
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designing for conjugate addition: an amine functionalised quinone anolyte for redox flow batteries†
Rajesh B.
Jethwa
 ,
Evan Wenbo
Zhao
,
Rachel N.
Kerber‡
,
Erlendur
Jónsson
,
Dominic S.
Wright
* and
Clare P.
Grey
*
,
Evan Wenbo
Zhao
,
Rachel N.
Kerber‡
,
Erlendur
Jónsson
,
Dominic S.
Wright
* and
Clare P.
Grey
*
Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: DSW1000@cam.ac.uk; CPG27@cam.ac.uk
First published on 19th May 2021
Abstract
Redox flow batteries (RFBs) are promising grid-level electrical storage systems. The key to this emerging technology is the development of cheap, highly soluble, and high energy-density inorganic and organic electrolytes. Although quinone-based electrolytes have been investigated previously as two-electron anolytes, their susceptibility to undergo conjugate addition is generally viewed as a limiting factor in their application in RFBs. The study of the amine functionalised benzoquinone, 2,5-(4-carboxy-anilino)-1,4-benzoquinone (CABQ), presented here shows, however, that the resulting dihydroxylation leads to stabilisation of the anolyte at a lower potential, bringing with it increased energy density. A combination of ex situ and in situ techniques is used both to identify the anolyte formed after nucleophilic attack and to help explain the cyclic voltammetry and galvanostatic cycling data. A proof-of-concept, lab-scale RFB cell was run against potassium ferrocyanide (voltage of approximately 1.1 V) in basic aqueous solution which showed high coulombic efficiencies (>99.5%).
Introduction
Redox flow batteries (RFBs) with their decoupled power and capacity scaling1–8 are attractive electrical energy storage systems. This is especially the case for grid storage because, for a given power density, the duration of power storage can be varied by changing the amount of electrolyte. This, coupled with the advantages of minimal physical changes to the electrode during cycling3,4,8 and the intrinsically safe architecture,2,3,5 has contributed to the considerable attention that RFBs have received in recent years.1,7,8 However, their low energy density3,8,9 and initial costs of the large quantities of electrolytes3,8 and their rapid degradation, currently limit the implementation of such systems.The redox active species has a significant effect on the cost of the RFB through its solubility, molar mass, number of electrons stored per molecule, material cost and cell potential,2 both cheaper materials and increased cell potentials leading to reduced overall costs.2 The U.S. Department of Energy's recommended capital cost for a system with a 4 h discharge at rated power2,10,11 is $150 per kW h, however, current systems are too expensive.2,6,12 Hence, new redox chemistries need to be introduced. In the literature, attempted solutions to these key issues have focused on both organic and inorganic electrolytes7 in both aqueous and non-aqueous solvents.4
Aqueous systems have a limited cell voltage due to the electrochemical stability window of water13,14 but are appealing due to the high conductivity of salts/ions in water, lower costs, high safety, easy processing and low volatility.1 Non-aqueous systems, on the other hand, generally have a larger stability window and consequently allow for higher cell voltages.1,11,14 Moreover, organic solvents generally have lower freezing temperatures and so are more suited for low-temperature operations where water can freeze.15 However, non-aqueous systems typically suffer from increased cost, reduced conductivity, flammability,11,14 moisture sensitivity and often toxicity.11 Additionally, non-aqueous systems suffer from a lower power density than aqueous RFBs despite the greater electrochemical stability windows offered by organic solvents.1,14 It is for these reasons that the targets for the active concentration of redox active materials for non-aqueous and aqueous systems are 5 M11,14 (ideally <$5 kg−1)11 and 2 M, respectively.11,14
Carbonyl groups are a common functional motif that can undergo reversible oxidation and reduction, and depending on the surrounding substituents, they are capable of storing one or more electrons.16 Organic carbonyl molecules are therefore often used as electroactive materials, benefitting from low cost and fast reaction kinetics.1 Further tailoring of electrochemical properties can be done by substitution or addition of functional groups to the conductive organic framework, where electron-withdrawing groups (EWGs) increase the potential and electron-donating groups (EDGs) reduce the potential.1,17 Three classes of carbonyl compounds have been investigated for flow battery use: those containing vicinal carbonyls that form enolates on reduction, those in which the carbonyl group is stabilised by conjugation into an aromatic system and quinone-based derivatives, where aromaticity is formed upon electrochemical reduction.16
Quinones have received a great deal of research interest as promising electrolytes for next-generation organic flow battery electrolytes. 2,5-Dihydroxy-1,4-benzoquinone (DHBQ, Scheme 1) was explored as an anolyte in basic aqueous media and demonstrated a 1.21 V cell potential when paired against potassium ferrocyanide with a capacity retention of 99.76% per cycle at 100 mA cm−2. In the work conducted by Yang et al.,18 the advantages of the very high solubility of DHBQ (4.31 M for the potassium salt in 1 M potassium hydroxide (KOH)) were counteracted by the high rate of crossover through the cation-conducting NAFION membrane. However, the experimentally determined crossover rate of 0.012% cycle−1 did not fully account for the diminishing full-cell performance (corresponding to a capacity fade of 0.24% cycle−1) and so the remaining capacity fade was attributed to molecular decomposition.18
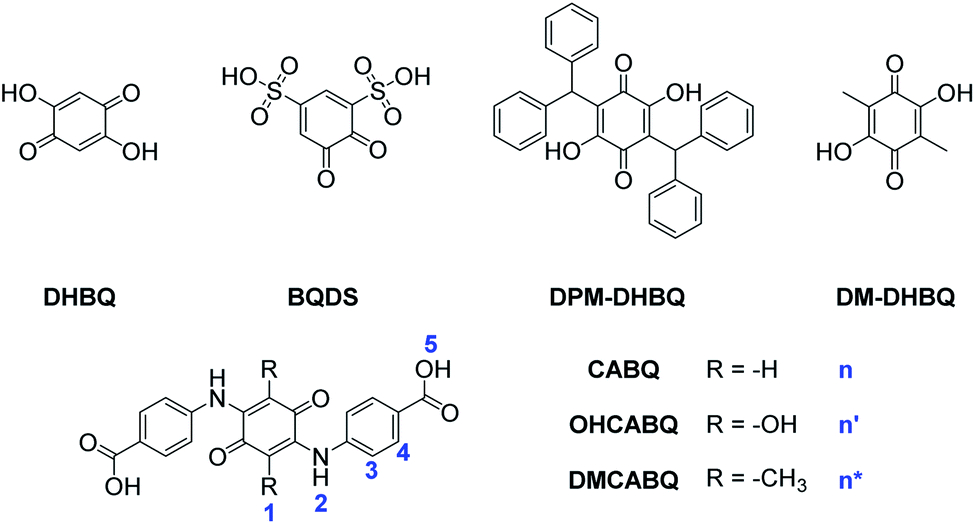 | ||
| Scheme 1 Derivatised benzoquinone electrolytes. DHBQ and DPM-DHBQ were explored in ref. 18, BQDS was explored in ref. 19 and 20, DM-DHBQ was explored in ref. 21, while CABQ and its derivatives, OHCABQ and DMCABQ are explored in this work. | ||
Benzoquinones are known to be susceptible to conjugate addition (1,4 addition to the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CO group of the quinone) and this was also observed when 1,2-benzoquinone-3,5-disulfonic acid (BQDS, Scheme 1) was investigated against anthraquinone-2-sulfonic acid22 and anthraquinone-2,6-disulfonic acid.19,22 In that system, the benzoquinone-based catholyte underwent addition of water, resulting in the addition of hydroxy groups to the initial quinone.19,23 Such addition has been shown to be mitigated by blocking the unsubstituted positions.23 For this reason, and with the knowledge that hydroxide ions are stronger nucleophiles than water at high pH, Yang et al. used diphenyl-methyl- (DPM-) groups to attempt to block conjugate addition in DHBQ by substitution of bulky aromatic rings on the 3 and 6 positions. The resulting species, DPM-DHBQ (Scheme 1), showed a similar redox potential to DHBQ under cyclic voltammetry (CV), but demonstrated slower kinetics with an increased peak-to-peak separation of the respective redox processes.18 Similarly sluggish kinetics and a large peak-to-peak separation (440 mV) were also observed with the dimethylated derivative (DM-DHBQ).21
