared powder is represented in green while the water-treated one is in blue. Note that the first reduction cycle was not included in the comparison as in this first step, species adsorbed on the sample surface other than oxygen are released. After being reduced in nitrogen at 480 °C, the as-prepared powder shows a higher oxidation kinetics rate (in air) compared with the powder, from the same synthesis batch, dipped in water prior to the TGA analysis. The degraded surface layer clearly affects the oxidation reaction rate. Two full redox cycles are presented in Fig. S14.† In this experiment the SCS powder was dipped in water for a longer time. Considering that the powder with a degraded surface was not completely oxidized before starting the reduction step, it can be concluded that the reduction kinetics rate is also slower for the SCS with a degraded surface. Therefore, whilst initially appearing to be a promising material for TCWS, substituted SCO phases are not exploitable for these applications. This finding also poses the problem of employing these materials in intermediate temperature SOFCs. A strict control of the cathode atmosphere (low water content) would be required to prevent rapid degradation of the IT-SOFC performance.
 |
| | Fig. 9 TGA analysis comparison between SCS powder as synthesized and after being dipped in water and dried. Only the oxygenation step after reduction is shown. TGA temperature was maintained at 480 °C during the redox process. | |
4 Conclusions
In this study the effect of water contact on substituted SCO materials was investigated in both thin film and powder form. At high temperatures (>600 °C), the substituted SCO reaction with water is macroscopic and appears as a phase decomposition of the B-site substituted SCO tetragonal phase (Sb or Mo). Substituted SCO decomposed to the more thermodynamically stable trigonal phase (Sr6Co5O15). By employing epitaxial dense SCS thin films as a model system, evidence of water surface reaction at room temperature was found by comparing XPS and LEIS analysis. The formation of a surface SrO-enriched layer and Sb2O3 after the SCS surface contact with water could be responsible for triggering the SCS phase decomposition and the appearance of Sr6Co5O15. Isotope exchange depth profiling experiments showed that the decomposed surface layer had an oxygen isotope self-diffusion constant, D*, of 5 × 10−17 cm2 s−1 at 400 °C. When the powder reaction with water is at RT, the forming degradation layer is likely confined to the first 5–10 nm of the material surface. This layer has a detrimental effect on the redox kinetics rate as demonstrated by TGA analysis.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors wish to acknowledge the EPSRC for funding from grant numbers EP/R024006/1, EP/R002010/1, EP/R023646/1 and EP/P003532/1 and the European Commission grant FETPROACT-2018-2020 “HARVERSTORE” 824072. Authors would like to thank Dr Sam Cooper from Imperial College London for the fruitful discussion.
References
- B. Koo, K. Kim, J. K. Kim, H. Kwon, J. W. Han and W. C. Jung, Sr Segregation in Perovskite Oxides: Why It Happens and How It Exists, Joule, 2018, 2, 1476–1499 CrossRef CAS.
- Y. Takeda, R. Kanno, T. Takada, O. Yamamoto, M. Takano and Y. Bando, Phase Relation and Oxygen Non-Stoichiometry of Perovskite-Like Compound SrCoOx (2.29 < x < 2.80), Z. Anorg. Allg. Chem., 1986, 540, 259–270 CrossRef.
- J. C. Grenier, S. Ghodbane, G. Demazeau, M. Pouchard and P. Hagenmuller, Strontium Cobaltite, Sr2Co2O5 Characterization and Magnetic Properties, Mater. Res. Bull., 1979, 14, 831–839 CrossRef CAS.
- J. Rodriguez, J. M. Gonzalezcalbet, J. C. Grenier, J. Pannetier and M. Anne, Phase Transitions in Sr2Co2O5 – A Neutron Thermodiffractometry Study, Solid State Commun., 1987, 62, 231–234 CrossRef CAS.
- W. T. A. Harrison, S. L. Hegwood and A. J. Jacobson, A Powder Neutron Diffraction Determination of the Structure of Sr6Co5O15, Formerly Described as the Low Temperature Hexagonal Form of SrCoO3−x, J. Chem. Soc., Chem. Commun., 1995, 1953–1954 RSC.
