
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanochemically synthesized Pb-free halide perovskite-based Cs2AgBiBr6–Cu–RGO nanocomposite for photocatalytic CO2 reduction†
Santosh
Kumar
 a,
Idil
Hassan
b,
Miriam
Regue
a,
Soranyel
Gonzalez-Carrero
c,
Eduardo
Rattner
a,
Mark A.
Isaacs
d and
Salvador
Eslava
*a
a,
Idil
Hassan
b,
Miriam
Regue
a,
Soranyel
Gonzalez-Carrero
c,
Eduardo
Rattner
a,
Mark A.
Isaacs
d and
Salvador
Eslava
*a
aDepartment of Chemical Engineering, Imperial College London, London, SW7 2AZ, UK. E-mail: s.eslava@imperial.ac.uk
bDepartment of Chemical Engineering, University of Bath, Claverton Down, Bath, BA2 7AY, UK
cDepartment of Chemistry, Imperial College London, White City Campus, London W12 0BZ, UK
dDepartment of Chemistry, University College London, London, WC1H 0AJ, UK
First published on 5th May 2021
Abstract
Pb-based halide perovskites have recently showed great potential in various applications such as solar cells, optoelectronics and photocatalysis. Despite their high performance, the Pb2+ toxicity along with poor stability hinders long term applications in photocatalysis. Herein, we report mechanochemically prepared Pb-free Cs2AgBiBr6 double perovskite nanoplates and their heterostructure with Cu-loaded reduced graphene oxide (Cu–RGO) for gas-phase photocatalytic CO2 reduction using water vapor as the proton source in the absence of a hole scavenger. The resulting Cs2AgBiBr6–Cu–RGO nanocomposite shows significant photocatalytic activity of 10.7 (±0.6) μmol CH4 g−1 h−1, 1.9 (±0.3) μmol CO g−1 h−1 and 1.0 (±0.2) μmol H2 g−1 h−1, with a CH4 selectivity of 93.0 (±0.5)% on an electron basis with 1 sun and a remarkable apparent quantum efficiency of 0.89 (±0.21)% at 590 nm. A further 32% enhancement in photocatalytic activity on an electron basis is achieved when the light intensity is doubled (2 suns). The high performance was attributed to their improved charge separation and suppressed electron–hole recombination, along with extended visible light absorption, better stability in a humid environment and improved CO2 adsorption. These findings support Cs2AgBiBr6 as a potential Pb-free alternative to conventional halide perovskites for photocatalytic solar-to-fuel conversion and CO2 utilization.
Introduction
Semiconductor photocatalysis has attracted great attention in recent years for various applications such as organic synthesis,1 pollutant degradation,2 N2 fixation,3 H2 production,4 and CO2 reduction.5 In particular, photocatalytic CO2 reduction, so called “artificial photosynthesis”, has the potential to produce carbon-based value-added fuels and chemicals as sustainable feedstocks for the chemical industry.6,7 This photocatalysis technology provides promising and viable routes to both tackle global climate change and deliver on renewable energy demand.8,9 Among different photocatalytic materials, halide perovskites have recently emerged as new potential candidates for photocatalytic energy applications such as H2 production and CO2 reduction, due to their tuneable bandgap, high absorption coefficient and high carrier mobility.10,11 For example, in recent years, the all-inorganic perovskite CsPbBr3 has greatly attracted attention for photocatalytic CO2 reduction. Hou et al. prepared CsPbBr3 quantum dots for photocatalytic reduction of CO2 to CO and CH4, with a production of 4.3 and 1.5 μmol g−1 h−1, respectively.12 Furthermore, CsPbBr3-based composites such as CsPbBr3 quantum dots (QD)-reduced graphene oxide (RGO),13 CsPbBr3 QD-ZIF-67,14 CsPbBr3-UiO-66(NH2),15 CsPbBr3@iron-based MOFs,16 and CsPbBr3-graphitic carbon nitride (g-C3N4) have been explored to protect the halide perovskite from