
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical performance and reaction mechanism investigation of V2O5 positive electrode material for aqueous rechargeable zinc batteries†
Qiang
Fu
 *a,
Jiaqi
Wang
a,
Angelina
Sarapulova
a,
Lihua
Zhu
a,
Alexander
Missyul
c,
Edmund
Welter
d,
Xianlin
Luo
a,
Ziming
Ding
ef,
Michael
Knapp
a,
Helmut
Ehrenberg
ab and
Sonia
Dsoke
ab
*a,
Jiaqi
Wang
a,
Angelina
Sarapulova
a,
Lihua
Zhu
a,
Alexander
Missyul
c,
Edmund
Welter
d,
Xianlin
Luo
a,
Ziming
Ding
ef,
Michael
Knapp
a,
Helmut
Ehrenberg
ab and
Sonia
Dsoke
ab
aInstitute for Applied Materials (IAM), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany. E-mail: qiang.fu@kit.edu; Fax: +49-721 608-28521; Tel: +49-721 608-41445
bHelmholtz Institute Ulm for Electrochemical Energy Storage (HIU), Helmholtzstrasse 11, 89081 Ulm, Germany
cCELLS-ALBA Synchrotron, E-08290 Cerdanyola del Valles, Barcelona, Spain
dDeutsches Elektronen-Synchrotron DESY, Notkestraße 85, D-22607 Hamburg, Germany
eInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
fTechnische Universität Darmstadt, 64289 Darmstadt, Germany
First published on 27th July 2021
Abstract
The electrochemical performance and reaction mechanism of orthorhombic V2O5 in 1 M ZnSO4 aqueous electrolyte are investigated. V2O5 nanowires exhibit an initial discharge and charge capacity of 277 and 432 mA h g−1, respectively, at a current density of 50 mA g−1. The material undergoes quick capacity fading during cycling under both low (50 mA g−1) and high (200 mA g−1) currents. V2O5 can deliver a higher discharge capacity at 200 mA g−1 than that at 50 mA g−1 after 10 cycles, which could be attributed to a different type of activation process under both current densities and distinct degrees of side reactions (parasitic reactions). Cyclic voltammetry shows several successive redox peaks during Zn ion insertion and deinsertion. In operando synchrotron diffraction reveals that V2O5 undergoes a solid solution and two-phase reaction during the 1st cycle, accompanied by the formation/decomposition of byproducts Zn3(OH)2V2O7·2(H2O) and ZnSO4Zn3(OH)6·5H2O. In the 2nd insertion process, V2O5 goes through the same two-phase reaction as that in the 1st cycle, with the formation of the byproduct ZnSO4Zn3(OH)6·5H2O. The reduction/oxidation of vanadium is confirmed by in operando X-ray absorption spectroscopy. Furthermore, Raman, TEM, and X-ray photoelectron spectroscopy (XPS) confirm the byproduct formation and the reversible Zn ion insertion/deinsertion in the V2O5.
1. Introduction
Aqueous rechargeable zinc batteries (ARZBs) have received much attention for application in large-scale energy storage because of their advantages, such as high safety, low cost, and sufficient abundance.1,2 Aqueous electrolytes have higher ionic conductivity (up to 1 S cm−1) than that of non-aqueous electrolytes (about 10 mS cm−1), resulting in a higher rate capability.1 Meanwhile, the utilization of aqueous electrolytes can lower the activation energy for charge transfer at the electrode/electrolyte interface. The manufacturing costs of ARZBs are expected to be low since water-based electrolytes are non-toxic, inflammable, and strict humidity control is not required during cell assembling.3,4 Moreover, metallic Zn has a high specific capacity (820 mA h g−1) and high volumetric capacity (5854 mA h cm−3), making it promising as a negative electrode. It also owns sufficiently high overpotentials with respect to the hydrogen evolution, overcompensating the negative value of −0.76 V vs. SHE that makes it usable in water5 and, therefore, can be directly used as the negative electrode in aqueous-based electrolytes. However, the lack of high-performance positive electrode materials, the heavy mass, and the large polarization of divalent Zn2+ hinder the practical applications of ARZBs.Many efforts have been made on the exploration of high-performance positive electrode materials for ARZBs, including the polymorphs of manganese oxide (α-, β-, γ-, δ-MnO2, and spinel-MnO2),4,6–17 Prussian blue analogues,18–21 vanadium-based oxides (Ca0.25V2O5·nH2O, Zn0.25V2O5·nH2O, Na0.33V2O5, V2O5·nH2O, LiV3O8, and H2V3O8),2,5,22–38 organic compounds,39,40 and other compounds such as Na3V2(PO4)3,41 Na3V2(PO4)2F3,42 VS2,43 and Mo6S8.44 Among them, vanadium-based oxides are very promising due to their open framework and relatively high capacity as well as operation voltage of around 0.6–1.0 V. For example, Kundu et al.5 reported Zn0.25V2O5·nH2O positive electrode, which delivers a high initial discharge/charge capacity of 282/278 mA h g−1 at C/6 rate, excellent rate capability, and high cycling stability with a capacity retention of 80% after 1000 cycles at 15C rate. Xia et al.29 demonstrated that Na0.33V2O5 nanowires have a high capacity of 367.1 mA h g−1 at 100 mA g−1 when used as the positive electrode in ARZBs. This material also exhibits good rate capability and high capacity retention of 93% after 1000 cycles. Pang et al.34 investigated H2V3O8/graphene composite, which shows a high capacity of 394 mA h g−1 at C/3, high rate capability, and excellent cycling stability with a capacity retention of 87% after 2000 cycles. Despite this significant progress, it is still urgently required to deeply understand the electrochemical reaction mechanism of positive electrode materials. Because it is more meaningful to the development of ARZBs by understanding the reaction mechanism behind good electrochemical performance than trying to improve performance empirically.
Because of its typical layered structure and relatively high capacity, V2O5 is considered a promising positive electrode for ARZBs.45–48 Despite its good electrochemical performance in ARZBs, the structural changes of V2O5 during electrochemical processes are still under debate. Three different viewpoints regarding the structural changes of V2O5 in ARZBs have been proposed so far:
(i) Zhou et al.46 compared the performance of V2O5 in different electrolytes (i.e. Zn(NO3)2, Zn(CH3COO)2, ZnCl2, and ZnSO4 aqueous-based electrolytes) and with different concentrations of ZnSO4-based electrolytes. In 3 M ZnSO4, V2O5 delivers the best performance with a high capacity of 224 mA h g−1 at 100 mA g−1 and good cycling stability at the high current densities of 1 and 2 A g−1, respectively. Ex situ X-ray diffraction (XRD) demonstrated the formation of a new phase of ZnxV2O5 upon Zn insertion into V2O5.
(ii) Zhang et al.45 also reported a V2O5 cathode material with a capacity of 470 mA h g−1 at 0.2 A g−1 and high capacity retention of 91.1% after 4000 cycles at 5 A g−1 in 3 M Zn(CF3SO3)2 electrolyte. They proved that this material can work in extreme conditions at both high (50 °C) and low (−10 °C) temperatures. The co-insertion of hydrated Zn ions into the V2O5 crystal structure was proposed based on ex situ XRD, X-ray photoelectron spectroscopy (XPS), and transmission electron microscopy (TEM).
(iii) Chen et al.47 proposed a phase transition reaction mechanism via ex situ XRD, Raman, and XPS. Orthorhombic V2O5 underwent a phase transition to zinc pyrovanadate during the first discharge, where the formed zinc pyrovanadate showed reversible Zn2+ (de)insertion during subsequent cycles.
