
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning of Pt–Co nanoparticle motifs for enhancing the HOR performance in alkaline media†
D. J.
Weber
ab,
C.
Dosche
 b and
M.
Oezaslan
*ab
b and
M.
Oezaslan
*ab
aTechnical Electrocatalysis Laboratory, Institute of Technical Chemistry, Technical University of Braunschweig, 38106 Braunschweig, Germany. E-mail: m.oezaslan@tu-braunschweig.de
bInstitute of Chemistry, University of Oldenburg, 26129 Oldenburg, Germany
First published on 2nd July 2021
Abstract
For a future sustainable hydrogen economy, the electrocatalysis of hydrogen oxidation/evolution reactions (HOR/HER) needs to be better understood. In order to describe the strong alteration of HOR/HER rates on platinum group metals (PGM) in acidic and alkaline media, at least two prominent descriptors based on a Pt–Had binding energy model and a bifunctional model are frequently discussed in the literature. A clear discrimination of these both descriptors for PGM-based multi-metallic materials, yet, remains a critical challenge. To solve this dilemma, we have made use of the oxophilic and structural properties of cobalt atoms in Pt-based nano-materials to tune their catalytic HOR performance in alkaline media. More precisely, we have designed PtxCo1−x nanoparticles with various structural motifs. Remarkably, the surface rearrangement of PtxCo1−x nanoparticles along the uniform alloy, Pt-rich shell, and Co-segregated core–shell motifs is controlled by the electrochemical environment. To identify the elemental distribution of Co and Pt atoms, a new descriptor is presented in this work, the so-called Q(Hupd)/Q(Coz+) ratio. The coverage of OHad species can be controlled by the coverage of oxophilic Co surface atoms to investigate the bifunctional mechanism on the HOR kinetics in alkaline media. Thus, we are able to correlate the HOR exchange current densities with tuneable Co coverage on the particle surface to mimic e.g. the coverage of “Co(hydr)oxide clusters”. On the other hand, the strain effect to modify the Pt–Had binding energy is solely investigated on a Pt-rich shell induced by a Co-rich particle core. Despite the separation of these critical model parameters, cobalt as an oxophilic component strongly bonds OHad species at the bimetallic particle surface and modifies the HOR kinetics. In contrast, the induced lattice strain in the Pt-enriched shell of dealloyed core–shell nanoparticles leads to an increase of the Pt surface area-based specific exchange current density j0spec by a factor of ∼2 compared to pure Pt. Based on our results, we provide deeper insights into the kinetics and mechanism of the HOR on PtxCo1−x catalysts with different structural nanoparticle motifs in an alkaline environment.
Introduction
The electrocatalysis of hydrogen oxidation/evolution reactions (HOR/HER) is the main key for the hydrogen economy to meet the demands of renewable and clean energy conversion and energy storage in the near future. The hydrogen electrocatalysis is not only a fundamental research in academia. Actually, there are numerous industrial technologies in the field of water and chlor-alkali electrolysis,1,2 fuel cells, metal deposition,3 and corrosion.4 Both hydrogen reactions have very recently experienced a renaissance. Although the HOR/HER have been studied over decades, their mechanism and kinetics are still poorly understood to date. In particular, the alteration of their reaction rate from acidic to alkaline environments is dramatic. Very low loadings of platinum group metals (PGM) are needed to accelerate the HOR kinetics in proton exchange membrane fuel cells,5 whereas the same reaction at higher pH becomes at least two order of magnitudes slower.6 Improvements in HOR/HER activities have recently been achieved by adding other metals such as ruthenium (Ru) or nickel (Ni) to platinum.7–12 The origin of the activity improvement especially in an alkaline environment is intensively and controversially discussed in the electrocatalysis community.One of the important descriptors for the HOR/HER rates is the M–Had binding energy on the metal electrode surface.6,13–15 Some studies have also reported that the higher Pt–Had binding energy in alkaline media is caused by the shortening of the Pt–Had bond distance compared to that in acidic media.16–18 Sheng et al.14 have shown a nearly linear dependence of the HOR/HER activity on the pH value of buffered electrolyte solutions. Interestingly, they correlated the shift of the underpotential deposition of hydrogen (Hupd) features measured in cyclic voltammetry (CV) directly with the strengthening/lowering of the Pt–Had binding energy at different pH values. The Pt–Had binding energy established from the CV profiles increases with pH, resulting in a decrease of the HOR/HER activity. The contribution of OH− species in the reaction path via adsorption has mainly been excluded by the authors. Durst et al.6 have also shown that the ratio of the HOR/HER rates at pH ≈ 0 and pH ≈ 13 varies between 120 and 200, which confirms the order of magnitude of the exchange current density difference in acidic and alkaline environments. If the H binding energy is linearly proportional to the activation energy (as described in the Brønsted–Evans–Polany relationship19), they assumed that the Volmer step is the rate limiting step for the HOR/HER on pure Pt in alkaline media.
Besides the theory of M–Had binding energy, an additional critical descriptor for HOR/HER rates is the interfacial structure of water molecules near the electrode surface reported by Ledezma-Yanez et al.20 They proposed that the interfacial water structure correlates with the pH value and thus affects the energy of hydrogen adsorption. In other words, the transfer of charge (H+ in an acid or OH− in a base) through the interfacial double layer depends on the extent to which the solvation environment can accommodate this charge migration. Thus, if the interfacial water can be easily rearranged, the transfer of charge through the double layer is fast. In contrast, a rigid and stiff interfacial water structure impedes and slows down the migration of charge. The extent of water reorganization is strongly related to the position of the potential of zero charge (pzc) as described by the interfacial electric field between the electrode and electrolyte. The pzc shifts from the Hupd region at low pH to a more anodic potential region of Pt (hydr)oxide formation at high pH.21 As a consequence, the strength of the electric field and the energy associated with the arrangement of interfacial water molecules are strongly influenced by the pH. Due to the pzc shift towards more anodic potentials on a Pt surface, the adsorption rate of hydrogen in the Hupd region is very sluggish in an alkaline environment. Ledezma-Yanez et al.20 have also shown that the introduction of Ni clusters on a Pt(111) surface is able to modify the electric field and results in a shift of the pzc free charge of −25 mV which is closer to the Hupd region. This shift of the pzc free charge leads to improved HER kinetics in alkaline media. Both descriptors, metal–hydrogen binding energy and rearrangement of interfacial water, are strongly linked to the electronic properties of the catalyst surface for the HOR/HER.
Another important descriptor of the HOR/HER rates is the bifunctional mechanism, in which reactive OHad species might promote the oxidation of Had on Pt sites according to the Volmer step (M–Had + OH− → M + H2O + e−). Strmcnik et al.22 suggested that the adsorption of reactive OH− species in the Hupd region might dictate the HOR/HER activity on a PGM surface. They have correlated the HOR/HER performance with the oxophilic properties of the PGM-based catalysts and shown that the HOR activity increases in the order of Pt < Pt0.5Ru0.5 ≪ Pt0.1Ru0.9 = polycrystalline Ir in an alkaline environment.22 Further studies from the same group have reported a critical interplay between the energetics of Pt–Had and Pt–OHad to control the HOR/HER rates.23–25 For instance, Danilovic et al.23 have shown that the modification of the Pt surface with Ni(OH)2 clusters improves the HER activity on Pt. They have suggested that the Ni(OH)2 clusters play an important role in accelerating the dissociative adsorption of water and in supplying the production of hydrogen and hydroxyl intermediates during the HER in alkaline media.
Altogether, different models like M–Had binding energy, an interfacial structure of water molecules and/or bifunctional mechanism by forming “reactive” OH species are drawn on to describe the changes in HOR/HER kinetics from an acidic toward an alkaline environment. It is noticed that most factors to describe the HOR/HER kinetics are very difficult to separate from each other and often occur together. Moreover, the diffusion of OH− species, mechanistic formation pathway of OHad, OHad–H/Had–OH bond formation, water desorption step,26 interactions of alkali cations27 and water at the electrode–electrolyte interface might also play a role in HOR/HER kinetics, but less studied or still unexplored to date.
In this work, PtxCo1−x catalysts along the uniform alloy, Pt-rich shell and Co-segregated core–shell nanoparticle motifs have been investigated for the HOR in alkaline media. The surface structure and re-arrangement of PtxCo1−x catalysts are controlled by the electrochemical environment. The changes in HOR exchange current densities on the PtxCo1−x catalysts in their different nanoparticle motifs were investigated and related to the strain effect (reduced Pt–Pt distances) and/or bifunctional effect. Due to the high oxophilic character of Co, the surface and subsurface layers of the bimetallic nanoparticles are systematically modified to control the coverage of OHad species. A new descriptor will be presented to determine the atomic composition and distribution of Co and Pt at the bimetallic particle surface. Very remarkably, in a series of different particle morphologies and compositions, the compressive strain as well as the bifunctional mechanism can be distinguished from each other and allow studying the HOR/HER kinetics in a unique manner. Despite the separation of these critical parameters, cobalt as an oxophilic component strongly bonds OHad species at the bimetallic particle surface. On the other hand, the induced lattice strain in the Pt-enriched shell of dealloyed core–shell nanoparticles causes only a moderate lowering of the Pt–H binding energy and thus improved HOR activity in alkaline media.
Experimental part
Synthesis of PtxCo1−x alloy nanoparticles supported on high surface area carbon (HSAC)
High surface area carbon (HSAC) supported Pt3Co, PtCo, and PtCo3 alloy nanoparticles were prepared by using a liquid impregnation–freeze drying–reductive thermal annealing technique.28,29 50 mg of commercially available 28.4 wt% Pt/HSAC (Tanaka Kikinzoku International Inc, TKK) was added to 35 mg, 105 mg, and 316 mg of Co(NO3)2·6H2O (99.999% metal basis, Alfa Aesar) for the preparation of PtxCo1−x with atomic Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co ratios of 3:1, 1:1, and 1:3, respectively. First, the cobalt precursor salt was dissolved in around 3 mL of highly purified water (18 MΩ cm at room temperature, Elga Purelab Classic). After horn-sonication for 5 minutes, the viscous black slurry was immediately frozen in liquid nitrogen for 20 minutes and then freeze-dried in a vacuum for two days. The dried powder was then annealed in a three-zone tube furnace in a 5/95 vol% H2/Ar atmosphere (quality 5.0, Air Liquide) with a flow rate of 200 mL min−1. The temperature program consisted of two plateaus at 250 °C for two hours followed by at 800 °C for six hours with a heating rate of 10 °C min−1, respectively.
:Co ratios of 3:1, 1:1, and 1:3, respectively. First, the cobalt precursor salt was dissolved in around 3 mL of highly purified water (18 MΩ cm at room temperature, Elga Purelab Classic). After horn-sonication for 5 minutes, the viscous black slurry was immediately frozen in liquid nitrogen for 20 minutes and then freeze-dried in a vacuum for two days. The dried powder was then annealed in a three-zone tube furnace in a 5/95 vol% H2/Ar atmosphere (quality 5.0, Air Liquide) with a flow rate of 200 mL min−1. The temperature program consisted of two plateaus at 250 °C for two hours followed by at 800 °C for six hours with a heating rate of 10 °C min−1, respectively.