decomposition and to enhance its photocatalytic activity for CO2 reduction to CO and CH4.17 More recently, we have mechanochemically prepared a composite of CsPbBr3 nanosheets with Cu-loaded reduced graphene oxide (Cu–RGO). This material showed significantly improved activity for CO2 photoreduction to CH4 with a selectivity of 98.5% on an electron basis and a remarkable apparent quantum efficiency of 1.1% at 523 nm with water as a proton source.18 Unfortunately, the well-known toxicity of Pb cations does not comply with sustainable design principles, hampering their development and application.Recently, lead-free halide perovskites have been studied to overcome the toxicity challenge. Examples include CsSnX3, CsSbX3 and various double perovskites (A2BB′X6).19–22 In particular Cs2AgBiBr6, with a band gap of 1.8–2.2 eV and a long carrier recombination lifetime, has shown better stability toward moisture, air, heat and light when compared to other Pb-free double perovskites.11 Because of this, bulk Cs2AgBiX6 (X = Cl, Br) has been studied to understand its intrinsic properties from both an experimental and a theoretical point of view.23 Very recently, Cs2AgBiX6 showed its potential for application in photocatalytic H2 production,24 CO2 reduction25,26 and dye degradation.27 For example, Zhou et al. prepared Cs2AgBiBr6 double perovskite nanocrystals with cubic shape via a solution-phase hot injection method. These nanocrystals showed an activity of 2.35 μmol g−1 h−1 CO and 1.6 μmol g−1 h−1 CH4 (both products on electron consumption basis) for the CO2 reduction reaction under simulated solar light, using ethyl acetate as both the solvent and proton source.25 The disadvantages of such a proton source are its high cost and the misleading formation of CO through oxidation.13 Therefore, it is vital to develop the use of water as a proton source and an organic-free medium for a sustainable application.
In this work, we have successfully prepared Cs2AgBiBr6 nanoplates and their nanocomposites with Cu–RGO by an efficient, scalable and reproducible mechanochemical method. More importantly, we further tested them for gas-phase photocatalytic CO2 reduction to CH4 (major) and CO (minor) with H2O vapor as a proton source in the absence of organic solvents, under simulated sunlight. The Cs2AgBiBr6–Cu–RGO heterostructure photocatalyst showed significant improvement when compared to the pristine double perovskite nanoplates. The photocatalytic CO2 reduction achieved 10.7 (±0.6) μmol CH4 g−1 h−1 and 1.9 (±0.3) μmol CO g−1 h−1 and 93.0 (±0.5)% CH4 selectivity, along with an apparent quantum efficiency of 0.89 (±0.21)% at 590 nm.
Experimental details
Materials and methods
CsBr (99.9%, Acros Organics), AgBr (99%, Sigma-Aldrich), BiBr3 (99%, Sigma-Aldrich), Cu(NO3)2·3H2O (99%, Sigma-Aldrich), ethylene glycol (99%, Acros Organics), tetrabutylammonium hexafluorophosphate (98%, Sigma-Aldrich), acetonitrile (99.9%, Sigma-Aldrich) and isopropanol (99.7%, VWR Chemicals) used in this work are of analytical grade and used as received.Cs2AgBiBr6 nanoplates were synthesized by mechanochemical synthesis. In detail, 2 mmol of CsBr, 1 mmol of AgBr, and 1 mmol of BiBr3 were added to a 45 mL zirconia bowl containing 180 zirconia balls of 5 mm under an Ar atmosphere in a glovebox (<0.5 ppm H2O, <0.5 ppm O2). The gas-tight zirconia bowl with zirconia balls and Cs, Ag and Bi bromide precursors was then placed in a ball mill (Planetary Mono Mill Pulverisette, Fritsch) and milled at 500 rpm for 60 min. This was the optimal time to achieve good crystallization and avoid excessive sintering. The as-prepared Cs2AgBiBr6 nanoplates were collected and stored in the glovebox.
