
Dual functions of CO2 molecular activation and 4f levels as electron transport bridges in erbium single atom composite photocatalysts therefore enhancing visible-light photoactivities†
Qiuyu
Chen‡
a,
Guoyang
Gao‡
a,
Yanzhou
Zhang
a,
Yini
li
a,
Hongyang
Zhu
*b,
Peifen
Zhu
 c,
Yang
Qu
a,
Guofeng
Wang
*a and
Weiping
Qin
d
c,
Yang
Qu
a,
Guofeng
Wang
*a and
Weiping
Qin
d
aKey Laboratory of Functional Inorganic Material Chemistry, Ministry of Education, School of Chemistry and Materials Science, Heilongjiang University, Harbin 150080, China. E-mail: 2010070@hlju.edu.cn
bSchool of Physics and Electronic Engineering, Linyi University, Linyi 276005, China. E-mail: zhuhongyang@lyu.edu.cn
cDepartment of Physics and Engineering Physics, The University of Tulsa, Tulsa, OK 74104, USA
dCollege of Electronic Science and Engineering, Jilin University, Changchun 130012, China
First published on 14th June 2021
Abstract
Only when the interfacial charge separation is enhanced and the CO2 activation is improved, can the heterojunction nanocomposite photocatalyst be brought into full play for the CO2 reduction reaction (CO2RR). Here, Er3+ single atom composite photocatalysts were successfully constructed based on both the special role of Er3+ single atoms and the special advantages of the SrTiO3:Er3+/g-C3N4 heterojunction in the field of photocatalysis for the first time. As we expected, the SrTiO3:Er3+/g-C3N4 (22.35 and 16.90 μmol g−1 h−1 for CO and CH4) exhibits about 5 times enhancement in visible-light photocatalytic activity compared to pure g-C3N4 (4.60 and 3.40 μmol g−1 h−1 for CO and CH4). In particular, the photocatalytic performance of SrTiO3:Er3+/g-C3N4 is more than three times higher than that of SrTiO3/g-C3N4. From Er3+ fluorescence quenching measurements, photoelectrochemical studies, transient PL studies and DFT calculations, it is verified that a small fraction of surface doping of Er3+ formed Er single-atoms on SrTiO3 building an energy transfer bridge between the interface of SrTiO3 and g-C3N4, resulting in enhanced interfacial charge separation. Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) and adsorption energy calculations demonstrated that the exposed Er single-atoms outside the interface on SrTiO3 preferentially activate the adsorbed CO2, leading to the high photoactivity for the CO2RR. A novel enhanced photocatalytic mechanism was proposed, in which Er single-atoms play dual roles of an energy transfer bridge and activating CO2 to promote charge separation. This provides new insights and feasible routes to develop highly efficient photocatalytic materials by engineering rare-earth single-atom doping.
1. Introduction
The photocatalytic CO2 reduction (CO2RR) with the use of solar energy has been extensively investigated over the past few years because it can not only solve the greenhouse effect of CO2 but also convert CO2 into fuels or other valuable chemical products.1–4 So far, various photocatalysts have been employed in different types of photocatalytic processes, including photodegradation, H2 evolution, CO2 reduction, etc.5–8 However, new photocatalytic materials with enhanced charge separation and CO2 activation are highly desired due to the comparatively low efficiency at present.9–11Graphitic carbon nitride (g-C3N4) as one of the metal-free polymeric semiconductors has the advantages of the appropriate bandgap for absorbing solar energy, very negative conduction band (CB) potential (−1.1 eV) for reduction, low cost and environmental friendliness.12–17 However, the high charge recombination rate prevents high photocatalytic activity. Additionally, because there is no metal, the surface catalytic capacity of CN is poor, especially for the CO2RR.18,19 Many approaches have been utilized to improve the photocatalytic performance of CN.20–22 Among them, coupling with other materials to construct type II heterojunction nanocomposites has been proven as one of the most efficient methods.23–27 However, it is an energy losing process for the photoelectrons in type II heterojunctions, although the charge separation is enhanced.28 Considering the maintenance of the thermodynamic energy of photoelectrons from CN, the CB of the coupled semiconductor should be as negative as possible. SrTiO3 (STO) with a negative CB (−0.71 eV) and high stability has been regarded as an ideal candidate for coupling with CN for the photocatalytic CO2RR.29 However, the mismatched interface of STO/CN suppresses the charge transfer across the heterogeneous interface and the poor CO2 activation further reduces the photoactivity.
As for improving the quality of the interface, an electron transfer bridge achieved by employing noble metals such as Au and Ag, and carbon materials like graphene is confirmed to be effective. However, the higher price, complicated operation and instability restrict their application in photocatalysis. In recent years, it has been widely considered that single-atom catalysts (SACs) play a huge role due to their maximized atom utilization and tunable electronic properties.30–35 In particular rare earth (RE) single-atom ones displayed promoted activity for the CO2RR due to their unique 4f electronic structure.36–38 Therefore, it is desirable to introduce RE single-atoms into the interface of the STO/CN nanocomposite. To the best of our knowledge, there is little work focused on this point which may due to the lack of an efficient method for preparing RE single-atoms.
