
Biomimetic photocatalysts for the conversion of aqueous- and gas-phase nitrogen species to molecular nitrogen via denitrification and ammonia oxidation
 *c
and
Wooyul
Kim
*a
*c
and
Wooyul
Kim
*a
Abstract
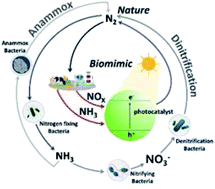
Denitrification and anaerobic ammonium oxidation (anammox) are important biological processes of the nitrogen cycle that help to preserve the global ecosystem. However, indiscriminate development and global population growth result in the discharge of large amounts of nitrogen species (e.g., via the Haber–Bosch process), particularly nitrogen oxides and ammonia, which cannot be fully digested by microorganisms and therefore accumulate in soil and water. Photocatalysts can promote the conversion of nitrogen oxides and ammonia to molecular nitrogen under the action of photogenerated electrons and holes, thus mimicking denitrifying and anammox bacteria, respectively. Herein, we review the biomimetic photocatalysts and photoelectrochemical cells used to convert aqueous and airborne nitrogen species to molecular nitrogen and shed light on the charge transfer mechanism that should be selectively controlled to favor the formation of molecular nitrogen over that of nitrogen-containing intermediates and by-products. Last but not least, we discuss the outlooks and perspectives of solar-powered molecular nitrogen recovery and suggest guidelines for the design of high-performance denitrification/anammox bacteria-like photocatalysts.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