C–CO group of the quinone) and this was also observed when 1,2-benzoquinone-3,5-disulfonic acid (BQDS, Scheme 1) was investigated against anthraquinone-2-sulfonic acid22 and anthraquinone-2,6-disulfonic acid.19,22 In that system, the benzoquinone-based catholyte underwent addition of water, resulting in the addition of hydroxy groups to the initial quinone.19,23 Such addition has been shown to be mitigated by blocking the unsubstituted positions.23 For this reason, and with the knowledge that hydroxide ions are stronger nucleophiles than water at high pH, Yang et al. used diphenyl-methyl- (DPM-) groups to attempt to block conjugate addition in DHBQ by substitution of bulky aromatic rings on the 3 and 6 positions. The resulting species, DPM-DHBQ (Scheme 1), showed a similar redox potential to DHBQ under cyclic voltammetry (CV), but demonstrated slower kinetics with an increased peak-to-peak separation of the respective redox processes.18 Similarly sluggish kinetics and a large peak-to-peak separation (440 mV) were also observed with the dimethylated derivative (DM-DHBQ).21
Possibly due to benzoquinone's tendency to act as an electrophile and undergo degradation, anthraquinones (AQs) have been the most widely studied as organic quinone-based electrolytes for RFBs. Due to the presence of the two additional aromatic rings, these molecules tend to have a greater chemical stability compared to benzoquinone (BQ) but are inherently insoluble in aqueous solution.1 Therefore, AQs are often functionalised with protic functional groups to increase their solubility.1 An example is the RFB based on 2,6-dihydroxy-AQ (DHAQ, >0.6 M) dissolved in 1 M KOH versus potassium ferrocyanide, which gives a voltage of 1.20 V, a current efficiency of >99% and an energy efficiency of 84%.24 The current benchmark in aqueous organic flow battery performance is the third generation electrolyte from Ji et al. based on 2,6-dihydroxyanthraquinone (DHAQ), ((((9,10-dioxo-9,10-dihydroanthracene-2,6-diyl)bis(oxy))bis(propane-3,1-diyl))bis(phosphonic acid)) (DPPEAQ) which forms a 1.0 V battery against potassium ferrocyanide, with a 0.75 M solubility at pH 9 and a capacity fade of 0.00036% per cycle.25
Considerable work has been carried out to increase the solubility of quinones and explore their degradation pathways. For DHBQ, LC-MS and ex situ NMR were used to identify some of the degradation products observed.18 Similar to the BQDS catholyte,19,23 conjugate addition was suspected to be the cause of degradation but no definitive mechanism was established.
The use of amines with additional functional groups, in principle, represents a facile route to increasing the stability and solubility of quinone molecules for flow battery use and motivates the current work. This work describes an exploration of an amine functionalised benzoquinone, 2,5-bis(4-carboxyanilino-)-1,4-benzoquinone (CABQ) as a potential anolyte/negolyte for use in organic aqueous flow batteries. Aminobenzoic acid was chosen as a candidate for derivatising the quinone, due to its carboxylic acid group and rigid aromatic spacer. The use of carboxylic acid groups was driven by the fact that by increasing the pH, the solubility could be increased. This coupled with the hope that the rigid aromatic spacer may sterically prevent the approach of any other nucleophilic species, following the logic proposed by Tabor et al., was the idea behind targeting CABQ as an anolyte.26
Here we use a powerful combination of cyclic voltammetry, in situ NMR27 and EPR28 spectroscopy alongside density functional theory (DFT) to investigate the behaviour and evolution of the amine functionalised benzoquinone electrolyte. We show that CABQ (Scheme 1) undergoes two conjugate additions in basic media and restabilises at a lower voltage once it is fully substituted, yielding a battery with a voltage of 1.2 V when paired against potassium ferrocyanide. Cyclic voltammetry is used to track the electrochemical profile of the molecule as it undergoes two instances of conjugate addition; this is then followed up by lab-scale full-cell testing of a flow battery using both NMR and EPR to observe the chemical signals of the evolving species and radical species formed during cycling. DFT is used to model the proposed intermediary species formed during the double-addition reaction and to identify the structures of the semiquinones formed as the battery is cycled as well as the degree of electron delocalisation that occurs within those species.
Cyclic voltammetry and ex situ NMR
Synthesis of CABQ was initially attempted using the protocol suggested by Nain-Perez et al.29 However, due to the difficulty found when attempting to isolate the target molecule, the protocol suggested for aminobenzoic acid functionalisation by Macgregor et al.30 was instead adopted. However, in our hands this protocol still produced a significant proportion of impurities, potentially complicating our analysis of the electrochemical properties of the system. Thus, the protocol was modified to use an increased excess of the amine and a greater reaction time (see ESI† for further details), leading to a higher crude yield. The crude product was then used for all electrochemical analyses as the electrochemical performance was found to be unaffected (Fig. S21–S22 and S30†) by the minor product which is likely to be the reduced, hydroquinone analogue.31 Also note that from here onwards, -BQ will refer to the oxidised benzoquinone species, -BSQ will refer to the singly reduced quinone and -BHQ will refer to the hydroquinone, i.e., the full reduction product.Electrochemical characterisation of CABQ was carried out using both aqueous cyclic voltammetry (CV, Fig. 1) and aqueous galvanostatic cycling within potential limits (GCPL) in a lab-scale full cell (further examples of both can be found in the ESI†). CABQ showed poor solubility in a range of organic solvents and although it is soluble in acetone, addition of the supporting salt (t-butylammonium hexafluorophosphate (TBAPF6)) caused precipitation (Fig. S13†), thereby precluding any further investigation in non-aqueous solvents. Moreover, CABQ is effectively insoluble in neutral water but is soluble in basic aqueous media up to 24.9 g l−1 (0.066 M) which would correspond to a charge capacity of 3.5 A h l−1 based on the 2e− capacity of the molecule.
 | ||
| Fig. 1 Cyclic voltammograms (CVs) of 1 mM CABQ dissolved in 1 M KOH/D2O. All CV experiments reported in this paper were run at ambient temperatures using a 3 mm diameter glassy carbon working electrode, a mercury–mercury oxide (1 M KOH) reference electrode and coiled platinum wire as the counter electrode. The scan rate was 20 mV s−1, scanning towards negative potentials first (as indicated by the arrow). | ||
Under basic conditions, CABQ initially exhibits a redox couple at −0.32 V(SHE) (α), but the molecule appears to react chemically giving rise to a second, primarily oxidative, couple b at −0.52 V(SHE) before re-stabilising at a lower voltage of −0.69 V(SHE) (γ) (Fig. 2). Due to the observed evolution in the electrochemical profile of CABQ, long-term CV was carried out to follow this process.