Depending on the electronic band structure, STO is the photoelectron accepter and the substrate of the consequent reduction for the CO2RR. Since the activation by photogenerated electrons occurs on STO, the activation and surface catalytic activity of CO2 are of significance. Coincidentally, owing to the abundant f electronic structure and strong affinity to CO2, RE single-atoms are regarded as great candidates for the CO2RR by the separated photogenerated electrons. Recently, our group has developed an in situ doping method for preparing RE single-atoms with tunable dispersion density, and promoted photocatalytic CO2RR activities were achieved, especially for Er single-atoms.36 Thus, modifying RE single-atoms like Er on STO may promote photocatalytic CO2RR activities. On the other side, if RE single-atoms can be uniformly prepared on STO, the side contact with CN will build an electronic transfer tunnel to facilitate interfacial charge separation, because the triazine structure of CN is easy to anchor Er.36,39 Moreover, the ones outside the interface on STO nanoparticles will play a significant role in the activation of CO2 molecules for accelerating the photocatalytic reaction.
Herein, this is the first report of single atom composite photocatalysts. Specifically, highly dispersed Er single-atom modified STO nanoparticles have successfully been obtained by the in situ doping method. The STO:Er3+ nanoparticles were then modified on the surface of CN nanosheets to prepare STO:Er3+/CN nanocomposites. The Er single-atoms were confirmed by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM). The as-prepared STO:Er3+/CN nanocomposites exhibited more than 5 times enhancement in the photocatalytic CO2RR compared to bulk g-C3N4 and SrTiO3. By means of Er3+ fluorescence quenching experiments, transient PL studies and DFT calculations, the promoted photoactivities were attributed to the dual-function role of the Er single-atoms which acted as interfacial electronic transfer bridges and active sites for CO2 activation, thus enhancing the charge separation.
2. Experimental
The chemicals and characterization details are shown in the ESI.†Synthesis of STO and STO:Er3+. For STO, 1 mmol Sr(NO3)2 was dissolved in 19.67 mL PEG200 under vigorous stirring. Subsequently, 0.33 mL Ti(OC4H9)4 and 22 mmol NaOH were added to the above solution, respectively. After being stirred, the mixture was transferred to an autoclave and maintained at 180 °C for 15 h. The final product was collected by centrifugation, washing with acetone, CH3COOH, and deionized water, and drying in an oven at 70 °C. The synthesis process of STO:Er3+ is the same as that of STO, except adding Er(NO3)3·6H2O into the Sr(NO3)2 solution in the initial reaction stage. And the prepared STO:Er3+ composite was labeled as STO:Er3+.
Synthesis of STO:Er3+/CN. The CN was obtained by annealing melamine at 550 °C for 2 h under an N2 atmosphere followed by a grinding treatment for subsequent experiments. A certain amount of CN was added into methanol and put in an ultrasonic bath for 3 h to make CN exfoliated completely. Then, different masses of STO:Er3+ are put into the above solution and ultrasonicated for 30 min, and finally stirred thoroughly until evaporation.
3. Results and discussion
3.1. Photocatalytic CO2 reduction mechanism
In the proposed composite catalyst system (Fig. 1), the photoelectrons of CN were modulated by coupling STO as an appropriate platform for photoreduction of CO2 by using visible light as an energy source. We achieved efficient photogenerated electron transfer from the conduction band of CN to STO and Er3+ ions are chosen as the ET bridge. As a bridge of ET, Er3+ ions must meet the following several conditions: (1) the energy level position of Er3+ ions as ET bridges should be in the middle of CN and STO conduction band positions. (2) The probability of nonradiative decay was found to be strongly dependent upon the proximity of lower energy levels. And thus, the position of the energy level as the ET bridge and the position of the lower level of rare-earth ions should be large enough to reduce the nonradiative transition probability inside energy levels of rare-earth ions. Here, it is noted that Er3+ is a very good upconversion activator (luminous center) rather than a good downconversion activator, so Er3+ is more suitable as an energy transfer bridge between STO and CN. (3) The radiation transition probability of inside energy levels of RE ions is as small as possible; thus, fluorescence quenching can occur. According to the bandgap structure of CN and STO, the 4G11/2 energy level of Er3+ ion energy is the most suitable ET bridge. See ESI† for the explanation of the effect of the fluorescence quenching effect of Er3+ ions on the electron transfer from CN to STO. Of course, the lattice distortion of STO (Fig. S1†) caused by Er3+ doping will also affect the CO2 adsorption capacity and catalytic performance of STO, which will be discussed in detail later. According to the mechanism proposed above, the synthesis route of the composite system for promoting electron separation is shown in Fig. S2,† where STO:Er3+ and CN were separately synthesized first, and then STO:Er3+/CN composites were obtained by chemical adsorption. The detailed synthesis information and sample abbreviation are stated in the ESI.† | ||
| Fig. 1 (a and b) The proposed mechanism for Er3+ ions as ET bridges to promote charge separation thereby enhancing photocatalytic efficiency based on the fluorescence quenching of Er3+ ions. | ||
3.2. Crystal structure and morphology properties
The particle size of the sample does not change obviously after being doped with Er3+ because the concentration of Er3+ (only 3 mol%) is relatively low. The average particle size of STO or SrTO3:Er3+ is about 10 nm, and the loading of small STO:Er3+ particles on the surface of CN was observed (Fig. 2, S3 and S4†). As shown in Fig. 2d–f, the formation of erbium single atomic sites in STO:Er3+/CN composite materials was confirmed by AC HAADF-STEM. The elemental maps further indicate that the single erbium atoms are uniformly distributed on the STO nanoparticle surface (Fig. 2g). The results of XRD patterns indicate that STO and STO:3% Er3+ were pure perovskite oxides (Fig. S5†), and CN was a pure hexagonal phase (JCPDS 87-1526). When the concentration of Er3+ is more than 5 mol%, the diffraction peaks of impurities were observed. The characteristic peaks of CN and STO were detected in the composite sample, proving a two-phase composition of CN and STO in the 5-STO:Er3+/CN sample. | ||
| Fig. 2 TEM images of (a) STO:Er3+ and (b) 5-STO:Er3+/CN. (c) HRTEM image of 5-STO:Er3+/CN. (d–f) The AC HAADF-STEM images of 5-STO:Er3+/CN. (g) HAADF-STEM image and EDX elemental maps of 5-STO:Er3+/CN. | ||
The bandgap energies (Eg) of the CN, 5-STO:Er3+/CN, 10-STO:Er3+/CN, and STO:Er3+ are determined as approximately 2.64, 2.67, 2.70, and 3.16 eV, respectively (Fig. 3a). CN can exhibit visible light absorption while STO:Er3+ can only absorb ultraviolet light, and thus, the visible light catalytic performance of the composite is due to the absorption of light by CN first, then the electron transport to STO with the help of Er3+ as the ET bridge, and finally the CO2 reduction reaction takes place. It is worth noting that two very weak absorption peaks at 522 nm and 655 nm in the inset of Fig. 3a were assigned to the transitions from the ground 4I15/2 state to the excited 2H11/2 and 4F9/2 states, respectively. The results show that the absorption of Er3+ ions is very weak and thus its contribution to the enhancement of visible light absorption of the catalyst can be neglected, which further proves our proposed hypothesis above. In addition, the specific surface area of sample 5-STO:Er3+/CN is increased compared to that of CN (Fig. 3b), showing that some molecules (such as CO2) are beneficial to be adsorbed on the composite catalytic system surface, which is consistent with the above theoretical results.