However, the electrochemical reaction mechanism of V2O5 in the above-reported works was investigated through ex situ techniques. The findings are in controversy with each other and a detailed investigation of the structural changes of V2O5 upon Zn-ion insertion/deinsertion is still missing. Moreover, non-equilibrium or intermediate species or states cannot be detected using ex situ studies, while in operando studies can provide a more reliable understanding of the structural evolution of a battery material in “real use”.
In addition, it has been frequently reported that a mass of complex byproducts was produced/decomposed during the discharge/charge cycling of vanadium oxides such as NaV3O8·1.5H2O,2 VO1.52(OH)0.77,24 Zn2V2O7,35 and NH4V4O10.49 Interestingly, byproducts were not observed in the three works related to the study of V2O5 reported above. To better understand the electrochemical mechanism of V2O5 in ARZBs in operando techniques like diffraction using synchrotron radiation are required. Due to their extremely bright, high flux, and tunable also high energy, synchrotron radiation-based characterization provides deep penetration into the sample, high-quality data, and real-time diffraction. Particularly, in operando synchrotron study can effectively avoid the unpredictable contamination and irreversible changes of highly reactive samples during material preparation, handling, and transportation, which can provide more reliable and precise data for analysis. Hence, in this work, orthorhombic V2O5 were prepared by a facile hydrothermal method and the detailed structure evolution and reaction mechanism of orthorhombic V2O5 in ARZBs are studied via in operando synchrotron diffraction and X-ray absorption spectroscopy (XAS) together with ex situ Raman, TEM, and X-ray photoelectron spectroscopy (XPS).
2. Experimental
2.1 Synthesis of V2O5 nanowires
V2O5 nanowires were prepared via a modified hydrothermal method followed by heat treatment.50,51 Briefly, 0.18 g of commercial V2O5 powder (Alfa Aesar, 99.99%) was added to 30 ml of deionized water under vigorous stirring for a few minutes to form a light orange suspension. Then, 2.5 ml 30% hydrogen peroxide (H2O2) was dropwise added to the above suspension and kept stirring for 30 min to get a transparent orange solution. The obtained solution was transferred to a 50 ml Teflon-lined stainless-steel autoclave and kept at 190 °C for 4 days. The precipitate was collected and washed with deionized H2O several times and dried at 80 °C for 12 h. Finally, the product was annealed at 400 °C for 2 h with a heating rate of 5 °C min−1 in air atmosphere2.2 Preparation of the electrolyte
The 1 M ZnSO4 electrolyte was prepared by dissolving a corresponding amount of ZnSO4·7H2O powder in distilled H2O with vigorous stirring at room temperature.2.3 Morphological and structural study
The morphology was studied with a Zeiss Supra 55 Scanning Electron Microscope (SEM) with primary energy of 15 keV. The structural characterization was performed using synchrotron radiation (λ = 0.4132 Å, 30 keV) at the Material Science and Powder Diffraction beamline (MSPD) of ALBA synchrotron (Barcelona, Spain).52 The powder was filled in 0.5 mm Ø boro-silicate capillary, and the diffraction pattern was collected in capillary geometry. A LabRam HR Evolution Raman microscope from Horiba Scientific equipped with HeNe laser (633 nm, 17 mW) and a CCD detector (Horiba) was used to collect the Raman scattering of the samples. Meanwhile, a 600 grating was used to split the measurement signal with a ×100 objective (NA 0.95) for all the pristine and cycled samples. The data were collected for 30 seconds with 4.25 mW of the laser and five measurements were added to reduce signal noise. Transmission Electron Microscopy (TEM) imaging and high angle annular dark field-scanning TEM (HAADF-STEM) electron dispersive X-ray (EDX) mapping were acquired by Themis 300 under 300 kV with Ceta camera and Super-X EDX detector, respectively. The last measured screen current for high-resolution TEM (HRTEM) imaging was 998 pA and for EDS mapping was 93.4 pA. X-ray photoelectron spectroscopy measurements were performed using a K-Alpha XPS spectrometer (ThermoFisher Scientific, East Grinstead, UK), applying a micro-focused, monochromated Al Kα X-ray source with a spot size of 400 μm. To prevent any localized charge buildup, the K-Alpha+ charge compensation system was employed during analysis, using electrons of 8 eV energy and low-energy argon ions. The Thermo Avantage software was used for data acquisition and processing.53 The spectra were fitted with one or more Voigt profiles (binding energy uncertainty: ±0.2 eV). All spectra were referenced to the O 1s peak of vanadium oxide at 530.0 eV binding energy. In addition, the discharged and charged V2O5 and discharged and charged Zn electrodes were sealed in an in situ Raman cell with a quartz window inside a glovebox. Note the “discharged Zn” refers to the counter electrode of V2O5 at the discharged state of 0.3 V from the same cell, while “charged Zn” refers to the counter electrode of V2O5 at the charged state of 1.6 V from the same cell.2.4 Electrochemical characterization
The electrode was prepared by mixing active material V2O5 nanowires with Super C65C (Timcal) and polyvinylidene difluoride (PVDF) binder in a weight ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:10 with N-methyl-2-pyrrolidone solvent. The dried electrode mixture with mass loading of ∼1.4 mg cm−2 was pressed (4 tons) on a stainless steel mesh of 12 mm and dried at 120 °C overnight under vacuum. CR2032-type coin cells for electrochemical measurements were assembled in air at room temperature. The cells were built with V2O5 positive electrode, Zn foil as the negative electrode, 1 M ZnSO4 as the electrolyte, and a piece of glass microfiber (Whatman) as the separator. The galvanostatic cycling with the potential limitation (GCPL) and cyclic voltammetry (CV) measurements were performed between 0.3 and 1.6 V (vs. Zn2+/Zn) with a VMP3 potentiostat (BioLogic) at 25 °C. GCPL was performed at different current densities ranging from 50 to 1600 mA g−1 to determine the rate capability of the battery.
:20:10 with N-methyl-2-pyrrolidone solvent. The dried electrode mixture with mass loading of ∼1.4 mg cm−2 was pressed (4 tons) on a stainless steel mesh of 12 mm and dried at 120 °C overnight under vacuum. CR2032-type coin cells for electrochemical measurements were assembled in air at room temperature. The cells were built with V2O5 positive electrode, Zn foil as the negative electrode, 1 M ZnSO4 as the electrolyte, and a piece of glass microfiber (Whatman) as the separator. The galvanostatic cycling with the potential limitation (GCPL) and cyclic voltammetry (CV) measurements were performed between 0.3 and 1.6 V (vs. Zn2+/Zn) with a VMP3 potentiostat (BioLogic) at 25 °C. GCPL was performed at different current densities ranging from 50 to 1600 mA g−1 to determine the rate capability of the battery.