 | ||
| Fig. 2 The qualitative peak currents as a function of cycle number for processes α, β, and γ, shown by the red, blue, and black lines, respectively. The top graph presents the oxidative current while the bottom graph presents the reductive current. Note: the currents are only qualitative due to the uncertainty in correcting for the charging current.32 | ||
The rate of conversion from CABQ to the species that gives rise to redox couple β is sufficiently slow that redox couple β is not noticeable in the profile of the polarogram until cycle 20, where there is clear formation of an oxidative peak at −0.52 V(SHE). This oxidative peak continues to grow in intensity whilst redox couple α becomes increasingly polarised and diminishes in current until it is almost completely lost at around cycle 2000. The corresponding reductive wave of β is not observed clearly until cycle 40 at a voltage of −0.56 V(SHE), due to its lower current when compared to the oxidative wave, giving β an E1/2 of −0.54 V(SHE). By cycle 100, this redox couple has become more prominent and can be observed at −0.53 V(SHE) and by cycle 500 it becomes more positive still, at −0.52 V(SHE). However, from this point onwards there does not appear to be a significant continued drift towards more oxidising potentials. By cycle 3000 the reductive wave of β can no longer be observed but the oxidative wave continues to be observable through to the end of the experiment, albeit with continually diminishing current. As β grows and α diminishes, γ can be observed to grow in intensity from cycle 200, becoming prominent from cycle 1000 onwards. As can be seen from Fig. 2, as α decays, γ grows with a corresponding rise and fall in the current response of β. The current observed for the lower voltage (γ) process is lower than that measured initially for the couple at −0.32 V(SHE). This suggests that either there is some net loss of the redox-active electrolyte that occurs during the conversion of the initial species to that responsible for the low potential couple, or that the effective area of the working electrode has been reduced. It is more likely that the latter is responsible for this effect.
Substitution of the quinone backbone by hydroxy groups has literature precedent in acidic media23 and was speculated to be responsible for the degradation and capacity fade of DHBQ.18 On this basis, a reaction pathway caused by the large excess of the nucleophile, the hydroxide ion, present in the electrolyte system is proposed in Scheme 2, where redox couple γ is assigned to OHCABQ (Scheme 1). Scheme 2, moving from top to bottom as the reaction proceeds, initially begins with CABQ (top-centre). CABQ can undergo reduction to CABHQ (top-left) and is assigned to be the redox couple α in Fig. 1. However, CABQ is also susceptible to nucleophilic attack, under both direct (1,2) and conjugate (1,4) modes. Direct attack will initially yield the gem-diol but is more likely to undergo elimination of the hydroxide after attack than it is to continue to form a new species. Conjugate attack, however, yields a species (M1, top-right) that upon keto-enol tautomerisation achieves aromaticity (I1, middle-right) and installation of a hydroxy group. For the mechanism to then proceed, I1 needs to be oxidised to form I2 (middle-centre). This step is assigned to redox couple β in Fig. 1. Once I2 has been formed, whilst it is able to undergo reduction to re-form I1, it is, importantly, also electrophilic, and therefore can undergo conjugate attack to generate M2 (middle-left). M2 is then able to undergo keto-enol tautomerisation to form OHCABHQ, which itself is then able to be oxidised to OHCABQ (γ in Fig. 1). OHCABQ has no remaining electrophilic sites and therefore is unable to undergo further conjugate attack but is still susceptible to direct attack by hydroxide.
 | ||
| Scheme 2 A potential reaction mechanism by which substitution of the vacant positions in benzoquinone by hydroxide occurs, similar to that reported by ref. 23 under acidic conditions. The process labelled β observed in the CV experiment is likely the conversion between I1 and I2. | ||
To provide supporting evidence for the reactions proposed in Scheme 2, ex situ NMR analysis of the CV solutions were performed before and after 4000 cycles (Fig. 3). Note that D2O was used as the solvent throughout rather than H2O to enable ex situ NMR analysis to be performed alongside the CV studies. Due to the high ionic conductivity and dilute nature of the sample, 13C-NMR could not be recorded but the 1H-NMR clearly showed that two processes had occurred. Firstly (process 1), the quinone-proton (δ 6.07 ppm, Fig. 3a – position 1 in Fig. 3b) disappears rapidly in basic media. This process occurs via H/D exchange with the solvent in these strongly basic solutions (1 M KOH, pH 14) since it occurs before the NMR spectrum can even be recorded at high pH. The absence of this process in near-neutral pH (see Fig. S10†) suggests that the hydroxide ions are responsible for the abstraction and loss of proton resonance 1.
 | ||
| Fig. 3 (a) Ex situ1H NMR spectrum of CABQ in basic deuterated media highlighting both H/D exchange and conjugate addition. Bottom-to-top: NMR spectrum of the as-synthesised CABQ taken in DMSO (black, DMSO), NMR spectrum of 1 mM CABQ dissolved in 1 M KOH/D2O taken before electrochemical cycling (red, START), NMR spectrum of the same control sample taken after 3 days (purple, CTRL, 3D), NMR spectrum taken after 4000 CV cycles in 1 M KOH/D2O solution (dark blue, END), NMR spectrum of the control (CTRL) sample left for 4 days in a sealed NMR tube, a time coinciding with the end of the CV experiment (light blue). (b) Labelled proton positions of CABQ2−, and (c) OHCABQ4−. | ||
The second process involves the loss of the two doublets for the aryl C–H protons of the benzoate group (δ 6.91 and 7.81 ppm, peaks 3 and 4, respectively in Fig. 3b). These are replaced by an upfield-shifted pair of doublets at δ 6.73 ppm and 7.63 ppm (peaks 3′ and 4′ respectively). Comparing with the CV, doublets 3 and 4 are assigned to the species responsible for redox couple α (−0.32 V(SHE)) while doublets 3′ and 4′ correspond to the species responsible for redox couple γ (−0.69 V(SHE)).
A second NMR experiment was performed to monitor the chemical reactions of CABQ in basic media (Fig. 4). After day 2, a triplet and an additional doublet can be clearly observed that likely stem from the species responsible for process β in the cyclic voltammetry. This process also results in complete loss of the proton position that gives rise originally to proton resonance 1, the reaction occurring via nucleophilic attack by hydroxide ions (process 2, M1 to M2 and M2 to OHCABQ in Scheme 2). The addition of hydroxy groups in the unsubstituted 3 and 6 positions of CABQ increases the shielding of the proton resonances.1
 | ||
| Fig. 4 Ex situ 1H NMR of a 10 mM CABQ solution in 1 M KOH/D2O. The sample was kept in a sealed J Young's NMR tube and NMR was recorded daily for the purpose of tracking the addition reaction. | ||
Substitution of the 3 and 6 positions has an appreciable driving force as the reaction readily occurs in solution, as demonstrated by the ex situ NMR presented in Fig. 4, and from DFT modelling of the reaction mechanism (Fig. S64–S68†). The calculations show that there is a negative Gibbs free energy change in the reaction going from CABQ to OHCABQ and in the absence of a driving current, it is likely that CABQ itself functions as the oxidant in this process, being correspondingly reduced to its hydroquinone analogue.33 The DFT further reinforces the observed trend in redox potential values, −0.15 V being predicted for α, to −0.16 V for β then −0.17 V for γ, which is consistent with the presence of increased numbers of electron donating groups surrounding the quinone core (c.f. −0.32, −0.52, and −0.69 V(SHE) experimentally).1,17
The loss of intensity of the color of a solution of CABQ in 1 M KOH over time in the absence of cycling is similar to that observed for the cycled sample and is consistent with the expected reduction in conjugation of the product OHCABQ compared to CABQ.
