
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular sieving using metal–polymer coordination membranes in organic media†
Rifan
Hardian
 a,
Peter
Pogany
b,
Young Moo
Lee
c and
Gyorgy
Szekely
*a
a,
Peter
Pogany
b,
Young Moo
Lee
c and
Gyorgy
Szekely
*a
aAdvanced Membranes and Porous Materials Center, Physical Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: gyorgy.szekely@kaust.edu.sa; Web: [http://www.szekelygroup.com] Web: https://twitter.com/SzekelyGroup
bDepartment of Inorganic & Analytical Chemistry, Budapest University of Technology and Economics, Szent Gellert ter 4, Budapest 1111, Hungary
cWCU Department of Energy Engineering, Hanyang University, Seoul 04763, Republic of Korea
First published on 10th June 2021
Abstract
Improving the chemical resistance of membranes without sacrificing their molecular sieving performance is highly challenging. Herein, a novel scalable methodology was developed for fabricating solvent-resistant nanofiltration membranes based on metal–polymer coordination (MPC) through a facile yet highly effective method. The controlled deposition of copper(I) iodide enabled the fine-tuning of the molecular sieving performance of MPC membranes by altering both their chemistry and morphology. Spectroscopic and morphological analyses were conducted to elucidate the microscopic and macroscopic properties of the membranes. The formation of coordination bonds between the metal and polybenzimidazole chains protected the membranes from dissolving in harsh organic solvents. Additionally, computational modeling was performed to reveal the stabilization energy and fractional free volume (FFV). Our work opens more sustainable avenues for robust membrane fabrication without conventional crosslinking, which requires reactive chemicals.
Introduction
Organic solvent nanofiltration (OSN) is a membrane technology that offers an energy-efficient alternative to traditional thermal separations. However, it needs materials that are stable in a wide range of organic solvents. Polybenzimidazole (PBI) is known to have good solvent-resistant properties toward many organic solvents, including ketones, ethers, alcohols, and non-polar solvents. Consequently, PBI has recently been explored for OSN.1–5 Nevertheless, PBI membranes are not stable in harsh polar aprotic solvents without crosslinking.To obtain solvent-resistant membranes, crosslinking is commonly required, usually by immersing membranes in a solution of reactive crosslinking agents, such as dibromoxylene,6 bisepoxide,7 triethylenetetramine,8 diamine,9 and acyl chloride.10 Metal derivatives,11,12 thermal rearrangement processes,13,14 and interpenetrating polymer networks,15 have recently been proposed for improving the stability of membranes. In addition, metal–organic coordination has been extensively explored for crosslinking polyphenols in the field of materials science.16–21 However, the fabrication of membranes based on metal–polymer coordination (MPC) is scarce.
Nunes et al.,22 reported a self-assembly and chelation assisted non-solvent induced phase separation technique for polysulfone films by applying strong acid hydrolysis and the addition of CuSO4 or FeCl3 into a coagulation bath. Complexation-induced phase separation has also been reported as an approach for fabricating metal–polymer composite membranes.23 Moreover, soft metal ions, such as silver and palladium, produced thinner dense layers than those produced by borderline metal ions such as cobalt and nickel. This method is dedicated to form a metal–polymer complex only on the outer parts of membrane layers. Consequently, the inner part of a membrane cannot resist the solvents that can dissolve polymers. The aforementioned phase inversion techniques22,23 resulted in crosslinked membranes, however the metal–polymer interactions were not investigated therein. Therefore, herein, we developed robust MPC membranes by molecular engineering through controlled Cu(I) metal deposition on polybenzimidazole (PBI) substrates (Fig. 1).
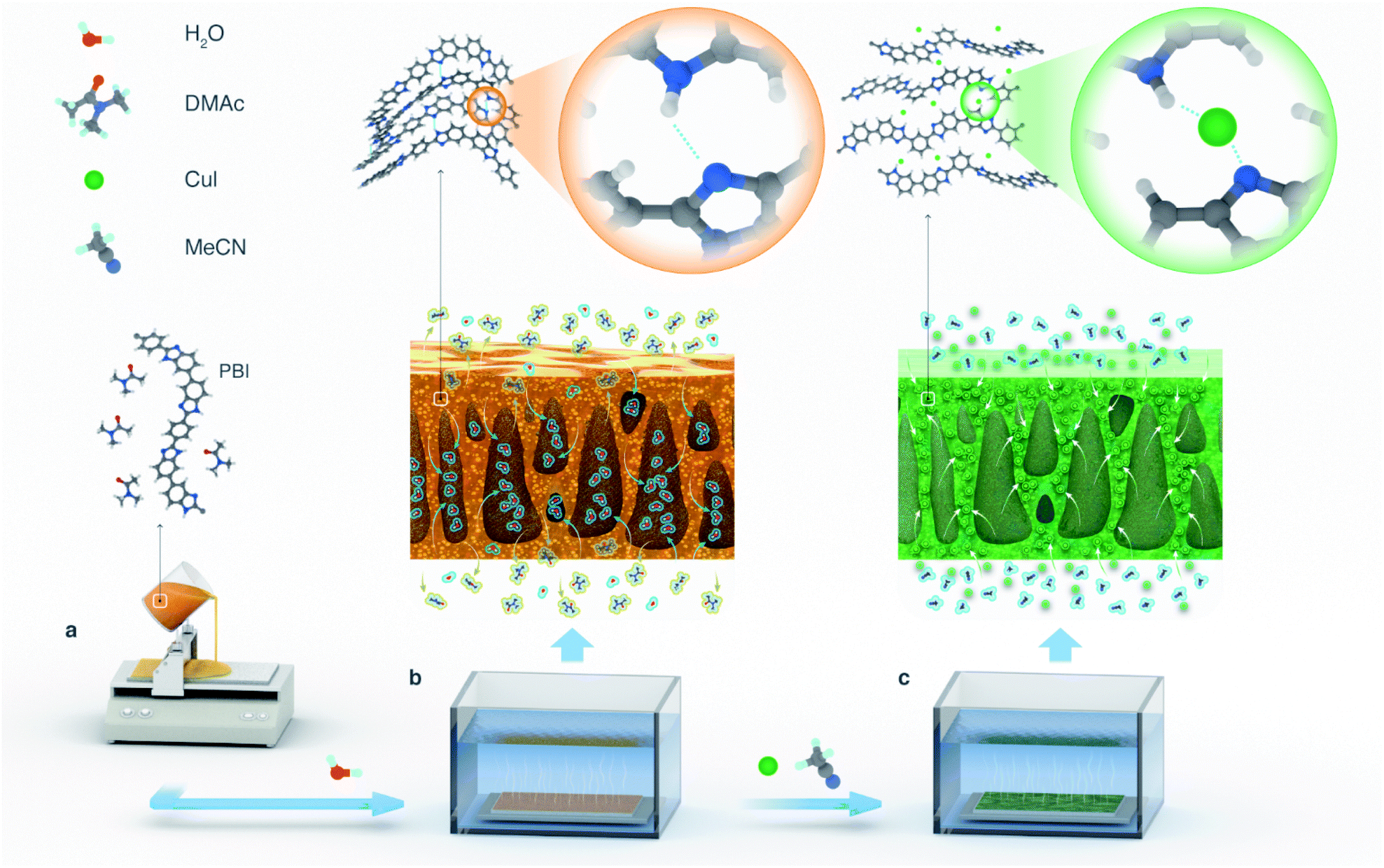 | ||
| Fig. 1 (a) Casting of a PBI dope solution in dimethylacetamide (DMAc). Membrane fabrication at macro- and microscale: (b) phase inversion of the cast polymer dope solution in water; (c) MPC fabrication through CuI treatment in acetonitrile. | ||
Owing to the thermal stability and complexation ability of benzimidazole moieties, polybenzimidazole (PBI) was selected in this study to fabricate MPC membranes. The direct formation of a coordination network between copper iodide and benzimidazole, which resulted in crystalline metal–organic frameworks, has been reported.24
The complexation ability of copper(I) with aromatic nitrogen moieties has been investigated in numerous studies, which reported diverse structures such as metal coordination spheres and cluster formations.25–27 In particular, copper(I) iodide complexes have emerged as useful materials for organic light emitting diodes, sensors, imaging technologies, and solar cells.27
In this research, we designed MPC membranes and investigated their structure–property relationship through thorough materials characterization, molecular modeling, and nanofiltration in organic media.