Electrochemical experiments using the thin-film rotating disc electrode (TF-RDE) technique
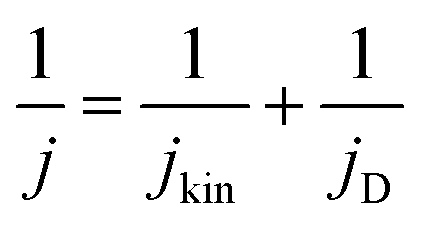 | (1) |
 | (2) |
485.3 A s mol−1], T – temperature [K], and R – gas constant [8.314 J mol−1 K−1].
Structural characterization
:Co ratio via EDX data.
Results and discussion
Structural characterization of pristine PtxCo1−x alloy nanoparticle electrocatalysts
The as-prepared PtxCo1−x nanoparticles supported on high surface area carbon (HSAC) were structurally characterized by using HR-TEM, XRD, and EDX. Fig. 1 displays the HR-TEM micrographs of all three PtxCo1−x catalysts, showing that the particles are well-dispersed on the carbon support material. Particle agglomeration was not observed from the TEM data. The particle size distribution and mean particle size of pristine PtxCo1−x catalysts (Fig. 1d–f) were established by analyzing more than 300 particles from several HR-TEM micrographs. In summary, the mean particle sizes are 3.8 ± 0.8 nm, 4.2 ± 0.9 nm, and 4.4 ± 1.0 nm for pristine Pt3Co, PtCo, and PtCo3, respectively. Based on the EDX analysis, the atomic Pt:Co ratios were found to be 72:28, 47:53 and 24:76 (standard deviations of 4–5 at%) and are in good agreement with the amount of Co used in the synthetic route for the preparation of bimetallic nanoparticles with desired ratios. This observation and our previous studies28–31 point out the high robustness and good reproducibility of the wet impregnation–freeze drying–annealing route to prepare uniform alloy nanoparticles.
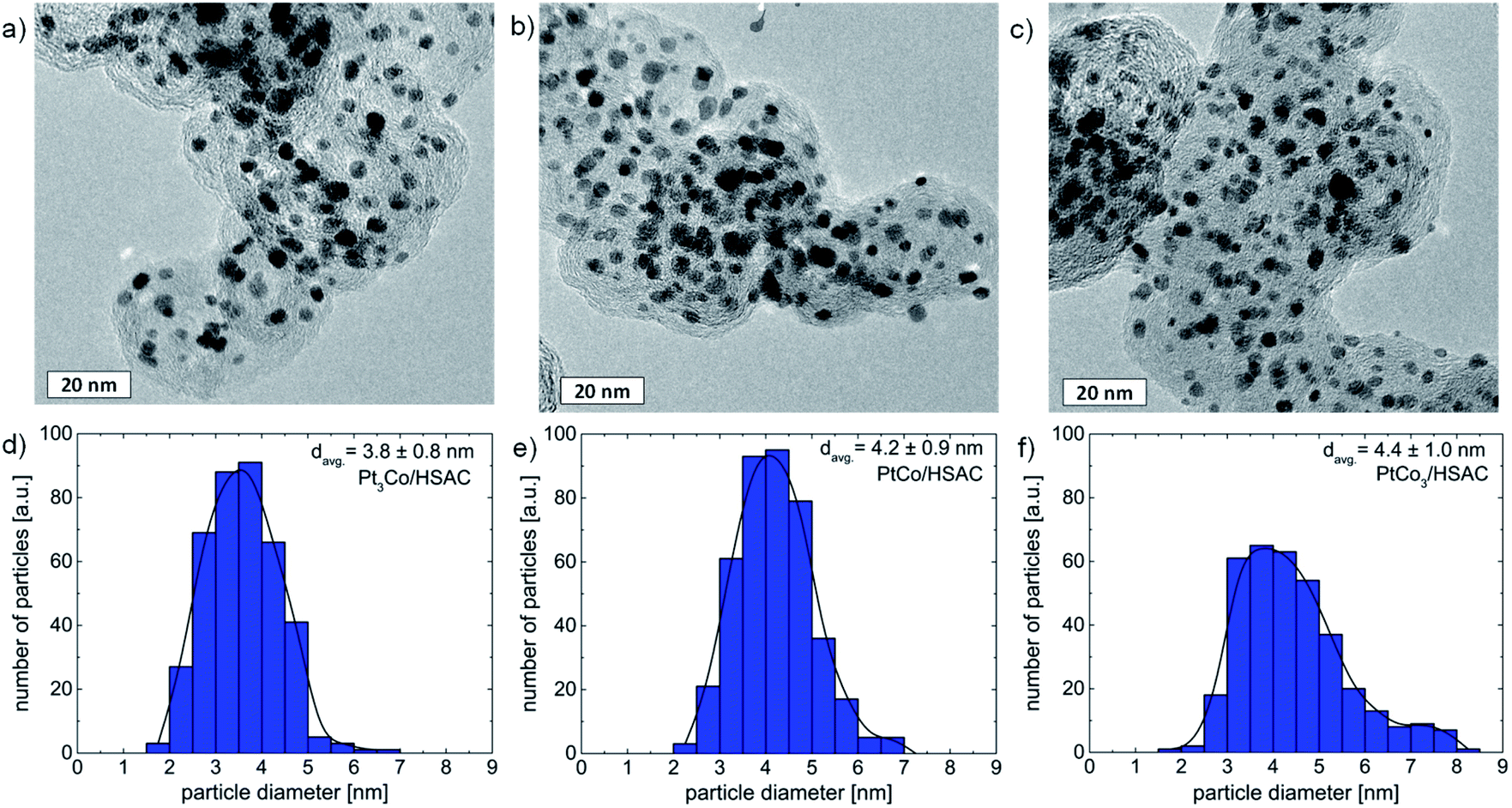 | ||
| Fig. 1 HR-TEM micrographs and particle size distributions for pristine Pt3Co (a and d), PtCo (b and e), and PtCo3 (c and f) prepared by the wet-impregnation–freeze drying–annealing route. The scale bar for all HR-TEM micrographs is 20 nm, respectively. | ||
The XRD patterns of PtxCo1−x catalysts are displayed in Fig. S1 of the ESI.† The reflexes at 2θ values of around 40°, 48°, 71°, and 86° correspond to the formation of a disordered face centered cubic (fcc) Pt–Co alloy phase with a space group of Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. Ordered alloy or oxide phases were not detected by XRD. The formation of ordered alloy phases can be suppressed by quenching during the annealing process from 800 °C to room temperature.31 The calculated crystallite sizes for PtxCo1−x are similar to the mean particle sizes obtained from the TEM data. Based on the Rietveld quantification, the pristine Pt3Co catalyst appears as a single disordered fcc alloy crystal phase with a stoichiometric composition of Pt84Co16 according to Vegard's law. However, the pristine PtCo and PtCo3 catalysts exhibit two crystal phases. The major crystal phase for the PtCo catalyst is a Pt-rich disordered fcc alloy phase, while the PtCo3 catalyst mainly consists of Pt53Co47 as the major phase. Table S1† summarizes the obtained crystallite sizes, lattice parameters, results from Rietveld quantification, and stoichiometric compositions for pristine PtxCo1−x alloy nanoparticles. It is noted that the EDX results reflect the average composition of particle ensembles, while the XRD analysis involves the structure and composition of each crystal phase. This leads to different results in the chemical composition of alloy nanoparticles.
m. Ordered alloy or oxide phases were not detected by XRD. The formation of ordered alloy phases can be suppressed by quenching during the annealing process from 800 °C to room temperature.31 The calculated crystallite sizes for PtxCo1−x are similar to the mean particle sizes obtained from the TEM data. Based on the Rietveld quantification, the pristine Pt3Co catalyst appears as a single disordered fcc alloy crystal phase with a stoichiometric composition of Pt84Co16 according to Vegard's law. However, the pristine PtCo and PtCo3 catalysts exhibit two crystal phases. The major crystal phase for the PtCo catalyst is a Pt-rich disordered fcc alloy phase, while the PtCo3 catalyst mainly consists of Pt53Co47 as the major phase. Table S1† summarizes the obtained crystallite sizes, lattice parameters, results from Rietveld quantification, and stoichiometric compositions for pristine PtxCo1−x alloy nanoparticles. It is noted that the EDX results reflect the average composition of particle ensembles, while the XRD analysis involves the structure and composition of each crystal phase. This leads to different results in the chemical composition of alloy nanoparticles.
Surface-rearrangement of PtxCo1−x electrocatalysts along the alloy, Pt-rich shell and Co-segregated core-shell nanoparticle motifs
The surface structure and chemical composition of PtxCo1−x alloy nanoparticles can be controlled by the (electro)chemical reaction environment. In principle, cobalt atoms are rapidly dissolved by exposure to a strong acid. On the other hand, cobalt can be (electro)chemically stabilized by forming cobalt (hydr)oxide species, such as Co(OH)2, Co3O4, CoOOH, and Co(OH)3, depending on the potential and pH applied. The potential stability together with the proton activity of the cobalt–water system is thermodynamically predicted in the Pourbaix diagram.32,33 Moreover, its enhanced oxophilic character tends to migrate internal Co atoms from the deeper subsurface layers towards the surface compared to platinum.34,35This unique electrochemical behaviour of cobalt inspired us to take advantage of the adsorbate-induced surface rearrangement of nano-sized PtxCo1−x alloys to tailor their catalytic HOR/HER performance in alkaline media. Fig. 2 illustrates our approach to tune the structure of bimetallic PtxCo1−x catalysts along the uniform alloy, Pt-rich shell and Co-segregated core–shell nanoparticle motifs controlled by the electrochemical conditions. Very importantly, the initial chemical surface composition and structure of pristine PtxCo1−x catalysts in the first surface layers can facilely be probed by recording in situ cyclic voltammetry (CV) profiles in alkaline media and ex situ X-ray photoelectron spectroscopy (XPS).
 | ||
| Fig. 2 Illustration of the surface rearrangement of PtxCo1−x electrocatalyst along the uniform alloy, Pt-rich shell and Co-segregated core–shell nanoparticle (NP) motifs controlled by the (electro)chemical environment. Pt and Co atoms are symbolized with grey and yellow spheres, respectively. | ||
Fig. 3a displays the CV profiles of pristine PtxCo1−x alloy catalysts in Ar-saturated 0.1 M KOH. The electrochemical features of pristine PtCo3 (blue line), PtCo (orange), and Pt3Co (green) show the appearance of both metal atoms at the particle surface. To better assign the features to the bimetallic surface, we will briefly describe the characteristics of a pure Pt surface (dashed lines, Fig. 3). The ad/desorption of hydrogen on a Pt surface referred to as underpotential hydrogen deposition (Hupd) occurs between 0.05 and 0.40 V vs. RHE, while at potentials above 0.65 V the Pt surface is electrochemically oxidized by forming (hydr)oxide species. The transition from the hydrogen coverage to (hydr)oxide takes place via the capacitive double-layer region (0.40–0.65 V vs. RHE). As Co atoms co-appear at the particle surface, additional electrochemical features become clearly visible. In Fig. 3a, broad current peaks exist between 0.6–0.7 V and 0.3–0.6 V vs. RHE in the anodic and cathodic scans, respectively. According to the Pourbaix diagram, Co(OH)2, CoO, Co3O4, and CoOOH are predicted in the investigated potential range at pH 13.32,33,36 It is noted that for bimetallic systems the reduction/dissolution potential of a metal A surrounded by a more noble metal B might be shifted to more negative potentials compared to its Nernst potential, referred to as underpotential metal deposition.37,38 Depending on the elemental surface distribution, the shift of the redox potential for Co (hydr)oxide species might be more or less pronounced. We also observed a broadening of these current peaks particularly in the cathodic direction, indicating an overlap of multiple electrochemical reduction processes of active redox species such as Co3O4/Co(OH)2, Co3O4/CoO and CoOOH/Co(OH)2. Although cobalt oxide materials have intensively been investigated as electrocatalysts for the oxygen evolution reaction (OER) from water under alkaline conditions, the accurate chemical states of Co (hydr)oxide species are dependent on several parameters like pH,39–42 potential,39,41–44 pre-treatment,45 kind of electrolyte,40,46etc.