Attempting to prevent conjugate addition
DMCABQ (Scheme 1), the 3,6-dimethyl analogue of CABQ, was also synthesized to test whether the conjugate addition could be prevented. By chemically blocking the unsubstituted sites with a functional group that was resistant to substitution, the aim was to stabilize the redox couple of DMCABQ and preclude the formation of redox waves corresponding to the products of such addition (Fig. 1, β and γ).Initially an asymmetric redox couple at −0.24 V(SHE) can be observed in the CV of DMCABQ (Fig. 5), in which the oxidative wave is broader, and therefore less intense, than the corresponding reductive peak. This suggests that the charge transfer coefficient for the redox process has a value of >0.5.34 Under continued cycling, an irreversible reductive process is observed at −0.53 V(SHE) which continues to grow at the expense of the redox couple at −0.24 V(SHE), the latter broadening and decreasing in intensity.
 | ||
| Fig. 5 Cyclic voltammograms of 1 mM of DMCABQ dissolved in 1 M KOH/H2O. The scan rate was 20 mV s−1, scanning towards negative potentials first (as indicated by the arrow). | ||
From this, substitution of the 3 and 6 positions of CABQ with methyl groups appeared to be successful in preventing conjugate addition. However, the presence of the methyl groups appears to facilitate alternate degradation pathways of the electrolyte, resulting in a lack of reversibility in the electrochemical activity of the new chemically formed molecule. This alternative degradation pathway may be due to the increased acidity of the aliphatic protons (Scheme 3) and may further suggest that substitution of a group without protons in that position could be more beneficial.
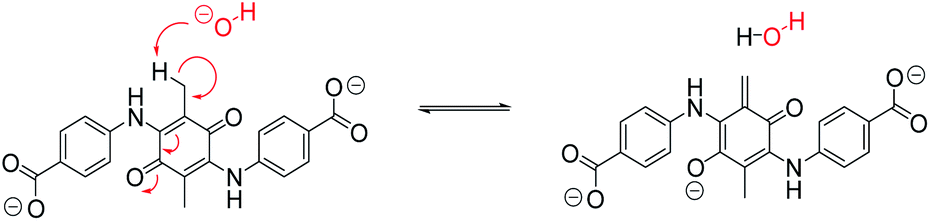 | ||
| Scheme 3 Reaction scheme demonstrating the charge stabilization mechanism available following deprotonation of the aliphatic methyl groups in the 3 and 6 positions of DMCABQ. The alkene formed on the right would be susceptible to conjugate attack and the enolate itself would serve as a suitable nucleophile for conjugate attack on another molecule. | ||
Moreover, the methyl groups were expected to cause a decrease in the initial potential of the redox wave due to their inductive donation effect. However, compared to CABQ where α sits at −0.32 V(SHE), a more oxidative shift is observed. This may be due to either a reduced ability of the aniline to π-donate to the quinone, as compared to its ability in CABQ, or this may be due to competitive conjugation of the methyl groups with the enone-like conjugated system of the quinone core in basic media. Overall, the increased potential of the redox couple relative to α implies a reduction in overall electron donation when compared to CABQ.
The behaviour of CABQ in less basic solutions was also investigated as a means of reducing the driving force for attack on the quinone backbone and to move to milder conditions. Using stoichiometric amounts of KOH (Fig. S20†) and triethylamine (Fig. S23–24†), CABQ's electrochemical performance in potassium chloride solution was investigated. However, the similar performance under these lower pH environments suggests that CABQ, and DMCABQ, decompose in a similar way – with a chemical reaction that forms a new species at a lower potential. This new species then undergoes an irreversible reduction to form an electrochemically inactive species, as the reverse oxidation is not observed.
Battery performance
Despite the generally poor performance of DMCABQ in basic solutions and CABQ at near neutral pH, CABQ at pH 14 performed relatively well under long-term cycling at a concentration of 1 mM within a CV cell, once conversion to OHCABQ was complete (Fig. S25†). However, at a greater concentration of 10 mM, the redox couple of OHCABQ becomes less reversible with a greater reductive current than oxidative current being observed (Fig. 7). As cycling progresses, the oxidative and reductive waves become increasingly polarized while the current intensity diminishes, and an additional redox couple is observed to form in the middle of the two major redox processes. This could suggest that a concentration dependent degradation mechanism exists for OHCABQ. However, no noticeable changes were observed in the ex situ1H NMR taken before and after CV cycling (Fig. S28–29 and S31–32,† respectively). Therefore, the change in electrochemical profile is more likely to stem from non-covalent intermolecular interactions between anolyte molecules as opposed to permanent chemical degradation.27A lab-scale redox flow battery against potassium ferrocyanide in 1 M KOH/D2O was used to test the molecule using galvanostatic cycling (Fig. 6). A suspension of CABQ was used in the first instance to ensure the presence of enough material during the in situ cycling of the battery (ESI Section 1.3.3†). The cycling data of the material exhibited three plateaus within the 0.4–1.7 V window, at roughly 1.00, 1.15 and 1.38 V. From the in situ1H NMR spectroscopic studies,27 it is apparent that the reaction between CABQ and hydroxide begins soon after dissolution.
