
Applications of MAX phases and MXenes as catalysts

Abstract
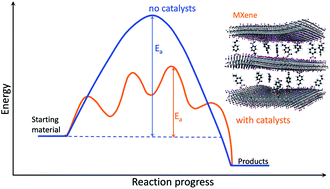
MAX phases and MXenes are important materials that have recently gained great popularity due to their special properties, which render them particularly useful in many applications, including catalytic ones. This can be seen in the large number of publications that appear annually on these materials and their applications. This review aims to evaluate MAX phases and MXenes as materials for heterogeneous, non-electrocatalytic, catalytic applications. The review begins with a brief introduction to the MAX phase and MXene properties that recommend them as potential materials for heterogeneous catalytic applications, followed by four sections grouped according to the processes in which they have already proven effective. These include supports to activate the C–H or C–O bonds in applications such as dehydrogenation of light or aromatic alkanes, methanol formation from CH4, dry reforming, and CO oxidation or the water gas shift reaction (Section 2), and their use in fine chemical reactions (Section 3) and in chemical degradation (Section 4). The last section deals with photocatalytic applications (Section 5). The review ends by highlighting the huge potential of these materials for a wide range of heterogeneous catalytic applications as well as the challenges ahead.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