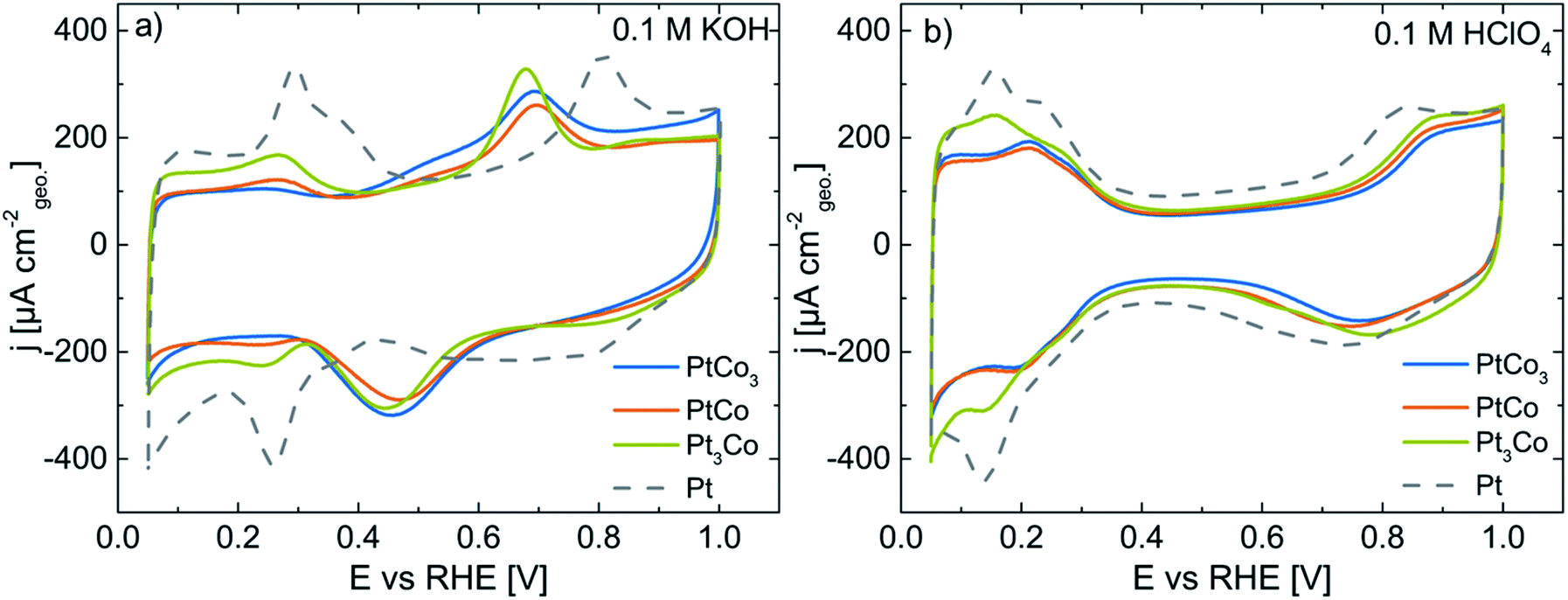 | ||
| Fig. 3 CV profiles of pristine PtCo3 (blue), PtCo (orange), Pt3Co (green), and Pt (grey dashed line) in Ar-saturated 0.1 M KOH (a) and after dealloying (0.05–1.00 V vs. RHE, 50 mV s−1, and 10 cycles) in Ar-saturated 0.1 M HClO4 (b). All CV profiles were recorded at 50 mV s−1 with a Pt loading of 8–10 μgPt cmgeo−2. The current density is normalized to the geometric surface area of the working electrode. | ||
Despite a similar particle size and metal loading (see Table 1), the decrease of the current density in the Hupd region with increasing Co content signifies a successive reduction of Pt surface atoms. To quantify this observation, we determined the electrochemically active Pt surface area (ECSA) via the Hupd method. In Table 2, the ECSA values are 2 ± 1, 4 ± 1, and 10 ± 2 m2 gPt−1 for pristine PtCo3, PtCo, and Pt3Co in 0.1 M KOH, respectively.
| Catalyst | Particle size (via TEM) [nm] | Crystallite size (via XRD) [nm] | Composition (via Vergard's law) [at%] | Composition (via EDX) [at%] | Pt loading (via TGA/EDX) [wt%] |
|---|---|---|---|---|---|
| PtCo3 | 4.4 ± 1.0 | 3.7 ± 0.1 | Pt53Co47 (61.6 wt%) | Pt24±5Co76±5 | 33.1 |
| 13.2 ± 0.5 | Pt21Co79 (38.4 wt%) | ||||
| PtCo | 4.2 ± 0.9 | 2.9 ± 0.1 | Pt73Co27 (69.8 wt%) | Pt47±4Co53±4 | 32.0 |
| 4.6 ± 0.1 | Pt52Co48 (30.2 wt%) | ||||
| Pt3Co | 3.8 ± 0.8 | 4.6 ± 0.1 | Pt84Co16 (100 wt%) | Pt72±5Co28±5 | 29.3 |
| Pt | 2.6 ± 0.9 | Pt100 | Pt100 | 28.4 |
| ECSAKOH, pristine [m2 gPt−1] | ECSAHClO4, core–shell [m2 gPt−1] | ECSAKOH, core–shell [m2 gPt−1] | ECSAKOH, Co-segr. [m2 gPt−1] | particle size via TEM [nm] | |
|---|---|---|---|---|---|
| a Co-segregated PtCo3 core–shell catalyst obtained after 1400 cycles between 0.05 and 1.0 V vs. RHE in 0.1 M KOH. | |||||
| PtCo3 | 2 ± 1 | 41 ± 4 | 27 ± 3 | 17 ± 2 | 4.4 ± 1.0 |
| 4 ± 1a | |||||
| PtCo | 4 ± 1 | 44 ± 3 | 30 ± 3 | 22 ± 3 | 4.2 ± 0.9 |
| Pt3Co | 10 ± 2 | 47 ± 5 | 37 ± 3 | 33 ± 3 | 3.8 ± 0.8 |
| Pt | 63 ± 4 | 77 ± 3 | 2.6 ± 0.9 | ||
Very remarkably, the CV profiles (Fig. 3a) show the suppression of Pt (hydr)oxide formation between 0.7 and 1.0 V vs. RHE with increasing Co content (compared with Pt/HSAC, grey dashed line). The disappearance of Pt (hydr)oxide in the CV profile is also supported by the concurrent decrease of Hupd and the preferred surface enrichment of Co (hydr)oxide species in 0.1 M KOH.
After pre-characterization, the PtxCo1−x catalysts were then transferred into Ar-saturated 0.1 M HClO4 and electrochemically treated between 0.05 and 1.00 V vs. RHE at 50 mV s−1 for 10 cycles by using the cyclic voltammetry (CV) method. This recipe causes a rapid electrochemical dissolution (dealloying) of Co atoms at the surface of alloy nanoparticles due to their negative Nernst potential. Thus, the obtained CV profiles of dealloyed PtxCo1−x catalysts resemble the electrochemical features of a clean Pt surface (grey dashed line), as shown in Fig. 3b. We wanted to emphasize that the decay of the Co content within a single particle is not only affected at the top layer, but also in the subsurface layers. Single crystal Pt–M (M = Ni, Co) studies in on ultra-high vacuum have revealed the structural and compositional fluctuations of the first three layers after Co leaching.47–49 In addition, our previous studies have reported the formation of core–shell nanoparticles with a Pt-rich shell of ∼1 nm (3–4 monolayers) and Co-rich particle core induced by dealloying.30,50 Throughout the paper, the electrochemically dealloyed PtxCo1−x nanoparticles will be referred to as core–shell nanoparticles. The enrichment of Pt surface atoms is also confirmed by the strong increase of the ECSA. Irrespective of the initial Co content, the ECSA values are very similar in 0.1 M HClO4 and are determined to be 41 ± 4, 44 ± 3 and 47 ± 5 m2 gPt−1 for dealloyed PtCo3, PtCo and Pt3Co core–shell catalysts, respectively. Although the absolute loss of the Co content for the pristine PtCo3 nanoparticles is significantly higher than those for the dealloyed PtCo and Pt3Co (see Table 3), which might result in an increased surface roughness, the ECSA of the dealloyed PtxCo1−x electrocatalysts is mainly influenced by the particle size. As large nanoparticles exhibit a lower surface-to-volume ratio compared to small nanoparticles, the highest ECSA value was obtained for pure Pt/HSAC with a particle size of 2.6 ± 0.9 nm.