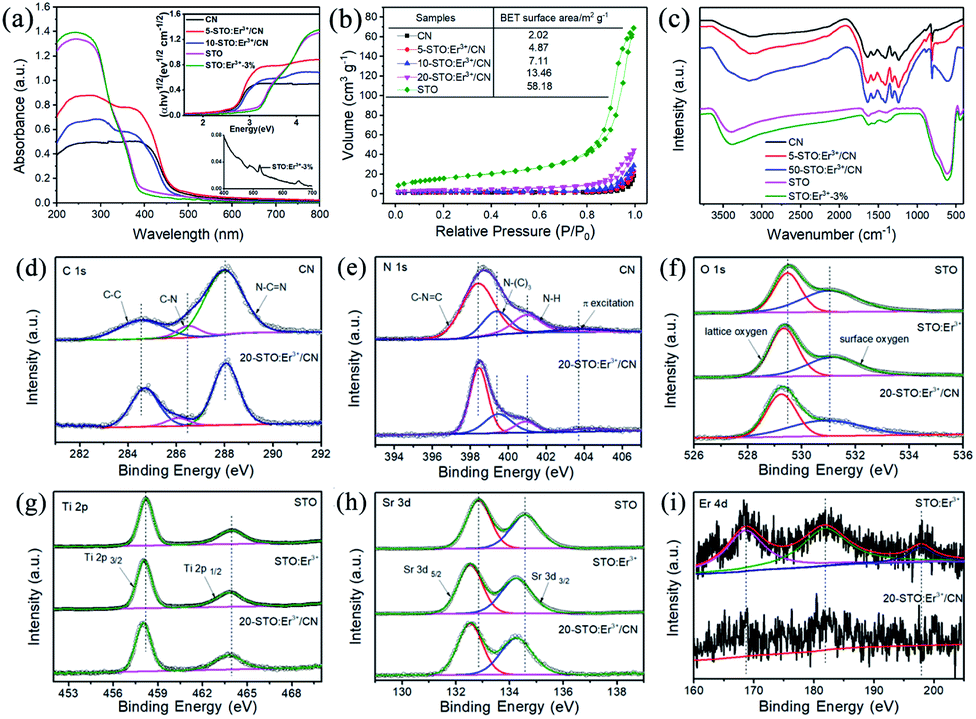 | ||
| Fig. 3 (a) UV-vis diffuse reflectance spectra and band gaps patterns (inset), (b) N2 adsorption–desorption isotherms, and (c) FT-IR spectra of different samples. (d–i) High-resolution XPS spectra of CN, STO, STO:Er3+, and 20-STO:Er3+/CN. | ||
The FT-IR and XPS spectroscopies were used to investigate the interactions between CN and STO:Er3+ in composite materials (Fig. 3c–i and S6–S8†). The peak at 813 cm−1 in the FT-IR spectrum of pure CN corresponds to the characteristic breathing vibration of the s-triazine ring. The pure CN also shows the bands between 1200 cm−1 and 1700 cm−1, which largely originate from the typical stretching vibration of CN heterocycles. The additional band at ∼3100 cm−1 is the stretching vibration modes of NH and NH2 groups, which result from incomplete calcination of melamine on the surface of CN. For the STO, the absorption peak at around 3440 cm−1 could be caused by the hydroxyl groups on the sample surface. The broadband in the region from 500 cm−1 to 700 cm−1 corresponds to the Ti–O stretching vibrations. Note that the vibration peak of STO in the composites weakens, widens, and shifts, meaning the surface doping of Er3+ on SrTiO3 as well as the existence of interactions in the nanocomposites.