2.5 In operando synchrotron diffraction and in operando X-ray absorption spectroscopy (XAS)
In operando synchrotron diffraction was performed at the Material Science and Powder Diffraction beamline (MSPD) at the ALBA synchrotron. The electrochemical cell consists of 2025-type coin cell with glass windows of 5 mm diameter for beam entrance. The positive electrode was prepared by pressing the dried electrode mixture (as described above) on a stainless steel mesh within a 5 mm hole in the center, a Zn foil with a 5 mm hole in the center was used as the negative electrode. In operando synchrotron diffraction was conducted with radiation λ = 0.4132 Å wavelength (30 keV) and the position-sensitive detector MYTHEN. Data in steps of 0.006° over an angular range of 1.8–42° in 2theta were gathered with an effective exposure time of 60 s during the 1.5 cycles with a current density of 50 mA g−1. The coin cell was continuously oscillated ±5° around the incoming beam direction to improve the powder averaging (i.e. increasing the number of crystallites fulfilling Bragg condition and contributing to the observed reflections). Diffraction data were analyzed by the Rietveld method using the Fullprof software package.54In operando XAS measurements were performed at beamline P65 at the synchrotron source PETRA III (DESY, Hamburg). XAS was carried out during the first charge/discharge process at the current of 50 mA g−1 in the same coin-cell configuration as above but with a Kapton window. X-ray absorption spectra of vanadium were recorded in quick-XAS (6 min per spectrum) mode in fluorescence geometry using a PIPS (passivated implanted planar silicon) diode. The V K-edge for V2O5 was investigated, and the energy was calibrated utilizing vanadium foil as commonly applied in XAS experiments. V2O3, VO2, and V2O5 were used as standard materials. All data were collected at room temperature with a Si(111) double crystal monochromator, and all spectra were processed using the DEMETER software package.553. Results and discussion
3.1 Structural and morphological characterization
The crystal structure of the prepared V2O5 nanowires was investigated by synchrotron diffraction and HRTEM imaging, as displayed in Fig. 1a. All reflections can be indexed to the orthorhombic V2O5 with space group Pmn21, and the lattice parameters are a = 11.515(1) Å, b = 4.374(1) Å, c = 3.566(1) Å, in good agreement with the previous work.56 The strong intensities of the reflections confirm the high crystallinity of the obtained V2O5 nanowires material. The SEM image (Fig. 1b) demonstrates that V2O5 material is composed of nanowire-like nanostructure with lengths up to several micrometers. TEM imaging further reveals the nanowire-like morphology of V2O5 while HRTEM image (Fig. 1c) displays the highly crystalline sample on the [010] zone axis with lattice fringe of 0.58 nm and 0.34 nm corresponding to the (200) and (110) plane of V2O5, respectively. This fits well with the result obtained from synchrotron diffraction. | ||
| Fig. 1 Rietveld refinement based on synchrotron diffraction data (a), SEM (b), and HRTEM image (c) of V2O5 nanowires, where Fast Fourier Transform (FFT) was done from the marked orange area. | ||
3.2 Electrochemical properties
Fig. 2a displays the discharge–charge profiles of V2O5 nanowires were at the specific currents of 50 mA g−1 in the potential range of 0.30–1.60 V vs. Zn2+/Zn. The open-circuit voltage (OCV) of the cell is 1.23 V. During the 1st discharge (Zn ions insertion), V2O5 nanowires electrode displays a flat plateau at around 1.00 V, followed by a sloping-like plateau at about 0.50 V. During the 1st charge process (Zn ions de-insertion), a slope and a flat plateau at 1.20 V together with a slope up to 1.6 V can be observed. Compared with the 1st discharge, the 2nd discharge profile shows a shorter plateau at around 1.00 V and similar sloping-like plateau at 0.50 V. In the 2nd charge process, a slope similar to that for the 1st charge is observed, but no clear plateau at 1.20 V can be seen. During cycling, the voltage profiles significantly change: the plateau at 1.20 V completely disappears, and only one slope and a sloping-like plateau at 0.50 V can be observed (see the 5th discharge profile). On the 5th charge profile, two sloping-like plateaus at 0.70 V and 1.0 V are observed. The V2O5 nanowires electrode delivers an initial discharge and charge capacity of 277 and 432 mA h g−1, respectively, at a current density of 50 mA g−1. The discharge capacity of 277 mA h g−1 almost reaches the theoretical one based on the insertion of 1 mol of Zn2+ in V2O5 (294 mA h g−1). The huge extra charge capacity might be attributed to side reactions such as O2 evolution from the aqueous-based electrolyte. The electrode delivers a discharge capacity of 302 mA h g−1 at the 2nd cycle, a value higher than that for the first cycle, which might be due to an activation of the active material (theoretical capacity of 442 mA h g−1 by considering insertion of 1.5 mol of Zn2+ into V2O5), accompanied by Zn insertion, as also reported by other previous studies.6,57 The V2O5 positive electrode exhibits a dramatic decrease of capacity during the following 20 cycles at 50 mA g−1 (94 mA h g−1 at the 22nd cycle) and delivers a discharge capacity of only 21 mA h g−1 after 100 cycles. Moreover, the cycling stability of V2O5 positive electrode was studied at a higher specific current of 200 mA g−1, as presented in Fig. 2b. An initial discharge capacity of 278 mA h g−1 is obtained, followed by a sudden capacity decline down to 180 mA h g−1 at the 2nd cycle. In the following four cycles, the behavior differs from the one of the electrode cycled at 50 mA g−1: it displays a capacity increase, indicating a long activation process of the active material, which is often observed in positive electrode materials for ARZBs.6,57 However, this activation phenomenon takes more cycles under high current density, demonstrating its possible correlation with the involved chemical ion kinetics during cycling.22 After 100 cycles, the capacity at 200 mA g−1 is higher (45 mA h g−1) than that delivered at 50 mA g−1. This could be attributed to a different type of activation process under different current densities. The side reactions (parasitic reactions) under low current have more relevance than at high current and, consequently, the degradation is faster. Moreover, a significant difference is observed between the two first charge profiles recorded at both current densities: the charge profile recorded at 200 mA g−1 displays a slope-like plateau (Fig. S1†); a clear flat plateau in the voltage profile is observed at the low current of 50 mA g−1. The difference of voltage profiles suggests a different mechanism reaction and therefore results in a type of activation process under high and low current.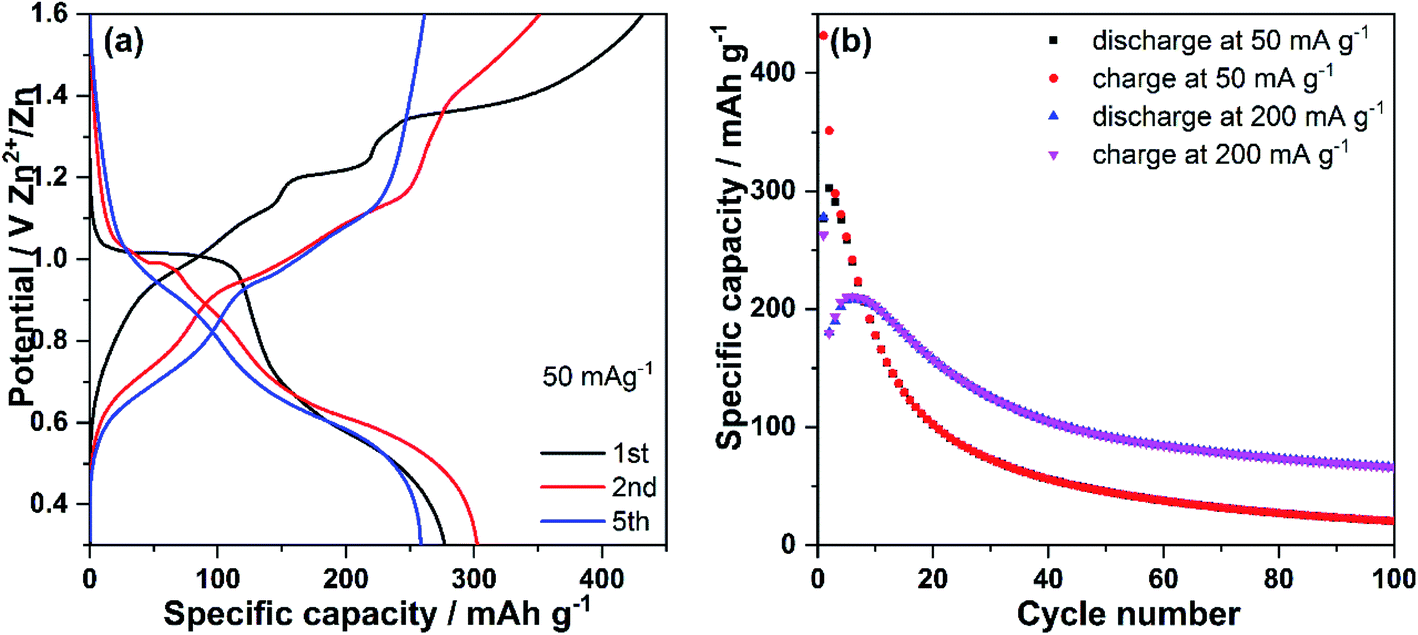 | ||
| Fig. 2 Discharge–charge profiles (a) and cycling performance (b) of V2O5 nanowires in 1 M ZnSO4. | ||
3.3 Electrochemical mechanism