Results and discussion
Metal–polymer coordination-driven membrane construction
The MPC membranes were prepared through the treatment of a PBI support with 0, 10, 100, and 1000 ppm copper(I) iodide, and they were denoted as M0, M1, M2, and M3, respectively. The structures of the membranes were investigated using spectroscopic techniques. The peaks of C 1s and N 1s were clearly visible for all the membranes in their wide scan (survey) XPS spectra (Fig. 2a). These two peaks correspond to the carbon and nitrogen governing the PBI structure. The presence of oxygen peaks in all the membrane series originated from the atmospheric water adsorbed on the membrane surface. The characteristic peaks of copper (between 930 and 960 eV) and iodine (ca. 619, 629, and 874 eV) were only observed for the membranes prepared with 100 and 1000 ppm copper(I) iodide (M2 and M3), indicating the negligible amount of copper(I) iodide incorporated into the membranes at 10 ppm concentration (M1). The copper XPS spectra of all the MPC membranes resembled the spectra of the Cu+ reference (Fig. 2b), indicating that the oxidation state of copper in the MPC membranes was maintained without transformation to Cu2+. There were no satellite peaks observed for any of the membranes at 940–945 eV, indicating the appearance of Cu2+.28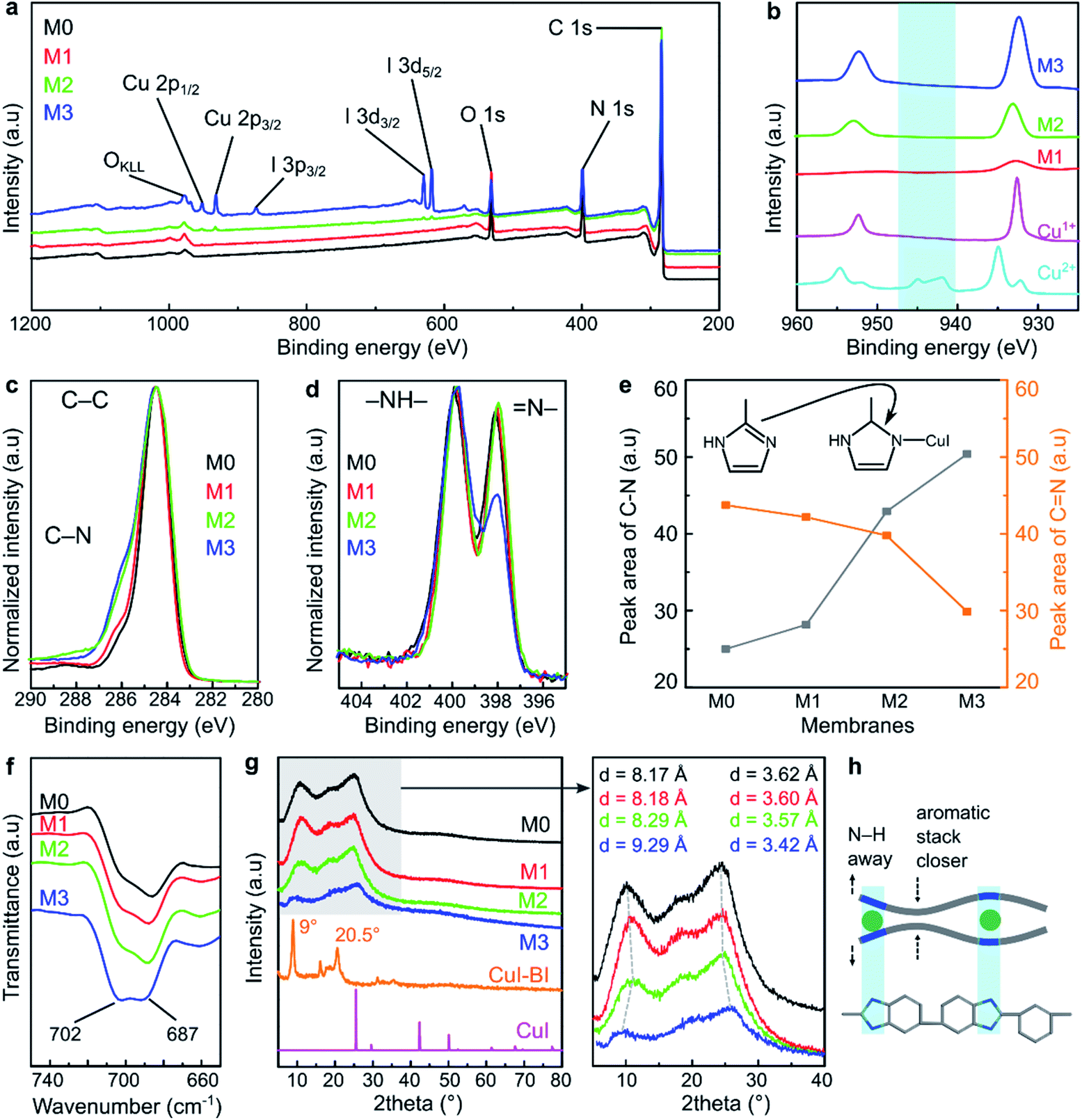 | ||
Fig. 2 Structural analysis of the MPC membranes. (a) Wide scan XPS spectra of the MPC membrane series. (b) Cu 2p XPS spectra of M1, M2, M3, Cu2+, and Cu+. C 1s and N 1s XPS spectra of the MPC membrane series (c and d). (e) The peak area corresponding to C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N from the deconvolution of the C 1s and N 1s XPS spectra. (f) FTIR spectra of the MPC membrane series. (g) XRD spectra of the MPC membrane series, CuI, and CuI–benzimidazole complex with the inset of the magnification of the XRD spectra for the MPC membrane series. (h) Proposed polymer chain arrangement in the PBI after the coordination with copper(I) iodide (h). N from the deconvolution of the C 1s and N 1s XPS spectra. (f) FTIR spectra of the MPC membrane series. (g) XRD spectra of the MPC membrane series, CuI, and CuI–benzimidazole complex with the inset of the magnification of the XRD spectra for the MPC membrane series. (h) Proposed polymer chain arrangement in the PBI after the coordination with copper(I) iodide (h). | ||
In the C 1s high resolution XPS spectra (Fig. 2c), two characteristic peaks at around 284.5 and 285.5 eV were observed for all the membranes, which can be attributed to the C–C and C–N binding energies, respectively.19 Interestingly, the spectra deconvolution (Fig. S1†) revealed that the peak area corresponding to the C–N bond increased as a function of the copper(I) iodide concentration (Fig. 2e), indicating the formation of new C–N bonds as a consequence of reducing the CN bond density in the imidazole ring. Indeed, the diminishing of the CN bond was also clearly observed from the XPS spectra of N 1s (Fig. 2d), which showed two characteristic peaks for –NH– and N– at around 400 and 398 eV, respectively.29,30 Moreover, the deconvolution of these spectra (Fig. S2†) showed that the peak corresponding to N– (at 398 eV) decreased as the copper(I) iodide concentration increased, whereas the –NH– (at 299 eV) peak remained quasi unchanged. This observation suggests the transformation of the double bond between the carbon and nitrogen to a single bond and the persistence of the –NH– bond in the imidazole ring. Fig. 2e shows the XPS peak area evolution and the mechanism of the copper–imidazole coordination.
Additional structural characterization using solid-state proton and carbon NMR were attempted, but the spectra were found to be too broad to reveal any additional information (Fig. S3†). Therefore, liquid-state proton NMR was performed on a model reaction between PBI, DMAc, and CuI. Following the reaction, the NH proton in PBI shifted from 13.51 to 13.35 ppm (Fig. S4†), indicating the effect of CuI coordination around the nitrogen atom, which supports the XPS results that the formation of coordination bonds between copper(I) iodide and PBI is facilitated through the breaking of the double bond in the imidazole ring without going through deprotonation.