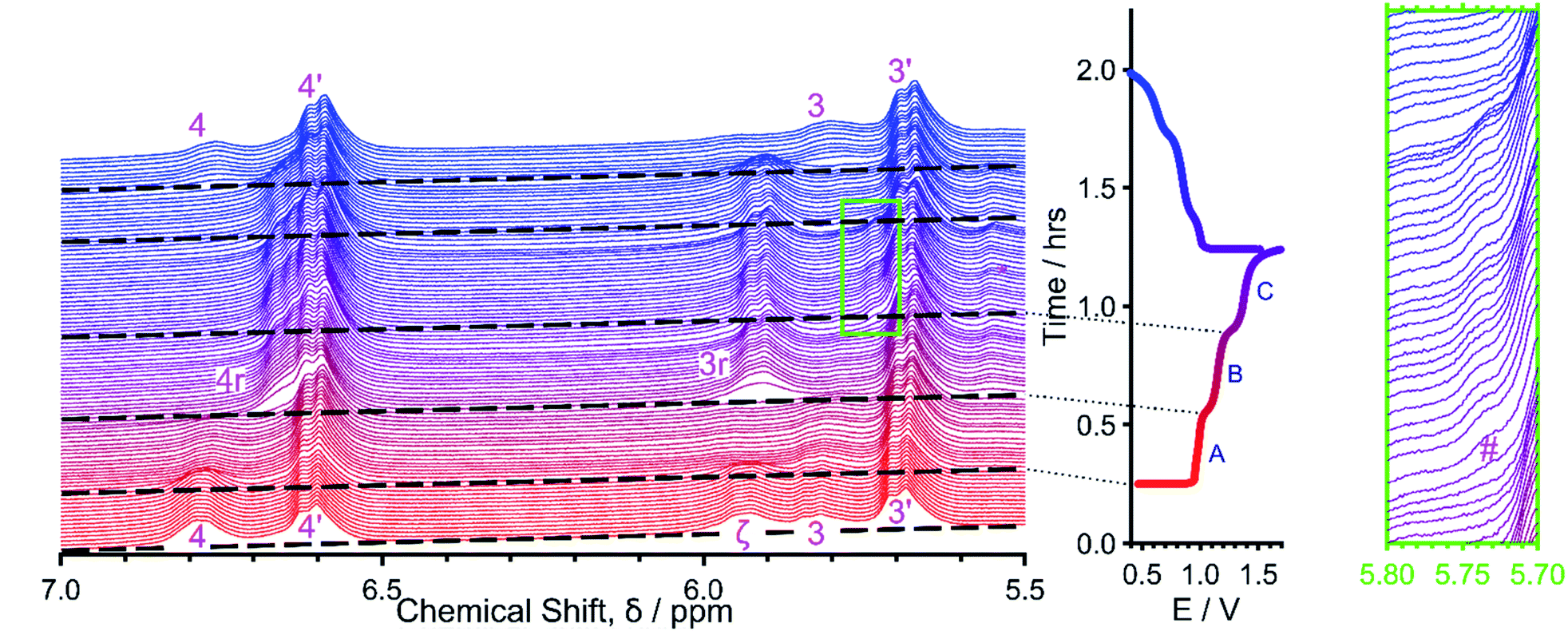 | ||
| Fig. 6 In situ 1H NMR data plotted as displaced/stacked spectra so that the changes in species can be observed (left). The first charge and discharge cycle of CABQ plotted as a potential–time graph (middle). Three plateaus can be observed during both charge and discharge. Expansion of the area highlighted by the green rectangle indicating the shoulder formed during the third (charging) plateau (right). | ||
 | ||
| Fig. 7 Cyclic voltammograms of 10 mM CABQ dissolved in 1 M KOH/D2O. The scan rate was 20 mV s−1, scanning towards negative potentials first (as indicated by the arrow). | ||
Before charging (Fig. 6), two doublets deriving from CABQ's (3, 4) and OHCABQ's (3′, 4′) aromatic sidearms can be observed along with an additional peak at ∼5.95 ppm (ζ), which is likely to be from an intermediate species along the reaction pathway (I1 or I2 in Scheme 2, spectra from day 2 in Fig. 4), as the intensity of the doublet 4 before charging does not match that of 3. This unidentified intermediate is characterized by two NMR resonances at ∼5.95 ppm (ζ) and ∼6.78 ppm (coincident with that of 4), corresponding to the 2 aromatic C–H environments from its aromatic side-arms. This intermediate is also observed in the ex situ NMR in Fig. 4. It is likely that in the in situ experiment, due to the transient nature of the experiment and the lower signal-to-noise ratio that these separate peaks were not resolved as clearly as in the ex situ experiment. An additional piece of evidence that supports this conjecture is the fact that when resonance 4 returns after the first charge and discharge cycle it sits at a slightly lower chemical shift than before any current has passed, implying that previously the overlap had shifted the center of the NMR peak.
Once charging begins, during the first plateau (A) at 1 V, the signals due to the doublets from CABQ (3, 4) are broadened and lost. This is due to the formation of the radical anion, CABSQ3−˙, which was confirmed by carrying out in situ EPR28 (Fig. 8a) in a separate GCPL experiment (ESI Section 1.3.3†). The peak at ∼5.95 ppm (ζ) also disappears upon charging. This may be due to the broadening effect of the paramagnetic radical species, but it may also be due to reduction of the species contributing to this peak resulting in loss of that proton signal. It is also possible that the accumulated intermediates responsible for the peak labelled ζ and the unequal intensities of 3 and 4 had been consumed by progressing further along the addition reaction and therefore this peak is far less intense. This makes sense if, in the presence of a charging current, the rate of conversion between I1 and I2 had increased.
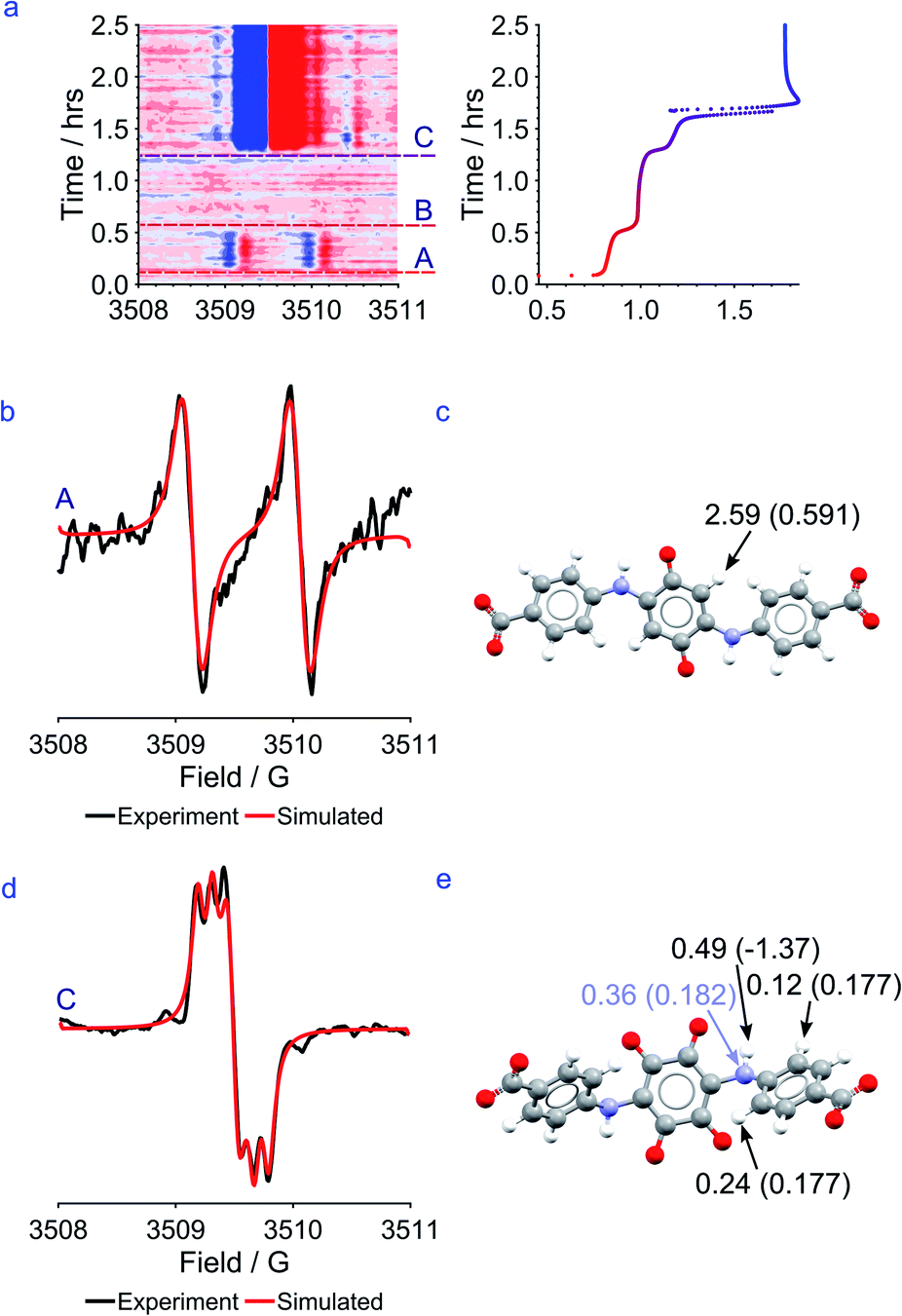 | ||
| Fig. 8 (a) Left: in situ EPR data showing the presence of unpaired electrons during the first and third plateaus of the cycle. Right: the first charge cycle of a full cell of CABQ vs. potassium ferrocyanide in 1 M KOH. (b) EPR data from the first plateau plotted against the fitted model assuming a single 1H coupling to the unpaired electron. (c) DFT model of CABSQ3−˙ labelled with the EPR-derived coupling constant (MHz). The value in brackets is the DFT derived Fermi-contact coupling of the proton indicated by an arrow; all other valid hyperfine constants are, by comparison, very small. (d) EPR data from the third plateau plotted against the fitted model assuming a single 14N and three 1H's coupling to the unpaired electron. (e) DFT model of OHCABSQ5−˙ labelled with the EPR-derived coupling constants (MHz) and DFT derived Fermi-contact couplings for the dominant interactions. | ||
During the second plateau (B) at 1.15 V, new peaks (3r, 4r) corresponding to the diamagnetic CABHQ4− are observed. Here the EPR data shows that the radicals formed from the first plateau are quenched as the hydroquinone is formed. The formation of the peak at ∼5.93 ppm during this second plateau (B), may suggest that the additional peak present before the cell is charged (ζ) is due to CABHQ. However, the slight difference in chemical shift further suggests that this is likely to be due to an intermediate species as opposed to the reduced quinone derivative.