In order to clarify the Zn-ion storage mechanism in the V2O5 material, CV was performed at a scan rate of 0.1 mV s−1 in the voltage range of 0.30–1.60 V (vs. Zn2+/Zn). Fig. 3 displays two peaks centered at 0.92 and 0.50 V during the 1st reduction process and a broad peak at 1.20 V with a shoulder at 1.05 V during the 1st oxidation process. In the following scans, three features are observed for both reduction and oxidation processes, respectively. The reduction peak at 0.92 V gradually shifts to higher potential, becomes weaker, and finally disappears. However, a new reduction peak at 0.88 V emerges and grows up and the reduction peak at 0.50 V gradually shifts to 0.57 V, indicating polarization (and resistance) decrease of this process. At the same time, a new oxidation peak appears at 0.74 V. The oxidation peak at 1.05 V shifts to 1.00 V after the first cycle and increases upon cycling. The oxidation peak at 1.20 V shifts to 1.12 V in the 3rd cycle; this shift is accompanied by decreasing in current intensity, indicating the decrease of polarization.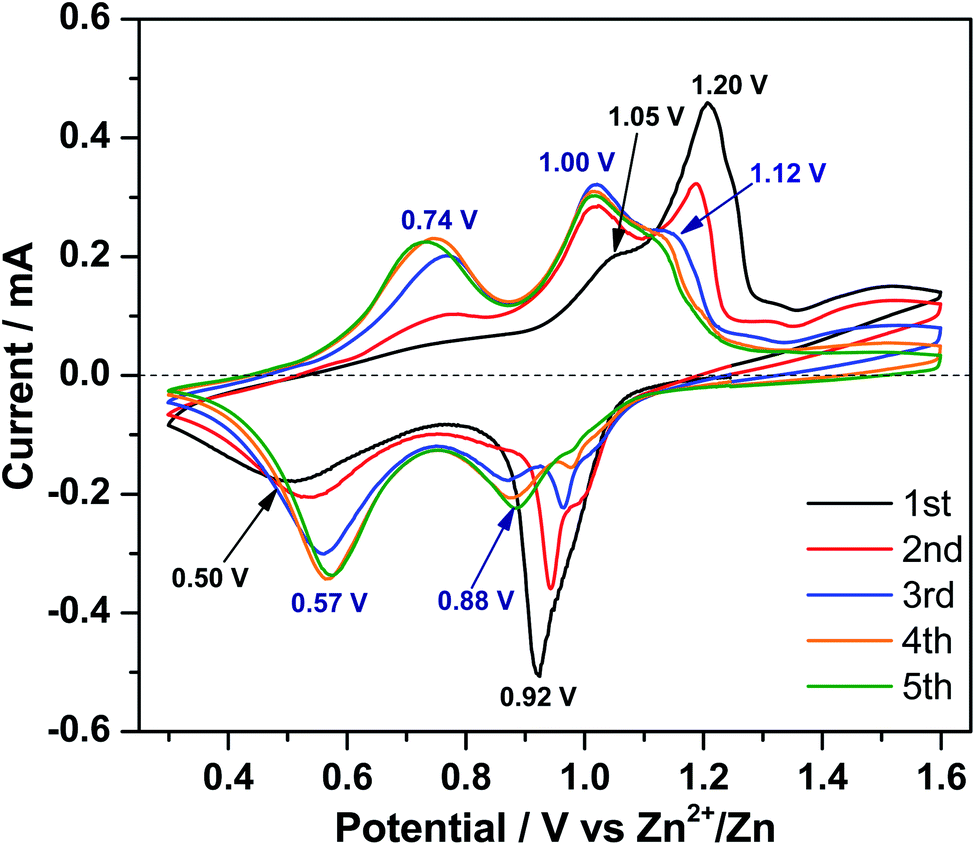 | ||
| Fig. 3 CV curves of V2O5 nanowires at a scan rate of 0.1 mV s−1 in 1 M ZnSO4. | ||
To investigate the structural evolution of V2O5 upon Zn-ion insertion/deinsertion, in operando synchrotron diffraction was performed during the initial 1.5 cycles. The contour maps of selected diffraction patterns and corresponding voltage–time profiles are provided in Fig. 4. Note that an electrochemical activation is observed at the high current of 200 mA g−1 during the first 5 cycles, with very interesting changes. Here we go deeper on understanding the very initial changes at low current and in the future, it would be interesting and necessary to study what is happening during the further cycles at higher current density.
 | ||
| Fig. 4 Contour maps of in operando synchrotron diffraction of V2O5 during the first one and half cycles and the corresponding voltage profile at a current density of 50 mA g−1. | ||
Before the discharge (at 1.2 V), all reflections of the parent material can be indexed on the orthorhombic V2O5 in the space group Pmn21 (see Fig. S2a† for the Rietveld refinement of pristine V2O5). At the beginning of the 1st discharge (Region I), most of the characteristic reflections of V2O5, such as ones at 200 (4.12°), 101 (6.96°), 400 (8.24°), 011 (8.58°), 301 (9.08°), 411 (11.90°), 600 (12.37°), 002 (13.31°), 012 (14.38°), 402 (15.67°), and 701(15.91°), continuously shift towards lower 2θ, accompanied by a slight reduction of their intensity. Therefore, a solid solution reaction upon Zn ions insertion into the V2O5 structure can be proposed up to the stoichiometry ZnxV2O5 (x = 0.22, 65 mA h g−1, black line at the end of Region I, Fig. S3†), where lattice parameters a and c increase and b decreases. At the same time, few reflections at 010 (5.42°), 020 (10.85°), and 120 (11.04°) slightly shift to high angles; few other reflections at 110 (5.80°), 320 (12.49°), 021 (12.73°), and 620 (16.46°) keep in the same positions. Upon total discharge capacity of 44 mA h g−1 from OCV (1.02 V, x = 0.15, cyan line in the middle of Region I, Fig. S3†), two small reflections at 3.29° and 13.23° emerge, which belong to a new phase of zinc pyrovanadate of 1.2%(wt/wt), Zn3+δ(OH)2V2O7·2H2O (see Fig. S2b† for the Rietveld refinement based on the 10th pattern recorded during discharge of V2O5). Other reflections related to this phase become visible at 4.49°, 7.78°, 7.99°, 8.46°, 9.57°, 10.20°, 11.16°, 15.97°, and 17.54° along with further Zn ions insertion. At the same time, other reflections at 3.81°, 5.67°, 7.01°, 7.61°, 11.42°, and 11.66° appear and grow (Region II). These reflections are attributed to the formation of the new Zn-rich phase ZnxV2O5 (x = 0.44) through a 2-phase reaction, corresponding to the capacity of 129 mA h g−1. Meanwhile, the reflections of the new Zn-rich phase Zn0.44V2O5 remain in their positions, together with the decrease of the intensities of the Zn0.22V2O5 phase. At the end of Region II (gray line in Fig. S3†), most of the reflections of Zn0.22V2O5 phase disappear, and the Zn-rich Zn0.44V2O5 phase grows up. It implies the 2-phase coexistence transition between Zn0.22V2O5 and Zn0.44V2O5 in Region II during the Zn ions insertion. The formation of Zn0.44V2O5 is accompanied by the formation of the byproduct Zn3(OH)2V2O7·2H2O. Note that the x value is determined without the consideration of the consumption of V2O5 to form the byproduct Zn3+δ(OH)2V2O7·2H2O and the possible dissolution of V2O5, where the dissolution and phase transformation mechanisms of V2O5 in ARZBs were newly reported.58 Therefore, the value of x = 0.44 is an estimate for the lower limit of the inserted Zn content. Upon further Zn ions insertion (Region III), some new reflections begin to appear at 2.16°, 4.34°, 9.26°, 9.35°, which are assigned to the byproduct of ZnSO4Zn3(OH)6·5H2O with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Once the byproduct is formed, all its Bragg reflections remain at the same 2θ-positions throughout the discharge process, but their intensities increase. Meanwhile, most of the reflections maintain their positions along with the increase of their intensities (Region III), while only two reflections of Zn0.44V2O5 at 3.81° and 7.01° shift to high and low angles, respectively. It indicates that a solid solution process happens in Region III to form a final phase Zn0.94V2O5 together with the above-mentioned two byproducts with a total discharge capacity of 277 mA h g−1 (see Fig. S2c† for the Rietveld refinement of 36th pattern at first fully discharged V2O5). During the 1st charge, the reflections do not evolve in a symmetric reverse backway, as indicated by the appearance and disappearance of a small reflection at 7.17°, where two 2-phase regions are observed (Region II). The reflections return back to their initial 2θ-positions for the pristine V2O5 state, but with much lower intensities (see Fig. S2d† for the Rietveld refinement, based on the 69th pattern at fully charged V2O5). This is possibly caused by the amorphization of the crystalline active material or by the vanadium dissolution in the electrolyte32,36 (see Fig. S4†). Interestingly, both byproducts disappear again along with the 1st charging process. The evolution of the reflections in the 2nd discharge is analogous to that during the 1st cycle. In Region I, the reflections show the same behavior as those for the first discharge process, suggesting a solid solution reaction. In Region II and III, the electrode undergoes the same process: a shorter two-phase transition and solid solution than in the first discharge, but without the appearance of the reflections related to the byproduct Zn3+δ(OH)2V2O7·2H2O (see Fig. S2e† for the Rietveld refinement, based on the 97th pattern at the 2nd fully discharged V2O5). The formation of the other byproduct ZnSO4Zn3(OH)6·5H2O is also detected again during the second discharge process. The Zn3+δ(OH)2V2O7·2H2O has an open layered structure and has been reported by Alshareef et al.26 as positive electrode material in ARZBs. The material has a capacity of 213 mA h g−1 at 50 mA g−1 and shows a shift of the reflection 001 during Zn-ion insertion and deinsertion.26 This means that the Zn3+δ(OH)2V2O7·2H2O byproduct, generated during cycling, still contributes to the overall capacity of the electrode. Recently, Chen et al.47 reported that Zn3(OH)2V2O7·2H2O is also formed via the 2-phase coexistence transition in an aqueous Zn–V2O5 battery, and then it functions as a host structure in the following cycles, where it shows a shift of reflection 001 during Zn ion insertion. However, in our case, the shift of the 001 reflection is not observed, which might suggest that Zn3+δ(OH)2V2O7·2H2O is not active or just the c-axis parameter does not change. Furthermore, our results directly show for the first time the formation and decomposition of both byproducts Zn3+δ(OH)2V2O7·2H2O and ZnSO4Zn3(OH)6·5H2O during cycling.