In the ATR-FTIR spectra (Fig. S5†), the characteristic peaks of the benzimidazole ring were observed at 1622 cm−1 (CN, CC stretching), 1435 cm−1 (in-plane ring vibration of benzimidazole), and 1284 cm−1 (imidazole ring breathing vibration).31 As shown in Fig. 2f, the FTIR peak at 687 cm−1 is assigned to the self-association of PBI through the hydrogen bonding between the polymer chains. The increasing (M1 → M2 → M3) peak intensity at 702 cm−1 is attributed to the incorporation of copper(I) iodide between the imidazole moieties of PBI as a result of the MPC induced alteration of the hydrogen bonds. This observations is in line with a previous report on PBI.5
The XRD analysis revealed that the structure of the MPC membranes is dominated by amorphous characteristics (Fig. 2g). The broad peak at 2θ (∼25°) was ascribed to the π–π stacking of the aromatic rings governing the partial structural organization of PBI.32 We found that the relative intensity of the XRD spectra decreased with the increase in the copper(I) iodide loading, particularly for M3, which was prepared at the highest concentration. This suggests that the polymer chain packing was disturbed by the incorporation of CuI; hence, the partially organized structure of PBI became more disordered. Moreover, the increase in the CuI concentration shifted the XRD peaks at 2θ (∼10° and 25°) to lower and higher angles, respectively. This phenomenon suggests that the distance (d-spacing) between the polymer chains increased and decreased in different parts of the polymer. The proposed polymer chain rearrangement before and after copper(I) iodide modification is schematically illustrated in Fig. 2h and S6.†
To further elucidate the coordination formation between CuI and PBI, a model reaction between CuI and benzimidazole was performed in acetonitrile at room temperature (Scheme S1 and Fig. S7†). The reaction product, which was a red solid crystal, showed Bragg's peaks at ∼9° and 20.5°, which differ from the PXRD peaks of copper(I) iodide (Fig. 2g). This observation suggests the formation of a new compound through the coordination between CuI and benzimidazole. This reaction product was found to be stable and insoluble in polar aprotic solvents, demonstrating the high strength of the coordination in the framework.
Indeed, the reaction between CuI and benzimidazole at elevated temperature has been reported to produce crystalline materials with an ordered coordination network.24 Nevertheless, our work shows that the appearance of a few observable Bragg's peaks in the reaction product at room temperature hints at an extended network formation that is at least partially ordered (crystalline). Therefore, in the case of the MPC membrane, it was reasonably speculated that the coordination between the PBI matrix and copper(I) iodide is governed by a random network with more disordered CuI–benzimidazole connections, as the treatment was performed at room temperature with less mobile species (solid polymer membrane).
By combining the findings of all the structural analyses together, we convincingly elucidated the formation of MPC membranes. The metal–polymer interaction was facilitated through the double-to-single bond transformation in the imidazole ring followed by a direct coordination formation between CuI and the nitrogen atoms of the imidazole ring. Similar XPS observation on the conversion of the nitrogen peak in imidazole unit, during the crosslinking of PBI with trimesoyl chloride, has been reported in the literature.10
Concentration and time dependent morphologies
The morphologies of the MPC membranes were investigated using electron and atomic force microscopes, as shown in Fig. 3. Macrovoids, which are typical for non-solvent induced phase inversion, were also observed in the cross-section of the membranes. The SEM analysis revealed a wavy surface for the control membrane (M0) in contrast to a flat surface for the MPC membranes, as shown in Fig. 3a and b, respectively. As previously discussed, the XRD analysis (Fig. 2g) revealed the coordination-induced rearrangement and altered packing of the polymer chains (Fig. 3e), which were also manifested in the flatter membrane surface (Fig. 3b).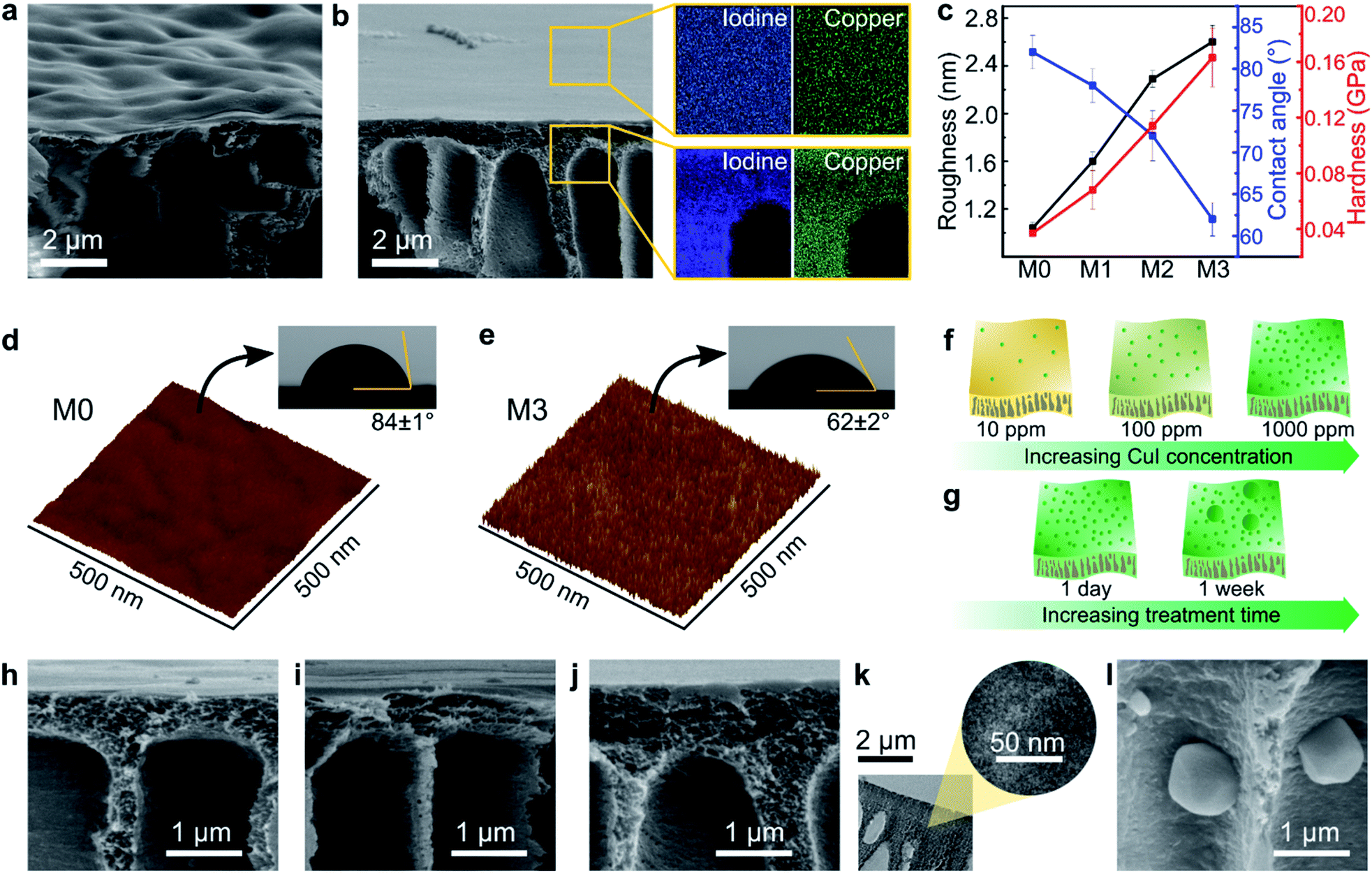 | ||
| Fig. 3 Morphologies of the MPC membranes. (a) SEM surface and cross-section images of M0. (b) SEM surface and cross-section images of M3 and the corresponding elemental mapping for iodine and copper on the surface and cross-section. (c) Roughness, contact angle, and hardness of M0–M3. AFM surface profiles and contact angles of M0 (d) and M3 (e). The proposed mechanism for the metal distribution as a function of the concentration (f) and treatment time (g). SEM cross-section images of M1 (h), M2 (i), and M3 (j) after treatment for 1 day. (k) TEM high magnification of M3 after treatment for 1 day. (l) SEM cross-section image of M3 after treatment for 1 week. | ||
The copper and iodine were homogeneously distributed not only on the surface but also through the cross-section of the MPC membranes (Fig. 3b, S8 and S9†). Unlike reported in a previous literature, where coordination only occurs on membrane surfaces,23 in our work, copper(I) iodide penetrated the membrane and diffused through the polymer matrix, resulting in solvent resistance for the entire membrane. The morphology of the membranes along with the evolution of the elemental mapping density as a function of the copper(I) iodide concentration are shown in Fig. S8.†
The surface of the MPC membranes became rougher owing to the increasing copper(I) iodide concentration, as revealed in the AFM analyses in Fig. 3c–e and S8q–t.† A more hydrophilic character in the MPC membrane was observed (Fig. 3c–e and S11†) because of the synergistic effect of copper iodide and the rougher surface topography. Copper iodide has a hydrophilic character (as measured by the water contact angle in Fig. S10†), and the rough surface on hydrophilic membranes tends to increase their hydrophilicity owing to the increased surface area in contact with water droplets.33 Nanoindentation revealed the hardening of MPC membranes with increasing CuI concentration (Fig. 3c). Because hardness implies resistance to plastic deformation,34 the increasing hardness of the MPC membranes prevented their plasticization and dissolution in various solvents.35
The bulk compositions of the copper and iodine in the MPC membranes were obtained through elemental analysis. Approximately 2.12 and 7.56 wt% copper in addition to ∼3.86 and 14.21 wt% iodine were incorporated into M2 and M3, respectively. The obtained values corresponded to the atomic ratio of copper to iodine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), indicating that the ratio of copper to iodine was maintained in the membranes (Table S1†). Negligible copper and iodine amounts were detected in M1, which was prepared at the lowest CuI concentration (10 ppm); this agrees well with our XPS results.