:Co ratios for PtxCo1−x catalysts along the pristine alloy, dealloyed, and Co-segregated core–shell nanoparticle motifs obtained from ex situ XPS and EDX data. The standard derivations are in the range of 3–5 at%
| Pristine | Core–shell | Co-segregated | ||||
|---|---|---|---|---|---|---|
| Pt:Co [at%] EDX |
Pt:Co [at%] XPS |
Pt:Co [at%] EDX |
Pt:Co [at%] XPS |
Pt:Co [at%] EDX |
Pt:Co [at%] XPS |
|
| PtCo3 | 24:76 |
37:63 |
51:49 |
59:41 |
55:45 |
52:48 |
| PtCo | 47:53 |
52:48 |
60:40 |
77:23 |
65:35 |
80:20 |
| Pt3Co | 72:28 |
75:25 |
78:22 |
91:9 |
81:19 |
89:11 |
In the next step, the dealloyed PtxCo1−x core–shell nanoparticles were transferred back to a fresh alkaline electrolyte solution. Fig. 4 displays the CV profiles of the dealloyed PtCo3, PtCo, and PtCo3 core–shell catalysts before (dashed line) and after 100 cycles (solid line) in Ar-saturated 0.1 M KOH. First, the initial CV profiles of dealloyed PtxCo1−x core–shell nanoparticles (dashed line) resemble the electrochemical characteristics of a pure Pt surface, signifying an enrichment of Pt atoms within the first at least three atomic subsurface layers. After exposure to 0.1 M KOH for 100 cycles, the CV profiles of dealloyed PtCo (Fig. 4b, orange) and PtCo3 (Fig. 4c, blue) have dramatically altered. The re-appearance of broad current peaks between 0.3 and 0.7 V vs. RHE is assigned to the Co (hydr)oxide species within the first surface layers (compared with Fig. 3a). Due to their oxophilic character, the Co atoms tend to segregate to the surface induced by increasing OH surface coverage e.g. controlled by the potential and pH. This observation evidences that the dealloyed PtxCo1−x core–shell nanoparticles can be further altered by modifying their (electro)chemical environment. As the particle size is still unchanged (see Fig. S2 of the ESI†), the gradual surface enrichment of Co atoms is simultaneously accompanied by the decrease of Pt surface atoms. To highlight this atomic re-arrangement, we denoted these modified nanoparticles as Co-segregated core–shell nanoparticles. As the electrochemical Pt features are further visible in the CV profiles, we suggest that both Co and Pt atoms are present at the particle surface. Only the dealloyed Pt3Co core–shell nanoparticles (Fig. 4a) remained unchanged after applying a similar electrochemical protocol in KOH, which can be explained by the inherently lower Co content compared to the initial PtCo and PtCo3 catalysts, as shown in Tables 1 and 3.
 | ||
| Fig. 4 CV profiles of dealloyed Pt3Co (a), PtCo (b), and PtCo3 (c) prior to (dashed line) and after (solid line) 100 potential cycles at 50 mV s−1 in Ar-saturated 0.1 M KOH. The Co enrichment process mainly reaches a steady-state after potential cycling for up to 1400 scans between 0.05 and 1.0 V vs. RHE (dash-point line) in 0.1 M KOH. The current density is normalized to the geometric surface area of the working electrode. | ||
The ECSA determination enables quantifying this adsorbate-induced re-arrangement process of Co and Pt atoms from the initial core–shell towards Co-segregated core–shell nanoparticles. Differences in the initial ECSA values measured in HClO4 and KOH come from the altered enthalpies for hydrogen ad/desorption in the presence of OH− and H+.14,51–53 Therefore, we only compared the ECSA values for dealloyed PtxCo1−x catalysts prior to and after 100 potential cycles in 0.1 M KOH. In Table 2, the ECSA values decrease from 27 ± 3 to 17 ± 2 m2 gPt−1 (−37%), from 30 ± 3 to 22 ± 3 m2 gPt−1 (−26%), and from 37 ± 3 to 33 ± 2 m2 gPt−1 (−10%) for the dealloyed PtCo3, PtCo, and Pt3Co catalysts, respectively. We wanted to stress that this dynamic re-arrangement process of Co and Pt atoms does not lead to a thermodynamically stable state of these bimetallic nanoparticles. Fig. 4c shows the CV profiles of the dealloyed PtCo3 nanoparticles as a course of various potential cycles (1, 100, and 1400) in 0.1 M KOH. The Co (hydr)oxide redox pair get more and more pronounced by additional potential cycling. However, a potential treatment for more than 1400 cycles does not result in an additional growth of the current densities for Co (hydr)oxide species, indicating that a “quasi-stable” state for Co-segregated PtCo3 core–shell nanoparticles has been reached. This “quasi-stable” CV profile shows still the presence of both Pt and Co atoms in the first surface layers. Very interestingly, a negative potential shift of the hydrogen adsorption (−15 mV) and a strong suppression of Pt (hydr)oxide formation are visible in the CV profile (Fig. 4c). This shift in the Hupd region is very likely caused by the increased coverage of strongly adsorbed OH species (OHad) on a Co-enriched bimetallic surface and/or changes in the potential of zero charge (pzc) due to its surface re-arrangement.
Advanced X-ray photoelectron spectroscopy (XPS) and energy-dispersive X-ray spectroscopy (EDX) analysis of various surface arrangements of PtxCo1−x nanoparticle motifs
Changes in the chemical composition and chemical state of PtxCo1−x catalysts along the pristine alloy, dealloyed, and Co-segregated core–shell nanoparticles were probed by ex situ XPS and EDX techniques. In Table 3, the EDX data show the highest loss of the Co content for PtxCo1−x catalysts after electrochemical dealloying in HClO4. The atomic Pt:Co ratios alter from 24:76 to 51:49 for PtCo3, from 47:53 to 60:40 for PtCo, and from 72:28 to 78:22 for Pt3Co (standard derivation of 4–5 at%), respectively. The subsequent potential cycling of dealloyed PtxCo1−x catalysts in KOH leads to almost unchanged composition, implying their high electrochemical stability in the applied potential range at pH 13. Hence, the surface re-arrangement of bimetallic nanoparticles from dealloyed towards the Co-segregated core–shell structure is not accompanied by electrochemical Co dissolution.
In this work, high-resolution XPS investigations were carried out to establish the chemical state of Co and Pt atoms and their atomic Pt:Co ratio for PtxCo1−x catalysts along the structural re-arrangements in detail. Fig. S3† displays the high-resolution XPS spectra of Pt 4f for PtxCo1−x catalysts along the pristine alloy (a), dealloyed (c), and Co-segregated (e) core–shell nanoparticle motifs. The Pt 4f XPS spectra were deconvoluted and fitted with double peaks for 4f7/2 and 4f5/2, respectively. From the Pt 4f7/2 peak, three species including metallic Pt0, Pt2+ and Pt4+ were identified at binding energies of 71.6, 72.6, and 73.5 eV (reference values54 of 71.2 ± 0.5 eV, 72.4 ± 0.8, and 74.9 ± 0.8 eV for metallic Pt0, Pt2+ and Pt4+, respectively). In the series of XPS spectra of Pt 4f, no changes in the chemical state of Pt species were observed in this work.
Generally, the XPS analysis of Co 2p is not trivial due to its multiplet spitting, plasmon loss features associated with Co metal55,56 and shake-up loss features associated with Co oxides.57 As shown in Fig. 5, the peak broadening is mainly based on the high pass energy of 100 eV (Table S2†) applied due to the very low residual Co content within the thin catalyst films after electrochemical experiments. The Co 2p3/2 signal was deconvoluted into three peaks at binding energies of ca. 778 eV, 781 eV, and 785 eV, which are assigned to metallic Co and oxidized Co species according to the literature.37,58 It is noted that an unambiguous allocation of various Co (hydr)oxide components, i.e. CoO, Co2O3, Co3O4, CoOOH, Co(OH)2, and Co(OH)3, is very difficult due to their similar binding energies (BE) and shake-up loss features.55,58–61 Thus, the oxidized Co species are only assigned to Coz+ without any accurate chemical state mentioned. The quantification was performed by analyzing the Co 2p3/2 and Pt 4f7/2 signals. As it is very difficult to deconvolve plasmon loss features and shake-up loss features from the signal at ∼785 eV, only the Co 2p XPS signals at around 778 eV and 781 eV were considered to establish the atomic Co0:Coz+ ratios in this work.
 | ||
| Fig. 5 (a, c and e) High-resolution XPS spectra of Co 2p for Pt3Co, PtCo, and PtCo3 along the pristine alloy (a), dealloyed (c) and Co-segregated (e) core–shell nanoparticle motifs. Experimental parameters like the step size, pass energy, dwell time and number of scans are listed in Table S2 of the ESI.† The experimental data are denoted as the grey line and the fit is symbolized by the black line. Red, blue and orange dashed lines denote the different Co species. (b, d and f) Bar diagrams display the atomic Co0:Coz+ ratios of the pristine and treated PtxCo1−x catalysts appearing in their various structural nanoparticle motifs. | ||
After synthesis, the XPS data show atomic Pt:Co ratios of 75:25, 52:48, and 37:63 for pristine Pt3Co, PtCo, and PtCo3, respectively, which is in good agreement with the elemental results obtained from EDX (see Table 3). As expected, the cobalt appears in a metallic and oxidized chemical state within the pristine alloy nanoparticles (Fig. 5). The content of oxidized Co in the as-prepared PtCo and PtCo3 catalysts is more than 50 at%, while the as-prepared Pt3Co catalyst shows a higher metallic cobalt content (Fig. 5a and b). The appearance of oxidized Co species very likely stems from non-reduced Co precursor salt or/and surface oxide immediately formed upon exposure of metallic cobalt to air. However, no residual CoNO3 precursor was detected from XPS. The XRD profiles (Fig. S1 and Table S1†) show no additional reflexes of Co oxide crystal phases. Based on the XRD and XPS data, we suggest that the observed Coz+ species largely exist as amorphous (hydr)oxides. In order to rule out the post-oxidation of Co within the electrochemically treated PtxCo1−x catalysts during the sample transfer, a control experiment was performed in an Ar-filled glovebox directly connected to a XPS chamber. Very remarkably, our control experiment in an oxygen-free glovebox reveals that the dealloyed PtCo core–shell catalyst shows a similar amount of Co oxide species (41 at%) compared to that from the RDE experiments (Coz+ 45 at%) (not shown, will be published in our next work). Therefore, we suggest that the Pt-enriched surface of dealloyed bimetallic nanoparticles protects the internal Co atoms for post-oxidation of metallic Co in air.
The electrochemical dealloying significantly alters the atomic Co0:Coz+ ratios of the PtCo and Pt3Co catalysts. Besides the loss of the total Co content, amorphous Co oxide species at the particle surface rapidly corrode under the harsh conditions (up to 1.0 V vs. RHE, pH 1), resulting in an increase of metallic Co species within the dealloyed nanoparticles. However, the dealloyed PtCo3 core–shell catalyst shows an opposite trend, namely an increase of the oxidized Co content. We suggest that the decay of the metallic cobalt content for pristine PtCo3 is very likely attributed to the Co-rich crystal phase detected by XRD (see Fig. S1 and Table S1†). The exchange to alkaline electrochemical conditions does not lead to a significant alteration of Co0:Coz+ ratios for dealloyed PtxCo1−x catalysts. Besides their almost stable chemical composition as discussed above and shown in Table 3, the atomic Co0:Coz+ ratios for dealloyed PtxCo1−x core–shell nanoparticles seem to be not influenced by the Co segregation process. This finding is very surprising because the content of oxidized cobalt species would significantly be increased if internal Co atoms migrate to the top layer and immediately form Co(OH)2 and Co3O4 species upon exposure to alkaline electrolyte solution. Actually, we observed a reverse effect that the additional potential cycling up to 1400 to achieve a steady-state CV profile (see Fig. 4c) leads to a transition of oxidized to metallic Co species, Fig. 5e and f. We suggest that this transformation is very likely related to the potential-dependent mobility of surface atoms and its dynamic changes in the coverage of adsorbates. As the surface-sensitive CV profile clearly shows the electrochemical features of redox-active Co (hydr)oxide species in Fig. 4c, we assume that the Co atoms tend to mainly enrich in the subsurface layers of the nanoparticles. But, the most interesting observation is the electrochemical resistance of amorphous Co oxide species within the bimetallic nanoparticles. We suggest that the amorphous Co oxide is a relic from the initial composition of the nanoparticles, which is formed during the fast surface passivation of the remaining noble metal, Pt, under these harsh conditions.
