
Peculiarly fast Li-ion conduction mechanism in a succinonitrile-based molecular crystal electrolyte: a molecular dynamics study†
 *ab
Makoto
Moriya,
cd
Yuki
Watanabe,
a
Kazunori
Nishio,
a
Taro
Hitosugi
a
and
Yoshitaka
Tateyama
*ab
*ab
Makoto
Moriya,
cd
Yuki
Watanabe,
a
Kazunori
Nishio,
a
Taro
Hitosugi
a
and
Yoshitaka
Tateyama
*ab
Abstract
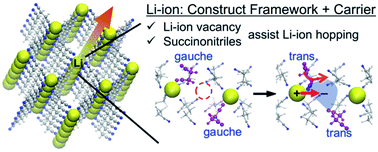
Li{N(SO2F)2}(NCCH2CH2CN)2 (Li(FSA)(SN)2) molecular crystals have been experimentally reported as a promising solid electrolyte for all-solid-state Li-ion battery applications because of their high Li-ion conductivity of ca. 10−4 S cm−1 at room temperature and an exceptionally low activation energy (Ea) of 28 kJ mol−1. However, the fast conduction mechanism remains unexplained because all the Li-ions are held in the crystal framework, and the distances between the constituent Li-ions are too long for hopping. Herein, molecular dynamics (MD) simulations were performed to clarify the mechanism of the extraordinarily fast Li-ion conduction in the succinonitrile (SN)-based molecular crystals. Atomistic MD simulations revealed that Li-ion vacancies can exist stably in Li(FSA)(SN)2 crystals and give rise to the one-dimensional Li-ion hopping pathway contrary to the conventional scenario in which the fast conduction is attributed to the three-dimensional pathway. The calculated Ea of 34 kJ mol−1 is in good agreement with the experimental value, which substantially supports the one-dimensional conduction. The low Ea is intimately connected with the motion of the SN molecules. Two SN molecules at the vacancy site change their conformation, following which one of the SN molecules creates an electronegative region, while the other carries the adjacent Li-ion to the electronegative region by the swing motion. The insights on the behavior of organic moieties and Li-ion conduction obtained from this study will promote the development of highly conductive molecular crystals.
 Please wait while we load your content...
Please wait while we load your content...