:1), indicating that the ratio of copper to iodine was maintained in the membranes (Table S1†). Negligible copper and iodine amounts were detected in M1, which was prepared at the lowest CuI concentration (10 ppm); this agrees well with our XPS results.
The effects of the CuI concentration and CuI treatment duration on the membrane's properties were investigated, and they gave insights into the possible mechanism of the MPC membrane formation. Increasing the CuI concentration from 10 to 1000 ppm during the 24 h treatment did not result in agglomerate formation, as shown by the SEM images in Fig. 3h–j. A high-resolution TEM was further performed to confirm the absence of agglomerates (Fig. 3k and S12†), which proved that the CuI coordinated with the PBI at molecular level. However, the longer treatment times at the 1000 ppm CuI concentration resulted in the formation of some agglomerates (Fig. 3l and S13†). Copper(I) iodide not only deposited on top of the membrane surface, but it also protruded inside the macrovoids across the cross-section of the membranes. Interestingly, the morphology of the agglomerates was smooth after one week (Fig. S14a†), whereas the one-month treatment resulted in a morphology similar to that of the desert rose selenite (Fig. S14b†).
The increase in the CuI concentration to up to 1000 ppm with the 24 h treatment increased the number of CuI molecules in coordination with the PBI matrix (Fig. 3f), whereas the prolonged treatment time resulted in an undesired growth of CuI nanoparticles (Fig. 3g). Neither the CuI concentration nor the treatment time influenced the membranes' top layer thickness (Fig. S19†). The chemical and morphological investigations allowed us to propose a possible mechanism for the formation of the MPC membranes and or preventing unwanted CuI agglomeration.
Stabilization of the metal–polymer interactions in the MPC membranes
The XPS and 1H NMR results confirmed that the nitrogen atom in the imine moiety of the imidazole ring (–N) formed a coordination bond with copper(I) iodide. To simulate the interaction between polybenzimidazole and CuI, initial screening was performed using the available JAWZUD crystal structure in Cambridge Crystallographic Data Centre, where copper directly binds to nitrogen in imidazole.24
The geometry optimization resulted in a crystal structure that showed the coordination interaction between the imine nitrogen and copper (interaction 2 in Fig. 4a and e). In addition, the crystal structure showed parallel-displaced π–π (interaction 3 in Fig. 4a and d) and T-shaped π–π interactions between the benzimidazole rings (interaction 4 in Fig. 4a and c) in addition to a tentative hydrogen–iodine interaction between iodine and benzimidazole (interaction 5 in Fig. 4a and e). The types of interactions are listed in Table 1 and illustrated in Fig. 4.
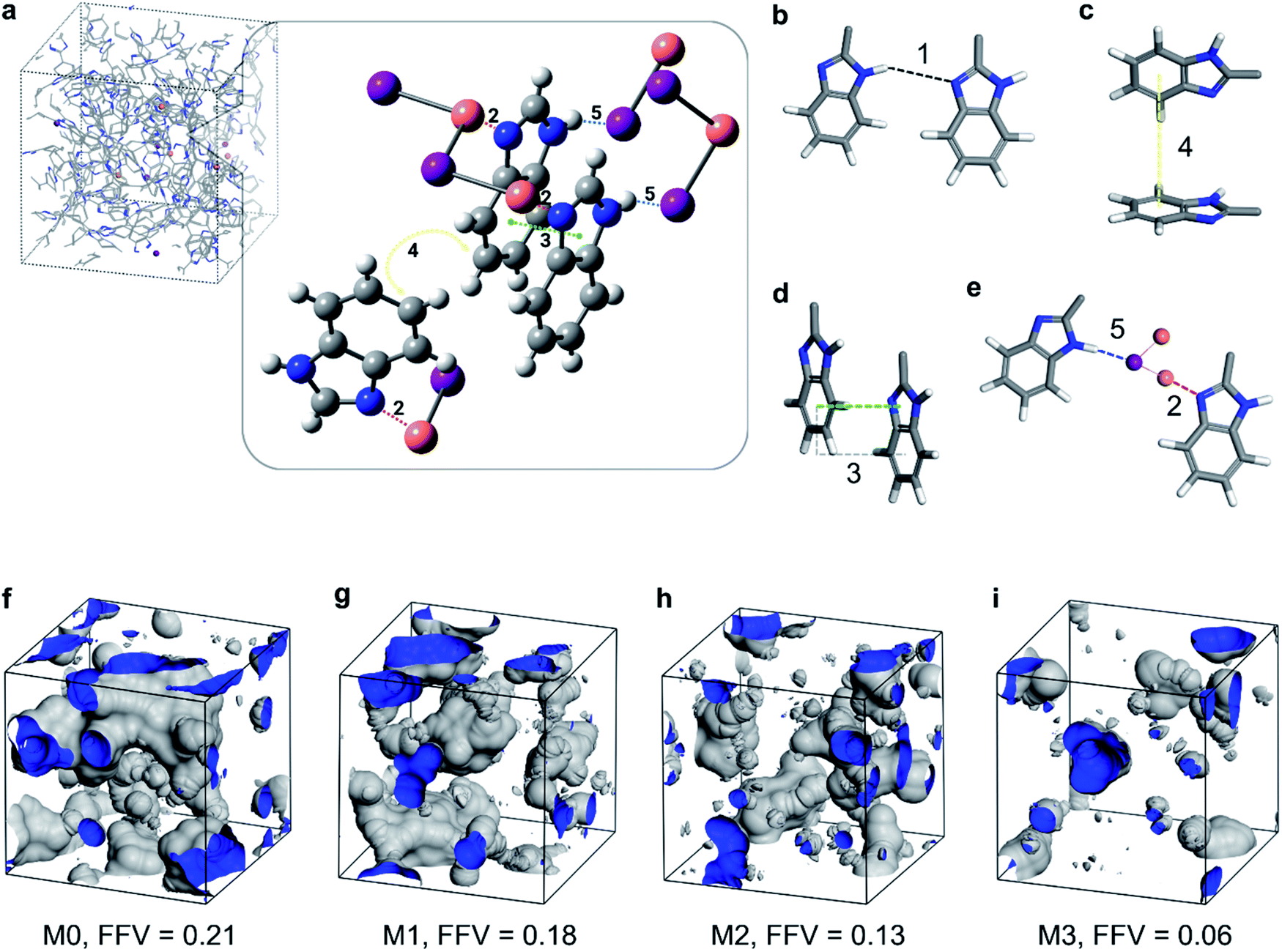 | ||
| Fig. 4 Molecular modeling of the MPC membrane. (a) Interactions appearing in the JAWZUD crystal structure that represents the interactions in the MPC membrane. The figure only shows the parts of the molecule that are important for depicting the interaction: copper (orange), iodine (purple), carbon (gray), and hydrogen (white). (b) Hydrogen bonding interaction between benzimidazoles. (c) Interaction between the benzimidazoles in the T-shaped π–π. (d) Interaction between the benzimidazoles in the parallel displaced π–π. (e) Interaction between the benzimidazoles and CuI through the N–Cu coordination (2) and NH–I coordination (5). Fractional free volume (FFV) of M0 (f), M1 (g), M2 (h), and M3 (i). | ||
| Entry | Type of interaction | Interaction energy [kJ mol−1] | Distance [Å] | Anglea [°] |
|---|---|---|---|---|
| a The angle is only defined in the case of the π–π interaction, and it denotes the angles defined between the planes of the π systems. b The benzimidazole – H-bond interaction was calculated through geometry optimization, which does not belong to a real crystal structure. c Owing to the crystal geometry, for each benzimidazole rings, five closest CuI moieties were kept to estimate the interaction energy more precisely. | ||||
| 1 | Benzimidazole – H-bondb | −45.8 | 1.926 | n.a. |
| 2 | Benzimidazole – 5 × CuI (N⋯Cu)c | −123.0 | 1.982 | n.a. |
| 3 | Benzimidazole – parallel-displaced π–π | −16.6 | 3.570 | 180 |