Although metallic Co tends to rapidly dissolve under corrosive conditions, the amorphous Co oxide species appear very stable up to 1.0 V vs. RHE at pH 1 (0.1 M HClO4). The high stability of crystalline Co oxide films or Co atoms incorporated in MnOx or PbOx crystals as a host substrate at potentials applied for the OER and low pH values (<2.5) has been very recently reported in the literature.62–64 An alternative explanation might come from the protecting role of a Pt-enriched particle surface to prevent the electrochemical dissolution of internal Co atoms. Very recently, nanoporous Pt–CoOx films prepared by a sequential magnetron-sputter process have shown a considerable Pt-based surface area and ORR performance.65 The origin of the superior ORR activity is based on the lattice strain in the Pt–Pt distance induced by the CoOx backbone.
In summary, we point out that the nature of the residual Co appears in two chemical states (metallic and oxidized) protected by the Pt-rich shell of dealloyed and Co-segregated PtxCo1−x catalysts. The question arises about the role of Co and Pt during the HOR mechanism and how the HOR performance can be tailored by the presence of metallic and oxidized Co.
HOR performance on various surface arrangements of PtxCo1−x nanoparticle motifs
The polarization measurements (a, c, and e) for Pt3Co (green), PtCo (orange), and PtCo3 (blue) catalysts as pristine alloy, dealloyed, and Co-segregated core–shell nanoparticles as well as the corresponding fits (dashed lines; b, d, and f) using the Butler–Volmer equation are shown in Fig. 6. Basically, three different regimes for the HOR polarization curves can be identified: kinetically controlled regime (0.0–0.015 V vs. RHE) followed by a mixed regime with contributions of kinetics and mass transport restrictions (0.015–0.15 V), and finally the mass transport-limiting regime (0.15–0.35 V). | ||
| Fig. 6 HOR/HER polarization curves (left) and Butler–Volmer plots (jkinvs. iR-corrected E, right) of PtxCo1−x electrocatalysts in the pristine alloy (a and b), dealloyed Pt-rich shell (c and d) and Co-segregated (e and f) core–shell nanoparticle motifs. The polarization curves were recorded from −0.05 to 0.4 V vs. RHE at 1600 rpm and 10 mV s−1 in H2-saturated 0.1 M KOH. The current density is normalized to the geometric surface area of the electrode, while the ikin extracted from the Butler–Volmer equation is normalized by the electrochemically active Pt surface area (ECSA). PtCo3 is coloured in blue, PtCo in orange, Pt3Co in green and pure Pt in black. | ||
First, we will discuss the HOR performance of pristine PtxCo1−x alloy catalysts. In Fig. 6a, pristine Pt3Co and pure Pt show a negative shift of the polarization curves and higher mass transport-limiting current densities compared to pristine PtCo and PtCo3, signifying improved HOR activities due to their Pt-enriched particle surface (large ECSA values, see Table 2 and Fig. 2). Fig. 6b displays a plot of Pt surface area-based kinetic current densities (jkin,spec) vs. iR-corrected potential to point out the increase of HOR performance in the order of: Pt < Pt3Co < PtCo < PtCo3, respectively. This opposite trend is very likely attributed to the different ECSA values of pristine PtxCo1−x catalysts which will be discussed in detail later.
In Fig. 6c, the HOR polarization curves for dealloyed PtxCo1−x core–shell nanoparticles are slightly shifted to negative potentials compared to pure Pt/C. The respective Butler–Volmer plots of jkin,specvs. iR-corrected potential (E) imply a slight activity improvement in the order of: Pt < Pt3Co < PtCo ≈ PtCo3 prepared by electrochemical dealloying in 0.1 M HClO4.
Fig. 6e displays the HOR/HER polarization curves of PtxCo1−x catalysts prepared by a sequence of dealloying in HClO4 and additional 100 potential cycles in 0.1 M KOH. The polarization curves are mainly on top of each other and are similar to those for dealloyed PtxCo1−x catalysts. We observed slight differences between these catalysts only in the mixed regime of the HOR polarization curves. More importantly, a direct comparison between Co-segregated PtCo3 core–shell nanoparticles before and after 1400 potential cycles in KOH exhibits a dramatic activity loss. The plots of jkin,specvs. iR-corrected E, yet, imply an opposite trend due to the critical normalization of jkin with a low ECSA value as discussed now.
To benchmark these PtxCo1−x catalysts along the pristine alloy, dealloyed, and Co-segregated core–shell nanoparticle motifs for the HOR activity in 0.1 M KOH, we also determined the intrinsic exchange current densities (j0) and transfer coefficients (α) extracted from the fits using the Butler–Volmer equation. The j0 values were then normalized with the initial Pt mass (denoted as j0mass), electrochemically active Pt surface area via the Hupd method (j0spec), and geometric surface area of the working electrode (j0geo) and are summarized in Tables 4–6.
| Pristine PtxCo1−x | j 0spec [mA cmPt−2] | j 0mass [A mgPt−1] | j 0geo [mA cmgeo−2] | α [a.u.] |
|---|---|---|---|---|
| PtCo3 | 4.12 ± 0.42 | 0.07 ± 0.01 | 0.66 ± 0.07 | 0.38 ± 0.05 |
| PtCo | 2.66 ± 0.17 | 0.12 ± 0.02 | 1.00 ± 0.07 | 0.39 ± 0.05 |
| Pt3Co | 2.02 ± 0.10 | 0.22 ± 0.04 | 2.10 ± 0.12 | 0.35 ± 0.03 |
| Pt | 0.55 ± 0.03 | 0.28 ± 0.02 | 2.40 ± 0.38 | 0.50 ± 0.02 |
The j0spec values were determined to be 4.12 ± 0.42, 2.66 ± 0.17, and 2.02 ± 0.10 mA cmPt−2 for pristine PtCo3, PtCo, and Pt3Co, respectively. In contrast, the j0mass increases in the opposite order of: PtCo3 (0.07 ± 0.01 A mgPt−1) < PtCo (0.12 ± 0.02 A mgPt−1) < Pt3Co (0.22 ± 0.04 A mgPt−1) < pure Pt (0.28 ± 0.02 A mgPt−1), respectively. The j0geo shows a similar trend to j0mass. Although the particle size and loading are nearly the same, this contrary trend between j0spec and j0mass (j0geo) might be caused by the limitation of the accurate ECSA determination particularly if the ECSA value is getting very small (see Table 2 for pristine PtCo3 and PtCo). Alternatively, CO stripping experiments were also applied to establish the ECSA of Pt. However, only the Hupd method has delivered reliable ECSA values for bimetallic Pt–Co surfaces under alkaline conditions, which will be discussed later. On the other hand, j0mass and j0geo dramatically decrease with increasing Co content for the pristine alloys, while the catalytic HOR performance of pristine Pt3Co is nearly comparable with that of pure Pt within the error range. Very importantly, the transfer coefficients (α) for all pristine PtxCo1−x catalysts are below 0.5, signifying an asymmetric behaviour between the HOR and HER branches and thus modification in reaction pathways on a Co-rich bimetallic surface. As the intrinsic catalytic reactivity of pure Co is very low, we can conclude that the preferred formation of OHad on a Co-rich Pt–Co surface dramatically alters the activity and reaction pathways for the HOR in alkaline media.
Very interestingly, the dealloyed PtxCo1−x catalysts with a Pt-rich particle shell exhibit similar HOR activities with respect to j0mass and j0geo and are comparable to pure Pt/HSAC, as listed in Table 5. Only the j0spec values increase in the order of: Pt(0.55 ± 0.03 mA cmPt−2) < Pt3Co(0.67 ± 0.06 mA cmPt−2) < PtCo(0.94 ± 0.09 mA cmPt−2) < PtCo3(1.04 ± 0.08 mA cmPt−2), respectively. Thus, the j0spec values for the dealloyed PtCo and PtCo3 core–shell catalysts improve by a factor of 1.5–1.9 compared to pure Pt/HSAC and dealloyed Pt3Co. Very recently, we have shown no influence of the particle size of polycrystalline and nano-sized Pt on the HOR activity in alkaline media.13,27 Thus, the improved j0spec for dealloyed PtCo and PtCo3 core–shell nanoparticles is very likely caused by the geometric effect in the Pt-enriched shell surrounded by the Pt–Co-CoOx particle core, lowering the Pt–H binding energy. Other research groups have recently reported an improved HOR activity on a strained Pt surface (reduced Pt–Pt distance) for Pt–Ni and Pt–Ru systems due to the modified hydrogen binding energy.7,9,10 Changes in Pt–H binding energy is often related to the potential shift of the hydrogen desorption in the Hupd region. As shown in Fig. S4,† a potential shift of the H desorption is not observed for all dealloyed PtxCo1−x core–shell nanoparticles irrespective of the initial Co content and possible characteristics in the formed Pt-enriched particle shell e.g. differences in the shell thickness and composition of the particle core. Only the reduction of the Pt (hydr)oxide current peak is significantly shifted to more negative potentials compared to that for pure Pt and dealloyed Pt3Co, implying a decrease of OHad coverage and thus an improved ORR performance. In fact, the compressive strain in the Pt shell only results in the improvement of j0spec, but the magnitude of HOR activity enhancement is much less pronounced compared to that for the ORR. The transfer coefficients (α) for all dealloyed PtxCo1−x core–shell catalysts were determined to be around 0.5, which is in very good agreement with that for pure Pt. The resulting symmetry for the HER and HOR branches implies similar reaction pathways on dealloyed core–shell catalysts with a Pt-rich shell.