- Y. Ito, R. F. Klie and N. D. Browning, Atomic resolution analysis of the defect chemistry and micro domain structure of brownmillerite-type strontium cobaltite, J. Am. Ceram. Soc., 2002, 85, 969–976 CrossRef CAS.
- C. de la Calle, A. Aguadero, J. A. Alonso and M. T. Fernández-Díaz, Correlation between reconstructive phase transitions and transport properties from SrCoO2.5 brownmillerite: A neutron diffraction study, Solid State Sci., 2008, 10, 1924–1935 CrossRef CAS.
- H. Kruidhof, H. J. M. Bouwmeester, R. H. E. v. Doorn and A. J. Burggraaf, Influence of order-disorder transitions on oxygen permeability through selected nonstoichiometric perovskite-type oxides, Solid State Ionics, 1993, 63–65, 816–822 CrossRef CAS.
- S. K. Jaiswal, V. K. Kashyap and J. Kumar, Correlation of structure, electrical conductivity, and oxygen permeability in strontium cobaltite ceramic membranes, J. Asian Ceram. Soc., 2020, 8(4), 1018–1026 CrossRef.
- Z. Q. Deng, W. S. Yang, W. Liu and C. S. Chen, Relationship between transport properties and phase transformations in mixed-conducting oxides, J. Solid State Chem., 2006, 179, 362–369 CrossRef CAS.
- A. Aguadero, C. de la Calle, J. A. Alonso, M. J. Escudero, M. T. Fernandez-Diaz and L. Daza, Structural and Electrical Characterization of the Novel SrCo0.9Sb0.1O3−δ Perovskite: Evaluation as a Solid Oxide Fuel Cell Cathode Material, Chem. Mater., 2007, 19(26), 6437–6444 CrossRef CAS.
- P. Zeng, R. Ran, Z. Chen, W. Zhou, H. Gu, Z. Shao and S. Liu, Efficient stabilization of cubic perovskite SrCoO3−δ by B-site low concentration scandium doping combined with sol–gel synthesis, J. Alloys Compd., 2008, 455, 465–470 CrossRef CAS.
- P. Zeng, Z. Shao, S. Liu and Z. P. Xu, Influence of M cations on structural, thermal and electrical properties of new oxygen selective membranes based on SrCo0.95M0.05O3−δ perovskite, Sep. Purif. Technol., 2009, 67, 304–311 CrossRef CAS.
- T. Nagai, W. Ito and T. Sakon, Relationship between cation substitution and stability of perovskite structure in SrCoO3−δ-based mixed conductors, Solid State Ionics, 2007, 177, 3433–3444 CrossRef CAS.
- V. V. Kharton, S. Li, A. V. Kovalevsky and E. N. Naumovich, Oxygen permeability of perovskites in the system SrCoO3−δ–SrTiO3, Solid State Ionics, 1997, 96, 141–151 CrossRef CAS.
- X. Xu, J. Zhao, M. Li, L. Zhuang, J. Zhang, S. Aruliah, F. Liang, H. Wang and Z. Zhu, Sc and Ta-doped SrCoO3−δ perovskite as a high-performance cathode
for solid oxide fuel cells, Composites, Part B, 2019, 178, 107491 CrossRef CAS.
- J. Wang, L. Jiang, X. Xiong, C. Zhang, X. Jin, L. Lei and K. Huang, A Broad Stability Investigation of Nb-Doped SrCoO2.5+δ as a Reversible Oxygen Electrode for Intermediate-Temperature Solid Oxide Fuel Cells, J. Electrochem. Soc., 2016, 163(8), F891–F898 CrossRef CAS.
- M. James, D. Cassidy, K. F. Wilson, J. Horvat and R. L. Withers, Oxygen vacancy ordering and magnetism in the rare earth stabilised perovskite form of “SrCoO3−δ”, Solid State Sci., 2004, 6, 655–662 CrossRef CAS.
