
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of novel, known, and low-valent transition metal phosphates via reductive phosphatization†
Niklas
Stegmann
 ,
Hilke
Petersen
,
Claudia
Weidenthaler
and
Wolfgang
Schmidt
*
,
Hilke
Petersen
,
Claudia
Weidenthaler
and
Wolfgang
Schmidt
*
Department of Heterogeneous Catalysis, Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470 Mülheim an der Ruhr, Germany. E-mail: schmidt@mpi-muelheim.mpg.de
First published on 18th August 2021
Abstract
Novel and known low-valent transition metal phosphates (TMPs) are accessible via a novel and facile pathway. The method allows syntheses of TMPs also with reduced oxidation states. The key feature of the new route is the reductive character of a hypophosphite salt melt, which acts as reaction medium and enables directing the oxidation state of the transition metal.
The number of compounds belonging to the class of transition metal phosphates (TMPs) has constantly been growing for decades due to persistent interest in their use as functional materials. Extensively investigated and applied in research and industry, TMP materials contribute to numerous present and future key technologies. Based on unique structural features, TMPs play an important role for clean and efficient power generation and energy storage in the 21st century. Rechargeable lithium-ion batteries apply TMPs as cathode materials. Especially lithium iron phosphate (LiFePO4) is commercially utilized, combining high power capability and reversibility with environmental sustainability and low costs.1–5 In next generation fuel cell technology, metal pyrophosphates (MP2O7, M = Ti, Zr, …) serve as proton conducting membrane materials, due to numerous structure-based proton bonding sites and transport pathways.6 In line with their high density of Brønsted acid sites, proton conducting TMPs are also serving as effective ion-exchangers.7 In the context of heterogeneous catalysis, TMPs are widely used materials for thermo-, electro- and photocatalytic applications due to their unique combination of redox and/or acidic properties. Among the class of TMP catalysts, the vanadyl pyrophosphate (VO)2(P2O7) is a prominent example, representing one of the most extensively studied catalysts in chemistry and the only commercially applied material for the conversion of butane to maleic anhydride.8–10 With respect to renewable energy storage via water splitting, using cobalt or nickel phosphates or mixed cobalt–nickel phosphate (NiPO) for the oxygen evolution reaction presents an interesting alternative to classical catalysts for electrochemical water splitting.11,12 The wide range of applications is complemented by the field of nonlinear optics (NLO), where TMP materials are prepared via crystallization from high-temperature melts of phosphate salts and used for the production of coherent deep-UV light with wavelengths below 200 nm.13
Synthetic phosphate salts are known for most transition metals. Considering the number of metal cations and the structural variety of condensed phosphate units, the sheer quantity of different phases is staggering. The variety of compounds formed by the class of TMP materials is primarily limited by the intrinsic chemical nature of the elementary ionic building units. However, it can be also restricted by availability of precursors and preparation methods. In literature, versatile preparation methods, such as hydrothermal procedures, molten salt processes, or simple precipitation, are established for the synthesis of highly crystalline bulk TMPs. Application of TMPs as functional materials often requires quite sophisticated preparation methods though. Structure directing techniques are used to optimize the performance of the materials, porosity is generated via nanocasting strategies, thin films are formed via gas phase deposition, and nanoparticles are synthesized by electrochemical approaches.12,14–18 No matter what kind of technique is used or what kind of application is envisaged, all methods have in common the use of phosphate-based precursor compounds, very different to the method presented here. In contrast to conventional syntheses, in our route a hypophosphite salt acts as precursor in combination with metal oxides. While the molten hypophosphite salt serves concurrently as reaction medium and phosphate precursor, the metal oxide provides the transition metal cation under reductive conditions. Due to the reducing character of the hypophosphite melt, the transition metal gets reduced during the TMP formation.