| Dealloyed PtxCo1−x core–shell | j 0spec [mA cmPt−2] | j 0mass [A mgPt−1] | j 0geo [mA cmgeo−2] | α [a.u.] |
|---|---|---|---|---|
| PtCo3 | 1.04 ± 0.08 | 0.24 ± 0.02 | 2.28 ± 0.15 | 0.53 ± 0.02 |
| PtCo | 0.94 ± 0.09 | 0.25 ± 0.01 | 2.38 ± 0.25 | 0.50 ± 0.03 |
| Pt3Co | 0.67 ± 0.06 | 0.26 ± 0.03 | 2.18 ± 0.28 | 0.52 ± 0.02 |
| Pt | 0.55 ± 0.03 | 0.28 ± 0.02 | 2.40 ± 0.38 | 0.50 ± 0.02 |
After 100 potential cycles in 0.1 M KOH, the influence of the re-appearance of Co atoms in the subsurface layers on the catalytic HOR performance is displayed in Fig. 6 and Table 6. Compared to the dealloyed catalysts with a Pt-rich particle shell (Fig. 6c, d and Table 5), the values of j0mass and j0geo for the Co-segregated PCo and PtCo3 core–shell nanoparticles are almost unchanged and are thus similar to those of pure Pt. However, the j0spec increases up to 2.6 particularly for the Co-segregated PtCo3 core–shell nanoparticles compared to pure Pt. To correlate the increase of j0spec with the Co segregation, it is necessary to compare dealloyed PtCo3 with dealloyed Pt3Co. First, the re-appearance of Co (hydr)oxide species is not observed for dealloyed Pt3Co after 100 potential cycles in 0.1 M KOH due to the inherently low Co content within the dealloyed nanoparticles. In this case, the improved j0spec for dealloyed Pt3Co core–shell nanoparticles is very likely related to the drop of the ECSA (from 37 ± 3 to 33 ± 3 m2 gPt−1, −11%, Table 2) due to the carbon corrosion associated with particle detachment as the main degradation process in strong alkaline media.66,67 In the other case, the j0mass and j0geo for the Co-segregated PtCo3 core–shell nanoparticles decrease with gradual enrichment of Co surface atoms induced by additional potential cycles. Thus, the increase of j0spec might be caused by the discrepancy to determine the accurate ECSA of Pt in the presence of stable Co atoms in the sub-surface layers. To the best of the authors' knowledge, the Hupd method seems to be the reliable and reproducible approach for ECSA determination of Pt surrounded by oxidized Co species under alkaline conditions. Similar trends for the HER performance are observed in this work (see Fig. S5†). Apart from the drastic activity drop, the transfer coefficient decreases from 0.47 ± 0.01 to 0.38 ± 0.02 with successive Co enrichment at the particle surface and reached values similar to those of pristine PtxCo1−x alloy catalysts.
| Co-segregated PtxCo1−x core–shell | j 0spec [mA cmPt−2] | j 0mass [A mgPt−1] | j 0geo [mA cmgeo−2] | α [a.u.] |
|---|---|---|---|---|
| a Co-segregated PtCo3 core–shell catalyst obtained after 1400 cycles between 0.05 and 1.0 V vs. RHE in 0.1 M KOH. | ||||
| PtCo3 | 1.43 ± 0.05 | 0.24 ± 0.02 | 2.10 ± 0.15 | 0.47 ± 0.01 |
| PtCo3_1400a | 2.61 ± 0.2 | 0.09 ± 0.01 | 0.80 ± 0.14 | 0.38 ± 0.02 |
| PtCo | 1.09 ± 0.09 | 0.22 ± 0.03 | 2.10 ± 0.33 | 0.46 ± 0.01 |
| Pt3Co | 0.80 ± 0.07 | 0.29 ± 0.03 | 2.40 ± 0.37 | 0.47 ± 0.01 |
| Pt | 0.55 ± 0.03 | 0.28 ± 0.02 | 2.40 ± 0.40 | 0.50 ± 0.02 |
Correlation between the HOR activities and Q(Hupd)/Q(Coz+) ratio
To evaluate the elemental distribution within the first surface layers and thus to point out the role of cobalt as an oxophilic component for the catalytic HOR performance, we developed a new descriptor by relating the electric charges yielded from the hydrogen ad/desorption and Co(hydr)oxide reduction/formation, the so-called Q(Hupd)/Q(Coz+) ratio (see Fig. S6†). According to the Pourbaix diagram, Co(OH)2, CoO, Co3O4, and CoOOH are predicted in the investigated potential range at pH 13.32,33 The respective redox potentials and the extended Pourbaix diagram of the cobalt–water system are given in Fig. S7 of the ESI.† It is noted that the redox pair of Co3O4/Co(OH)2 and CoOOH/Co(OH)2 are controversially discussed in the literature, which might be based on the pre-treatment,45 electrochemical conditions36,39–42,68 and nature44,69 of Co oxide-based materials as well as the discrimination of different meta-stable Co(hydr)oxide species40,41,44,46 on the electrode surface. According to the Pourbaix diagram32,33 and other research studies,41,43,46,63,68,70 we consider the redox pair of Co3O4/Co(OH)2 with the following electrochemical reaction: 3Co(OH)2 + 2OH− → Co3O4 + 4H2O + 2e−. If the value of the Q(Hupd)/Q(Coz+) ratio is larger than or below one, the particle surface consists of an excess of Pt or Co to accelerate or block the kinetics of the HOR by forming OHad species. To illustrate the modification of HOR performance by Pt–H binding energy and coverage of OHad, Fig. 7 shows a sketch of the 2D atomic arrangement of Pt and Co within the nanoparticles. Due to the chemical properties of Co, this bimetallic catalyst system with different atomic arrangements enables discriminating between the Pt–Had binding energy model and bifunctional model. The coverage of OHad species can be modified on these different particle motifs (disordered alloy vs. Co-segregated core–shell) and different Co surface contents. In other words, the concentration of Co surface atoms strongly correlates with the coverage of OHad species which play a critical role in the bifunctional mechanism for the HOR in alkaline media. | ||
| Fig. 7 Illustration of the different surface arrangements of platinum and cobalt in a metallic or oxidized state to tailor the HOR performance in an alkaline environment. Top panel: Metal–H bond mechanism to lower/strengthen the bond of adsorbed hydrogen on a strained Pt top layer surrounded by metallic Co (left) or CoO (right) underlay. Bottom panel: Bifunctional mechanism, where the coverage of OHad species is attributed to the enrichment of Co and Co oxide species near the surface. The here developed new descriptor, Q(Hupd)/Q(Coz+) ratio, expresses the elemental distribution of Co and Pt in the first surface layers. Grey, yellow and white balls denote Pt, Co and O, respectively. | ||
It is noted that the enhanced oxophilic character of the Co-segregated PtCo3 core–shell catalyst compared to a Pt-enriched shell is also evident from CO oxidation. Fig. S8† displays the CO stripping experiments of Pt-enriched and Co-segregated core–shell catalysts, signifying a broad oxidation peak at ∼0.43 V vs. RHE. The second CV profile signifies an irreversible process due to its disappearance. Based on the CO stripping experiments, we also tried to establish the ECSA of Pt. By integrating the charge of the oxidation of the adsorbed CO monolayer from 0.38 and 0.79 V vs. RHE, the ECSA value became extremely large for Co-segregated core–shell nanoparticles. This observation cannot be explained by the changes in the particle size (same volume-to-surface ratio) and leads to an overestimation of the ECSA of Pt.
In Table 7, the values of Q(Hupd)/Q(Coz+) ratios for as-prepared PtxCo1−x increases in the order of: PtCo3 (0.14 ± 0.03) < PtCo (0.25 ± 0.02) < Pt3Co (1.21 ± 0.07), respectively. This trend shows a successive enrichment of Pt surface atoms with decreasing Co content. On the other hand, the values of Q(Hupd)/Q(Coz+) ratios were determined to be 6.4 ± 0.8 and 20.0 ± 2.0 for the Co-segregated PtCo3 and PtCo core–shell catalysts. In fact, the pristine PtCo and PtCo3 catalysts indicate a higher contribution of Co surface atoms compared to the Co-segregated PtCo and PtCo3 core–shell nanoparticles. We wanted to emphasize that the observed changes in the value of Q(Hupd)/Q(Coz+) ratios are very likely related to the modification of the chemical surface composition and re-arrangement of the bimetallic nanoparticles instead of changes in the particle size (compare Fig. 1 with Fig. S2†). Almost no changes in electrochemical features for dealloyed Pt3Co were observed after 100 potential cycles in KOH (see Fig. 4a), which might be explained by the inherently low Co content after dealloying in HClO4.
Fig. 8 shows the relationship between the obtained Pt surface area-based specific (j0spec, panel a), Pt mass-based (j0mass, panel b) and geometric-normalized (j0geo, panel c) HOR exchange current densities as a function of the Q(Hupd)/Q(Coz+) ratio for pristine PtxCo1−x and Co-segregated PtCo and PtCo3 core–shell catalysts. The dashed lines in Fig. 8 denote the respective activities of pure Pt/HSAC and dealloyed PtCo3 core–shell catalysts with a Pt-rich particle shell. As mentioned above and shown in Fig. 7, the lattice strain in the Pt-enriched shell has less influence in weakening the Pt–H bond. The induced strain in the Pt-rich shell is originated from the sublayers consisting of Co and Co oxide species, resulting in a moderate weakening of the Pt–H bond. On the other hand, the Pt-rich top layer protects the further dissolution of internal Co and Co oxide species in the sub-layers. A detailed study for obtaining information about the arrangement and distribution of metallic and oxidized cobalt atoms is currently in progress. Due to the moderate modification of the Pt–H bond induced by the compressive strain in the Pt-rich shell, we focus now on the bifunctional mechanism, in which reactive OHad might promote the oxidation of Had on Pt sites according to the Volmer step (M–Had + OH− → M + H2O + e−).
 | ||
| Fig. 8 Relationship between the geometric-normalized (j0geo, panel a), Pt mass-based (j0mass, panel b) and Pt surface area-based specific (j0spec, panel c) HOR exchange current densities as a function of the Q(Hupd)/Q(Coz+) ratio for pristine PtxCo1−x and Co-segregated PtCo and PtCo3 core–shell catalysts. The pristine alloy catalysts are denoted with a hollow circle (○), while the Co-segregated core–shell catalysts are symbolized with a hollow triangle (Δ). The Co-segregated PtCo3 core–shell catalyst after 1400 potential cycles is denoted by a hollow inverted triangle. Even though no segregation of Co was observed, we decided to include the dealloyed Pt3Co core–shell catalyst as a reference in these plots. Pure Pt/HSAC (black dashed lines) and dealloyed PtCo3 core–shell catalysts (orange solid line) with a Pt-rich shell were also taken as reference materials. The grey dashed lines denote the standard deviation of the HOR activity of pure Pt/HSAC. | ||
First, we will discuss the correlation between the HOR performance and Q(Hupd)/Q(Coz+) ratio for pristine PtxCo1−x alloy catalysts. In Fig. 8a, the j0spec increases with increasing initial Co content, when j0 is normalized by the number of catalytically active Pt sites. This suggests that the nearby oxophilic Co atoms accelerate the kinetics of the HOR on the Pt surface based on the bifunctional mechanism. However, in Fig. 8b and c a reverse effect is observed for j0mass and j0geo, namely that the increased appearance of Co surface atoms reduces dramatically the kinetics of the HOR by blocking the surface with strongly OHad species. Only the pristine Pt3Co catalyst with a value of the Q(Hupd)/Q(Coz+) ratio which is larger than one shows similar j0mass and j0geo to pure Pt/HSAC and dealloyed PtCo3 core–shell catalysts.