- V. V. Kharton, L. Shuangbao, A. V. Kovalevsky, A. P. Viskup, E. N. Naumovich and A. A. Tonoyan, Oxygen permeability and thermal expansion of SrCo(Ti)O3−δ perovskites, Mater. Chem. Phys., 1998, 53(1), 6–12 CrossRef CAS.
- F. Wang, Q. Zhou, T. He, G. Li and H. Ding, Novel SrCo1−yNbyO3−δ cathodes for intermediate-temperature solid oxide fuel cells, J. Power Sources, 2010, 195(12), 3772–3778 CrossRef CAS.
- C. A. Hancock and P. R. Slater, Synthesis of silicon doped SrMO3 (M = Mn, Co): stabilization of the cubic perovskite and enhancement in conductivity, Dalton Trans., 2011, 40(20), 5599–5603 RSC.
- A. Aguadero, D. Pérez-Coll, J. A. Alonso, S. J. Skinner and J. Kilner, A New Family of Mo-Doped SrCoO3−δ Perovskites for Application in Reversible Solid State Electrochemical Cells, Chem. Mater., 2012, 24, 2655–2663 CrossRef CAS.
- Y. Zhu, Z.-G. Chen, W. Zhou, S. Jiang, J. Zou and Z. Shao, An A-Site-Deficient Perovskite offers High Activity and stability for Low-Temperature Solid-Oxide Fuel Cells, ChemSusChem, 2013, 6, 2249–2254 CrossRef CAS PubMed.
- C. Yang, Y. Gan, M. Lee, C. Ren, K. S. Brinkman, R. D. Greenc and X. Xue, Structural evolution, electrochemical kinetic properties, and stability of A-site doped perovskite Sr1−xYbxCoO3−δ, J. Mater. Chem. A, 2020, 8, 10450 RSC.
- W. Kobayashi, S. Ishiwata, I. Terasaki, M. Takano, I. Grigoraviciute, H. Yamauchi and M. Karppinen, Room-temperature ferromagnetism in Sr1−xYxCoO3−δ (0.2 ≤ x ≤ 0.25), Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 104408 CrossRef.
- R. L. Withers, M. James and D. J. Goosens, Atomic ordering in the doped rare earth cobaltates Ln0.33Sr0.67CoO3−δ (Ln = Y3+; Ho3+ and Dy3+), J. Solid State Chem., 2003, 174, 198–208 CrossRef CAS.
- S. Hébert, A. Maignan, V. Caignaert, V. Pralong, D. Pelloquin and B. Raveau, Ferromagnetism and metallicity in the modulated Sr1−xThxCoO3−δ oxygen deficient perovskites with x ∼ 0.1, Solid State Commun., 2005, 134, 815–820 CrossRef.
- S. Ya. Istomin, J. Grins, G. Svensson, O. A. Drozhzhin, V. L. Kozhevnikov, E. V. Antipov and J. P. Attfield, Crystal Structure of the Novel Complex Cobalt Oxide Sr0.7Y0.3CoO2.62, Chem. Mater., 2003, 15, 4012 CrossRef CAS.
- Q. T. Wei, R. S. Guo, F. H. Wang and H. L. Li, Structure and electrical properties of SrCoO3−δ doped by CeO2, J. Mater. Sci., 2005, 40, 1317–1319 CrossRef CAS.
- J. Wang, L. Jiang, X. Xiong, C. Zhang, X. Jin, L. Lei and K. Huang, A Broad Stability Investigation of Nb-Doped SrCoO2.5+δ as a Reversible Oxygen Electrode for Intermediate-Temperature Solid Oxide Fuel Cells, J. Electrochem. Soc., 2016, 163, F891–F898 CrossRef CAS.
- W. Lee, J. W. Han, Y. Chen, Z. Cai and B. Yildiz, Cation size mismatch and charge interactions drive dopant segregation at the surfaces of manganite perovskite, J. Am. Chem. Soc., 2013, 135, 7909–7925 CrossRef CAS PubMed.