The molten salt method is thus a simple and versatile approach, which can be used for the synthesis of known and novel first row transition metal phosphates with low oxidation states at moderate temperatures (300–500 °C). This approach was tested for a series of first row transition metals (Ti, V, Cr, Mn, Fe) under low (300 °C) and high (500 °C) temperature conditions. The results, listed in Table 1, indicate that low temperature conditions tend to form ammonium TMPs, while higher temperatures lead to the formation of ammonium-free TMPs with condensed metaphosphate structures. In almost all cases, the reaction was accompanied by a reduction of the transition metal.
| Precursor | Product (300 °C) | Product (500 °C) | |||
|---|---|---|---|---|---|
| TM oxide | o. s | TMP | o. s | TMP | o. s |
| TiO2 | 4+ | Ti(III)p | 3+ | Ti(PO3)3 | 3+ |
| V2O5 | 5+ | V(PO3)3 | 3+ | V(PO3)3 | 3+ |
| Cr2O3 | 3+ | Cr(NH4)HP3O10 | 3+ | Cr2(P6O18) | 3+ |
| MnO2 | 4+ | — | — | Mn2(P4O12) | 2+ |
| Fe2O3 | 3+ | Fe(II)p | 2+ | Fe2(P4O12) | 2+ |
In a typical reaction, a metal oxide powder, such as TiO2, V2O5, Cr2O3, MnO2, or Fe2O3, is mixed with a surplus of hypophosphite (NH4H2PO2) and heated briefly in a tube furnace under inert gas flow. Then the melt is cooled down and washed with water to yield the pure TMPs (Fig. S1 in ESI†). Detailed synthesis parameters for a series of transition metal compounds are reported in the ESI.† The process window of the method is determined by the temperature and time between the melting point and the thermal decomposition of the hypophosphite precursor (Fig. S2†). The hypophosphite melt mediates the reaction with the dispersed or dissolved metal oxides.
Due to the thermodynamic instability of the hypophosphite anion, the disproportionation into phosphane gas and phosphates limits the reaction. The phosphane acts as spectator under the presented reaction conditions. Excess phosphate compounds, as formed on the crystal surfaces, can easily be removed by washing with water. Formation of metal phosphides via reaction of phosphane and metal oxides, as often described in literature, was not observed under the reaction conditions used here.