The Co-segregated PtCo core–shell catalyst shows an order of magnitude higher value of the Q(Hupd)/Q(Coz+) ratio compared to the Co-segregated PtCo3 catalyst, but both values are significantly larger than one, indicating a large number of available Pt surface atoms. We suggest that this difference also reflects the coverage of OHad species at the particle surface and is therefore much lower than those for pristine alloys. In a broader sense, the Co-segregated core–shell nanoparticles simulate a Pt-enriched surface with “isolated less noble metal (hydr)oxide clusters”. With this picture in mind and illustrated in Fig. 7, our results reveal that a variation of OHad species for the Pt–Co system does not cause an enhancement of HOR activity in an alkaline environment. Unlike, the comparison of the dealloyed PtCo3 core–shell catalyst after 100 and 1400 potential cycles shows a dramatic decrease of j0mass and j0geo due to the additional enrichment of Co atoms within the first surface layers.
Based on our new descriptor, a broad range of Co-containing bimetallic surfaces were investigated in this work. Actually, it was expected that different Co-containing particle surfaces allow tailoring the coverage of OHad species to facilitate the oxidation of Had at low potentials (bottom panel of Fig. 8). This approach very likely mimics a single Pt(111) crystal surface decorated with Ni (hydr)oxide clusters to generate reactive OHad species for the HOR reported by Markovic and coworkers.23 This so-called bifunctional mechanism to enhance the HOR reactivity is not observed for pristine PtxCo1−x alloy and Co-segregated PtCo and PtCo3 core–shell catalysts. In this work, a reserve effect is highlighted, when the Co surface content increases in Pt-based nanoparticles. These findings are very likely related to the formation of “unreactive” OHad species in the Hupd region, which strongly block the catalytically active Pt sites for the oxidation of Had. The blockage of catalytically active Pt sites with oxophilic Co neighboring atoms is associated with the suppression of formation/reduction of Pt (hydr)oxide species, as shown in Fig. 4. Altogether, cobalt as an oxophilic component strongly binds the OHad species, resulting in a lowering of the HOR activity on the Pt-based surface in an alkaline environment.
Conclusions
In this work, we presented a comprehensive study about PtxCo1−x catalysts along the uniform alloy, Pt-rich shell, and Co-segregated core–shell nanoparticle motifs towards enhancing HOR performance in alkaline media. The atomic re-arrangement within the bimetallic nanoparticles can be controlled by the (electro)chemical environment like potential and pH. The oxophilic cobalt is used to tailor the coverage of OHad species, which play a critical role in the bifunctional mechanism. But on the other hand, the alloying of Pt with Co modifies the electronic and geometric structure of the Pt-enriched shell, which might weaken or strengthen the Pt–H bond. Very remarkably, the XPS analysis signifies not only a metallic but also an amorphous Co oxide species inside the dealloyed and Co-segregated PtxCo1−x core–shell catalysts. We assume that the amorphous Co oxide stemmed from the synthesis and air-storage is protected by the Pt-enriched shell under these harsh conditions. Depending on whether metallic or amorphous Co oxide species, the induced lattice strain in the Pt-enriched shell causes only a moderate lowering of the Pt–H binding energy, resulting in an increase of the Pt surface area specific exchange current density j0spec by a factor of ∼2 compared to pure Pt. Our second approach to control the coverage of OHad species is realized by comparing different Co surface concentrations (pristine alloys vs. Co-segregated). Tailoring the coverage of OHad species enables promoting the oxidation of Had on the Pt sites, referred to as a bifunctional mechanism. In this context, we developed a new descriptor, namely the Q(Hupd)/Q(Coz+) ratio, to evaluate the atomic distribution of Co and Pt at the particle surface.Our results indicate that the cobalt strongly adsorbs OHad species at low potentials and thus block the catalytically active Pt sites for the HOR in alkaline media. The blockage of catalytically active Pt sites with strongly oxophilic Co neighboring atoms is associated with the suppression of formation/reduction of Pt (hydr)oxide species. It is noted, that there is still a lack of methods to determine the real electrochemically active Pt surface area on bimetallic surfaces consisting of Pt and Co atoms.
Despite the separation of these critical parameters, cobalt as an oxophilic component strongly bonds OHad species at the particle surface and the induced lattice strain in the Pt-enriched shell causes only a moderate lowering of the Pt–H binding energy to enhance the kinetics of the HOR in an alkaline environment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
DJW and MO are gratefully acknowledged the Bundesministerium für Bildung und Forschung (BMBF, FKZ 03SF0539) for financial support. Furthermore, we thank the DFG for funding the JEOL JEM2100F TEM (INST 184/106-1 FUGG), ESCALAB 250 Xi XPS spectrometer (INST 184/144-1 FUGG), and Empyrean Theta-Theta diffractometer Panalytical (INST 184/154-1 FUGG).References
- J. Moorhouse, Modern Chlor-Alkali Technology, Wiley, 2007 Search PubMed.
- K. Zeng and D. Zhang, Recent progress in alkaline water electrolysis for hydrogen production and applications, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS.
- S. S. Djokic, Electrodeposition – Theory and Practice, Springer-Verlag, New York, 2010, ch. XVII, vol. 48, p. 295 Search PubMed.
- R. W. Revie, Uhlig's Corrosion Handbook, 2011, pp. i–xxvii Search PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- J. Durst, A. Siebel, C. Simon, F. Hasche, J. Herranz and H. A. Gasteiger, New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- J. N. Schwämmlein, B. M. Stühmeier, K. Wagenbauer, H. Dietz, V. Tileli, H. A. Gasteiger and H. A. El-Sayed, Origin of Superior HOR/HER Activity of Bimetallic Pt–Ru Catalysts in Alkaline Media Identified via Ru@Pt Core-Shell Nanoparticles, J. Electrochem. Soc., 2018, 165, H229–H239 CrossRef.
- Y. Wang, G. Wang, G. Li, B. Huang, J. Pan, Q. Liu, J. Han, L. Xiao, J. Lu and L. Zhuang, Pt–Ru catalyzed hydrogen oxidation in alkaline media: oxophilic effect or electronic effect?, Energy Environ. Sci., 2015, 8, 177–181 RSC.
- S. Lu and Z. Zhuang, Investigating the Influences of the Adsorbed Species on Catalytic Activity for Hydrogen Oxidation Reaction in Alkaline Electrolyte, J. Am. Chem. Soc., 2017, 139, 5156–5163 CrossRef CAS.
- K. Elbert, J. Hu, Z. Ma, Y. Zhang, G. Chen, W. An, P. Liu, H. S. Isaacs, R. R. Adzic and J. X. Wang, Elucidating Hydrogen Oxidation/Evolution Kinetics in Base and Acid by Enhanced Activities at the Optimized Pt Shell Thickness on the Ru Core, ACS Catal., 2015, 5, 6764–6772 CrossRef CAS.
- J. Li, S. Ghoshal, M. K. Bates, T. E. Miller, V. Davies, E. Stavitski, K. Attenkofer, S. Mukerjee, Z.-F. Ma and Q. Jia, Experimental Proof of the Bifunctional Mechanism for the Hydrogen Oxidation in Alkaline Media, Angew. Chem., Int. Ed. Engl., 2017, 56, 15594–15598 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Trends in activity for the water electrolyser reactions on 3d M(Ni,Co,Fe,Mn) hydr(oxy)oxide catalysts, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, Hydrogen Oxidation and Evolution Reaction Kinetics on Platinum: Acid vs. Alkaline Electrolytes, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS.
- W. Sheng, Z. Zhuang, M. Gao, J. Zheng, J. G. Chen and Y. Yan, Correlating hydrogen oxidation and evolution activity on platinum at different pH with measured hydrogen binding energy, Nat. Commun., 2015, 6, 5848 CrossRef CAS PubMed.
- J. Durst, C. Simon, A. Siebel, P. J. Rheinländer, T. Schuler, M. Hanzlik, J. Herranz, F. Hasché and H. A. Gasteiger, (Invited) Hydrogen Oxidation and Evolution Reaction (HOR/HER) on Pt Electrodes in Acid vs. Alkaline Electrolytes: Mechanism, Activity and Particle Size Effects, ECS Trans., 2014, 64, 1069–1080 CrossRef CAS.
- A. B. Anderson, R. A. Sidik, J. Narayanasamy and P. Shiller, Theoretical Calculation of Activation Energies for Pt + H + (aq) + e - (U) ↔ Pt–H: Activation Energy-Based Symmetry Factors in the Marcus Normal and Inverted Regions, J. Phys. Chem. B, 2003, 107, 4618–4623 CrossRef CAS.
- Y. Cai and A. B. Anderson, The Reversible Hydrogen Electrode: Potential-Dependent Activation Energies over Platinum from Quantum Theory, J. Phys. Chem. B, 2004, 108, 9829–9833 CrossRef CAS.
- T. Zhang and A. B. Anderson, Hydrogen Oxidation and Evolution on Platinum Electrodes in Base: Theoretical Study, J. Phys. Chem. C, 2007, 111, 8644–8648 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, S. Bahn, L. B. Hansen, M. Bollinger, H. Bengaard, B. Hammer, Z. Sljivancanin, M. Mavrikakis, Y. Xu, S. Dahl and C. J. H. Jacobsen, Universality in Heterogeneous Catalysis, J. Catal., 2002, 209, 275–278 CrossRef.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Interfacial water reorganization as a pH-dependent descriptor of the hydrogen evolution rate on platinum electrodes, Nat. Energy, 2017, 2, 972 CrossRef.
- R. Rizo, E. Sitta, E. Herrero, V. Climent and J. M. Feliu, Towards the understanding of the interfacial pH scale at Pt(1 1 1) electrodes, Electrochim. Acta, 2015, 162, 138–145 CrossRef CAS.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Improving the hydrogen oxidation reaction rate by promotion of hydroxyl adsorption, Nat. Chem., 2013, 5, 300–306 CrossRef CAS PubMed.
- R. Subbaraman, N. Danilovic, D. Strmcnik, K.-C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Enhancing the Alkaline Hydrogen Evolution Reaction Activity through the Bifunctionality of Ni(OH) 2/Metal Catalysts, Angew. Chem., 2012, 124, 12663–12666 CrossRef.
- D. Strmcnik, P. P. Lopes, B. Genorio, V. R. Stamenkovic and N. M. Markovic, Design principles for hydrogen evolution reaction catalyst materials, Nano Energy, 2016, 29, 29–36 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Enhancing Hydrogen Evolution Activity in Water Splitting by Tailoring Li+-Ni(OH)2-Pt Interfaces, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- Y. Cong, B. Yi and Y. Song, Hydrogen oxidation reaction in alkaline media: From mechanism to recent electrocatalysts, Nano Energy, 2018, 44, 288–303 CrossRef CAS.
- D. J. Weber, M. Janssen and M. Oezaslan, Effect of Monovalent Cations on the HOR/HER Activity for Pt in Alkaline Environment, J. Electrochem. Soc., 2019, 166, F66–F73 CrossRef CAS.