- Y. Yu, K. F. Ludwig, J. C. Woicik, S. Gopalan, U. B. Pal, T. C. Kaspar and S. N. Basu, Effect of Sr content and strain on Sr surface segregation of La1−xSrxCo0.2Fe0.8O3−δ as cathode material for solid oxide fuel cells, ACS Appl. Mater. Interfaces, 2016, 8, 26704–26711 CrossRef CAS PubMed.
- J. Y. Koo, H. Kwon, M. Ahn, M. Choi, J.-W. Son, J. W. Han and W. Lee, Suppression of cation segregation in (La,Sr)CoO3−δ by elastic energy minimization, ACS Appl. Mater. Interfaces, 2018, 10, 8057–8065 CrossRef CAS PubMed.
- H. Kwon, W. Lee and J. W. Han, Suppressing cation segregation on lanthanum-based perovskite oxides to enhance the stability of solid oxide fuel cell cathodes, RSC Adv., 2016, 6, 69782–69789 RSC.
- B. Koo, H. Kwon, Y. Kim, H. G. Seo, J. W. Han and W. Jung, Enhanced oxygen exchange of perovskite oxide surfaces through strain-driven chemical stabilization, Energy Environ. Sci., 2018, 11, 71–77 RSC.
- N. Tsvetkov, Q. Lu, L. Sun, E. J. Crumlin and B. Yildiz, Improved chemical and electrochemical stability of perovskite oxides with less reducible cations at the surface, Nat. Mater., 2016, 15(9), 1010–1016 CrossRef CAS PubMed.
- Z. Cai, M. Kubicek, J. Fleig and B. Yildiz, Chemical heterogeneities on La0.6Sr0.4CoO3−δ thin films-correlations to cathode surface activity and stability, Chem. Mater., 2012, 24, 1116–1127 CrossRef CAS.
- E. J. Crumlin, E. Mutoro, Z. Liu, M. E. Grass, M. D. Biegalski, Y.-L. Lee, D. Morgan, H. M. Christen, H. Bluhm and Y. Shao-Horn, Surface strontium enrichment on highly active perovskites for oxygen electrocatalysis in solid oxide fuel cells, Energy Environ. Sci., 2012, 5, 6081–6088 RSC.
- E. Mutoro, E. J. Crumlin, M. D. Biegalski, H. M. Christen and Y. Shao-Horn, Enhanced oxygen reduction activity on surface-decorated perovskite thin films for solid oxide fuel cells, Energy Environ. Sci., 2011, 4, 3689–3696 RSC.
- Y. Chen, H. Téllez, M. Burriel, F. Yang, N. Tsvetkov, Z. Cai, D. W. McComb, J. A. Kilner and B. Yildiz, Segregated chemistry and structure on (001) and (100) surfaces of (La1−xSrx)2CoO4 override the crystal anisotropy in oxygen exchange kinetics, Chem. Mater., 2015, 27, 5436–5450 CrossRef CAS.
- E. J. Crumlin, E. Mutoro, W. T. Hong, M. D. Biegalski, H. M. Christen, Z. Liu, H. Bluhm and Y. Shao-Horn, In situ ambient pressure X-ray photoelectron spectroscopy of cobalt perovskite surfaces under cathodic polarization at high temperatures, J. Phys. Chem. C, 2013, 117, 16087–16094 CrossRef CAS.
- M. Niania, R. Podor, T. Ben Britton, C. Li, S. J. Cooper, N. Svetkov, S. Skinner and J. Kilner,
In situ study of strontium segregation in La0.6Sr0.4Co0.2Fe0.8O3−δ in ambient atmospheres using high-temperature environmental scanning electron microscopy, J. Mater. Chem. A, 2018, 6, 14120–14135 RSC.
- R. J. Chater, S. Carter, J. A. Kilner and B. C. H. Steele, Development of a novel SIMS technique for oxygen self-diffusion and surface exchange coefficient measurements in oxides of high diffusivity, Solid State Ionics, 1992, 53–56, 859–867 CrossRef CAS.
- H. H. Brongersma, M. Draxler, M. de Ridder and P. Bauer, Surface Composition Analysis By Low-Energy Ion Scattering, Surf. Sci. Rep., 2007, 62, 63–109 CrossRef CAS.