A representative example is the reaction of titanium(IV) oxide (P25) and ammonium hypophosphite, resulting in novel and known crystalline titanium(III) phosphate compounds. At 300 °C an unknown crystalline ammonium titanium(III) phosphate compound (NH4Ti(III)P2O7), denoted as Ti(III)p, is formed from the melt as illustrated by the respective XRD pattern in Fig. 1a and S3.† The product is a pyrophosphate as evidenced by a characteristic band at 764 cm−1 in its Raman spectrum. Increasing the reaction temperature to 500 °C yields a known titanium(III) trimetaphosphate, Ti(PO3)3 (Fig. 1c). In this structure the isolated TiO6 octahedra are linked through infinite chains of PO4 tetrahedra.19
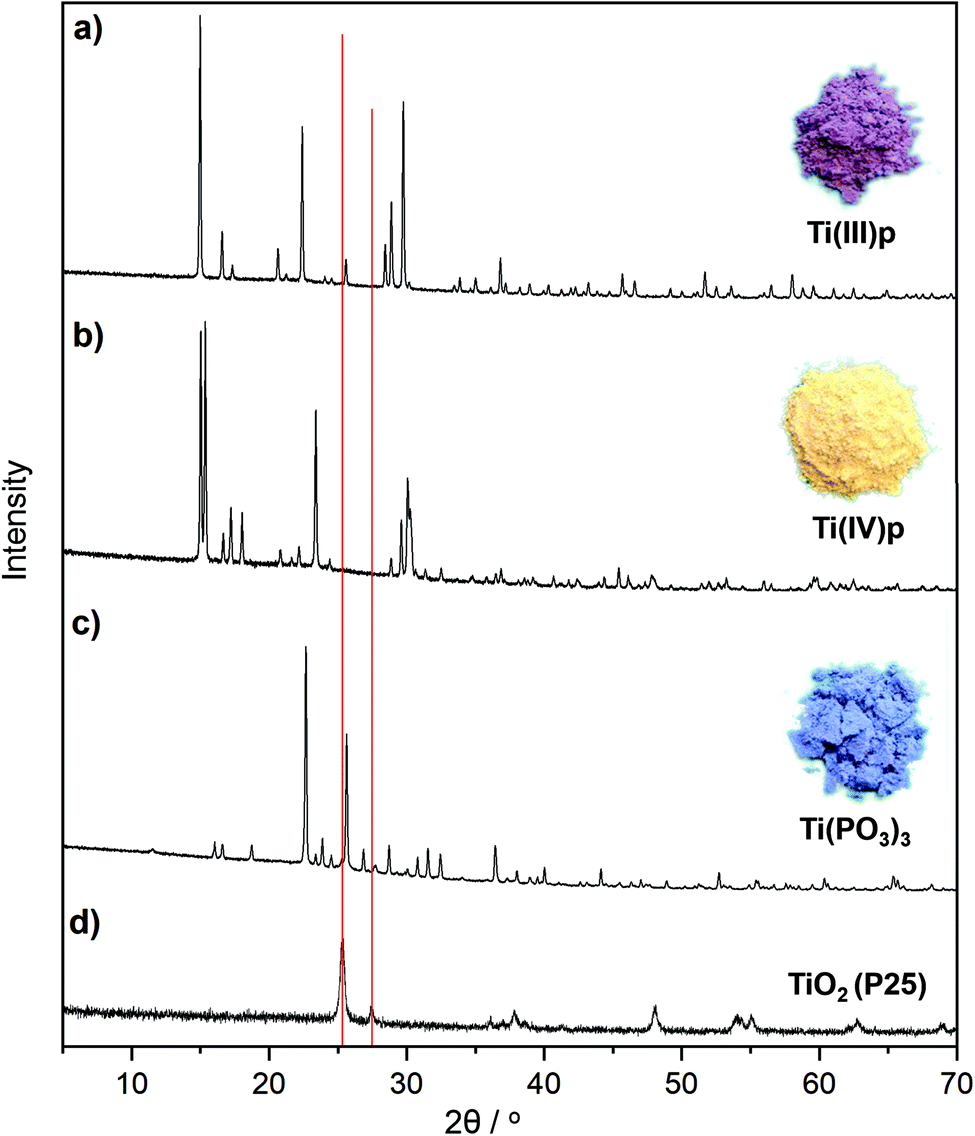 | ||
| Fig. 1 XRD patterns of two novel titanium phosphate phases (a) Ti(III)p, (b) Ti(IV)p and (c) Ti(PO3)3 synthesized via reductive phosphatization of (d) TiO2 (P25) in a melt of ammonium hypophosphite. Red lines indicate the main reflections of rutile and anatase phases of the TiO2 (P25) precursor. Images on the right illustrate the colors of the novel and known product phases. | ||
The formation of a melt by using an excess of hypophosphite is indispensable for complete conversion of the TiO2 and high crystallinity of the product. Syntheses of Ti(PO3)3 with increasing weight ratios of hypophosphite precursor result in different phase compositions as documented by the XRD patterns in Fig. 2. Lower ratios of the hypophosphite precursor (1/1–1/2) are apparently not sufficient to convert the TiO2 quantitatively. However, also these conditions cause partial reduction and the formation of stable titanium(III) oxide species on the surfaces of the titania particles as indicated by the characteristic black coloration and confirmed by XPS spectra (Fig. 3d). Unknown intermediate phases are formed by increasing hypophosphite ratios (1/3–1/5 in Fig. 2) before phase pure Ti(PO3)3 is obtained from the melt (ratio ≥1/6 in Fig. 2).