| 4 | Benzimidazole – T-shaped π–π | −9.6 | 3.743 | 61 |
| 5 | Benzimidazole – 5 × CuI (NH⋯I)c | −33.9 | 3.006 | n.a. |
The −45.8 kJ mol−1 hydrogen bonding complexation energy under entry #1 in Table 1 is favorable. However, this energy might be overestimated owing to the geometry optimization performed on the system containing only two benzimidazole moieties. This might lead to a shorter hydrogen bonding distance (thus a stronger bond) for the isolated system compared with the PBI polymer. The calculations showed that the benzimidazole–Cu complexation (entry #2) results had significantly larger stabilization than those derived from either the hydrogen bonding (entry #1) or the different π–π interactions (entries #3 and #4). This favored energetics enabled CuI to form a coordination bond with the PBI chains, resulting in the stronger stabilization of the membrane system.
By calculating the binding energies for the oriented 1 benzimidazole fragment and 5 CuI moieties as in the JAWZUD crystal structure, it was found that there existed −123.0 kJ mol−1 stabilization owing to the interaction between benzimidazole nitrogen and the CuI moieties, wherein the closest interaction was with Cu at a distance of 1.982 Å from the nitrogen. If the CuI moieties were considered on the other side of the benzimidazole, an interaction between the aromatic –NH– and iodine would have also been favorable, causing a stabilization of −33.9 kJ mol−1. The two interactions clearly outperformed the hydrogen bonding interaction between the benzimidazole moieties, making the insertion of CuI layers between benzimidazoles favorable.
Furthermore, having measured the geometric density of the membranes (M0 = 1.132 g cm−1, M1 = 1.145 g cm−1, M2 = 1.252 g cm−1, and M3 = 1.431 g cm−1), the fractional free volume (FFV) was calculated and visualized, as shown in Fig. 4f–i. The incorporation of copper(I) iodide into the PBI matrix resulted in denser membranes and subsequently lower FFV. This densification is manifested from the coordination-induced rearrangement and the altered packing of the polymer chains, which is in line with the previously discussed XRD analysis (Fig. 2g–h).
Molecular sieving performance
A stability test was performed by immersing the membranes in organic solvents (Table S2†). The M0, M1, and M2 membranes could resist many common organic solvents, and the results matched with the insoluble nature of PBI. Although these membranes were dissolved in harsh polar aprotic solvents, such as DMSO, DMF, and DMAc, the membrane with the highest copper(I) iodide content (M3) demonstrated excellent solvent resistance under these harsh environments.The stability of the MPC membranes was also tested by immersing the membranes in acidic (HCl) and basic (NaOH) conditions at various concentrations between 0.1 mM and 100 mM (Fig. S16†) for 1 day. The copper content in the membranes was checked by EDX, and it was found to gradually decrease with the increase in acid concentration. The quasi-complete removal of the copper suggests that the PBI could be recovered unlike with conventional chemical crosslinking. On the contrary, the copper content somewhat decreased and then remained constant in the membrane even at high NaOH concentrations. This observation suggests that the MPC membrane is stable under basic conditions.
The molecular sieving effects of the membranes were tested in a cross-flow nanofiltration system. The membranes became tighter as evidenced by the decrease in the molecular weight cut-off (MWCO) as a result of increasing the copper(I) iodide content (M0 → M3). This observed trend in the molecular sieving performance is in line with the evolution of the d-spacing, polymer chain packing (XRD), and decreased FFV.
The pore diameter distribution analysis revealed that the mean pore radius was around 0.94, 0.65, 0.30, and 0.34 nm for the M0, M1, M2, and M3 membranes, respectively (Fig. 5b). The details of the pore size calculation were shown in the ESI.† Typically, the MWCO values showed good correlation with the pore size distribution. Nonetheless, the pore diameter of M3 was slightly higher than that of M2. This can be attributed to the compromised effect in which some parts of the polymeric chain moved closer to one another, whereas the other parts drifted further away from each other, as previously explained by the XRD analysis (Fig. 2g–h).