Graphene oxide (GO) was prepared by using a modified Hummer's method previously reported by our group.28 RGO and Cu–RGO were prepared as we previously described.18 Briefly, a mixture of Cu(NO3)2·6H2O and GO was reduced in ethylene glycol at 180 °C for 24 h. Different Cu loading of 1, 3, 5, 7 and 10 wt% Cu–RGO were prepared by varying the amount of Cu(NO3)2·6H2O. Actual loadings determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES) were 0.94, 3.06, 4.91, and 9.87 wt%. RGO was synthesized in the same manner from GO, just without the addition of Cu(NO3)2·6H2O. Composites of Cs2AgBiBr6 and either Cu–RGO or RGO were prepared by adding 5 mL of either Cu–RGO or RGO isopropanol suspensions (2.4 mg mL−1) to the mixture of precursors before ball milling. RGO was approximately 1 wt% in all the Cs2AgBiBr6–RGO composites, which is known to be optimal in RGO-photocatalyst composites.29 A physical mixture of Cs2AgBiBr6 and Cu–RGO was also prepared by simply mixing the powders in the same mass ratio. Cs2AgBiBr6 is sometimes abbreviated in legends as DP for double perovskite.
Characterization
Powder X-ray diffraction (XRD) was carried out in a Bruker-AXS D8 ADVANCE diffractometer operated at 40 kV voltage and 40 mA current using Cu Kα (λ = 0.15418 nm) radiation in the 2θ range 5–70°. The coherent diffraction domain size of Cs2AgBiBr6 was calculated using the Scherrer equation on the 022, 004, and 044 diffraction peaks. X-ray photoelectron spectroscopy (XPS) analysis was done using a Kratos Axis HSi spectrometer and monochromated Al Kα X-ray source operated at 90 W. Furthermore, the binding energies were referenced relative to adventitious carbon at 284.6 eV. The XPS data were processed using CasaXPS version 2.3.16. The same XPS tool was used to carry out ultraviolet photoelectron spectroscopy (UPS). The morphology and crystallinity of the as-prepared samples were examined by high-resolution transmission electron microscopy (HRTEM) using a JEOL JEM-2100Plus microscope operating at an accelerating voltage of 200 kV. The surface mapping analysis was done using energy dispersive X-ray spectroscopy (EDX) with an Oxford INCA X-ray analyzer. Light absorption of the as-prepared samples was measured by UV-visible diffuse reflectance spectroscopy (UV-Vis DRS) on a Cary 100 spectrophotometer (Agilent Technologies) using an integrating sphere for diffuse reflectance and KBr as a standard. The baseline was corrected using pure KBr and then 10 mg of sample were mixed with KBr for analysis. Furthermore, the band gap energy (Eg) of all the Cs2AgBiBr6 samples was determined using Tauc plots of [F(R)hν]1/ηversus hν and drawing a tangential line at the onset of the linear response,30 where F(R), hν, and η are the Kubelka–Munk function, energy of light, and a variable that depends on the nature of the optical transition during photon absorption (equal to 2 in this case), respectively. F(R) was calculated as F(R) = (1 − R)2(2R)−1, where R is the reflectance of the sample.30 The photoluminescence (PL) emission kinetics of powder samples were measured on a DeltaFlex TCSPC equipment (Horiba Scientific). The samples were excited by using an LED at 470 nm (Horiba NanoLED) and the photons were detected with a photomultiplier detector (Horiba PPD-650). Each PL spectrum was individually normalized by the mass of the sample. Photocurrent measurements were carried out in a three-electrode PEC cell with quartz window, working electrode, Pt wire counter electrode, Ag/AgCl reference electrode, and 0.1 M tetrabutylammonium hexafluoro-phosphate in acetonitrile as electrolyte solution. A 300 W Xe lamp equipped with an AM1.5G solar simulator filter (LOT Quantum Design) was used with 100 mW cm−2 (1 sun) determined by the distance to the working electrode and this was chopped with an automatic shutter. An external potential of 0.0 V vs. Ag/AgCl reference electrode was applied with a potentiostat (Ivium CompactStat). Working electrodes were prepared by spin coating on fluorine-doped tin oxide coated glass the Cs2AgBiBr6 samples previously dispersed in dimethyl sulfoxide. Nitrogen and CO2 adsorption measurements were carried out with a Quantachrome Autosorb IQ2 porosimeter. Surface areas were calculated over the nitrogen relative pressure range 0.01–0.2 using the Brunauer–Emmett–Teller (BET) model.Photocatalysis