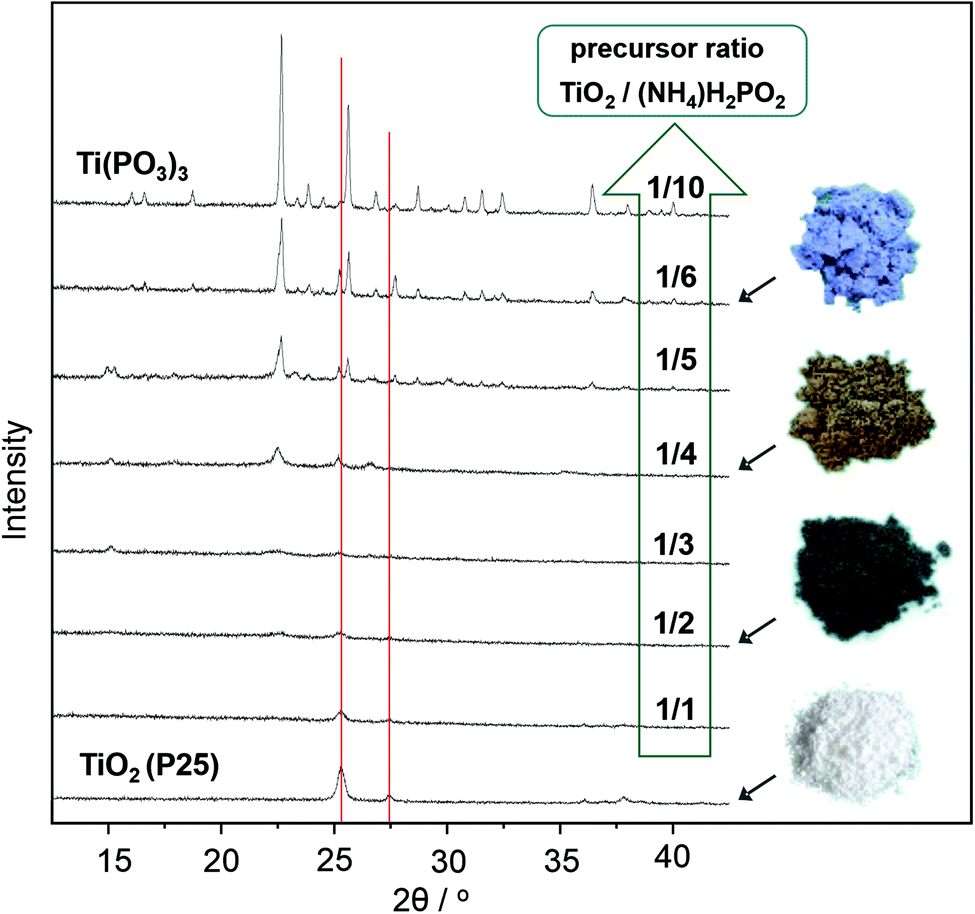 | ||
| Fig. 2 XRD patterns obtained by reductive phosphatization of TiO2 (P25) with increasing (NH4)H2PO2 weight ratios from 1/1 up to 1/10 (green arrow) at 500 °C in argon. Red lines indicate the main reflections of rutile and anatase phases of the TiO2 (P25) precursor. Images on the right illustrate the different colors of reduced products. | ||
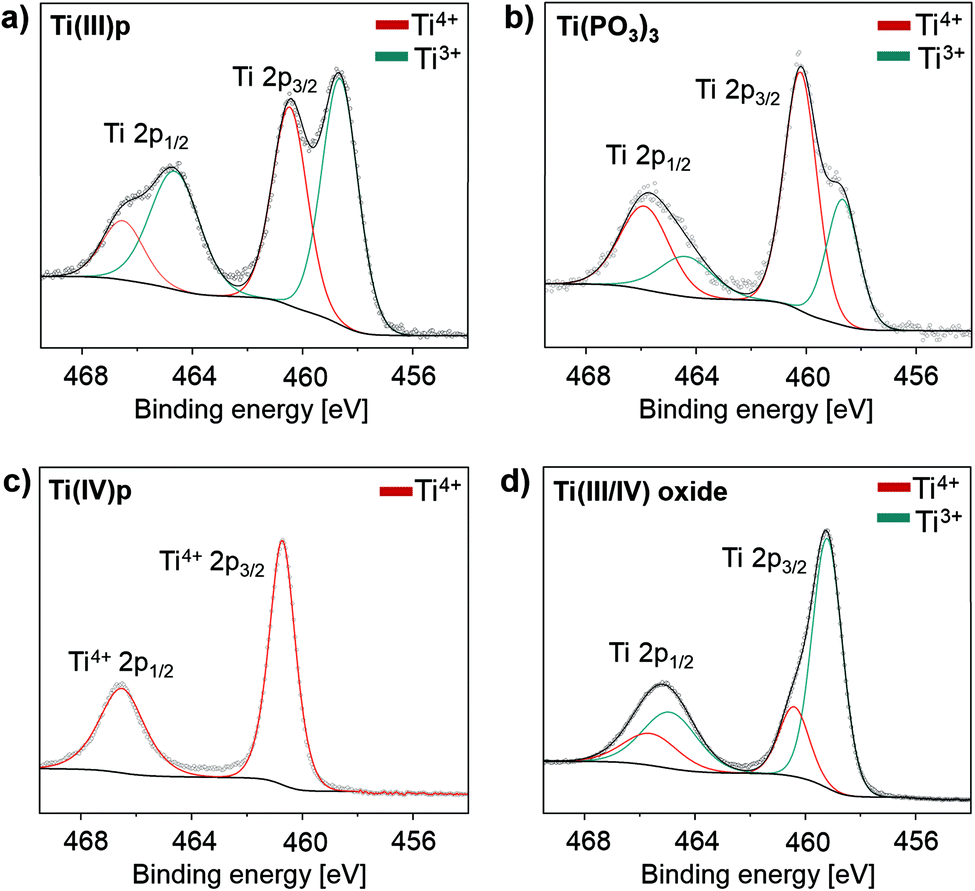 | ||
| Fig. 3 XPS spectra of (a) Ti(III)p, (b) Ti(PO3)3, (c) Ti(IV)p and (d) reductively phosphated Ti(III/IV) oxide synthesized from a mixture of TiO2 and NH4H2PO2 with a weight ratio of 1/2. | ||
In a proposed redox reaction mechanism, the precursors can be considered as oxygen donor and acceptor pairs. While the hypophosphite acts as oxygen acceptor to form stable phosphate compounds, the metal oxide donates oxygen atoms under reductive conditions. Provided that sufficient hypophosphite is available, all oxygen of the metal oxide will be consumed and the reduced cations are free to form crystalline TMP phases. Reports on low-valent titanium(III) phosphates are rather rare in literature, likely due to their strong tendency to oxidize in air.20 XPS spectra show the presence of two different titanium species on the crystal surfaces of Ti(III)p and Ti(PO3)3 as shown in Fig. 3a and b. They can be attributed to Ti(III) and Ti(IV) indicating that the Ti(IV) species exist on the crystal surface in both compounds, likely as the result of oxidation of Ti(III) on the surface by ambient air. XPS analysis probes surface-near regions of the particles, thus, the Ti(IV) signal is caused by surface species. Crystalline Ti(PO3)3 is known to be a pure Ti(III) compound.19