. Once the byproduct is formed, all its Bragg reflections remain at the same 2θ-positions throughout the discharge process, but their intensities increase. Meanwhile, most of the reflections maintain their positions along with the increase of their intensities (Region III), while only two reflections of Zn0.44V2O5 at 3.81° and 7.01° shift to high and low angles, respectively. It indicates that a solid solution process happens in Region III to form a final phase Zn0.94V2O5 together with the above-mentioned two byproducts with a total discharge capacity of 277 mA h g−1 (see Fig. S2c† for the Rietveld refinement of 36th pattern at first fully discharged V2O5). During the 1st charge, the reflections do not evolve in a symmetric reverse backway, as indicated by the appearance and disappearance of a small reflection at 7.17°, where two 2-phase regions are observed (Region II). The reflections return back to their initial 2θ-positions for the pristine V2O5 state, but with much lower intensities (see Fig. S2d† for the Rietveld refinement, based on the 69th pattern at fully charged V2O5). This is possibly caused by the amorphization of the crystalline active material or by the vanadium dissolution in the electrolyte32,36 (see Fig. S4†). Interestingly, both byproducts disappear again along with the 1st charging process. The evolution of the reflections in the 2nd discharge is analogous to that during the 1st cycle. In Region I, the reflections show the same behavior as those for the first discharge process, suggesting a solid solution reaction. In Region II and III, the electrode undergoes the same process: a shorter two-phase transition and solid solution than in the first discharge, but without the appearance of the reflections related to the byproduct Zn3+δ(OH)2V2O7·2H2O (see Fig. S2e† for the Rietveld refinement, based on the 97th pattern at the 2nd fully discharged V2O5). The formation of the other byproduct ZnSO4Zn3(OH)6·5H2O is also detected again during the second discharge process. The Zn3+δ(OH)2V2O7·2H2O has an open layered structure and has been reported by Alshareef et al.26 as positive electrode material in ARZBs. The material has a capacity of 213 mA h g−1 at 50 mA g−1 and shows a shift of the reflection 001 during Zn-ion insertion and deinsertion.26 This means that the Zn3+δ(OH)2V2O7·2H2O byproduct, generated during cycling, still contributes to the overall capacity of the electrode. Recently, Chen et al.47 reported that Zn3(OH)2V2O7·2H2O is also formed via the 2-phase coexistence transition in an aqueous Zn–V2O5 battery, and then it functions as a host structure in the following cycles, where it shows a shift of reflection 001 during Zn ion insertion. However, in our case, the shift of the 001 reflection is not observed, which might suggest that Zn3+δ(OH)2V2O7·2H2O is not active or just the c-axis parameter does not change. Furthermore, our results directly show for the first time the formation and decomposition of both byproducts Zn3+δ(OH)2V2O7·2H2O and ZnSO4Zn3(OH)6·5H2O during cycling.
In order to investigate the variation of the oxidation state and the local electron environment of vanadium during the discharge/charge (i.e. Zn insertion/deinsertion) process, in operando XAS was performed on the V2O5 nanowires as positive electrode within an in operando coin cell. Fig. 5 shows the normalized V K-edge spectra collected during the initial discharging and charging processes and reference spectra of standard vanadium oxides, where V2O5, VO2, and V2O3 have the oxidation state of +5, +4, and +3, respectively. The edge position of the V K-edge in the pristine V2O5 positive electrode overlaps with that of the standard V2O5 reference, indicating that the oxidation state of V in V2O5 is +5. Moreover, an intense pre-edge peak for the V K-edge of pristine V2O5 is observed, which is ascribed to the transitions between the 1s and bound p-hybridized d-states.59,60 Along with progressive discharging, the main absorption edge shifts towards lower binding energies, confirming the reduction of the oxidation state of vanadium upon the Zn-ion insertion. Meanwhile, the pre-peak (A in Fig. 5a) also shifts gradually to lower binding energy with the simultaneous decrease of intensity, confirming the reduction of V and the deformation of the local V environments during Zn-ion insertion. This is due to the co-existence of a distorted tetragonal pyramid and VO6 octahedra. The edge resonance (B in Fig. 5a) displays distinct changes in both intensity and shape, which is caused by the absorption of photons accompanied by core-electron excitations.60,61 During the discharge process, two broad peaks centered at 5494 eV and 5507 eV shift to lower energy and with a decrease of their intensities, before they converge into one very broad peak centered at 5500 eV from the initial stage to 0.99 V (peak B in Fig. 5a). After that, the formed broad peak centered at 5500 eV and the peak at 5486.5 eV continuously shift to lower energy accompanied by an increases of both intensities (from 0.99 V to 0.3 V). Two distinct isosbestic points62 at ∼5474 eV and ∼5502 eV (red arrows in Fig. 5a and b) are observed during both discharge (from 0.99 V to 0.3 V) and charge (from 0.3 V to 1.32 V) processes. This confirms the 2-phase coexistence transitions upon Zn-ion insertion/deinsertion into/from V2O5 structure, as already proposed from in operando synchrotron diffraction. At the fully discharged state at 0.30 V, the observed energy of the V K-edge lies almost in the middle of those of the V2O5 and V2O3 reference spectra. This means that the oxidation state of V is very close to V4+, in good agreement with the electrochemical data (see Fig. S5†). However, the energy of the edge does not completely overlap with the spectrum of VO2, reflecting structural differences between the discharged state with x = 1.00 in ZnxV2O5 and the reference material VO2. During the charging process (Zn-ion deinsertion), a completely reversible behavior can be observed. Pre-peak and edge resonance (A′ and B′ in Fig. 5a and b) prove a reversible process. At the fully charged state of ∼1.6 V, the V spectrum returns back to its initial state, indicating that the V ions are fully oxidized to the oxidation state of +5. The evolution of V K-edge spectra reveals that the V ions are reduced and reversibly oxidized during the Zn-ion insertion and deinsertion, respectively, accompanied by the local structural changes around the V ions and in full agreement with in operando synchrotron diffraction.