The XPS spectrum of C 1s for CN can be split into three peaks at 284.6, 286.5, and 288.0 eV attributed to the C–C, C–N, and N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N (Fig. 3d). However, the peaks at 284.7, 286.1, and 288.1 eV were observed for 20-STO:Er3+/CN, indicating the increased electronic density of the C surface and the existence of an interaction between STO:Er3+ and CN. For the N 1s XPS spectra of CN, the observed four peaks at 398.4, 399.4, 401.0, and 403.8 eV were ascribed to the sp2-hybridized nitrogen atoms (CN–C), bridging nitrogen atoms (C3–N), the N–H bonding and the charging effects caused by π-excitations. For 20-STO:Er3+/CN, the peaks of N–(C)3 and π-excitations were shifted to 399.5 and 403.1 eV, proving the existence of an interaction between STO:Er3+ and CN (Fig. 3e). The peaks at 529.5 and 531.1 eV in the O 1s spectrum of STO correspond to the lattice oxygen O2− species and surface oxygen, respectively (Fig. 3f), while the combined energy of lattice oxygen O2− species shifts to a low energy direction after Er3+ doping or CN coupling. The Ti 2p spectrum can be divided into two parts with peaks at 458.2 eV and 464.0 eV in STO (Fig. 3g), which corresponds to Ti 2p3/2 and Ti 2p1/2, respectively. In 20-STO:Er3+/CN and STO:Er3+ samples, the two peaks are located at 458.0 eV and 463.8 eV, respectively. The Sr spectrum in pure STO is primarily composed of two binding energy peaks of Sr 3d5/2 and Sr 3d3/2 at 132.9 eV and 134.6 eV (Fig. 3h). Compared with STO, the binding energy of Sr 3d peaks of 20-STO:Er3+/CN and STO:Er3+ shifts to a lower energy direction, which is due to the fact that Er3+ ion doping changes the local electronic structure. The peaks in Fig. 3i are attributed to Er 4d. The XPS valence band of STO and STO:Er3+ is at 2.19 and 2.13 eV, respectively (Fig. S7†). The position shift of binding energy in STO proves the change of electronic density as well as the existence of an interaction between STO:Er3+ and CN. The Er3+ concentration in the STO:Er3+ and 20-STO:Er3+/CN samples is 2.5816% and 0.3293% mg L−1, respectively, which was measured by using ICP-OES technology.
C–N (Fig. 3d). However, the peaks at 284.7, 286.1, and 288.1 eV were observed for 20-STO:Er3+/CN, indicating the increased electronic density of the C surface and the existence of an interaction between STO:Er3+ and CN. For the N 1s XPS spectra of CN, the observed four peaks at 398.4, 399.4, 401.0, and 403.8 eV were ascribed to the sp2-hybridized nitrogen atoms (CN–C), bridging nitrogen atoms (C3–N), the N–H bonding and the charging effects caused by π-excitations. For 20-STO:Er3+/CN, the peaks of N–(C)3 and π-excitations were shifted to 399.5 and 403.1 eV, proving the existence of an interaction between STO:Er3+ and CN (Fig. 3e). The peaks at 529.5 and 531.1 eV in the O 1s spectrum of STO correspond to the lattice oxygen O2− species and surface oxygen, respectively (Fig. 3f), while the combined energy of lattice oxygen O2− species shifts to a low energy direction after Er3+ doping or CN coupling. The Ti 2p spectrum can be divided into two parts with peaks at 458.2 eV and 464.0 eV in STO (Fig. 3g), which corresponds to Ti 2p3/2 and Ti 2p1/2, respectively. In 20-STO:Er3+/CN and STO:Er3+ samples, the two peaks are located at 458.0 eV and 463.8 eV, respectively. The Sr spectrum in pure STO is primarily composed of two binding energy peaks of Sr 3d5/2 and Sr 3d3/2 at 132.9 eV and 134.6 eV (Fig. 3h). Compared with STO, the binding energy of Sr 3d peaks of 20-STO:Er3+/CN and STO:Er3+ shifts to a lower energy direction, which is due to the fact that Er3+ ion doping changes the local electronic structure. The peaks in Fig. 3i are attributed to Er 4d. The XPS valence band of STO and STO:Er3+ is at 2.19 and 2.13 eV, respectively (Fig. S7†). The position shift of binding energy in STO proves the change of electronic density as well as the existence of an interaction between STO:Er3+ and CN. The Er3+ concentration in the STO:Er3+ and 20-STO:Er3+/CN samples is 2.5816% and 0.3293% mg L−1, respectively, which was measured by using ICP-OES technology.