The bulk of the novel Ti(III)p compound offers a good thermal and chemical stability as indicated by XRD data. It is longtime-stable in acidic aqueous solution even at elevated temperature (H3PO4, pH = 1, 80 °C, 72 h) and also thermally stable in ambient air up to 250 °C. At higher temperature in air, the material undergoes a phase transformation to a known titanium(IV) pyrophosphate (TiP2O7). Under non-oxidative conditions, thermal treatment of Ti(III)p yields another novel crystalline Ti(IV) phosphate (Ti(IV)P2O7), denoted as Ti(IV)p, showing exclusively Ti(IV) species (Fig. 3c). TG/DSC-MS (Fig. S4 and S5†) reveals that the phase transformation is accompanied by release of ammonia, hydrogen, and water. The XRD pattern of the novel Ti(IV)p is presented in Fig. 1b and S6.† Reproducibility and purity of the novel crystalline diamagnetic Ti(IV)p compound was confirmed by 31P MAS NMR spectra (Fig. S7†) showing no additional crystalline phase or any amorphous phase in the product (see ESI,† for details). SEM images show a homogenous distribution of micrometer sized crystals for pristine Ti(III)p and Ti(IV)p (Fig. S8†).
Overall, the conversion of TiO2 with ammonium hypophosphite offers reaction pathways to several known titanium phosphate compounds as well as two novel phases as sketched in Fig. 4. As representative example, the reductive phosphatization of Ti(IV) to Ti(III) shows the reductive feature and variability of the hypophosphite route.