 | ||
| Fig. 5 In operando V K-edge XANES spectra during the 1st discharge (a) and charge (b) in 1 M ZnSO4; the isosbestic points are pointed out by red arrows. | ||
Raman spectra were collected to investigate vibration modes as fingerprints of the short-range structure of the samples during Zn-ion insertion/deinsertion, as displayed in Fig. 6 and Table S1.† In the pristine V2O5, oxygen atoms lie in four distinct sites in a [VO5] pyramid unit, denoted as O(1)–O(4). The stretching mode of V–O(1) bond is located at 994 cm−1 and its bending vibrations are located at 405 and 284 cm−1. Raman peaks at 482 cm−1 and 701 cm−1 are ascribed to the bending vibration of the V–O(3) and the stretching vibration of the V–O(2), respectively. The peaks at 527 and 304 cm−1 are attributed to the stretching and bending vibrations of the V–O(4) bond, respectively. The peaks at lower wavenumbers, 145 and 197 cm−1 are assigned to weak van der Waals interaction between adjacent [VO5] slabs. These results are consistent with previous works.63,64 After Zn-ion insertion, significant changes are observed and some peaks related to V2O5 at 994, 701, 405, 304, and 284 cm−1 become weak and broad, while those at 527, 482, and 197 cm−1 completely disappear. Meanwhile, some broad and distinct new peaks appear at 1129, 967, 876, 610, and 450 cm−1 for discharged V2O5, where peaks at 1129, 967, and 610 cm−1 are attributed to the v3, v1, v4 of SO42− vibration bands of ZnSO4Zn3(OH)6·nH2O65,66 and peaks at 876 and 450 cm−1 are related to the V–O and Zn–O bonds in ZnxV2O5 and Zn3(OH)2V2O7·2H2O.67 Therefore, the coexistence of ZnxV2O5 and byproducts, ZnSO4Zn3(OH)6·5H2O and Zn3(OH)2V2O7·2H2O, is concluded in the discharged V2O5 electrode. On the other hand, for the Zn counter electrode in a discharged cell, several peaks at 1129, 967, 566, 440, and 398 cm−1 are observed. The peaks appearing at 440 and 566 cm−1 are assigned to the Zn–O vibration of Zn1+xO on the surface of Zn metal,65,68 while 1129, 967, and 398 are assigned to the v3, v1, v4 of SO42− vibration in the ZnSO4Zn3(OH)6·nH2O.65,66 Upon charging, the spectrum of fully charged V2O5 at 1.6 V returns back to its main original features with less intensity, indicating the disappearance of ZnxV2O5 and the byproducts, ZnSO4Zn3(OH)6·5H2O and Zn3+δ(OH)2V2O7·2H2O. This demonstrates the reversible reaction during Zn-ion insertion and deinsertion as identified by in operando synchrotron diffraction and XAS. Moreover, an additional broad peak at 875 cm−1 related to ZnxV2O5 is observed for the counter electrode Zn from a charged cell, compared with that from a discharged one. This indicates that the byproduct ZnSO4Zn3(OH)6·5H2O does not completely disappear on the surface of Zn metal. Dissolved ZnxV2O5 can pass through the separator and reaches the surface of Zn metal. This contributes to ongoing degradation and limits the cycling stability.
 | ||
| Fig. 6 Raman spectra of pristine V2O5, the 1st discharged and charged state of V2O5 and of the Zn counter electrode from a discharged and charged cell. | ||
TEM and elemental mapping were carried out to further study the structural and morphology evolution of V2O5 during cycling. It is unable to observe the nanowire-like feature of ZnxV2O5 on the fully discharged electrode, whereas a sheet-like morphology can be clearly seen (Fig. 7a). Fig. S6† shows that the O, S, V, and Zn elements are uniformly distributed in the sheet-like material. The sheet-like material is probably attributed to the byproducts of ZnSO4Zn3(OH)6·5H2O, as demonstrated by synchrotron diffraction. Fig. 7b confirms the disappearance of sheet-like morphology and the recovery of nanowire-like feature of V2O5 after the charge process, demonstrating the decomposition of byproducts.
 | ||
| Fig. 7 TEM images of discharged (a) and charged V2O5 (b). | ||
XPS was applied to investigate the surface chemistry and surface elemental composition of pristine V2O5, first discharged V2O5, and first recharged V2O5. As displayed in Fig. 8, the V 2p spectrum of pristine V2O5 can be fitted with two doublets: a main one with V 2p3/2 at 517.6 eV and a second one with weak intensity at 516.3 eV, which demonstrates that V exists mainly in the oxidation state +5 with a minor contribution of vanadium +4. It can be seen that the O 1s spectrum of pristine V2O5 can be fitted with one peak at 530.3 eV, corresponding to V–O group. The V 2p spectrum in the discharged state is not visible anymore due to the formation of the byproduct ZnSO4Zn3(OH)6·5H2O on the surface of the electrode, which blocks photoelectrons. While the V 2p spectrum of the charged state exhibits two V 2p3/2 components at 517.7 and 516.4 eV, the spectrum returns back to the one of pristine V2O5 after charge. For the discharged state of V2O5, a symmetric O 1s spectrum at 532.0 eV is obtained, assigning to the OH− and SO42− group of the byproduct ZnSO4Zn3(OH)6·5H2O, which was formed on the surface of the electrode and makes the O 1s spectrum from buried V–O groups invisible. After charging, the O 1s spectrum of the V–O group is visible again, accompanied by a minor contribution of OH− on the surface of the electrode.