3.3. Photocatalytic activity and charge separation
To confirm the photocatalytic mechanism described above, the performance of photocatalytic reduction of CO2 for STO, STO:Er3+, CN, and STO:Er3+/CN has been studied in detail (Fig. 4a–d). As shown in Fig. 4a, STO:Er3+ exhibits a higher CO and CH4 evolution rate than STO under ultraviolet light. This result shows that the exposed Er3+ ions on the surface can effectively activate CO2 to improve photocatalytic activity. Next, we studied the CO2 reduction performance of the photocatalyst under visible light irradiation. As shown in Fig. 4b, 5-STO:Er3+/CN shows a CO-evolution rate of 22.35 μmol g−1 h−1 and a CH4-evolution rate of 16.90 μmol g−1 h−1 in a mixed system of acetonitrile, water and triethanolamine. And compared with pure CN, the maximum production rate of CO and CH4 can be increased by about 5 times. The performance comparison of all rare earth single atom photocatalysts reported is summarized in Table 1. It is noted that the yield of CH4 is more important than that of CO during the process of CO2RR, so the performance of 5-STO:Er3+/CN in this work is quite good. Of course, it is noted that not all RE ions are suitable for charge transfer bridges because the role of RE ions is not only related to the structure of RE ions but also related to the properties of SrTiO3 and g-C3N4. So, there is still a lot of room for improvement in the design of RE single atom composite photocatalysts. We also further studied the photocatalytic CO2 reduction ability of CN, 5-STO/CN and x-STO:Er3+/CN (x = 1, 3, 5, 7, and 10) photocatalysts in the pure water system. As predicted, the 5-STO:Er3+/CN sample shows an excellent CO evolution rate of 5.68 μmol h−1 g−1 and CH4 production rate of 2.30 μmol g−1 h−1, which is approximately 2 fold higher than that of pure CN (Fig. 4c). However, the introduction of excessive STO:Er3+ will cause a decrease in photocatalytic CO2 reduction activity, indicating that the introduction of STO:Er3+ can improve the photocatalytic activity of CN, but the appropriate amount of STO:Er3+ is important because STO can only absorb UV light and excess STO will lead to the decrease of CN. The photoactivity of 5-STO/CN in the absence of Er3+ is lower compared to the corresponding samples containing Er3+, and this further proves that partial Er3+ ions can act as an energy transfer bridge between STO and CN to promote charge separation. In addition, after three consecutive photocatalytic tests, 5-STO:Er3+/CN still maintained excellent CO2 photoreduction stability (Fig. 4d). To further confirm the source of the produced CH4, we carried out an isotopic experiment using CO2 under identical photocatalytic reaction conditions. The photocatalytic evolution of CH4 was analyzed by gas chromatography mass (GC-MS), of which the peak m/z = 17 is assigned to CH4 and the fragment ion of CH4 is seen as well as shown in Fig. S9.† | ||
| Fig. 4 (a) The photocatalytic production amount of STO, STO:Er3+-1%, and STO:Er3+-3% under ultraviolet light irradiation (pure water system). (b) The photocatalytic production amount of CN, 5-STO/CN, and 5-STO:Er3+/CN under visible light irradiation with TEOA as the sacrificial agent for photocatalytic reaction. (c) The photocatalytic production amount of CN, x-STO:Er3+/CN (x = 1, 3, 5, 7, and 10), and 5-STO/CN under visible light irradiation (pure water system). (d) Evaluation of the stability of 5-STO:Er3+/CN under visible light irradiation (pure water system). (e) Photocurrent responses, (f) EIS spectra, (g) fluorescence spectra related to the produced hydroxyl radicals under visible-light irradiation, (h) PL spectra under the excitation wavelength of 350 nm, and (i) luminescence decay curves. | ||
The photocurrent generation behavior (Fig. 4e) shows that much higher photocurrent is obtained from the 5-STO:Er3+/CN sample than that of pristine CN, indicating that 5-STO:Er3+ can greatly improve the separation efficiency of photogenerated carriers.40 Moreover, the photocurrents of the two samples are reproducible and stable, indicating that the photostability of the samples is good. The electrochemical impedance spectra (EIS) Nyquist plots of pristine CN, STO/CN, and 5-STO:Er3+/CN are shown in Fig. 4f and the smallest arc radius was obtained from 5-STO:Er3+/CN. This means that photogenerated electron–hole pairs are more effectively separated and charges more efficiently transfer across the electrode/electrolyte interface in 5-STO:Er3+/CN. Thus 5-STO:Er3+/CN can greatly enhance the photogenerated electron–hole separation and transfer. Moreover, both the separated photogenerated electrons and holes increase hydroxyl radicals (˙OH) via redox of H+ and H2O, and thus the electron–hole separation can also be evaluated by measuring the concentration of ˙OH.41 We measured the fluorescence spectra of luminescent 7-hydroxycoumarin produced from the reaction between and ˙OH and coumarin by using the coumarin fluorescence method. As a general rule, more hydroxyl radical generation results in a stronger fluorescent signal. We observed the highest intensity from the 5-STO:Er3+/CN sample as shown in Fig. 4g, which means introducing STO:Er3+ significantly improved photogenerated charge separation. Furthermore, the PL intensity has been widely used as a measure of charge recombination rate and the higher PL intensity indicates a higher recombination rate. We observe lower PL intensity from 5-STO:Er3+/CN compared to that of pristine CN as shown in Fig. 4h. This further proves that introducing STO:Er3+ can effectively reduce electron–hole recombination. In addition, the emission peaks of Er3+ cannot be detected, indicating that the fluorescence quenching effect of Er3+ ions was observed (Fig. 1a). The average lifetime of the photogenerated carrier for CN and 5-STO:Er3+/CN was calculated as 6157 ns and 6270 ns, respectively, which further proves it is more effective to generate and separate electron–hole pairs in the 5-STO:Er3+/CN sample (Fig. 4i).