In the final charging plateau (C), at 1.38 V, OHCABQ (3′, 4′) undergoes a single electron reduction to form OHCABSQ5−˙, as shown by EPR. However, the radical does not seem to cause a broadening effect on the aromatic doublets and so these signals persist in the NMR, in which OHCABSQ5−˙ is observed as a shoulder at ∼5.75 ppm which grows in during plateau 3 (Fig. 6, green rectangle, #). The low concentration of OHCABSQ5−˙, compared to OHCABQ, observed in the NMR and EPR spectra could conceivably stem from this plateau being a two-electron process followed by a comproportionation reaction to the semiquinone.27 However, were this to be the case, one would expect additional NMR signals to be observed for OHCABHQ. It seems therefore more likely that this plateau is a one-electron transfer process where the low concentration of radicals is due to the parasitic side-reactions that result in self-discharge of OHCABSQ5−˙ back to OHCABQ4−. These self-discharge mechanisms may stem from oxygen impurities that leached into the cell or from reduction of the surrounding solvent to produce hydrogen gas.27
Modelling of the EPR signals suggests that in the first plateau, the electron in CABSQ3−˙ is more localized and the EPR spectrum is dominated by a hyperfine interaction with a single hydrogen (Fig. 8b) while in the third plateau a hyperfine coupling to one 14N environment and three proton environments in OHCABSQ5−˙ is seen (Fig. 8c). This implies that the mechanism of stabilization of the radical has changed and that post-hydroxylation, the substituted amine side-arms are more involved in charge stabilization.
Taking together the EPR signals and the DFT models it seems likely that for CABSQ3−˙, the delocalization of the electron occurs between the carbonyl carbon and the adjacent, unsubstituted carbon in the ring unit. The DFT calculated SOMO (Fig. 9a) shows that there is a node between the CC of the quinone core and the amine substituent, as well as between the nitrogen and the benzene ring of the benzoic acid itself. This may help to explain why no nitrogen coupling is observed for CABSQ3−˙.
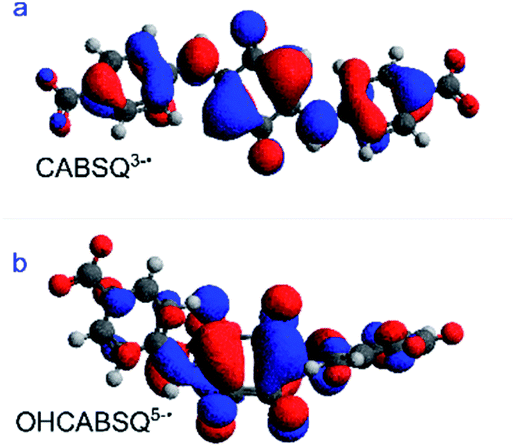 | ||
| Fig. 9 DFT optimized SOMOs of (a) CABSQ3−˙ and (b) OHCABSQ5−˙. | ||
Similarly, for OHCABSQ5−˙ the unpaired electron must be largely located on the nitrogen of the amine side-arm. It is only in this position that it can interact with the nitrogen and 3 protons. This is reinforced by the fact that the SOMO (Fig. 9b) shows orbital contributions from the nitrogen to the CC of the quinone ring.
During both the NMR and EPR experiments, the second reduction potential of OHCABQ was outside of the electrochemical window used and so was not observed. This was likely caused by overpotentials present in the assembled battery, which was reflected in the low voltaic efficiency observed (61.9%, Fig. S33†). From CV experiments, the expected voltage of the two-electron plateau of OHCABQ should be 1.2 V when compared against ferricyanide (0.5 V(SHE)).24 However, as opposed to the single redox couple in the CV at a concentration of 1 mM, in the GCPL, both at 0.066 M and at 1 mM two single-electron reduction events are observed. This may suggest that the semi-quinone is stabilized or that the second reduction event has sluggish kinetics. The fact that both the EPR experiment and the CV experiment were carried out at 1 mM concentrations suggests that the change in electrode material (glassy carbon for CV, carbon felt for GCPL) may be responsible for this change in electrochemical behaviour.
Aging of the molecule in basic solution (Fig. S38†) causes complete loss of the first two plateaus, assigned to CABQ reduction, leaving only one plateau assigned to the single electron reduction of OHCABQ. This is in line with the previously discussed CV behaviour (Fig. S25†).
Use of carbon felt as the electrode with CABQ resulted in a significant overpotential in the battery performance (61.9%, Fig. S33†). As a result, low Voltaic efficiencies (VE) and consequently low energy efficiencies (EE) were observed. To allow for greater utilization of the active material, the carbon felt was replaced with baked carbon paper. Baking carbon paper over 24 hours at 400 °C has been shown to improve the wettability and consequently reduce the contact resistance associated with the electrode.24 This new carbon electrode, coupled with an increased flow rate, to further reduce any system-based resistances, was used in a third lab scale battery test (see Fig. S46†). The experiment showed a CE of 98.64 ± 0.03%, a high first cycle VE of 84% and a first cycle capacity of 2.8 A h l−1. However, the rate of capacity fade was also high resulting in poor capacity retention. In the first cycle, three plateaus were observed (0.88, 1.05 and 1.25 V) on charging and four during discharge (1.15, 0.95, 0.75 and 0.55 V) where the dQ/dV profiles can be found in the ESI (Fig. S49–S50†). The differential profiles match the form of the CV, as expected, though the additional plateau (0.55 V) present during discharge could be a sign of electrolyte degradation or it could derive from the catholyte. Nonetheless, along with the rapid capacity fade, loss of electrochemical processes was also observed during cycling. The 0.55 V plateau was lost after cycle 30, the 0.75 V plateau after cycle 35 and then the 0.95 V plateau was lost after cycle 50. Due to loss of these processes, the final profile was a singular plateau during both charge and discharge with a low capacity of <0.1 A h l−1. Ex situ NMR was carried out at the end of cycling to inspect the degree of crossover and new peaks were observed in the catholyte tank, confirming crossover and potential degradation under oxidising conditions (see Fig. S51†). Although it may be possible for species to have formed that are invisible to 1H NMR due to the highly deuterated environment, it is likely that the capacity loss of the cell can primarily be attributed to crossover, which then led to subsequent degradation. The increased pump-rate is likely to have increased the cross-over rate by increasing any potential pressure differences between the two electrolyte flow paths.
To reduce the rate of crossover, the same experiment was then carried out with two layers of NAFION 212, instead of the previous single layer, and with a lower flow rate. A slower crossover rate was observed as well as a slower rate of capacity fade (two layers, Fig. 10versus single layer, Fig. S46†). The need for two layers of membrane, as opposed to one, reinforces the previously reported issue of currently poor membrane compatibility of benzoquinone-based species, where a thicker membrane improves battery stability at the cost of lower efficiency.18 This is not only reflected in the starting values for VE for the single-layered (86.79%) and double-layered (70.8%) experiments, but also in the respective dQ/dV profiles (Fig. S54–S55†) where an increased polarization is observed for the double-layered membrane experiment, corresponding to an increased resistance through the membrane of approximately 350 mΩ.
 | ||
| Fig. 10 [Top] Galvanostatic cycling data of 0.066 M CABQ (15 ml) in 1 M KOH/D2O cycled at 50 ml min−1 at ambient temperature against a 0.15 M: 0.0375 M solution of potassium ferrocyanide: potassium ferricyanide (30 ml), [Bottom] coulombic and voltaic efficiency versus cycle number (left axis) and capacity versus cycle number (right axis). | ||
Encouragingly, the battery utilizing two layers of NAFION 212 showed a CE of 99.724 ± 0.003% with a stable VE of 70.39 ± 0.03%. However, 0.0226 mA h of capacity was still observed to be lost per cycle. Ex situ NMR (Fig. S56†) showed additional peaks in both the catholyte and anolyte tanks, which confirmed that crossover had taken place. It is evident also that this crossover causes some decomposition of the anolyte. The experiment showed 3 plateaus during charge and 3 during discharge. This is consistent with the previously described charge profiles with reduction of CABQ2− occurring first to CABSQ3−˙ and then to CABHQ4− followed by reduction of OHCABQ4− to OHCABSQ5−˙. The capacity achieved was lower than the theoretical value for the mass of the sample used implying that the full reduction of OHCABQ was not achieved. From Fig. 10, 80.5% of the theoretical 191C (53.1 mA h) capacity can be seen to be achieved, translating to an average of 1.61 electrons being stored per molecule (a similar result was obtained from a combination of the OHCABQ Randles–Sevcik data (Fig. S26) and the DOSY derived diffusion data shown in Fig. S11 and S12. For more details, see ESI Section 2.2†).
Finally, to confirm whether the electrolyte degradation taking place was caused by cross-over, a symmetric cell experiment was carried out (Fig. 11). Following from the logic above, two NAFION membranes were used alongside baked carbon paper electrodes. Potassium ferrocyanide was used to charge CABQ2− to CABHQ4− under a constant-current-constant-voltage charging profile to 1.9 V (see Fig. S60†). The ferrocyanide tank was then replaced by a fresh solution of CABQ and the cell was left to run for 600 cycles (see Fig. S61–S62†). A steady capacity fade of 0.066 mA h per cycle was observed. Ex situ NMR (see Fig. S63†) was carried out on both the capacity limiting and non-limiting CABQ reservoirs, but no new NMR signals were observed. Moreover, no significant amount of crossover was observed to have occurred in this symmetric system with the volumes of the tanks remaining relatively constant throughout cycling. This implies that the capacity fade most likely stems from capacity slippage as the conjugate addition reaction proceeds, rather than from electrolyte degradation. Furthermore, the lack of any additional NMR peaks also implies that the previously observed degradation depends on the presence of ferrocyanide and that in its absence CABQ/OHCABQ is relatively stable.
 | ||
| Fig. 11 [Top] Galvanostatic cycling data of 0.066 M CABQ (20 ml) in 1 M KOH/D2O cycled at 50 ml min−1 at ambient temperature against 0.066 M CABQ (30 ml) in 1 M KOH/D2O, [Bottom] coulombic efficiency versus cycle number (left axis) and capacity versus cycle number (right axis). Note: electro-impedance spectroscopy was run every 100th cycle to monitor the change in conductivity of the battery. | ||
Conclusions
Work described in this paper has demonstrated a novel organic negolyte (2,5-(4-carboxyanilino)-1,4-benzoquinone) that undergoes a conjugate addition under basic conditions to yield a dihydroxylated analogue (3,6-dihydroxy-2,5-(4-carboxyanilino)-1,4-benzoquinone). The quinone form of the electrolyte shows a solubility of 0.066 M in 1 M KOH aqueous solution corresponding to a charge capacity of approximately 3.5 A h l−1. We have used long-term cyclic voltammetry to observe the change in electrochemical response that goes alongside the conjugate addition reaction and have observed that the final species is stable in aqueous basic solution. We have also demonstrated that moving towards lower pH regimes yields less-reversible behaviour and that chemically blocking the electrophilic sites with aliphatic groups in basic media introduces alternate degradation pathways that cause the molecule to react to form a new species that is then irreversibly reduced.We have tracked and confirmed the stability of CABQ/OHCABQ using both ex situ and in situ NMR and have identified the processes occurring during each plateau using in situ EPR. The EPR signal further suggests that functionalisation using amine-based functional groups is beneficial towards stabilising the reduced species, supported by DFT. DFT also affirmed the spontaneous reactions in the hydroxylation mechanism and reflected the trends in reduction potentials exemplified in the CV.
Ultimately, the work has demonstrated that CABQ can be used against potassium ferrocyanide in lab-scale redox flow batteries and that the molecule demonstrates a high Coulombic efficiency even in a non-optimised system; the observed Voltaic efficiency can be improved significantly by reducing the resistances in the full-cell setup. One of the key impacts of the current study is that it provides a potential roadmap towards improved electrolytes for flow batteries, by illustrating that the instability of quinone electrolytes to conjugate addition may not necessarily be a roadblock to their development for application in RFBs. With the increase in electron donation, the hydroxylated products often have a lower redox potential, leading to a greater energy density. This, coupled with the new hydroxy groups occupying the remaining electrophilic sites, will lead to a greater stability of the hydroxylated products due to a reduced proclivity to react with any nucleophiles present in the solution. Together, these properties could lead to electrolytes with greater stability and greater energy density for use in redox flow battery systems.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. C. P. G. and D. S. W. supervised the project. R. B. J., D. S. W and C. P. G. conceived the idea. R. B. J. performed the synthesis and electrochemical experiments. R. B. J. and E. W. Z. performed the in situ NMR experiment. E. W. Z. performed the in situ E. P. R. experiment. R. B. J. and R. N. K. performed the DFT calculations of the reactions, radical species, and their proton hyperfine couplings. R. B. J. and E. J. performed the CV peak extraction and fitting analysis. All authors contributed to project discussions. R. B. J., D. S. W., and C. P. G. wrote the manuscript with input from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. B. J., E. W. Z., and C. P. G. acknowledge support from the EPSRC and Shell via I-Case studentship EP/R511870/1. E. W. Z. and C. P. G. acknowledge support from the Centre of Advanced Materials for Integrated Energy Systems (CAM-IES), via EPSRC grant no. EP/P007767/1. E. W. Z. acknowledges support from the Manifest exchange program via EPSRC grant no. EP/N032888/1. E. J. and C. P. G. were supported by EC H2020 grant no. 835073. R. N. K. was supported by EPSRC Grant no. EP/M009521/1 DJR00640. Calculations were carried out on the in-house Odyssey HPC cluster and the authors are grateful for the calculation time used therein. We thank the EPSRC EPR National Research Facility (NS/A000055/1) for access to their EPR spectrometers. The authors thank the Yusuf Hamied Department of Chemistry's NMR service for running and analysing the DOSY 1H NMR experiment. We thank Dr Peter A. A. Klusener (Shell Global Solutions International B. V.), Dr Elizabeth Castillo-Martinez (Universidad Complutense de Madrid) and Dr Javier Carretero-Gonzalez (ICTP-CSIC) for many fruitful discussions.Notes and references
- Y. Ding, C. Zhang, L. Zhang, Y. Zhou and G. Yu, Chem. Soc. Rev., 2018, 47, 69–103 Search PubMed.
- J. D. Milshtein, R. M. Darling, J. Drake, M. L. Perry and F. R. Brushett, J. Electrochem. Soc., 2017, 164, A3883–A3895 Search PubMed.
- W. Wang, Q. Luo, B. Li, X. Wei, L. Li and Z. Yang, Adv. Funct. Mater., 2013, 23, 970–986 Search PubMed.
- M. L. Perry and A. Z. Weber, J. Electrochem. Soc., 2016, 163, A5064–A5067 Search PubMed.
- R. Dmello, J. D. Milshtein, F. R. Brushett and K. C. Smith, J. Power Sources, 2016, 330, 261–272 Search PubMed.
- P. Leung, A. A. Shah, L. Sanz, C. Flox, J. R. Morante, Q. Xu, M. R. Mohamed, C. Ponce de León and F. C. Walsh, J. Power Sources, 2017, 360, 243–283 Search PubMed.
- J. Noack, N. Roznyatovskaya, T. Herr and P. Fischer, Angew. Chem., Int. Ed., 2015, 54, 9776–9809 Search PubMed.
- R. A. Potash, J. R. McKone, S. Conte and H. D. Abruña, J. Electrochem. Soc., 2016, 163, A338–A344 Search PubMed.
- P. Alotto, M. Guarnieri and F. Moro, Renewable Sustainable Energy Rev., 2014, 29, 325–335 Search PubMed.
- I. Gyuk, M. Johnson, J. Vetrano, K. Lynn, W. Parks, R. Handa, L. Kannenberg, S. Hearne, K. Waldrip and R. Braccio, Grid Energy Storage, Washington DC, 2013 Search PubMed.
- R. M. Darling, K. G. Gallagher, J. A. Kowalski, S. Ha and F. R. Brushett, Energy Environ. Sci., 2014, 7, 3459–3477 Search PubMed.
- J. D. Milshtein, L. Su, C. Liou, A. F. Badel and F. R. Brushett, Electrochim. Acta, 2015, 180, 695–704 Search PubMed.
- B. Li, J. Liu, Z. Nie, W. Wang, D. Reed, J. Liu, P. McGrail and V. Sprenkle, Nano Lett., 2016, 16, 4335–4340 Search PubMed.
- G. L. Soloveichik, Chem. Rev., 2015, 115, 11533–11558 Search PubMed.
- K. Gong, Q. Fang, S. Gu, S. F. Y. Li and Y. Yan, Energy Environ. Sci., 2015, 8, 3515–3530 Search PubMed.
- B. Häupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1402034 Search PubMed.
- K. M. Pelzer, L. Cheng and L. A. Curtiss, J. Phys. Chem. C, 2017, 121, 237–245 Search PubMed.
- Z. Yang, L. Tong, D. P. Tabor, E. S. Beh, M. A. Goulet, D. De Porcellinis, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Adv. Energy Mater., 2018, 8, 1702056 Search PubMed.
- B. Yang, L. Hoober-Burkhardt, S. Krishnamoorthy, A. Murali, G. K. S. Prakash and S. R. Narayanan, J. Electrochem. Soc., 2016, 163, A1442–A1449 Search PubMed.
- Y. Chen, E. Trzop, A. Makal, Y.-S. Chen and P. Coppens, Dalton Trans., 2014, 43, 3839–3841 Search PubMed.
- P. Sun, Y. Liu, Y. Li, M. A. Shehzad, Y. Liu, P. Zuo, Q. Chen, Z. Yang and T. Xu, Ind. Eng. Chem. Res., 2019, 58, 3994–3999 Search PubMed.
- B. Yang, L. Hoober-Burkhardt, F. Wang, G. K. S. Prakash, S. R. Narayanan, G. K. Surya Prakash and S. R. Narayanan, J. Electrochem. Soc., 2014, 161, A1371–A1380 Search PubMed.
- L. Hoober-Burkhardt, S. Krishnamoorthy, B. Yang, A. Murali, A. Nirmalchandar, G. K. S. Prakash and S. R. Narayanan, J. Electrochem. Soc., 2017, 164, A600–A607 Search PubMed.
- K. Lin, Q. Chen, M. R. Gerhardt, L. Tong, S. B. Kim, L. Eisenach, A. W. Valle, D. Hardee, R. G. Gordon, M. J. Aziz and M. P. Marshak, Science, 2015, 349, 1529–1532 Search PubMed.
- Y. Ji, M. Goulet, D. A. Pollack, D. G. Kwabi, S. Jin, D. Porcellinis, E. F. Kerr, R. G. Gordon and M. J. Aziz, Adv. Energy Mater., 2019, 9, 1900039 Search PubMed.
- D. P. Tabor, R. Gómez-Bombarelli, L. Tong, R. G. Gordon, M. J. Aziz and A. Aspuru-Guzik, J. Mater. Chem. A, 2019, 7, 12833–12841 Search PubMed.
- E. W. Zhao, T. Liu, E. Jónsson, J. Lee, I. Temprano, R. B. Jethwa, A. Wang, H. Smith, J. Carretero-González, Q. Song and C. P. Grey, Nature, 2020, 579, 224–228 Search PubMed.
- E. W. Zhao, E. Jónsson, R. B. Jethwa, D. Hey, D. Lyu, A. Brookfield, P. A. A. Klusener, D. Collison and C. P. Grey, J. Am. Chem. Soc., 2021, 143, 1885–1895 Search PubMed.
- A. Nain-Perez, L. C. A. Barbosa, M. C. Picanço, S. Giberti and G. Forlani, Chem. Biodiversity, 2016, 13, 1008–1017 Search PubMed.
- K. A. Macgregor, M. K. Abdel-Hamid, L. R. Odell, N. Chau, A. Whiting, P. J. Robinson and A. McCluskey, Eur. J. Med. Chem., 2014, 85, 191–206 Search PubMed.
- V. K. Tandon, S. Kumar, N. N. Mishra and P. K. Shukla, Eur. J. Med. Chem., 2012, 56, 375–386 Search PubMed.
- A. J. Bard and L. R. Faulkner, in Electrochemical Methods: Fundamentals and Applications, Wiley, 2nd edn, 2001, pp. 239–244 Search PubMed.
- J. B. Gerken, A. Stamoulis, S.-E. Suh, N. D. Fischer, J. Kim, I. A. Guzei, S. S. Stahl and R. Li, Chem. Commun., 2020, 56, 1199 Search PubMed.
- R. S. Nicholson, Anal. Chem., 1965, 37, 1351–1355 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta02870g |
| ‡ Present address: Johnson Matthey Technology Centre, Core-Science Physical and Chemical Modelling Group, Blounts Ct Rd, Sonning Common, Reading, RG4 9NH, UK. |
| This journal is © The Royal Society of Chemistry 2021 |