 | ||
| Fig. 8 V 2p and O 1s X-ray photoelectron spectra of pristine V2O5, together with the 1st discharged and the 1st charged state. | ||
4. Conclusion
In summary, orthorhombic V2O5 nanowires were prepared via a facile hydrothermal approach. In the 1 M ZnSO4 electrolyte, V2O5 nanowires deliver an initial discharge/charge capacity up to 277 and 432 mA h g−1, respectively, at a current density of 50 mA g−1, which almost reaches the theoretical capacity based on 1 Zn2+ insertion with two electrons per formula unit V2O5 (294 mA h g−1). The V2O5 positive electrode exhibits a dramatic decrease of capacity during the following 20 cycles at 50 mA g−1 (94 mA h g−1 for 22nd) and delivers a very low discharge capacity of 21 mA h g−1 after 100 cycles. Moreover, it delivers an initial discharge capacity of 278 mA h g−1 at 200 mA g−1, followed by an activation process of the material. The capacity is higher for 200 mA g−1 than for 50 mA g−1, which could be attributed to a different type of activation process under both current densities and different resulting degrees of side reactions (parasitic reactions). CV displays in the first scan two reduction peaks, centered at 0.92 and 0.50 V, and a broad oxidation peak at 1.20 V with a shoulder at 1.05 V. In the following four scans, the CV curves indicate significant changes in both reduction and oxidation peaks. In operando synchrotron diffraction focused on the first 1.5 cycles, reveals that V2O5 first undergoes a solid solution and 2-phase coexistence transitions upon Zn-ion insertion. It also confirms the formation of two byproducts, Zn3+δ(OH)2V2O7·2(H2O) and ZnSO4Zn3(OH)6·5H2O during the Zn-ion insertion. The electrode undergoes a reversible process upon Zn-ion deinsertion with the decomposition of both byproducts. The V2O5 electrode goes in the 2nd discharge process through the same two-phase reaction as that in the 1st discharge without the formation of the byproduct Zn3+δ(OH)2V2O7·2(H2O). In operando XAS confirms the reduction/oxidation of vanadium during the Zn insertion/deinsertion. Moreover, ex situ Raman and XPS also prove the reversibility of the reactions during cycling. The electrochemical performance of V2O5 can be improved by electrode engineering such as surface coating with carbon/graphene oxide and electrolyte optimization with more concentrated salt and water–organic solvent.Author contributions
Q. F. conceived the idea and discussed with J. W., A. S., L. Z., A. M., E. W., X. L., Z. D., M. K., H. E., and S. D.; Q. F. and J. W. performed material synthesis, sample preparation, characterizations, electrochemical measurements, and analyzed the data. L. Z. performed Raman and analyzed the data. Q. F., J. W., A. S., A. M., E. W., M. K., and H. E. conducted in operando measurements and analyzed the data. X. L. performed XPS measurements and analyzed the XPS data. Z. D. carried out TEM and analyzed the data. Q. F. wrote the preliminary draft with input from J. W.; Q. F., J. W., M. K., H. E., and S. D., discussed the results and revised the manuscript. All authors contributed to interpreting the findings, reviewing, and commenting on the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work contributes to the research performed at CELEST (Center for Electrochemical Energy Storage Ulm-Karlsruhe) and is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Project ID 390874152 (POLiS Cluster of Excellence). Our research work has gained benefit from beamtime allocation at BL04-MSPD at ALBA Synchrotron (2018022738-qfu), Barcelona, Spain, and at PETRA-III beamline P65 (I-20180402) at DESY, Hamburg, Germany. The K-Alpha instrument was supported by the German Federal Ministry of Economics and Technology (grant 03ET6040) based on a decision by the German Bundestag. The authors acknowledge the support of Karlsruhe Nano Micro Facility (KNMF) for the usage of TEM. We thank the help of Dr Francois Fauth from Experiments Division at ALBA. We thank Mrs Bettina Hunzinger and Mr Udo Geckle for their contributions to SEM measurements. We appreciate Dr Georgian Melinte (HIU) for the helpful discussion regarding TEM result.References
- H. Kim, J. Hong, K.-Y. Park, H. Kim, S.-W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed.
- F. Wan, L. Zhang, X. Dai, X. Wang, Z. Niu and J. Chen, Nat. Commun., 2018, 9, 1656 CrossRef PubMed.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., 2012, 51, 933–935 CrossRef CAS PubMed.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- M. H. Alfaruqi, V. Mathew, J. Gim, S. Kim, J. Song, J. P. Baboo, S. H. Choi and J. Kim, Chem. Mater., 2015, 27, 3609–3620 CrossRef CAS.
- B. K. Wu, G. B. Zhang, M. Y. Yan, T. F. Xiong, P. He, L. He, X. Xu and L. Q. Mai, Small, 2018, 14, 1703850 CrossRef PubMed.
- M. H. Alfaruqi, J. Gim, S. Kim, J. Song, J. Jo, S. Kim, V. Mathew and J. Kim, J. Power Sources, 2015, 288, 320–327 CrossRef CAS.
- B. Lee, C. S. Yoon, H. R. Lee, K. Y. Chung, B. W. Cho and S. H. Oh, Sci. Rep., 2014, 4, 6066 CrossRef CAS PubMed.
- J. Lee, J. B. Ju, W. I. Cho, B. W. Cho and S. H. Oh, Electrochim. Acta, 2013, 112, 138–143 CrossRef CAS.
- C. Xu, S. W. Chiang, J. Ma and F. Kang, J. Electrochem. Soc., 2013, 160, A93–A97 CrossRef CAS.
- W. Sun, F. Wang, S. Hou, C. Yang, X. Fan, Z. Ma, T. Gao, F. Han, R. Hu, M. Zhu and C. Wang, J. Am. Chem. Soc., 2017, 139, 9775–9778 CrossRef CAS PubMed.
- M. H. Alfaruqi, J. Gim, S. Kim, J. Song, D. T. Pham, J. Jo, Z. Xiu, V. Mathew and J. Kim, Electrochem. Commun., 2015, 60, 121–125 CrossRef CAS.
- S. Islam, M. H. Alfaruqi, V. Mathew, J. Song, S. Kim, S. Kim, J. Jo, J. P. Baboo, D. T. Pham, D. Y. Putro, Y. K. Sun and J. Kim, J. Mater. Chem. A, 2017, 5, 23299–23309 RSC.
- M. H. Alfaruqi, S. Islam, J. Gim, J. Song, S. Kim, D. T. Pham, J. Jo, Z. Xiu, V. Mathew and J. Kim, Chem. Phys. Lett., 2016, 650, 64–68 CrossRef CAS.
- B. Lee, H. R. Lee, H. Kim, K. Y. Chung, B. W. Cho and S. H. Oh, Chem. Commun., 2015, 51, 9265–9268 RSC.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- R. Y. Wang, C. D. Wessells, R. A. Huggins and Y. Cui, Nano Lett., 2013, 13, 5748–5752 CrossRef CAS PubMed.
- K. Lu, B. Song, Y. X. Zhang, H. Y. Ma and J. T. Zhang, J. Mater. Chem. A, 2017, 5, 23628–23633 RSC.
- R. Trócoli and F. La Mantia, ChemSusChem, 2015, 8, 481–485 CrossRef PubMed.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Adv. Energy Mater., 2015, 5, 1400930 CrossRef.
- M. H. Alfaruqi, V. Mathew, J. Song, S. Kim, S. Islam, D. T. Pham, J. Jo, S. Kim, J. P. Baboo, Z. Xiu, K.-S. Lee, Y.-K. Sun and J. Kim, Chem. Mater., 2017, 29, 1684–1694 CrossRef CAS.
- P. He, Y. L. Quan, X. Xu, M. Y. Yan, W. Yang, Q. Y. An, L. He and L. Q. Mai, Small, 2017, 13, 1702551 CrossRef PubMed.
- J. H. Jo, Y.-K. Sun and S.-T. Myung, J. Mater. Chem. A, 2017, 5, 8367–8375 RSC.
- C. Xia, J. Guo, P. Li, X. Zhang and H. Alshareef, Angew. Chem., Int. Ed., 2018, 57, 3943–3948 CrossRef CAS PubMed.
- C. Xia, J. Guo, Y. Lei, H. Liang, C. Zhao and H. Alshareef, Adv. Mater., 2018, 30, 1705580 CrossRef PubMed.
- X. Dai, F. Wan, L. Zhang, H. Cao and Z. Niu, Energy Storage Mater., 2018, 17, 143–150 CrossRef.