3.4. DFT calculations and discussion
To further study the rationality of the charge separation system designed, suitable lattice structures of STO, ErTiO3 (ETO), and CN were selected and optimized by using DFT (Fig. 5, S10 and Table S1†). Both the STO and ETO are semiconductors with an indirect bandgap energy of 3.16 and 2.30 eV, and a direct bandgap energy of 3.54 and 2.71 eV, respectively. Thus, the bandgap energy of Er-doped STO could be engineered by tuning the ratio of Er to Sr, which is consistent with our experimental results. For both SrTiO3 and ErTiO3, the CBs are mainly contributed by the orbitals of Ti atoms, and the VBs are mainly contributed by the orbitals of O atoms. The replacement of Sr with Er significantly lowered the CB minimum while no obvious change of VB maximum was observed.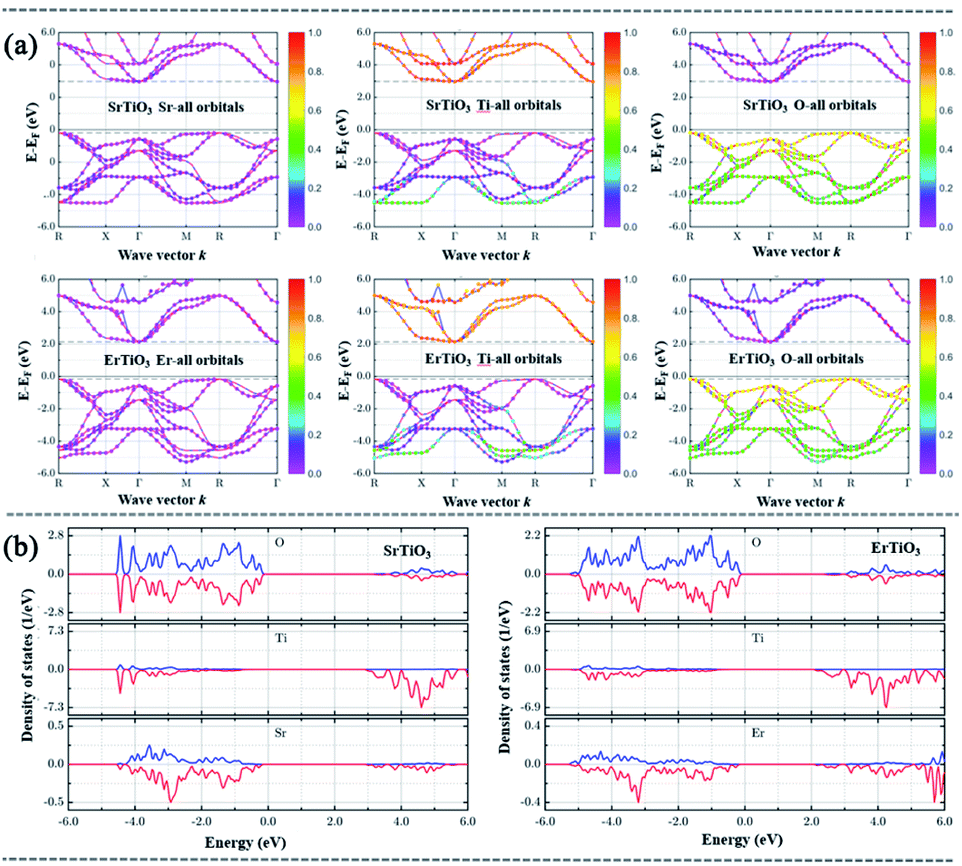 | ||
| Fig. 5 (a) The band structure (BS) and (b) the density of states (DOS) contributed by each element of SrTiO3 and ErTiO3. | ||
The theoretical simulation results of the differential charge density and electron localization function (ELF) in Fig. 6 showed that the net charge on Ti and O is 2.65 and −1.44, respectively in SrTiO3, while the net charge on Ti and O is 2.40 and −1.26, respectively in ErTiO3. This indicates that the replacement of Sr with Er affects the interactions between Ti and O. Compared with SrTiO3, the interaction between Ti and O is stronger, and Ti loses fewer electrons, and therefore, the bandgap of ErTiO3 is smaller than that of SrTiO3. The doping of Er into SrTiO3 introduced additional energy levels between the CB minimum of CN and SrTiO3 and these additional energy levels serve as energy transfer bridges for the ease of electron transfer from CN to SrTiO3.
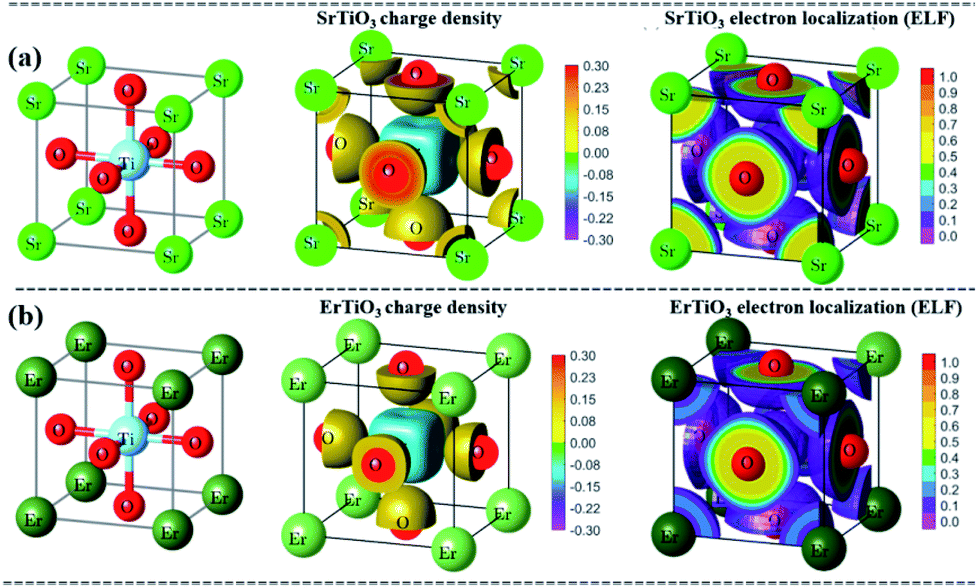 | ||
| Fig. 6 Theoretical simulation results of the charge density and electron localization: (a) SrTiO3 and (b) ErTiO3. | ||
The results of theoretical simulation in Fig. S11† show that the electronic structure and optical properties of SrTiO3 and ErTiO3 are different. The absorption coefficient of ErTiO3 is larger than that of SrTiO3 in the visible region, which indicates that doping Er into SrTiO3 might also increase the light absorption capability. Furthermore, the optical conductivity of ErTiO3 shows better optical conductivity than that of SrTiO3. Thus, doping Er into SrTiO3 also increases the charge transfer and separation. Therefore, the substitution of Er3+ for the Sr2+ lattice in SrTiO3 will give some novel properties to the SrTiO3, and is expected to improve the photocatalytic activity of SrTiO3.