- M. Oezaslan, F. Hasché and P. Strasser, Oxygen Electroreduction on PtCo3, PtCo and Pt3Co Alloy Nanoparticles for Alkaline and Acidic PEM Fuel Cells, J. Electrochem. Soc., 2012, 159, B394 CrossRef CAS.
- M. Oezaslan and P. Strasser, Activity of dealloyed PtCo3 and PtCu3 nanoparticle electrocatalyst for oxygen reduction reaction in polymer electrolyte membrane fuel cell, J. Power Sources, 2011, 196, 5240–5249 CrossRef CAS.
- M. Oezaslan, M. Heggen and P. Strasser, Size-Dependent Morphology of Dealloyed Bimetallic Catalysts: Linking the Nano to the Macro Scale, J. Am. Chem. Soc., 2012, 134, 514–524 CrossRef CAS PubMed.
- M. Oezaslan, F. Hasché and P. Strasser, In Situ Observation of Bimetallic Alloy Nanoparticle Formation and Growth Using High-Temperature XRD, Chem. Mater., 2011, 23, 2159–2165 CrossRef CAS.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, Nat'L Assoc. of Corrosion, Oxford, New York, 1974 Search PubMed.
- J. Chivot, L. Mendoza, C. Mansour, T. Pauporté and M. Cassir, New insight in the behaviour of Co–H2O system at 25–150 °C, based on revised Pourbaix diagrams, Corros. Sci., 2008, 50, 62–69 CrossRef CAS.
- F. H. B. Lima, J. F. R. de Castro, L. G. R. A. Santos and E. A. Ticianelli, Electrocatalysis of oxygen reduction on carbon-supported Pt–Co nanoparticles with low Pt content, J. Power Sources, 2009, 190, 293–300 CrossRef CAS.
- K. J. J. Mayrhofer, V. Juhart, K. Hartl, M. Hanzlik and M. Arenz, Adsorbate-Induced Surface Segregation for Core–Shell Nanocatalysts, Angew. Chem., Int. Ed., 2009, 48, 3529–3531 CrossRef CAS PubMed.
- W. A. Badawy, F. M. Al-Kharafi and J. R. Al-Ajmi, Electrochemical behaviour of cobalt in aqueous solutions of different pH, J. Appl. Electrochem., 2000, 30, 693–704 CrossRef CAS.
- I. G. Casella and M. R. Guascito, Anodic electrodeposition of conducting cobalt oxyhydroxide films on a gold surface. XPS study and electrochemical behaviour in neutral and alkaline solution, J. Electroanal. Chem., 1999, 476, 54–63 CrossRef CAS.
- S. R. Brankovic, Electrochemical Deposition as Surface Controlled Phenomenon: Fundamentals and Applications, J. Electrochem. Soc., 2016, 163, Y21 CrossRef CAS.
- N. Schubert, M. Schneider and A. Michealis, The mechanism of anodic dissolution of cobalt in neutral and alkaline electrolyte at high current density, Electrochim. Acta, 2013, 113, 748–754 CrossRef CAS.
- J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, Electrochemical Water Oxidation with Cobalt-Based Electrocatalysts from pH 0–14: The Thermodynamic Basis for Catalyst Structure, Stability, and Activity, J. Am. Chem. Soc., 2011, 133, 14431–14442 CrossRef CAS PubMed.
- A. Minguzzi, F.-R. F. Fan, A. Vertova, S. Rondinini and A. J. Bard, Dynamic potential–pH diagrams application to electrocatalysts for water oxidation, Chem. Sci., 2012, 3, 217–229 RSC.
- J. Chen and A. Selloni, First Principles Study of Cobalt (Hydr)oxides under Electrochemical Conditions, J. Phys. Chem. C, 2013, 117, 20002–20006 CrossRef CAS.
- T. R. Jayaraman, V. K. Venkatesan and H. V. K. Udupa, Cyclic voltammetric studies of electroless cobalt in NaOH, Electrochim. Acta, 1975, 20, 209–213 CrossRef CAS.
- D. Totir, Y. Mo, S. Kim, M. R. Antonio and D. A. Scherson, In Situ Co K-Edge X-Ray Absorption Fine Structure of Cobalt Hydroxide Film Electrodes in Alkaline Solutions, J. Electrochem. Soc., 2000, 147, 4594 CrossRef CAS.
- L. Gragnaniello, S. Agnoli, G. Parteder, A. Barolo, F. Bondino, F. Allegretti, S. Surnev, G. Granozzi and F. P. Netzer, Cobalt oxide nanolayers on Pd(100): The thickness-dependent structural evolution, Surf. Sci., 2010, 604, 2002–2011 CrossRef CAS.
- K. M. Ismail and W. A. Badawy, Electrochemical and XPS investigations of cobalt in KOH solutions, J. Appl. Electrochem., 2000, 30, 1303–1311 CrossRef CAS.
- Y. Gauthier, Y. Joly and R. Baudoing, Surface-sandwich segregation on nondilute bimetallic alloys: Pt50Ni50 and Pt78Ni22 probed by low-energy electron diffraction, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 6216–6218 CrossRef CAS.
- Y. Gauthier, Pt-metal alloy surfaces: Systematic trends, Surf. Rev. Lett., 1996, 3, 1663–1689 CrossRef CAS.
- U. Bardi, A. Atrei, E. Zanazzi, G. Rovida and P. N. Ross, Study of the reconstructed (001) surface of the Pt80Co20 alloy, Vacuum, 1990, 41, 437–440 CrossRef CAS.
- M. Heggen, M. Oezaslan, L. Houben and P. Strasser, Formation and Analysis of Core-Shell Fine Structures in Pt Bimetallic Nanoparticle Fuel Cell Electrocatalysts, J. Phys. Chem. C, 2012, 116, 19073–19083 CrossRef CAS.
- X. Chen, I. T. McCrum, K. A. Schwarz, M. J. Janik and M. T. M. Koper, Co-adsorption of Cations as the Cause of the Apparent pH Dependence of Hydrogen Adsorption on a Stepped Platinum Single-Crystal Electrode, Angew. Chem., Int. Ed. Engl., 2017, 56, 15025–15029 CrossRef CAS.
- I. T. McCrum and M. J. Janik, pH and Alkali Cation Effects on the Pt Cyclic Voltammogram Explained Using Density Functional Theory, J. Phys. Chem. C, 2016, 120, 457–471 CrossRef CAS.
- X. Yang, J. Nash, N. Oliveira, Y. Yan and B. Xu, Understanding the pH Dependence of Underpotential Deposited Hydrogen on Platinum, Angew. Chem., Int. Ed. Engl., 2019, 58(49), 17718–17723 CrossRef CAS PubMed.
- F. Şen and G. Gökaǧaç, Different Sized Platinum Nanoparticles Supported on Carbon: An XPS Study on These Methanol Oxidation Catalysts, J. Phys. Chem. C, 2007, 111, 5715–5720 CrossRef.
- A. P. Grosvenor, S. D. Wik, R. G. Cavell and A. Mar, Examination of the bonding in binary transition-metal monophosphides MP (M = Cr, Mn, Fe, Co) by X-ray photoelectron spectroscopy, Inorg. Chem., 2005, 44, 8988–8998 CrossRef CAS PubMed.
- N. S. McIntyre and M. G. Cook, X-ray photoelectron studies on some oxides and hydroxides of cobalt, nickel, and copper, Anal. Chem., 1975, 47, 2208–2213 CrossRef CAS.
- D. B. Mitton, J. Walton and G. E. Thompson, An XPS and AES study of the ageing of a Co–20%Ni metal-evaporated tape, Surf. Interface Anal., 1993, 20, 36–42 CrossRef CAS.
- T. L. Barr, An ESCA study of the termination of the passivation of elemental metals, Journal of Physical Chemistry, 1978, 82, 1801–1810 CrossRef CAS.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, Synthesis and Characterization of Cobalt Hydroxide, Cobalt Oxyhydroxide, and Cobalt Oxide Nanodiscs, J. Phys. Chem. C, 2010, 114, 111–119 CrossRef CAS.
- D. Briggs and V. A. Gibson, Direct observation of multiplet splitting in 2P photoelectron peaks of cobalt complexes, Chem. Phys. Lett., 1974, 25, 493–496 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- M. Chatti, J. L. Gardiner, M. Fournier, B. Johannessen, T. Williams, T. R. Gengenbach, N. Pai, C. Nguyen, D. R. MacFarlane, R. K. Hocking and A. N. Simonov, Intrinsically stable in situ generated electrocatalyst for long-term oxidation of acidic water at up to 80 °C, Nat. Catal., 2019, 2, 457–465 CrossRef CAS.
- J. S. Mondschein, J. F. Callejas, C. G. Read, J. Y. C. Chen, C. F. Holder, C. K. Badding and R. E. Schaak, Crystalline Cobalt Oxide Films for Sustained Electrocatalytic Oxygen Evolution under Strongly Acidic Conditions, Chem. Mater., 2017, 29, 950–957 CrossRef CAS.
- M. Huynh, T. Ozel, C. Liu, E. C. Lau and D. G. Nocera, Design of template-stabilized active and earth-abundant oxygen evolution catalysts in acid, Chem. Sci., 2017, 8, 4779–4794 RSC.
- G. W. Sievers, A. W. Jensen, J. Quinson, A. Zana, F. Bizzotto, M. Oezaslan, A. Dworzak, J. J. K. Kirkensgaard, T. E. L. Smitshuysen, S. Kadkhodazadeh, M. Juelsholt, K. M. Ø. Jensen, K. Anklam, H. Wan, J. Schäfer, K. Čépe, M. Escudero-Escribano, J. Rossmeisl, A. Quade, V. Brüser and M. Arenz, Self-supported Pt–CoO networks combining high specific activity with high surface area for oxygen reduction, Nat. Mater., 2020, 20, 208–213 CrossRef PubMed.
- M. L. Perry, T. W. Patterson and C. Reiser, Systems strategies to mitigate carbon corrosion in fuel cells, ECS Trans., 2006, 3, 783–795 CrossRef CAS.
- J. Kim, J. Lee and Y. Tak, Relationship between carbon corrosion and positive electrode potential in a proton-exchange membrane fuel cell during start/stop operation, J. Power Sources, 2009, 192(2), 674–678 CrossRef CAS.
- W. K. Behl and J. E. Toni, Anodic oxidation of cobalt in potassium hydroxide electrolytes, J. Electroanal. Chem. Interfacial Electrochem., 1971, 31, 63–75 CrossRef CAS.
- B. S. Yeo and A. T. Bell, Enhanced Activity of Gold-Supported Cobalt Oxide for the Electrochemical Evolution of Oxygen, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- M. Bajdich, M. García-Mota, A. Vojvodic, J. K. Nørskov and A. T. Bell, Theoretical Investigation of the Activity of Cobalt Oxides for the Electrochemical Oxidation of Water, J. Am. Chem. Soc., 2013, 135, 13521–13530 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta02067f |
| This journal is © The Royal Society of Chemistry 2021 |