- G. M. Rupp, H. Tellez, J. Druce, A. Limbeck, T. Ishihara, J. Kilner and J. Fleig, Surface chemistry of La0.6Sr0.4CoO3−δ thin films and its impact on the oxygen surface exchange resistance, J. Mater. Chem. A, 2015, 3, 22759 RSC.
- R. P. Vasquez, X-ray photoelectron spectroscopy study of Sr and Ba compounds, J. Electron Spectrosc. Relat. Phenom., 1991, 56, 217–240 CrossRef CAS.
- E. Mutoro, E. J. Crumlin, M. D. Biegalski, H. M. Christen and Y. Shao-Horn, Enhanced oxygen reduction activity on surface-decorated perovskite thin films for solid oxide fuel cells, Energy Environ. Sci., 2011, 4, 3689 RSC.
- M. I. Sosulnikov and Y. A. Teterin, X-ray photoelectron studies of Ca, Sr and Ba and their oxides and carbonates, J. Electron Spectrosc. Relat. Phenom., 1992, 59, 111–126 CrossRef CAS.
- P. A. W. Van Der Heide, Systematic x-ray photoelectron spectroscopic study of La1−xSrx-based perovskite-type oxides, Surf. Interface Anal., 2002, 33, 414–425 CrossRef CAS.
- J. J. Tunney, P. Whitfield, X. Du and M. L. Post, Pulsed laser deposition, characterization and thermochemical stability of SrFeyCo1−yOx thin films, Thin Solid Films, 2003, 426, 221–231 CrossRef CAS.
- W. E. Morgan, W. J. Stec and J. R. Van Wazer, Inner-Orbital Binding-Energy Shifts of Antimony and Bismuth Compounds, Inorg. Chem., 1973, 12(4), 953–955 CrossRef CAS.
- W. C. Chueh,
et al., High-Flux Solar-Driven Thermochemical Dissociation of CO2 and H2O Using Nonstoichiometric Ceria, Science, 2010, 330(6012), 1797–1801 CrossRef CAS PubMed.
- M. Kubicek, A. H. Bork Ac and J. L. M. Rupp, “Perovskite oxides – a review on a versatile material class for solar-to-fuel conversion processes, J. Mater. Chem. A, 2017, 5, 11983–12000 RSC.
- A. Aguadero, J. A. Alonso, D. Pérez-Coll, C. de la Calle, M. T. Fernández-Díaz and J. B. Goodenough, SrCo0.95Sb0.05O3−δ as Cathode Material for High Power Density Solid Oxide Fuel Cells, Chem. Mater., 2010, 22(3), 789–798 CrossRef CAS.
- A. Aguadero, D. Pérez-Coll, J. A. Alonso, S. J. Skinner and J. Kilner, A New Family of Mo-Doped SrCoO3−δ Perovskites for Application in Reversible Solid State Electrochemical Cells, Chem. Mater., 2012, 24(14), 2655–2663 CrossRef CAS.
- ICSD – 155311, K. Iwasaki, T. Ito, T. Matsui, T. Nagasaki, S. Ohta and K. Koumoto, Mater. Res. Bull., 2006, 41, 732 CrossRef CAS.
- W.-W. Zhang, M. Chen, E. Povoden-Karadeniz and P. V. Hendriksen, Thermodynamic modeling of the Sr–Co–Fe–O system, Solid State Ionics, 2016, 292, 88–97 CrossRef CAS.
- R. E. Usiskin, T. C. Davenport, R. Y. Wang, W. Guan and S. M. Haile, Bulk Properties of the Oxygen Reduction Catalyst SrCo0.9Nb0.1O3−δ, Chem. Mater., 2016, 28, 2599–2608 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta04174f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
George E.
Wilson
,
Gwilherm
Kerherve
,
Eleonora
Cali
,
Celeste A. M.
van den Bosch
,
Paul
Boldrin
*,
George E.