As mentioned above, the lattice parameters, work functions, adsorption capacity of CO2 on the surface of STO, and charge density difference are changed through Er3+ doping (Fig. 7, S12 and S13†). The thickness of the vacuum layer is 10 Å. The work functions of STO (001), STO:Er3+ (001), STO (110), STO:Er3+ (110), and CN (001) are 3.64, 2.74, 5.82, 5.97, and 4.22 eV, respectively. The work function of CN (001) is greater than that of STO (001) and STO:Er3+ (001) while less than that of STO (110) and STO:Er3+ (110). The adsorption energy of CO2 on the surface of STO (110) and CN (001) is 5.82 and 4.22 eV, respectively. The charge differential density shows that Er3+ not only can result in the change of the density of the surrounding electron cloud but also can better absorb CO2 and play the role of an activation center.33 All these theoretical results further prove that the composite system proposed is conducive to promoting charge separation and improving photocatalytic activity.
 | ||
| Fig. 7 (a–c) Side and top views of the optimized crystal structure and CO2 adsorption on the different surfaces of STO, STO:Er3+, and CN. (d) The calculated work function and adsorption energy (the Fermi energy is used as the zero of the energy scale). (e) The effect Er3+ on the charge density difference around Er3+ in STO:Er3+ (001) as well as CO2 in CO2-STO:Er3+ (001). In (e), the yellow and cyan surfaces correspond to the charge gain and charge loss, respectively. | ||
4. Conclusions
In conclusion, the mechanism proposed in this work is based on fully considering the characteristics of narrow half peak width and low luminous efficiency of rare-earth single-atoms, breaking through the traditional view that RE ions are used as light conversion agents, and realizing the dual effects of erbium ions activating CO2 and acting as an energy transfer bridge to promote charge separation. The formation of Er single-atoms in the STO:Er3+/CN composite material was confirmed by AC HAADF-STEM. The results of UV-vis DRS, EIS, and XPS show that the surface doping of single-atoms Er3+ on SrTiO3 favors building an energy transfer bridge between SrTiO3 and g-C3N4, resulting in enhanced interfacial charge separation. A series of calculation results of band structure, work function, and CO2 adsorption energy prove that the STO:Er3+ surface plays dual functional roles of charge separation and CO2 activation to promote the photocatalytic activity of the CO2RR. As expected, the experimental results indicate that STO:Er3+/CN exhibits much higher photocatalytic performance under visible light irradiation compared to that of pristine g-C3N4 and STO/CN. This provides a new way of thinking and perspective for the development of highly efficient photocatalytic materials modified by RE single-atoms.Author contributions
Q. C. and G. G. analyzed the experimental data and wrote the original draft. Q. C., G. G., Y. Z. and Y. L. performed major experiments. G. W., H. Z., Q. C. and G. G. designed the concept of this work. G. W., H. Z. and P. Z. performed the computational study. G. W., H. Z., W. Q. and Y. Q revised the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21871079 and 11774128), the National Science Foundation (No. 1945558), the Outstanding Youth Project of Natural Science Foundation of Heilongjiang Province (YQ2019B006 and LBH-Q19052), and the Natural Science Foundation of Shandong Province (ZR2018JL003, 2019KJJ003).Notes and references
- J. Ran, M. Jaroniec and S. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- G. Wang, C. He, R. Huang, J. Mao, D. Wang and Y. Li, J. Am. Chem. Soc., 2020, 142, 19339–19345 CrossRef CAS PubMed.
- X. Jiao, K. Zheng, Z. Hu, Y. Sun and Y. Xie, ACS Cent. Sci., 2020, 6, 653–660 CrossRef CAS PubMed.
- C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC.
- L. Ji, L. Li, X. Ji, Y. Zhang, S. Mou, T. Wu, Q. Liu, B. Li, X. Zhu, Y. Luo, X. Shi, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2020, 59, 758–762 CrossRef CAS PubMed.
- H. Zhang, P. Zhang, M. Qiu, J. Dong, Y. Zhang and X. W. Lou, Adv. Mater., 2019, 31, 1804883 Search PubMed.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, J. Am. Chem. Soc., 2017, 139, 7217–7223 CrossRef CAS PubMed.
- H. Mao, Z. Jin, F. Zhang, H. He, J. Chen and Y. Qian, Ceram. Int., 2018, 44, 17535–17538 CrossRef CAS.
- Y. Jiang, H. Chen, J. Li, J. Liao, H. Zhang, X. Wang and D. Kuang, Adv. Funct. Mater., 2020, 30, 2004293 CrossRef CAS.
- Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C. Sun, Z. Cai, J. R. Guest, Y. Ren, V. Stamenkovic, L. A. Curtiss, Y. Liu and T. Rajh, Nat. Energy, 2019, 4, 957–968 CrossRef CAS.
- H. Zhang, Y. Wang, S. Zuo, W. Zhou, J. Zhang and X. W. Lou, J. Am. Chem. Soc., 2021, 143, 2173–2177 CrossRef CAS PubMed.
- J. Shen, Y. Li, H. Zhao, K. Pan, X. Li, Y. Qu, G. Wang and D. Wang, Nano Res., 2019, 12, 1931–1936 CrossRef CAS.