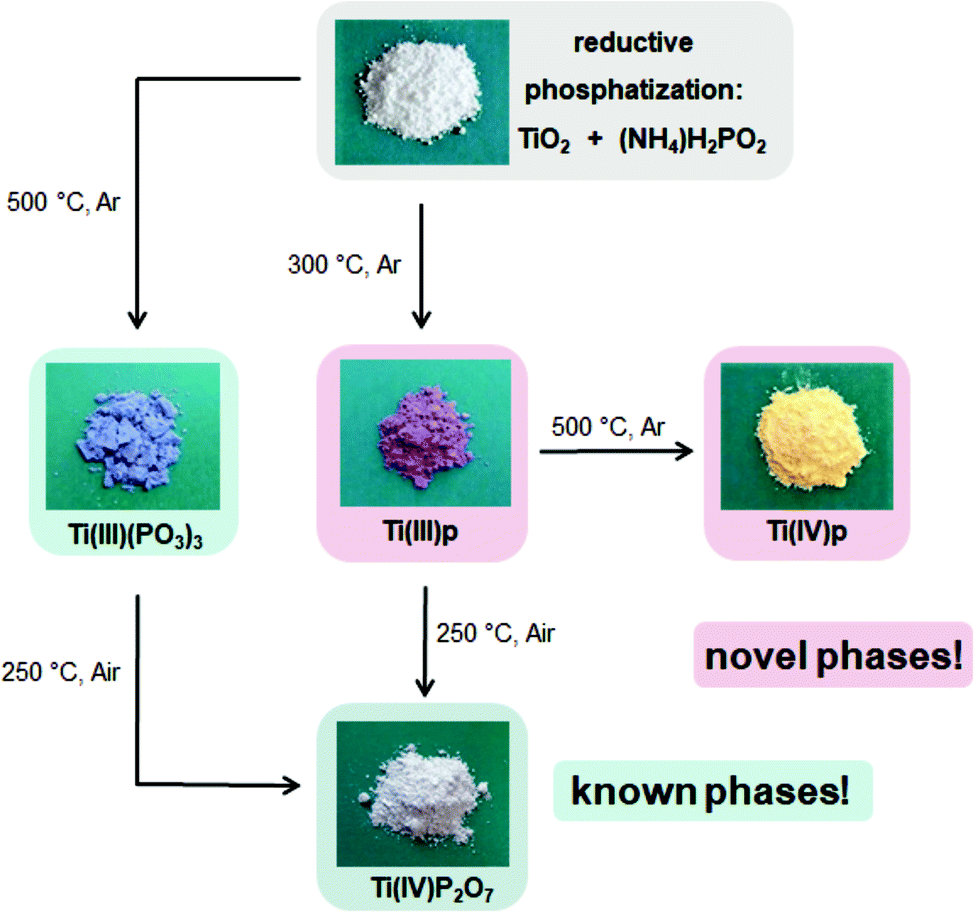 | ||
| Fig. 4 Reaction pathways for the preparation of novel and known titanium phosphate phases accessible by reductive phosphatization of TiO2 in a melt of ammonium hypophosphite. | ||
Formation of low-valent TMPs, as reported here for the titanium compounds, is observed also for other transition metal compounds. Via identical synthesis procedures V(PO3)3, Cr(NH4)HP3O10, Cr2(P6O18), Mn2(P4O12), Fe(II)p (new phase), and Fe2(P4O12) were readily obtained (see ESI,† for details). In literature, low oxidation states of TMPs are generally reported to be accessible by using metal powders or low-valent metal compounds as precursors. In comparison, the new route offers the possibility to direct the oxidation state of the transition metal via the hypophosphite, which results in quite moderate reaction conditions.
In summary, first-row transition metal oxides are converted in a hypophosphite melt to crystalline metal phosphates. The facile method operates at moderate temperatures (300–500 °C) and offers access to a number of novel, known and, very specifically, low-valent TMPs. The reducing feature of the hypophosphite precursor is exemplified by the reaction with TiO2 resulting in a number of known and novel Ti(III) and Ti(IV) phosphates. Reaction of the hypophosphite with other transition metal oxides (Cr, V, Mn, Fe) revealed comparable results, i.e., formation of transition metal phosphates with reduced and stabilized oxidation states of the metal cation. Overall, the synthesis of transition metal phosphates via a hypophosphite melt provides a highly versatile route towards products with rich structural and compositional variety for a class of materials that is key for various future technologies, including energy storage, power generation, catalysis and optical applications.
Author contributions
N. S. conceived and performed the synthetic procedures and pathways reported here. H. P. worked on the structural characterization of the new materials. C. W. evaluated the spectroscopic data for determination of oxidation states of the novel materials. W. S supervised collection and interpretation of experimental results and conceptually designed the present work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank S. Leiting for measuring XPS spectra, B. Zibrowius for supporting us with NMR spectra, B. Mienert and E. Bill (MPI for Chemical Energy Conversion) for Mößbauer spectra, and S. Palm for providing SEM images of selected samples. Financial support by the Max Planck Society (MPG) is greatly acknowledged. Open Access funding provided by the Max Planck Society.References
- D. Jugović and D. Uskoković, J. Power Sources, 2009, 190, 538–544 CrossRef.
- W.-J. Zhang, J. Power Sources, 2011, 196, 2962–2970 CrossRef CAS.
- H. Li, S. Xing, Y. Liu, F. Li, H. Guo and G. Kuang, ACS Sustainable Chem. Eng., 2017, 5, 8017–8024 CrossRef CAS.
- L. Hong, L. Li, Y. K. Chen-Wiegart, J. Wang, K. Xiang, L. Gan, W. Li, F. Meng, F. Wang, J. Wang, Y. M. Chiang, S. Jin and M. Tang, Nat. Commun., 2017, 8, 1194 CrossRef PubMed.
- S. Yang, P. Y. Zavalij and M. S. Whittingham, Electrochem. Commun., 2001, 3, 505–508 CrossRef CAS.
- Y. Jin, Y. Shen and T. Hibino, J. Mater. Chem., 2010, 20, 6214–6217 RSC.
- A. Clearfield, Chem. Rev., 1988, 88, 125–148 CrossRef CAS.
- P. L. Gai and K. Kourtakis, Science, 1995, 267, 661–663 CrossRef CAS PubMed.
- G. Centi, Catal. Today, 1993, 16, 5–26 CrossRef CAS.
- G. Busca, F. Cavani, G. Centi and F. Trifiro, J. Catal., 1986, 99, 400–414 CrossRef CAS.
- P. Bhanja, Y. Kim, B. Paul, J. Lin, S. M. Alshehri, T. Ahamad, Y. V. Kaneti, A. Bhaumik and Y. Yamauchi, ChemCatChem, 2020, 12, 2091–2096 CrossRef CAS.
- N. L. W. Septiani, Y. V. Kaneti, K. B. Fathoni, K. Kani, A. E. Allah, B. Yuliarto, Nugraha, H. K. Dipojono, Z. A. Alothman, D. Golberg and Y. Yamauchi, Chem. Mater., 2020, 32, 7005–7018 CrossRef CAS.
- S. Zhao, P. Gong, S. Luo, L. Bai, Z. Lin, C. Ji, T. Chen, M. Hong and J. Luo, J. Am. Chem. Soc., 2014, 136, 8560–8563 CrossRef CAS PubMed.
- R. Lin and Y. Ding, Materials, 2013, 6, 217–243 CrossRef CAS PubMed.
- D.-H. Kuo and W.-C. Tseng, Mater. Chem. Phys., 2005, 93, 361–367 CrossRef CAS.
- A. F. Holdsworth, H. Eccles, A. M. Halman, R. Mao and G. Bond, Sci. Rep., 2018, 8, 13547 CrossRef PubMed.
- T. Dobbelaere, F. Mattelaer, J. Dendooven, P. Vereecken and C. Detavernier, Chem. Mater., 2016, 28, 3435–3445 CrossRef CAS.
- N. Syed, A. Zavabeti, J. Z. Ou, M. Mohiuddin, N. Pillai, B. J. Carey, B. Y. Zhang, R. S. Datta, A. Jannat, F. Haque, K. A. Messalea, C. Xu, S. P. Russo, C. F. McConville, T. Daeneke and K. Kalantar-Zadeh, Nat. Commun., 2018, 9, 3618 CrossRef PubMed.
- J. M. Rojo, J. L. Mesa, R. Calvo, L. Lezama, R. Olazcuaga and T. Rojo, J. Mater. Chem., 1998, 8, 1423–1426 RSC.
- H. Assi, G. Mouchaham, N. Steunou, T. Devic and C. Serre, Chem. Soc. Rev., 2017, 46, 3431–3452 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta03782j |
| This journal is © The Royal Society of Chemistry 2021 |