Wilson
,
Gwilherm
Kerherve
,
Eleonora
Cali
,
Celeste A. M.
van den Bosch
,
Paul
Boldrin
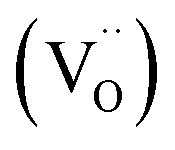 accumulation at the LSC surface and strontium segregation.36 The relatively positively charged oxygen vacancies electrostatically attract the negatively charged A-site dopants, described in the Kröger–Vink notation as
accumulation at the LSC surface and strontium segregation.36 The relatively positively charged oxygen vacancies electrostatically attract the negatively charged A-site dopants, described in the Kröger–Vink notation as  . Theoretical and experimental results proved that this could also be a cause for cation segregation. Due to the complex nature of the problem, both elastic and electrostatic driving forces concurrently affect cation segregation, and each contribution is not easily separable. The third and less explored aspect of the segregation is the reaction of the material with the gas atmosphere, in particular with H2O and CO2. These chemical reactions are ultimately responsible for the appearance of reactant species such as hydroxide, oxide or carbonates at the cathode surfaces.37–42 Niania et al. studied the effect of different gas atmospheres at high temperature on surface strontium segregation of a La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) bulk material and determined that a water enriched atmosphere had a much greater effect upon particle growth compared to air or oxygen atmospheres.42 In this study, we investigated the surface SrO-formation in the B-site substituted SCO perovskite (Sb, Mo). The reactivity of the B-site substituted SCO phase towards water has been explored both at room temperature and at high temperature by two different experimental approaches. The first method employed dense epitaxial SCS films grown by pulsed laser deposition (PLD) on different single crystals: SrTiO3 (001) (STO), (LaAlO3)0.3(Sr2TaAlO6)0.7 (001) (LSAT) and NdGaO3 (110) (NGO). The effect of water cleaning at room temperature before thermal treatment was investigated. The water reaction with the SCS surface was studied by comparative XPS and LEIS experiments. In addition, the quantification of the oxygen diffusion in the SCS surface phase after the water reaction was attempted using the isotope exchange depth profile technique (IEDP). In our complementary experiments we replicated the study with SrCo0.095Sb0.05O3−δ and SrCo0.095Mo0.05O3−δ (SCM) powder samples due to their commercial interest for water splitting application. Finally, transmission electron microscopy (TEM) and thermogravimetric analysis (TGA) were employed to explore both the material surface phase change and the redox kinetics behaviour after the materials' reaction with water.
. Theoretical and experimental results proved that this could also be a cause for cation segregation. Due to the complex nature of the problem, both elastic and electrostatic driving forces concurrently affect cation segregation, and each contribution is not easily separable. The third and less explored aspect of the segregation is the reaction of the material with the gas atmosphere, in particular with H2O and CO2. These chemical reactions are ultimately responsible for the appearance of reactant species such as hydroxide, oxide or carbonates at the cathode surfaces.37–42 Niania et al. studied the effect of different gas atmospheres at high temperature on surface strontium segregation of a La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) bulk material and determined that a water enriched atmosphere had a much greater effect upon particle growth compared to air or oxygen atmospheres.42 In this study, we investigated the surface SrO-formation in the B-site substituted SCO perovskite (Sb, Mo). The reactivity of the B-site substituted SCO phase towards water has been explored both at room temperature and at high temperature by two different experimental approaches. The first method employed dense epitaxial SCS films grown by pulsed laser deposition (PLD) on different single crystals: SrTiO3 (001) (STO), (LaAlO3)0.3(Sr2TaAlO6)0.7 (001) (LSAT) and NdGaO3 (110) (NGO). The effect of water cleaning at room temperature before thermal treatment was investigated. The water reaction with the SCS surface was studied by comparative XPS and LEIS experiments. In addition, the quantification of the oxygen diffusion in the SCS surface phase after the water reaction was attempted using the isotope exchange depth profile technique (IEDP). In our complementary experiments we replicated the study with SrCo0.095Sb0.05O3−δ and SrCo0.095Mo0.05O3−δ (SCM) powder samples due to their commercial interest for water splitting application. Finally, transmission electron microscopy (TEM) and thermogravimetric analysis (TGA) were employed to explore both the material surface phase change and the redox kinetics behaviour after the materials' reaction with water.