- X. Liu, S. Xu, H. Chi, T. Xu, Y. Guo, Y. Yuan and B. Yang, Chem. Eng. J., 2019, 359, 1352–1359 CrossRef CAS.
- Y. Dai, C. Li, Y. Shen, T. Lim, J. Xu, Y. Li, H. Niemantsverdriet, F. Besenbacher, N. Lock and R. Su, Nat. Commun., 2018, 9, 60 CrossRef PubMed.
- T. Xiong, W. Cen, Y. Zhang and F. Dong, ACS Catal., 2016, 6, 2462–2472 CrossRef CAS.
- Z. Jiang, W. Wan, H. Li, S. Yuan, H. Zhao and P. K. Wong, Adv. Mater., 2018, 30, 1706108 CrossRef PubMed.
- F. Yang, X. Chua, J. Sun, Y. Zhang, Z. Li, H. Liu, L. Bai, Y. Qu and L. Jing, Chin. Chem. Lett., 2020, 31, 2784–2788 CrossRef CAS.
- R. Kuriki, M. Yamamoto, K. Higuchi, Y. Yamamoto, M. Akatsuka, D. Lu, S. Yagi, T. Yoshida, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2017, 56, 4867–4871 CrossRef CAS PubMed.
- B. Ma, G. Chen, C. Fave, L. Chen, R. Kuriki, K. Maeda, O. Ishitani, T.-C. Lau, J. Bonin and M. Robert, J. Am. Chem. Soc., 2020, 142, 6188–6195 CrossRef CAS PubMed.
- J. Fu, B. Zhu, C. Jiang, B. Cheng, W. You and J. Yu, Small, 2017, 13, 1603938 CrossRef PubMed.
- H. Pan, Y.-W. Zhang, V. B. Shenoy and H. Gao, ACS Catal., 2011, 1, 99–104 CrossRef CAS.
- W. J. Ong, L. L. Tan, Y. H. Ng, S.-T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- Y.-C. Pu, H.-C. Fan, T.-W. Liu and J.-W. Chen, J. Mater. Chem. A, 2017, 5, 25438–25449 RSC.
- H. Shi, G. Chen, C. Zhang and Z. Zou, ACS Catal., 2014, 4, 3637–3643 CrossRef CAS.
- L. Jiang, K. Wang, X. Wu and G. Zhang, Sol. RRL, 2021, 5, 2000326 CrossRef CAS.
- Y. Wang, L. Guo, Y. Zeng, H. Guo, S. Wan, M. Ou, S. Zhang and Q. Zhong, ACS Appl. Mater. Interfaces, 2019, 11, 30673–30681 CrossRef CAS PubMed.
- P. Ding, H. Zhao, T. Li, Y. Luo, G. Chen, S. Gao, X. Shi, S. Lu and X. Sun, J. Mater. Chem. A, 2020, 8, 21947–21960 RSC.
- Z. Zhang, L. Bai, Z. Li, Y. Qu and L. Jing, J. Mater. Chem. A, 2019, 7, 10879–10897 RSC.
- T. Kanagaraj and S. Thiripuranthagan, Appl. Catal., B, 2017, 207, 218–232 CrossRef CAS.
- Q. Yang, C. Yang, C. Lin and H. Jiang, Angew. Chem., Int. Ed., 2019, 58, 3511–3515 CrossRef CAS PubMed.
- C. Gao, S. Chen, Y. Wang, J. Wang, X. Zheng, J. Zhu, L. Song, W. Zhang and Y. Xiong, Adv. Mater., 2018, 30, 1704624 CrossRef PubMed.
- Y. Li, B. Li, D. Zhang, L. Cheng and Q. Xiang, ACS Nano, 2020, 14, 10552–10561 CrossRef CAS PubMed.
- J. Wang, T. Heil, B. Zhu, C.-W. Tung, J. Yu, H. M. Chen, M. Antonietti and S. Cao, ACS Nano, 2020, 14, 8584–8593 CrossRef CAS PubMed.
- R. Sun, Y. Liao, S.-T. Bai, M. Zheng, C. Zhou, T. Zhang and B. F. Sels, Energy Environ. Sci., 2021, 14, 1247–1285 RSC.
- C. Chen, Y. Li, D. Cheng, H. He and K. Zhou, ACS Appl. Mater. Interfaces, 2020, 12, 40415–40425 CrossRef CAS PubMed.
- S. Ji, Y. Qu, T. Wang, Y. Chen, G. Wang, X. Li, J. Dong, Q. Chen, W. Zhang, Z. Zhang, S. Liang, R. Yu, Y. Wang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 10651–10657 CrossRef CAS PubMed.
- P. Chen, B. Lei, X. a. Dong, H. Wang, J. Sheng, W. Cui, J. Li, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 15841–15852 CrossRef PubMed.
- J. Liu, X. Kong, L. Zheng, X. Guo, X. Liu and J. Shui, ACS Nano, 2020, 14, 1093–1101 CrossRef CAS PubMed.
- X. Li, K. Pan, Y. Qu and G. Wang, Nano Res., 2018, 11, 1322–1330 CrossRef CAS.
- H. Dong, X. Zhang, Y. Lu, Y. Yang, Y. Zhang, H. Tang, F. Zhang, Z. Yang, X. Sun and Y. Feng, Appl. Catal., B, 2020, 276, 119173 CrossRef CAS.
- J. Sun, J. Bian, J. Li, Z. Zhang, Z. Li, Y. Qu, L. Bai, Z. Yang and L. Jing, Appl. Catal., B, 2020, 277, 11919 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta02926f |
| ‡ Qiuyu Chen and Guoyang Gao contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |