
Real-time monitoring of electrochemical carbon corrosion in alkaline media†
Sang Gu
Ji
a,
Haesol
Kim
 a,
Woong Hee
Lee
b,
Hyung-Suk
Oh
b and
Chang Hyuck
Choi
*a
a,
Woong Hee
Lee
b,
Hyung-Suk
Oh
b and
Chang Hyuck
Choi
*a
aSchool of Materials Science and Engineering, Gwangju Institute of Science and Technology (GIST), Gwangju 61005, Republic of Korea. E-mail: chchoi@gist.ac.kr
bClean Energy Research Center, Korea Institute of Science and Technology (KIST), Hwarang-ro 14-gil 5, Seongbuk-gu, Seoul, 02792, Republic of Korea
First published on 11th May 2021
Abstract
Carbon materials have been used as supporting substrates for electrocatalytically active species in many important reactions due to their high electrical conductivity and chemical inertness. To secure durable electrocatalysis, the fundamental understanding and prevention of corrosion are pivotal. The real-time monitoring of evolved CO2 enabled by differential electrochemical mass spectroscopy (DEMS) has provided potential-resolved information about carbon corrosion. However, this approach is typically limited to acidic media and it is unavailable in alkaline media because of the carbonation of the CO2 tracer. In this study, we investigate carbon corrosion using carbon monoxide (CO) as a corrosion marker. The electrochemical oxidation of selected model substrates (i.e., activated carbon, graphite, and Pt/C) is analyzed under dynamic and static potential excursions using DEMS. The results demonstrate that the CO tracer successfully monitored carbon corrosion in both acidic and alkaline media. The corrosion in alkaline media is qualitatively analogous to that in acidic media, but it is quantitatively alleviated, as confirmed by the decreased DEMS intensity and the decreased faradaic charge flowed.
Introduction
Owing to their high electrical conductivity and high surface area, carbon materials have been widely adopted in electrochemical research studies.1–4 Although carbon is thermodynamically unstable under various electrochemical reaction conditions—i.e., relatively low equilibrium potentials of 0.207 and 0.518 VRHE for its oxidation to CO2 and CO, respectively—its slow kinetics renders the carbon electrochemically inert. Carbon typically provides a large stable potential window, e.g., −0.5 to 1.5 VRHE for glassy carbon,5 within which many important electrochemical reactions can be investigated.6–8 Therefore, carbon materials have not only been used as a working electrode for electrochemical analyses, but also employed as a catalyst support for the efficient dispersion of active metal species and fast electron transport from/to the catalytically active species.9–11Despite the slow kinetics, the electrochemical oxidation of carbon materials, i.e., carbon corrosion, can unintentionally occur at certain electrochemical operating events. For instance, fuel starvations and repetitive start-up/shut-down processes during fuel cell operations can impose a highly anodic potential of >1.5 VRHE on the electrodes.12–14 Unfortunately, these anomalous events induce carbon corrosion on the electrodes, causing the undesirable loss and agglomeration of active species and disconnection of electrical paths, and thereby hampering the durable electrocatalysis.15–17
Additionally, several physicochemical parameters can affect the electrochemical corrosion of carbon materials. One representative example is a Pt/C fuel cell catalyst, in which the Pt nanoparticles supported on carbon catalyze carbon corrosion reactions and effectively lower their kinetic barriers.18–20 Increase in the temperature and decrease in the crystallinity of carbon materials also promote electrochemical oxidation of the carbon materials.21–23 Therefore, considerable efforts have been made to understand the electrochemical reactions of carbon oxidation and eventually to develop synthetic and systematic strategies preventing the carbon corrosion.24,25
Typically, the corrosion behavior of carbon materials has been investigated by monitoring CO2 gas evolution during electrochemical operations.26,27 Gas chromatography (GC) and differential electrochemical mass spectroscopy (DEMS) techniques enable the quantitative detection of CO2 evolved from carbon corrosion,28,29 successfully clarifying how the experimental conditions (potential, temperature, operational protocol, etc.) and physicochemical properties of carbon materials (crystallinity, catalyst, etc.) affect the overall kinetics of carbon corrosion. However, these approaches, which investigate the CO2 tracer, are restrictively applicable to acidic electrolytes, and cannot be used to study carbon corrosion in alkaline (and neutral) electrolytes. Upon CO2 evolution at high electrolyte pH, the carbonation of CO2 forms (bi)carbonate anions,30,31 which cannot be detected by GC and DEMS. Therefore, as compared to the large number of the previous research studies on acidic electrolytes,26–28 carbon corrosion in alkaline electrolytes has rarely been investigated,29–32 leaning on the indirect observations of either electrochemical currents or oxygen functional groups generated during the corrosion events.
In this study, we have investigated carbon corrosion in both acidic and alkaline media by employing CO as a corrosion tracer. DEMS coupled to an electrochemical flow cell (EFC) identified an identical trend of competitive CO evolution with that of CO2 evolution in an acidic electrolyte, enabling the potential-resolved monitoring of carbon corrosion in an alkaline electrolyte using the new tracer. The effects of carbon crystallinity and the presence of Pt nanoparticles on the corrosion were analyzed under dynamic and stationary potential excursions, verifying that carbon corrosion in alkaline electrolyte occurs in a qualitatively similar manner to that in an acidic electrolyte, but quantitatively mitigated.
Results and discussion
One of the most widely applied carbon substrates, Vulcan XC-72R (denoted as ‘Vulcan’), was first used to investigate electrochemical carbon corrosion. Carbon corrosion was monitored using an online EFC-DEMS by tracking the ionic current signals of CO2 (m/z = 44) and CO (m/z = 28), which evolved during electrochemical operations. Note that the CO2 contribution to the signal at m/z = 28, induced by its fragmentation during ionization, was corrected by an 11% signal subtraction from m/z = 44. The electrochemical operations comprised four slow cyclic voltammetries (CVs; scan rate = 5 mV s−1) with different upper potential limits (UPLs) of 1.0–1.6 VRHE (denoted as ‘CV protocol’), and subsequent four potential holds (chronoamperometry, CA) at identical UPLs (denoted as ‘CA protocol’; Fig. 1a).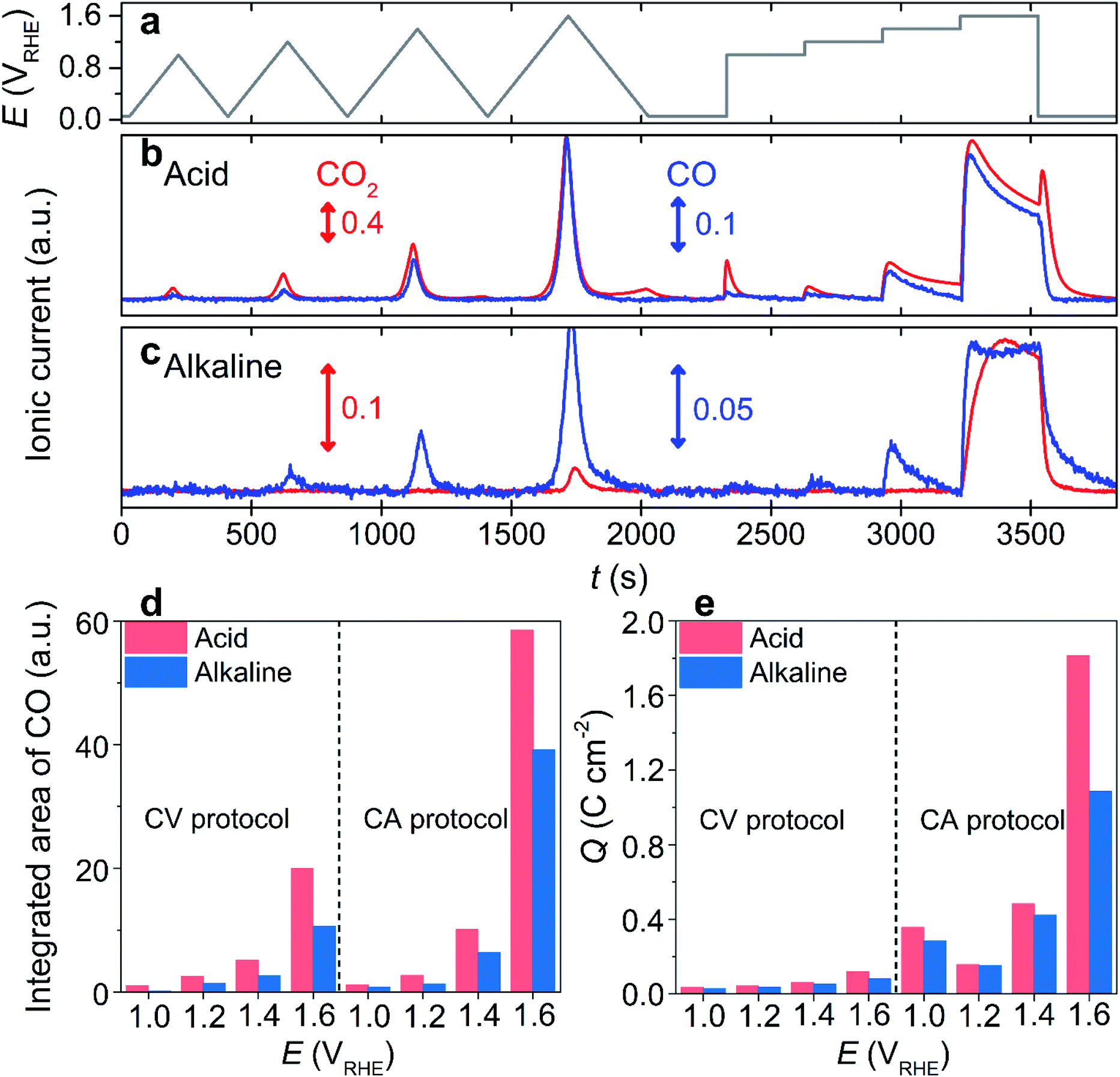 | ||
| Fig. 1 Online DEMS analysis using Vulcan as a model catalyst. (a) The potential program consisting of four CVs with different UPLs (1.0, 1.2, 1.4, and 1.6 VRHE) and four CAs at 1.0–1.6 VRHE. (b and c) The DEMS signals of CO2 (m/z = 44) and CO (m/z = 28) monitored in an Ar-saturated 0.1 M HClO4 (b) and KOH (c) electrolytes. (d) The integrated area of DEMS signal at m/z = 28, and (e) the charge density that flowed during the CV and CA protocols. | ||
In an acidic electrolyte, 0.1 M HClO4, the DEMS study during the CV protocol revealed a CO2 evolution at a potential above ca. 0.80 VRHE, and it increased with an increase in the applied potential (Fig. 1b and S1†). Additionally, a non-negligible amount of CO2 evolution was confirmed during the cathodic sweep at the end of each CV, and the cathodic carbon corrosion intensified with the increase in UPL (Fig. S2†). Similarly, the DEMS study of the subsequent CA protocol confirmed that carbon corrosion increased as the UPL increased. The intensity of the CO2 signal gradually decreased during the CA protocol, probably due to surface passivation as corroborated by the decreased current (Fig. S3†).33 Further, cathodic corrosion was identified with an intense CO2 signal that evolved once the potential jumped from 1.6 to 0.05 VRHE (Fig. S4†). These observations corresponded with those in the literature,34 where the cathodic corrosion was attributed to the alternation of the carbon surface composition, e.g., the decarboxylation of the oxygenated carbon surface.
Contrary to the successful investigations on acidic electrolyte using a CO2 tracer, the carbon corrosion study in an alkaline electrolyte, 0.1 M KOH, verified a highly suppressed CO2 ionic current during the identical potential excursion (Fig. 1c). Despite the discernible oxidation current at a potential >1.2 VRHE (Fig. S5†), the CO2 signal was almost inappreciable at a potential below 1.4 VRHE in the CV and CA protocols (Fig. 1c and S6†), which is much higher than that in an acidic electrolyte (>0.8 VRHE). The poor sensitivity of the CO2 tracer in an alkaline electrolyte was due to its rapid conversion into (bi)carbonate anions, i.e., pH-dependent chemical equilibriums known as a Bjerrum plot,35 which cannot be extracted by vacuum, and consequently, it cannot be detected by DEMS. Hence, DEMS can monitor carbon corrosion only if the CO2 evolution rate is faster than its conversion to (bi)carbonate anions, as shown at potentials higher than 1.4 VRHE.
However, in acidic electrolytes, we observed that the trend of the CO ionic current was similar to that of CO2 (Fig. 1b and S1†). Although the absolute intensities of their ionic currents were different, i.e., stronger CO2 intensity inferred that the predominant CO2 production rather than CO, their ratios were almost identical for the CV and CA protocols (Fig. S7†). Additionally, regardless of the type of tracer, the onset potential of the carbon corrosion, determined by the onset potential of DEMS signals for the tracers, was similar. The erroneous subtraction of CO2 contribution to CO in DEMS signals, which can lead to the identical trends in ionic currents at m/z = 44 and 28, could be ruled out because of the absence of CO ionic current during the cathodic carbon corrosion, indicating that cathodic carbon corrosion produced CO2 only. Therefore, these results allowed us to reasonably suggest that CO can be used as an alternative tracer, which enables the online observation of carbon corrosion over a wide pH range of the electrolyte. Despite the failure in detecting the cathodic corrosion using the CO tracer, we believe that this approach is still effective because carbon corrosion at a high potential range is more dominant than cathodic corrosion, and the cathodic corrosion accompanies the precedent anodic corrosion (i.e., no cathodic corrosion without precedent anodic corrosion; Fig. 1b).
To verify our hypothesis, the corrosion of Vulcan in the alkaline electrolyte was monitored using a CO tracer (Fig. 1c). Unlike the poor sensitivity of the CO2 tracer, that was observed in the CV protocol only with the highest UPL of 1.6 VRHE, the CO tracer identified carbon corrosion signals in the CV protocol with a UPL of >1.0 VRHE. The onset potential of carbon corrosion, determined by the CO tracer in the CV protocol, was ca. 1.0 VRHE (Fig. S6†). Further, carbon corrosion was discernible during the CA protocol at potentials >1.0 VRHE. The ionic current of CO during the potential protocols in the alkaline electrolyte was qualitatively comparable to that in the acidic electrolyte (Fig. S8†). However, the absolute intensity of the CO signal in the alkaline electrolyte was weaker than that in the acidic electrolyte (Fig. 1d). The mitigated carbon corrosion in the alkaline electrolyte was also corroborated by the lower oxidation current that flowed during the protocols in the alkaline electrolyte than that in the acidic electrolyte (Fig. 1e and S9†). Note that the faradaic efficiencies toward oxygen evolution were less than 2 and 4% in acidic and alkaline electrolytes (Fig. S10†), respectively, indicating that the oxidation current was mainly attributed to carbon corrosion. Further, Raman spectroscopy and identical location energy-dispersive X-ray spectroscopy (IL-EDX) of the Vulcan before and after corrosion identified the enhanced carbon corrosion in the acidic electrolyte, while its disordered domains were preferentially oxidized regardless of electrolyte pH (Fig. S11 and S12†). Therefore, all the results validated that CO can be successfully used in DEMS as a tracer for carbon corrosion investigations in a wide range of electrolyte pH values.
After the confirmation, the proof of concept for using CO as a carbon corrosion tracer in DEMS was further investigated using other carbon substrates having different crystallinity and carbon substrate supported with Pt nanoparticles, which are important parameters to determine the extent of carbon corrosion.22,23,27,30,36,37 Commercially-available substrates in the sequence of greater disordered structure, Ketjen Black EC 600JD (denoted as ‘Ketjen’), Vulcan, and graphite were employed (Fig. S13†).38 During the identical potential protocol (Fig. 2a), CO evolution was observed at a potential above ca. 1.0 VRHE in the acidic and alkaline electrolytes regardless of the type of carbon substrates (Fig. 2b and S14†). However, Ketjen showed the strongest intensity of CO ionic current, which was approximately 2 and 1.5 times higher than that of graphite and Vulcan in the acidic electrolyte, respectively (Fig. 2c). A similar tendency was observed in the alkaline electrolyte (Fig. 2d). Therefore, the enhancement of the corrosion rate in relation to the reduced crystallinity of the carbon subtracted, a well-known phenomenon previously verified using a CO2 tracer in an acidic electrolyte,33 was also successfully identified using the CO tracer in an alkaline electrolyte (Fig. S13†).
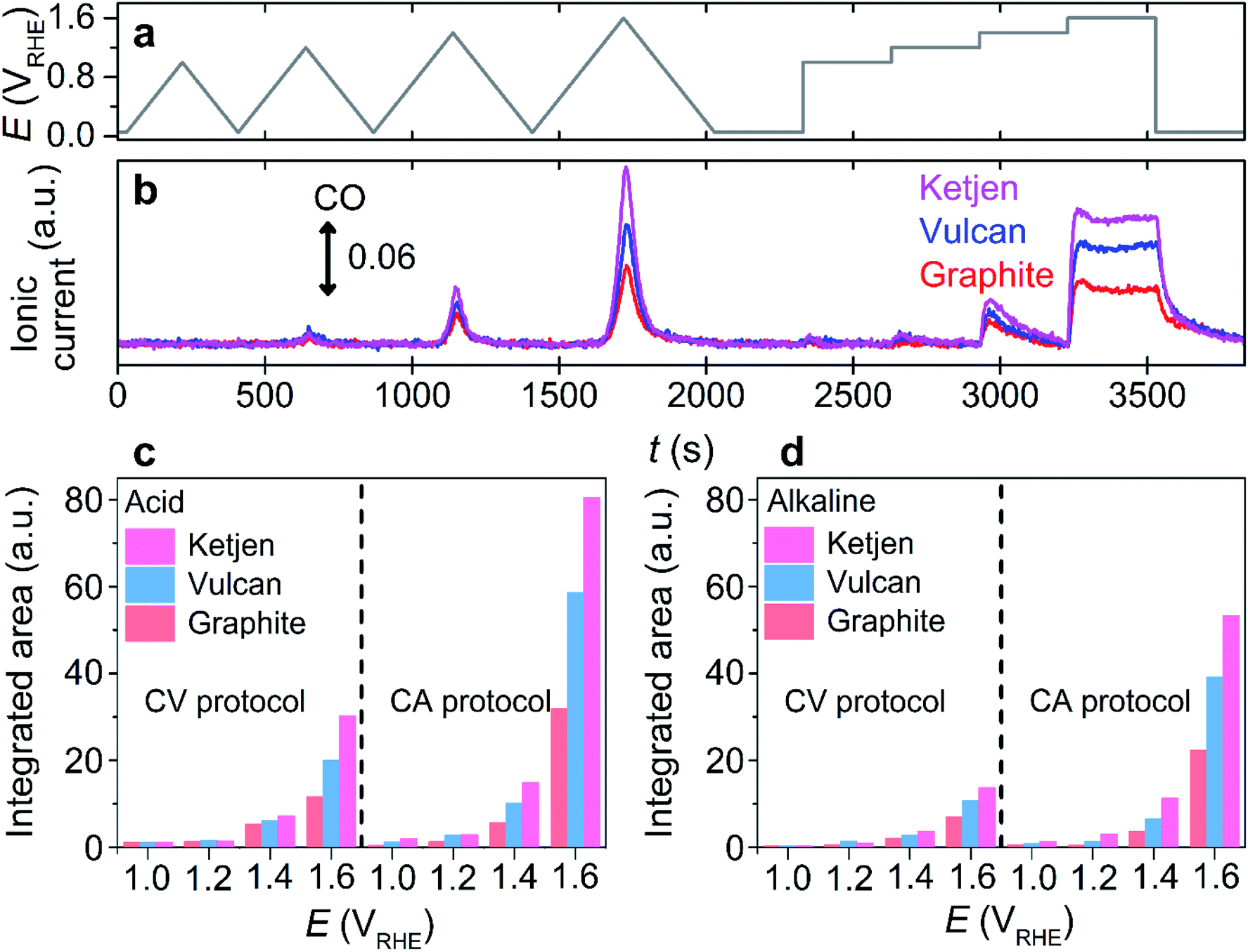 | ||
| Fig. 2 Online DEMS analysis of Ketjen, Vulcan, and graphite substrates with a CO tracer. (a) The potential program consisting of four CVs with different UPLs (1.0, 1.2, 1.4, and 1.6 VRHE) and four CAs at 1.0–1.6 VRHE. (b) The DEMS signal of CO (m/z = 28) monitored in an Ar-saturated 0.1 M KOH electrolyte. (c and d) The integrated area of DEMS signal at m/z = 28 in Ar-saturated 0.1 M HClO4 (c) and KOH (d) electrolytes during the CV and CA protocols. | ||
Afterward, we investigated the electrochemical corrosion of a carbon substrate supported with 5 wt% of Pt nanoparticles, i.e., Pt/C, which is a more reliable material in electrocatalysis than carbon substrates without catalytic species. The adopted EFC-DEMS experimental conditions that were similar to those for carbon only, but the potential protocol (one CV from 0.05 to 1.6 VRHE and consecutive CAs at 1.2, 1.4, and 1.6 VRHE) and catalyst loading (400 μgcarbon cm−2) were slightly modified to avoid the vigorous evolution of gaseous/volatile species (e.g., CO2, CO, and O2) and obtain clear DEMS signals (Fig. 3a). The EFC-DEMS study using a CO2 tracer confirmed the enhanced carbon corrosion of the Pt-decorated carbon substrate in the acidic electrolyte compared to that of the Pt-free carbon substrate (Fig. S15†), but no clear CO2 signal was obtained in the alkaline electrolyte (Fig. 3b). However, using CO as an alternative tracer, we monitored the electrochemical corrosion of Pt/C in an alkaline electrolyte. Compared to that of Pt-free Vulcan, the carbon corrosion of the Pt/C catalyst was almost doubled, verifying the Pt-catalyzed carbon corrosion even in an alkaline electrolyte.
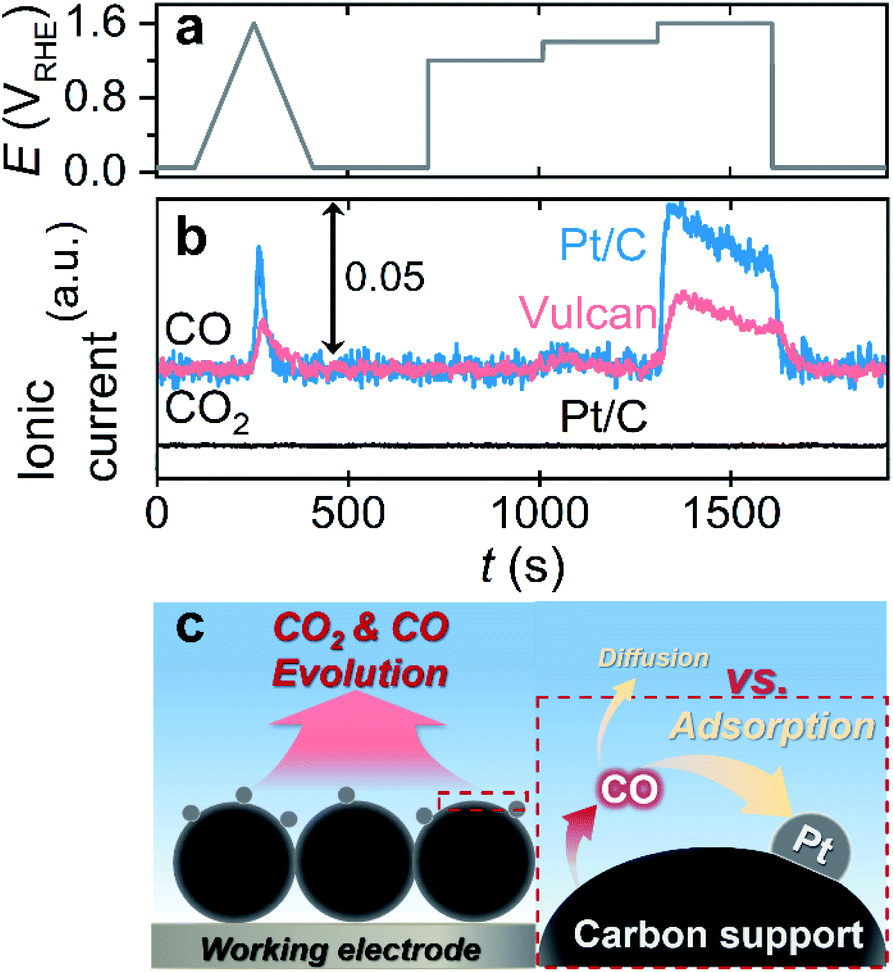 | ||
| Fig. 3 Online DEMS analysis of Pt/C with a CO tracer. (a) Potential protocol consisting of one CV from 0.05 to 1.6 VRHE and three CAs at 1.2, 1.4, and 1.6 VRHE. (b) The DEMS signals of CO2 (m/z = 44; black line) and CO (m/z = 28; blue line) monitored in an Ar-saturated 0.1 M KOH electrolyte. For comparison, the CO signal of Vulcan measured in the 0.1 M KOH electrolyte was shown (red line). (c) A scheme describing a relatively high probability of adsorption and oxidation of evolved CO to CO2 on the nearby Pt surfaces before its diffusion to bulk electrolyte with an increased Pt loading. | ||
Despite the great availability of the CO tracer in DEMS investigation in alkaline electrolytes, as shown by several control experiments, the same experiments with higher Pt loading on the carbon substrate revealed a critical drawback of this approach. The depreciation of the CO signal intensity was observed with an increase in Pt loading on the carbon substrate (10 and 20 wt%) in the acidic and alkaline electrolytes (Fig. S16†). Since CO molecules have a strong binding affinity with the Pt surface, it is reasonably presumed that evolved CO can easily be adsorbed to the Pt surface and oxidized to CO2 before its diffusion to the bulk electrolyte and DEMS (Fig. 3c). Consequently, when CO is used as the corrosion tracer in DEMS investigation, optimization of the loading amount of the active materials on the carbon substrate would be needed prior to the experiments to minimize the undesirable electrochemical oxidation of CO to CO2 and maximize the CO signal in DEMS, if the active materials adsorb CO strongly.
Conclusions
In this study, we demonstrated that CO can be used as a corrosion marker in alkaline media. Using Vulcan as a model substrate, almost identical trends of CO and CO2 evolutions were identified in acidic environments, indicating that not only CO2 but CO can also be used to track the corrosion behavior in DEMS. Despite the qualitatively similar profiles for the CO mass signal, corrosion in the alkaline electrolyte was less intense than that in the acidic electrolyte, confirming the mitigated carbon corrosion. Additionally, the carbon corrosion behavior in an alkaline electrolyte was investigated using various carbon substrates having different crystallinities and Pt nanoparticles. Analogous to the conventional belief that has been developed in acidic electrolytes, promoted carbon corrosion was demonstrated in alkaline electrolytes with an increase in the disordered structure of the carbon or the presence of the Pt catalysts. Thus, this work highlights that the CO marker can be effectively used to monitor corrosion behavior using online DEMS, which can be versatilely exploited to evaluate the stability of carbon-based electrocatalysts under a wide range of pH conditions.Experimental methods
The carbon substrates, graphite (Merck), Vulcan XC-72R (Cabot), and Ketjen Black EC 600JD (Mitsubishi Chemical), were used as received. Commercial Pt/C catalysts with different Pt loadings, 5% (Alfa Aesar), 10% (HiSPEC 2000, Johnson-Matthey), and 20% (HiSPEC 3000, Johnson-Matthey), were used as received. An EFC connected to a mass spectrometer (Max 300 LG, Extrel) was used for the DEMS analysis. The scheme of the online EFC-DEMS and the detailed experimental methods are provided in Fig. S17 and the ESI Note,† respectively. A glassy carbon disk electrode with a diameter of 3 mm (A-011169, Bio-Logic) was used as the working electrode, on which ink (for Pt-free carbon, 10 mg catalyst + 590 μL water + 80 μL 5 wt% Nafion solution) was dropped. The carbon loading on the electrode was set to 1 mg cm−2 and 400 μgcarbon cm−2 for the DEMS study of Pt-free carbon and Pt/C, respectively. The inlet and outlet of the EFC were connected to an Ag/AgCl reference electrode and a graphite tube counter electrode, respectively. 0.1 M HClO4 and KOH electrolytes were prepared using deionized (DI) water (>18.2 MΩ, Arium® Mini, Sartorius) with concentrated HClO4 (70%, Merck) and KOH (99.99% trace metals basis, Merck), respectively. The electrolytes continuously flowed into the EFC at a flow rate of 70 μL min−1. For the DEMS study of Pt-free carbon substrates, the electrochemical operations comprised four CVs at a scan rate of 5 mV s−1 from 0.05 VRHE to different UPLs of 1.0–1.6 VRHE, and then four CAs at 1.0, 1.2, 1.4, and 1.6 VRHE for 5 min each. For the DEMS study of Pt/C, the electrochemical operations comprised one CV from 0.05 to 1.6 VRHE and three CAs at 1.2, 1.4, and 1.6 VRHE for 5 min each. Before the measurements, the electrolytes were deaerated by purging with Ar (5N, Daedeok). The reference electrode was calibrated in H2-saturated (5N, Daedeok) electrolytes using a polycrystalline Pt wire. The DEMS signals were represented in an arbitrary unit due to incertitude in its calibration accuracy.39 The CO2 contribution to the signal at m/z = 28 was corrected by an 11% signal subtraction from m/z = 44. IrO2 (Alfa Aesar) with a catalyst loading of 100 μg cm−2 was used to calibrate the faradaic efficiency of oxygen. Raman spectra were obtained using micro-Raman spectroscopy (Renishaw) with 514.5 nm laser excitation. IL-EDX was performed using a Talos F200X (FEI) at an operating voltage of 200 kV. The Raman spectra and IL-EDX images of carbon samples were obtained before and after 5000 CV cycles (from 1.2 to 1.6 VRHE, 200 mV s−1) at 50 °C in 0.1 M HClO4 and KOH electrolytes. Samples for the Raman spectroscopy study were prepared by spraying carbon ink on a fluorine-doped tin oxide (FTO)-coated glass slide. The IL-EDX samples were prepared with a gold finder grid coated with a holey carbon film (AGS147AH7, Agar Scientific), on which a diluted catalyst ink was drop cast. The grid was electrically contacted on a GC disk electrode (diameter = 5 mm) and held by a homemade polyether ether ketone (PEEK) holder.Author contributions
C. H. C. conceived and directed the project. S. G. J. conducted most of experiments. H. K. contributed to part of the DEMS analyses. W. H. L. and H.-S. O. contributed to part of IL-EDX analyses. C. H. C. and S. G. J. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) Grant funded by the Ministry of Science and ICT (No. NRF-2019M3D1A1079309) and by the KIST Institutional Program.References
- E. Frackowiak and F. Beguin, Carbon, 2001, 39, 937–950 Search PubMed.
- P. Trogadas, T. F. Fuller and P. Strasser, Carbon, 2014, 75, 5–42 Search PubMed.
- Y. Liu, C. Ji, W. Gu, J. Jorne and H. A. Gasteiger, J. Electrochem. Soc., 2011, 158, B614–B621 Search PubMed.
- R. Rizo, D. Sebastián, J. L. Rodríguez, M. J. Lázaro and E. Pastor, J. Catal., 2017, 348, 22–28 Search PubMed.
- J. D. Benck, B. A. Pinaud, Y. Gorlin and T. F. Jaramillo, PLoS One, 2014, 9, e107942 Search PubMed.
- W. E. V. d. Linden and J. W. Dieker, Anal. Chim. Acta, 1980, 119, 1–24 Search PubMed.
- K. M. Sundberg, L. Atanasoska, R. Atanasoski and W. H. Smyrl, J. Electroanal. Chem., 1987, 220, 161–168 Search PubMed.
- M. Noel and P. N. Anantharaman, Surf. Coat. Technol., 1986, 28, 161–179 Search PubMed.
- Y. X. Tuo, L. J. Shi, H. Y. Cheng, Y. A. Zhu, M. L. Yang, J. Xu, Y. F. Han, P. Li and W. K. Yuan, J. Catal., 2018, 360, 175–186 Search PubMed.
- P. Ehrburger, O. P. Mahajan and P. L. W. Jr, J. Catal., 1976, 43, 61–67 Search PubMed.
- H. Huang and X. Wang, J. Mater. Chem. A, 2014, 2, 6266–6291 Search PubMed.
- A. Taniguchi, T. Akita, K. Yasuda and Y. Miyazaki, J. Power Sources, 2004, 130, 42–49 Search PubMed.
- K. H. Lim, W. H. Lee, Y. Jeong and H. Kim, J. Electrochem. Soc., 2017, 164, F1580–F1586 Search PubMed.
- G. Lee, H. Choi and Y. Tak, Nanotechnology, 2018, 30, 085402 Search PubMed.
- M. F. Labata, G. Li, J. Ocon and P. Y. A. Chuang, J. Power Sources, 2021, 487, 1–10 Search PubMed.
- J. Willsau and J. Heitbaum, J. Electroanal. Chem., 1984, 161, 93–101 Search PubMed.
- Y. Shao, G. Yin and Y. Gao, J. Power Sources, 2007, 171, 558–566 Search PubMed.
- N. Linse, L. Gubler, G. G. Scherer and A. Wokaun, Electrochim. Acta, 2011, 56, 7541–7549 Search PubMed.
- S. Maass, F. Finsterwalder, G. Frank, R. Hartmann and C. Merten, J. Power Sources, 2008, 176, 444–451 Search PubMed.
- F. Maillard, A. Bonnefont and F. Micoud, Electrochem. Commun., 2011, 13, 1109–1111 Search PubMed.
- H. S. Oh, J. H. Lee and H. Kim, Int. J. Hydrogen Energy, 2012, 37, 10844–10849 Search PubMed.
- P. L. Antonucci, L. Pino, N. Giordano and G. Pinna, Mater. Chem. Phys., 1989, 21, 495–506 Search PubMed.
- S. Ye, M. Hall and P. He, ECS Trans., 2008, 16, 2101–2113 Search PubMed.
- S. W. Lee, S. R. Choi, J. Jang, G. G. Park, S. H. Yu and J. Y. Park, J. Mater. Chem. A, 2019, 7, 25056–25065 Search PubMed.
- J. E. Owejan, P. T. Yu and R. Makharia, ECS Trans., 2007, 11, 1049–1057 Search PubMed.
- S. J. Ashton and M. Arenz, Electrochem. Commun., 2011, 13, 1473–1475 Search PubMed.
- L. Castanheira, W. O. Silva, F. H. B. Lima, A. Crisci, L. Dubau and F. Maillard, ACS Catal., 2015, 5, 2184–2194 Search PubMed.
- N. Linse, G. G. Scherer, A. Wokaun and L. Gubler, J. Power Sources, 2012, 219, 240–248 Search PubMed.
- S. Möller, S. Barwe, J. Masa, D. Wintrich, S. Seisel, H. Baltruschat and W. Schuhmann, Angew. Chem., Int. Ed., 2020, 59, 1585–1589 Search PubMed.
- C. Lafforgue, F. Maillard, V. Martin, L. Dubau and M. Chatenet, ACS Catal., 2019, 9, 5613–5622 Search PubMed.
- C. Lafforgue, A. Zadick, L. Dubau, F. Maillard and M. Chatenet, Fuel Cells, 2018, 18, 229–238 Search PubMed.
- A. Zadick, L. Dubau, N. Sergent, G. Berthome and M. Chatenet, ACS Catal., 2015, 5, 4819–4824 Search PubMed.
- L. C. Colmenares, A. Wurth, Z. Jusys and R. J. Behm, J. Power Sources, 2009, 190, 14–24 Search PubMed.
- F. Maillard, W. O. Silva, L. Castanheira, L. Dubau and F. H. B. Lima, ChemPhysChem, 2019, 20, 3106–3111 Search PubMed.
- D. A. Wolf-Gladrow, R. E. Zeebe, C. Klaas, A. Körtzinger and A. G. Dickson, Mar. Chem., 2007, 106, 287–300 Search PubMed.
- L. Castanheira, L. Dubau, M. Mermoux, G. Berthomé, N. Caqué, E. Rossinot, M. Chatenet and F. Maillard, ACS Catal., 2014, 4, 2258–2267 Search PubMed.
- Z. Zhao, L. Castanheira, L. Dubau, G. Berthomé, A. Crisci and F. Maillard, J. Power Sources, 2013, 230, 236–243 Search PubMed.
- N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, J. Am. Chem. Soc., 2013, 135, 15443–15449 Search PubMed.
- S. G. Ji, H. Kim, C. Park, W. Kim and C. H. Choi, ACS Catal., 2020, 10, 10773–10783 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional EFC-DEMS results and electrochemical responses. See DOI: 10.1039/d1ta01748a |
| This journal is © The Royal Society of Chemistry 2021 |