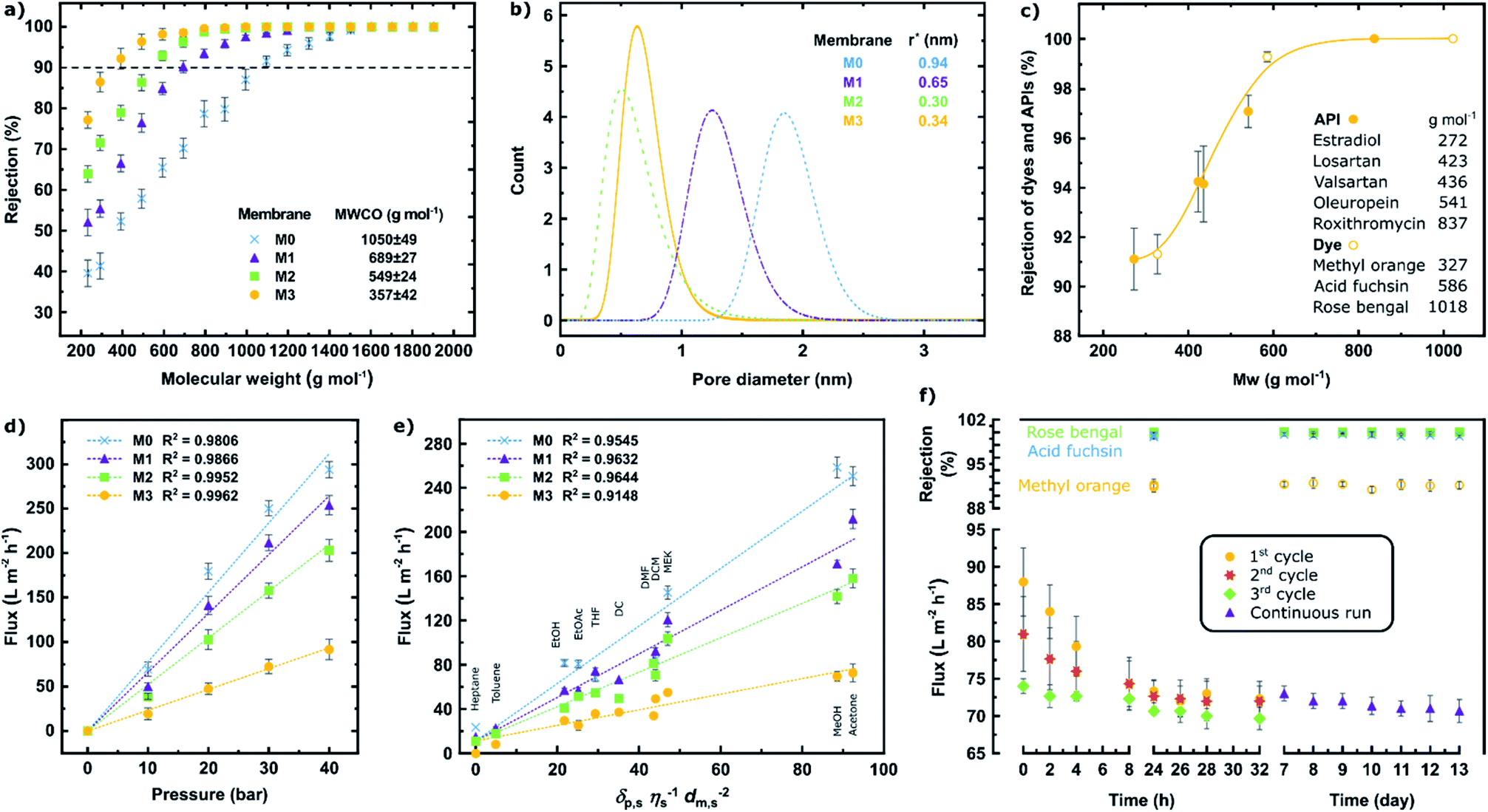 | ||
| Fig. 5 Nanofiltration performance of the MPC membranes. (a) Polystyrene rejection profiles and MWCO values. (b) Pore size distribution. (c) API and dye rejection profile of the M3 membrane. Solvent flux as a function of the applied pressure (d), and solvent solubility parameter (e) at 30 bar. (f) Flux decline and long-term stability test of the membrane performance. The data in panels a–d and f were obtained in acetone at 30 bar. | ||
The separation performance of M3 was further tested for the separation of five active pharmaceutical ingredients (API) and three dye molecules with various molecular weights ranging from 272 to 1018 g mol−1 (Fig. 5c). A small shift in the MWCO curve to lower molecular weights can be observed. In other words, the API and dye solutes, which have various functional groups and structures, exhibited higher rejection than the more uniform and inert polystyrene series. The MWCO value for M3 (using polystyrene markers) was 357 g mol−1 (Fig. 5a), whereas this value decreased to less than 300 g mol−1 (Fig. 5c) (using diverse solutes). The rejection and flux values were tabulated in the ESI.†
The increase in pressure resulted in increasing the solvent flux, indicating that the membranes were not blocked upon applying high pressure (Fig. 5d), although membrane compaction was observed (Fig. S18†). Eleven different solvents with varying polarity values were used to assess the performance of the membranes, and a linear correlation between the solvent flux and solubility parameter was observed (Fig. 5e). The solubility parameter comprises the solvent solubility (δp,s), viscosity (η), and molar diameter (dm,s).36
The tightest membrane (M3) was selected to assess the long-term stability during the pressure cycles and continuous filtration (Fig. 5f). The rejections of the rose bengal (99.9%), acid fuchsin (99.5%), and methyl orange (91.6%) dyes were found to be stable over 13 days of continuous nanofiltration in acetone at 30 bar. A considerable flux decline was observed during the three pressure cycles with a decrease in the decline rate itself after each cycle (17% → 11% → 6%). During the subsequent 13 days, continuous nanofiltration and a negligible 3% flux-decline with an average of 72 L m−2 h−1 acetone flux were observed. The filtration performance of M3 was also compared to the literature on membranes fabricated via complexation of polymers with metals,23,37 and the results are tabulated in Table S7.†
The molecular sieving results demonstrated that metal–polymer coordination improves the solvent resistance of the membranes, and that their separation performance can be tailored resulting in different MWCO values. It was also found that the fabricated membranes exhibit stable performance over long-term continuous operation.
Conclusions
Herein, we successfully fabricated a metal–polymer coordination (MPC) membrane using copper(I) iodide and polybenzimidazole. The conducted structural investigations revealed the successful formation of metal–polymer coordination through double-to-single bond transformation in the imidazole ring without going through deprotonation. Additionally, the performed molecular modeling revealed the stabilization effect of copper(I) iodide on the polymer matrix, resulting in more stable membranes. The formation of metal–polymer coordination not only led to increased solvent resistance but also improved the mechanical properties of the membranes and allowed the tailoring of the molecular sieving performance. The membranes also exhibited long-term stability over 13 days of continuous nanofiltration processes at 30 bar. The simple, yet effective method for preparing metal–polymer coordination membranes provides new avenues for exploring solvent-resistant membranes without the need for covalent crosslinking. We envisage diverse applications for such membranes, including conductive membranes, catalytic membranes, and electrochemical and fuel cell membranes.Experimental
Materials
Polybenzimidazole dope solution (PBI, 26% in N,N-dimethylacetamide) was purchased from PBI Performance Products, Inc. (Charlotte, USA). Copper(I) iodide (CuI) powder (99%) was purchased from Fischer Scientific. Rose bengal (98%), methyl orange (97%), and acid fuchsin (95%) (dyes) were purchased from Alfa Aesar, Sigma Aldrich, and Sungyoung Chemical, respectively. Estradiol (98%), losartan (97%), valsartan (98%), and oleuropein (active pharmaceutical ingredients) were purchased from Sigma Aldrich, Acros Organics, TCI Chemicals, and Biopurify Phytochemicals, respectively. Roxithromycin (98%) was provided by Hovione PharmaScience. Deionized water with a resistivity of 18.20 MΩ cm at 25.8 °C was generated from Milli-Q. N,N-Dimethylacetamide (99%), N,N-dimethylformamide (99%), dimethyl sulfoxide (synthesis grade), and dimethyl carbonate (99%) (solvents) were supplied by Alfa Aesar. Acetonitrile (99%) (solvent) was bought from Fischer Scientific. Heptane (synthesis grade), toluene (synthesis grade), ethanol (synthesis grade), acetone (technical grade), methanol (synthesis grade), and ethyl acetate (synthesis grade) (solvents) were purchased from Alfa Aesar. Tetrahydrofuran (synthesis grade) (solvent) was bought from Sigma Aldrich. Dichloromethane (synthesis grade) and methylethylketone (synthesis grade) (solvents) were purchased from Merck.Membrane preparation
Homogeneous PBI dope solutions were realized by overhead stirring the components at 50 rpm at room temperature for 20 h. Then, degassing was performed under a nitrogen atmosphere in an incubator shaker at 200 rpm and at 25 °C for 4 h to ensure that the dope solution was free from any entrapped gas. The membranes were cast onto a Novatexx 2471 non-woven polypropylene support using an Elcometer 4340 film applicator with a casting knife, which was set to a thickness of 250 μm, and its casting speed was 5 cm s−1. The membranes were formed by precipitating the films in a coagulation bath containing deionized water at 23 °C. The deionized water was replaced after 0.5 h with fresh deionized water for an additional 24 h to ensure complete solvent exchange. The membranes (M0) were washed with acetonitrile prior to the copper(I) iodide treatment.The MPC membranes (M1, M2, and M3) were fabricated by immersing the prepared PBI membrane (M0) into 10, 100, and 1000 ppm of copper(I) iodide in an acetonitrile solution, respectively. To study the effect of the treatment time, the immersion time was varied as follows: 24 h, 1 week, and 1 month.