- J. Ding, Z. Du, L. Gu, B. Li, L. Wang, S. Wang, Y. Gong and S. Yang, Adv. Mater., 2018, 30, 1800762 CrossRef PubMed.
- P. He, G. Zhang, X. Liao, M. Yan, X. Xu, Q. An, J. Liu and L. Mai, Adv. Energy Mater., 2018, 8, 1702463 CrossRef.
- P. Hu, T. Zhu, X. Wang, X. Wei, M. Yan, J. Li, W. Luo, W. Yang, W. Zhang, L. Zhou, Z. Zhou and L. Mai, Nano Lett., 2018, 18, 1758–1763 CrossRef CAS PubMed.
- D. Kundu, S. Hosseini Vajargah, L. Wan, B. Adams, D. Prendergast and L. F. Nazar, Energy Environ. Sci., 2018, 11, 881–892 RSC.
- M. Yan, P. He, Y. Chen, S. Wang, Q. Wei, K. Zhao, X. Xu, Q. An, Y. Shuang, Y. Shao, K. Mueller, L. Mai, J. Liu and J. Yang, Adv. Mater., 2018, 30, 1703725 CrossRef PubMed.
- J. S. Park, J. H. Jo, Y. Aniskevich, A. Bakavets, G. Ragoisha, E. Streltsov, J. Kim and S. T. Myung, Chem. Mater., 2018, 30, 6777–6787 CrossRef CAS.
- Q. Pang, C. Sun, Y. Yu, K. Zhao, Z. Zhang, P. Voyles, G. Chen, Y. Wei and X. Wang, Adv. Energy Mater., 2018, 0, 1800144 CrossRef.
- B. Sambandam, V. Soundharrajan, S. Kim, M. H. Alfaruqi, J. Jo, S. Kim, V. Mathew, Y. K. Sun and J. Kim, J. Mater. Chem. A, 2018, 6, 3850–3856 RSC.
- B. Sambandam, V. Soundharrajan, S. Kim, M. H. Alfaruqi, J. Jo, S. Kim, V. Mathew, Y. K. Sun and J. Kim, J. Mater. Chem. A, 2018, 6, 15530–15539 RSC.
- C. Shen, X. Li, N. Li, K. Xie, J.-g. Wang, X. Liu and B. Wei, ACS Appl. Mater. Interfaces, 2018, 10, 25446–25453 CrossRef CAS PubMed.
- V. Soundharrajan, B. Sambandam, S. Kim, M. H. Alfaruqi, D. Y. Putro, J. Jo, S. Kim, V. Mathew, Y. K. Sun and J. Kim, Nano Lett., 2018, 18, 2402–2410 CrossRef CAS PubMed.
- D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid, E. Tervoort, A. Pasquarello and M. Niederberger, Chem. Mater., 2018, 30, 3874–3881 CrossRef CAS.
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 CrossRef PubMed.
- L. Li, S. Peng, H.-Y. Chen, X. Han, F. Cheng, M. Srinivasan, S. Adams, S. Ramakrishna and J. Chen, Nano Energy, 2016, 19, 307–317 CrossRef CAS.
- W. Li, K. Wang, S. Cheng and K. Jiang, Energy Storage Mater., 2018, 15, 14–21 CrossRef.
- P. He, M. Yan, G. Zhang, R. Sun, L. Chen, Q. An and L. Mai, Adv. Energy Mater., 2017, 7, 1601920 CrossRef.
- Y. Cheng, L. Luo, L. Zhong, J. Chen, B. Li, W. Wang, S. X. Mao, C. Wang, V. L. Sprenkle, G. Li and J. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 13673–13677 CrossRef CAS PubMed.
- N. Zhang, Y. Dong, M. Jia, X. Bian, Y. Wang, M. Qiu, J. Xu, Y. Liu, L. Jiao and F. Cheng, ACS Energy Lett., 2018, 3, 1366–1372 CrossRef CAS.
- J. Zhou, L. Shan, Z. Wu, X. Guo, G. Fang and S. Liang, Chem. Commun., 2018, 54, 4457–4460 RSC.
- X. Chen, L. Wang, H. Li, F. Cheng and J. Chen, J. Energy Chem., 2019, 38, 20–25 CrossRef.
- D. M. Xu, H. W. Wang, F. Y. Li, Z. C. Guan, R. Wang, B. B. He, Y. S. Gong and X. L. Hu, Adv. Mater. Interfaces, 2019, 6, 1801506 CrossRef.
- B. Y. Tang, J. Zhou, G. Z. Fang, F. Liu, C. Y. Zhu, C. Wang, A. Q. Pan and S. Q. Liang, J. Mater. Chem. A, 2019, 7, 940–945 RSC.
- G. Li, S. Pang, L. Jiang, Z. Guo and Z. Zhang, J. Phys. Chem. B, 2006, 110, 9383–9386 CrossRef CAS PubMed.
- T. Zhai, H. Liu, H. Li, X. Fang, M. Liao, L. Li, H. Zhou, Y. Koide, Y. Bando and D. Golberg, Adv. Mater., 2010, 22, 2547–2552 CrossRef CAS PubMed.
- F. Fauth, I. Peral, C. Popescu and M. Knapp, Powder Diffr., 2013, 28, S360–S370 CrossRef CAS.
- K. L. Parry, A. G. Shard, R. D. Short, R. G. White, J. D. Whittle and A. Wright, Surf. Interface Anal., 2006, 38, 1497–1504 CrossRef CAS.
- J. Rodríguez-Carvajal and Commission on Powder Diffraction (IUCr), in Newsletter, 2001, vol. 26, pp. 12–19 Search PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- V. Shklover, T. Haibach, F. Ried, R. Nesper and P. Novák, J. Solid State Chem., 1996, 123, 317–323 CrossRef CAS.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894–12901 CrossRef CAS PubMed.
- K. Zhu, T. Wu and K. Huang, Chem. Mater., 2021, 33, 4089–4098 CrossRef CAS.
- T. Tanaka, H. Yamashita, R. Tsuchitani, T. Funabiki and S. Yoshida, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2987–2999 RSC.
- J. Wong, F. W. Lytle, R. P. Messmer and D. H. Maylotte, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 30, 5596–5610 CrossRef CAS.
- H. D. Yoo, Y. Liang, H. Dong, J. Lin, H. Wang, Y. Liu, L. Ma, T. Wu, Y. Li, Q. Ru, Y. Jing, Q. An, W. Zhou, J. Guo, J. Lu, S. T. Pantelides, X. Qian and Y. Yao, Nat. Commun., 2017, 8, 339 CrossRef PubMed.
- X. Liu, D. Wang, G. Liu, V. Srinivasan, Z. Liu, Z. Hussain and W. Yang, Nat. Commun., 2013, 4, 2568 CrossRef PubMed.
- R. Baddour-Hadjean, J. P. Pereira-Ramos, C. Navone and M. Smirnov, Chem. Mater., 2008, 20, 1916–1923 CrossRef CAS.
- G. Gershinsky, H. D. Yoo, Y. Gofer and D. Aurbach, Langmuir, 2013, 29, 10964–10972 CrossRef CAS PubMed.
- H. Marchebois, S. Joiret, C. Savall, J. Bernard and S. Touzain, Surf. Coat. Technol., 2002, 157, 151–161 CrossRef CAS.
- J. Kasperek and M. Lenglet, Rev. Metall., 1997, 94, 713–719 CrossRef CAS.
- S. Ni, G. Zhou, S. Lin, X. Wang, Q. Pan, F. Yang and D. He, Mater. Lett., 2009, 63, 2459–2461 CrossRef CAS.
- R. Zhang, P.-G. Yin, N. Wang and L. Guo, Solid State Sci., 2009, 11, 865–869 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta03518e |
| This journal is © The Royal Society of Chemistry 2021 |