Model reaction
100 mg (0.525 mmol) of copper(I) iodide were dissolved in 15 mL of acetonitrile, followed by the addition of 62 mg (0.525 mmol) of benzimidazole. The reaction mixture was sonicated for 5 min and then kept at room temperature for 24 h. The product was centrifuged at 7000 rpm for 20 min, followed by washing with acetonitrile, centrifuging three times, and then drying under vacuum overnight, yielding red powder.Acid–base stability test
For the acid stability test, 1 cm2 of each membrane (M0, M1, M2, and M3) were cut and placed in a glass vials containing 2 mL of 0.1, 1, 10, and 100 mM HCl solution for 24 h. The base stability test was performed in the same manner, by placing 1 cm2 of each membrane (M0, M1, M2, and M3) in a glass vials containing 2 mL of 0.1, 1, 10, and 100 mM NaOH solution for 24 h. The copper content in the membranes before and after test were quantified by using EDX analysis.Nanofiltration performance test
A cross-flow nanofiltration skid having three membrane cells with an active area of 52.8 cm2, a back pressure regulator, a high-pressure pump and a microannular gear pump was used for membrane separation. The flow rate of the microannular gear pump was maintained at a constant rate of 100 L h−1 to minimize concentration polarization. The membranes were conditioned in a solvent–solute mixture at 30 bar for 24 h prior to taking permeate and retentate samples for the rejection determination (eqn (1)). Polystyrene standards having 1 g L−1 PS580 and PS1300, 0.1 g L−1 methyl styrene dimer (236 g mol−1), 10 μM dyes, and 10 μM active pharmaceutical ingredients (API) were used as solutes in the feed stream. The volume of the solvent permeating through the membrane (V) at a certain time (t) over the active membrane surface area (A) was used to calculate the flux (eqn (2)). The reported membrane performance values are the average values of three independently prepared membranes.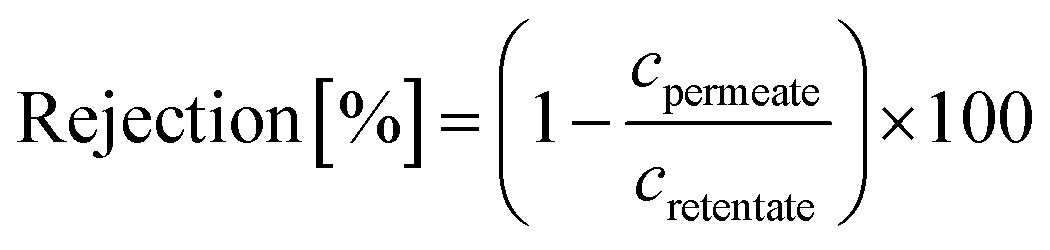 | (1) |
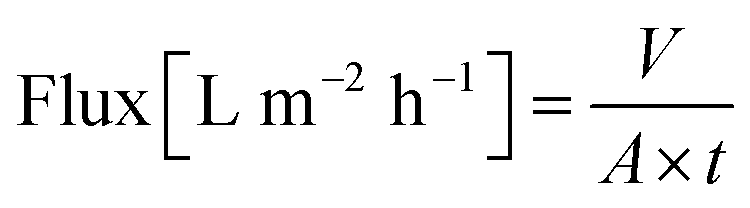 | (2) |
A long-term stability experiment was performed involving three pressure cycles followed by a week of continuous filtration. Each pressure cycle comprised of nanofiltration at 30 bar for 32 h, followed by depressurization of the skid. The system was allowed to stand overnight before initiating the next cycle. Upon the completion of the third cycle on day 7, continuous nanofiltration at 30 bar for 7 days was performed. Flux measurements and rejection determination occurred at given time intervals.
Computational details
The theoretical calculations were performed with the code Gaussian16 using the B3LYP-D3 and 6-31+G** levels of theory. Geometry optimization with vibrational frequency calculation was only performed to assess the hydrogen bonding interaction between two imidazole rings as this interaction was not detected in the case of the crystal structures containing copper. In all the other cases, the interaction energies were calculated using the crystal structure orientation of the molecules by removing the unnecessary parts using a crystal structure with the code JAWZUD.24 The interaction energies were calculated using the counterpoise method of the basis set superposition error (BSSE).38–40The FFV of the membranes was calculated using Materials studio after determining the geometric density of each membrane. The geometric densities of M0, M1, M2, and M3 were determined by measuring the mass of each membrane and then dividing it by its geometric volume (length × width × thickness). The COMPASS II force field was applied to assign force field parameters to the simulations. Amorphous cell membrane systems were constructed using an amorphous module by mixing PBI chains (each chain has 5 repeat units) with various numbers of copper(I) iodide molecules. A probe radius of 1.6 Å was used to calculate the FFV using eqn (3).
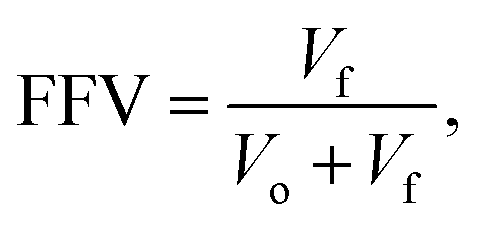 | (3) |
X-ray photoelectron spectroscopy (XPS)
XPS experiments were performed using a Kratos Axis Ultra DLD spectrometer equipped with a monochromatic Al Ka X-ray source (hν = 1486.6 eV) operating at 150 W, a multichannel plate, and a delay line detector under a vacuum of ∼10–9 mbar. All the spectra were recorded using an aperture slot of 300 × 700 μm. Survey spectra were collected using a pass energy of 160 eV and a step size of 1 eV, whereas a pass energy of 20 eV and a step size of 0.1 eV were used for the high-resolution spectra. For the XPS analysis, samples were mounted in the floating mode to avoid differential charging. Charge neutralization was required for all the samples.X-ray diffraction spectroscopy (XRD)
The powder XRD data were collected using a Cu Kα Bruker D8 Advance diffractometer in the angular range containing the peaks of interest: 2θ 5–40° with an increment of 0.02° and a scanning speed of 10° min−1. The samples were prepared by peeling off the dried membranes from the polypropylene support, and the small pieces were ground into powder. Prior to the data collection, the samples were placed on a zero-background XRD sample holder and then flattened using a glass plate.Fourier transform infrared spectrometry (FTIR)
An attenuated total reflectance (ATR) mode was applied using a Bruker FTIR spectrometer (VERTEX 70/70v) over the range of 600–4000 cm−1 with a total of 64 scans for each sample.Solid-state and liquid-state NMR
The solid-state spectra were recorded on a Bruker Avance III 400 MHz spectrometer at room temperature. The samples were prepared by peeling off the dried membranes from the polypropylene support, and the small pieces were ground into powder. Then, the obtained powder was placed inside the rotors. The spectra were acquired in a 4 mm MAS probe operating at a spinning speed of 10–11 kHz. The liquid-state spectra of the product of the model reaction were recorded using a Bruker AVANCE-III spectrometer at a frequency of 500 MHz in ppm. Deuterated dimethylsulfoxide (DMSO-d6) was used as a solvent.Scanning and transmission electron microscopies (SEM, TEM)
A scanning electron microscope (NovaNano) was employed to examine the top membrane surface and cross-sectional morphologies. The same instrument was used to perform an energy dispersive X-ray spectroscopy (EDX) analysis for elemental mapping on the membrane surface. First, the membranes were dried at room temperature for 1 day. Then, they were placed in a vacuum oven at 25 °C for 1 day. The dried membranes were then peeled off from the polypropylene support, and the fractures were used for a cross-sectional analysis. The prepared membranes were attached to the SEM stub using carbon tape and were then coated with 5 nm iridium using an ion sputtering device prior to the SEM analysis. Images were acquired at 5 kV high tension and 28 pA current with a working distance of 5 mm. TEM images were obtained using an FEI Titan 80-300 CT operated at 300 kV. Cross-sectional samples were prepared for TEM by embedding the membranes in a low-viscosity epoxy resin (Agar R1165) and then by sectioning them using a Leica EM UC6 ultramicrotome.Atomic force microscopy (AFM)
Atomic force microscopy (AFM, Bruker Dimension ICON) with was used a tapping mode (Acoustic AC) at room temperature. For each sample, an area of 5 × 5 μm was scanned at a rate of 1 Hz. Then, the scanned results were analyzed using NanoScope Analysis (version 1.5). The obtained mean roughness (Ra) was used to quantify the surface roughness.Nanoindentation
Nanoindentation was performed using a NanoTest Vantage instrument with a pyramidal diamond indenter. The hardness was measured using the average of four indentions per specimen. The membrane samples were cut into 1 cm2 parts and then stuck on the silicon wafer.Water contact angle
The water contact angles of the dried membranes were measured using a contact angle goniometer (Kruss EasyDrop) by applying the sessile drop method. Four measurements were performed at different locations to obtain the average value for each sample.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Rachid Sougrat for performing TEM characterization. Fig. 1 and Table of Content illustrations were created by Heno Hwang, scientific illustrator at King Abdullah University of Science and Technology (KAUST). The research reported in this publication was supported by funding from KAUST.References
- I. B. Valtcheva, S. C. Kumbharkar, J. F. Kim, Y. Bhole and A. G. Livingston, J. Membr. Sci., 2014, 457, 62–72 CrossRef CAS.
- G. Székely, I. B. Valtcheva, J. F. Kim and A. G. Livingston, React. Funct. Polym., 2015, 86, 215–224 CrossRef.
- B. Zhao, G. M. Shi, K. Y. Wang, J.-Y. Lai and T.-S. Chung, Sep. Purif. Technol., 2021, 255, 117702 CrossRef CAS.
- D. Y. Xing, S. Y. Chan and T.-S. Chung, Green Chem., 2014, 16, 1383–1392 RSC.
- V. Loianno, K. P. Bye, M. Galizia and P. Musto, J. Polym. Sci., 2020, 58, 2547–2560 CrossRef CAS.
- I. B. Valtcheva, P. Marchetti and A. G. Livingston, J. Membr. Sci., 2015, 493, 568–579 CrossRef CAS.
- S. Chisca, G. Falca, V. E. Musteata, C. Boi and S. P. Nunes, J. Membr. Sci., 2017, 528, 264–272 CrossRef CAS.
- M.-L. Liu, J.-L. Guo, S. Japip, T.-Z. Jia, D.-D. Shao, S. Zhang, W.-J. Li, J. Wang, X.-L. Cao and S.-P. Sun, J. Mater. Chem. A, 2019, 7, 3170–3178 RSC.
- C. Van Goethem, M. M. Magboo, M. Mertens, M. Thijs, G. Koeckelberghs and I. F. J. Vankelecom, J. Membr. Sci., 2020, 611, 118274 CrossRef CAS.
- M. H. Davood Abadi Farahani and T.-S. Chung, Sep. Purif. Technol., 2019, 209, 182–192 CrossRef CAS.
- H. M. Tham, S. Japip and T.-S. Chung, J. Membr. Sci., 2019, 588, 117219 CrossRef CAS.
- E. K. McGuinness, F. Zhang, Y. Ma, R. P. Lively and M. D. Losego, Chem. Mater., 2019, 31, 5509–5518 CrossRef CAS.
- T.-D. Lu, L.-L. Zhao, W. F. Yong, Q. Wang, L. Duan and S.-P. Sun, Chem. Eng. J., 2021, 409, 128206 CrossRef CAS.
- J. H. Kim, S. J. Moon, S. H. Park, M. Cook, A. G. Livingston and Y. M. Lee, J. Membr. Sci., 2018, 550, 322–331 CrossRef CAS.
- D. Zhao, J. F. Kim, G. Ignacz, P. Pogany, Y. M. Lee and G. Szekely, ACS Nano, 2019, 13, 125–133 CrossRef CAS PubMed.
- Md. A. Rahim, H. Ejima, K. L. Cho, K. Kempe, M. Müllner, J. P. Best and F. Caruso, Chem. Mater., 2014, 26, 1645–1653 CrossRef CAS.
- T. Chakrabarty, L. Pérez-Manríquez, P. Neelakanda and K.-V. Peinemann, Sep. Purif. Technol., 2017, 184, 188–194 CrossRef CAS.
- R. Wang, X. Zhao, Y. Lan, L. Liu and C. Gao, J. Membr. Sci., 2020, 615, 118566 CrossRef CAS.
- H. Wu, J. M. Ang, J. Kong, C. Zhao, Y. Du and X. Lu, RSC Adv., 2016, 6, 103390–103398 RSC.
- S. Chen, Y. Xie, T. Xiao, W. Zhao, J. Li and C. Zhao, Chem. Eng. J., 2018, 337, 122–132 CrossRef CAS.
- J. Feng, S. Xiong, Z. Wang, Z. Cui, S.-P. Sun and Y. Wang, J. Membr. Sci., 2018, 550, 246–253 CrossRef CAS.
- Y. Xie, N. Moreno, V. M. Calo, H. Cheng, P.-Y. Hong, R. Sougrat, A. R. Behzad, R. Tayouo and S. P. Nunes, Polym. Chem., 2016, 7, 3076–3089 RSC.
- L. F. Villalobos, M. Karunakaran and K.-V. Peinemann, Nano Lett., 2015, 15, 3166–3171 CrossRef CAS PubMed.
- T. Wu, D. Li and S. W. Ng, CrystEngComm, 2005, 7, 514–518 RSC.
- S. C. Bete, C. Würtele and M. Otte, Chem. Commun., 2019, 55, 4427–4430 RSC.
- J.-C. Li, H.-X. Li, H.-Y. Li, W.-J. Gong and J.-P. Lang, Cryst. Growth Des., 2016, 16, 1617–1625 CrossRef CAS.
- M. D. Kessler, T. Duston, M. Parker and R. D. Pike, Inorg. Chim. Acta, 2020, 509, 119706 CrossRef CAS.
- T. M. Ivanova, K. I. Maslakov, A. A. Sidorov, M. A. Kiskin, R. V. Linko, S. V. Savilov, V. V. Lunin and I. L. Eremenko, J. Electron Spectrosc. Relat. Phenom., 2020, 238, 146878 CrossRef CAS.
- A. Naderi, A. Asadi Tashvigh, T.-S. Chung, M. Weber and C. Maletzko, J. Membr. Sci., 2018, 563, 726–733 CrossRef CAS.
- G. Nesher, G. Marom and D. Avnir, Chem. Mater., 2008, 20, 4425–4432 CrossRef CAS.
- B. Zhao, L. Cheng, Y. Bei, S. Wang, J. Cui, H. Zhu, X. Li and Q. Zhu, Eur. Polym. J., 2017, 94, 99–110 CrossRef CAS.
- A. Asadi Tashvigh and T.-S. Chung, J. Membr. Sci., 2019, 572, 580–587 CrossRef CAS.
- J. Wang, Y. Wu, Y. Cao, G. Li and Y. Liao, Colloid Polym. Sci., 2020, 298, 1107–1112 CrossRef CAS.
- B. Bhushan, Tribology and Mechanics of Magnetic Storage Devices, Springer-Verlag, New York, 2nd edn, 1996 Search PubMed.
- K. Rezzadori, F. M. Penha, M. C. Proner, G. Zin, J. C. C. Petrus and M. D. Luccio, Chem. Eng. Technol., 2019, 42, 2700–2708 CrossRef CAS.
- S. Karan, Z. Jiang and A. G. Livingston, Science, 2015, 348, 1347–1351 CrossRef CAS PubMed.
- Y. Xie, B. Sutisna and S. P. Nunes, Chem. Commun., 2017, 53, 6609–6612 RSC.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef CAS.
- P. Salvador and M. Duran, J. Chem. Phys., 1999, 111, 4460–4465 CrossRef CAS.
- S. Simon, M. Duran and J. J. Dannenberg, J. Phys. Chem. A, 1999, 103, 1640–1643 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: For detailed experimental characterizations, pore size calculation, and membrane performance. See DOI: 10.1039/d1ta02601a |
| This journal is © The Royal Society of Chemistry 2021 |