Photocatalytic CO2 reduction experiments of the as-prepared powder photocatalysts were carried out at room temperature in a gas-tight 125 mL glass photoreactor with a top quartz light window. The light intensity at the photocatalyst surface was adjusted to 1 sun (100 mW cm−2) using an AM1.5G mass filtered 300 W Xe source at an appropriate distance. 50 mg of powder sample were placed in a glass holder within the photoreactor. Prior to irradiation, the reactor was purged with He gas to remove air from the system and then purged with CO2 for 1 h at 5 mL min−1 and humidified by injecting water at 1 mL h−1 simultaneously with a high-precision pump. The observed relative humidity was 60–65%, measured by a dew point meter HT-6292 from Landtek. Finally, the gas inlet and outlet in the reactor were closed before the CO2 reduction experiments were carried out for 4 h under 1 sun. The formed products were analyzed by gas chromatography (GCMS, Shimadzu GC-2030 Plus) with He as a carrier gas and a barrier ionization discharge (BID) detector. Reusability experiments were carried out for three successive experimental runs on the used powder sample by each time degassing with He, followed by purging with fresh CO2 and water, and then irradiating for 4 h (12 h of total irradiation) under 1 sun. The error bars in graphs or ± symbol in the text indicate standard deviation. The selectivity (S) of the obtained products was calculated on a total electron consumption basis as given in the following equations:31,32 | (1) |
 | (2) |
 | (3) |
 | (4) |
Further details of the AQE calculations were given in a previous publication.18
Results and discussion
A surfactant/ligand-free mechanochemical strategy was used to prepare Cs2AgBiBr6 nanocrystals and their Cu–RGO composites (Scheme 1). The kinetic energy of the balls is transferred to Cs, Ag and Bi bromide precursors during the milling process, allowing the collision between reactant molecules which then results in a chemical reaction to form Cs2AgBiBr6 nanocrystals, as shown below:33| 2CsBr + AgBr + BiBr3 → Cs2AgBiBr6 | (5) |
 | ||
| Scheme 1 Schematic diagram of the Cs2AgBiBr6 nanoplates and Cs2AgBiBr6–Cu–RGO nanocomposites by a surfactant/ligand-free mechanochemical synthesis method. | ||
The morphology and crystal structure were examined by TEM and HRTEM. Cs2AgBiBr6 nanocrystals show nanoplate, that is slab shape, with a thickness of 80–100 nm and a lateral size of a few hundred nm (Fig. 1a). HRTEM lattice fringes show an interplanar spacing of 0.29 nm which is assigned to the 004 plane of the Cs2AgBiBr6 phase (Fig. 1b).25 A selected angle electron diffraction (SAED) pattern further confirms their crystallinity and pure Cs2AgBiBr6 phase (Fig. S1†). The as-prepared RGO shows a layered structure with typical wrinkled surfaces of re-stacked nanosheets due to van der Waals (vdW) interlayer interactions in dried samples (Fig. S2a†). After decorating the surface with Cu nanoparticles (Cu–RGO), the typical wrinkled surface remains unchanged but a rougher surface can be appreciated (Fig. S2b†). The size range of uniformly dispersed Cu nanoparticles is 3–5 nm. More interestingly, after hybridising Cs2AgBiBr6 (DP) with Cu–RGO, the Cs2AgBiBr6 nanoplates appear to be highly dispersed on the Cu–RGO and show relatively smaller dimensions, with a thickness of 20–40 nm and lateral size of 60–70 nm (Fig. 1c and S2c†). HRTEM micrographs also show intimate contact between the Cs2AgBiBr6 nanoplates and Cu–RGO (Fig. 1d) and clear lattice fringes for both Cs2AgBiBr6 (Fig. 1e) and Cu (Fig. 1f). Furthermore, EDX mapping confirms the ubiquitous presence of Cs, Ag, Bi, Br, Cu and C in DP–Cu–RGO (Fig. S3†). The percentage of Cu is 0.25 wt% as calculated by EDX, whereas RGO is 1 wt% based on the loaded amount during mechanochemical synthesis.
 | ||
| Fig. 1 TEM and HRTEM micrographs of (a and b) DP and (c–f) DP–Cu–RGO. | ||
XRD patterns of Cs2AgBiBr6 nanoplates, DP–RGO and DP–Cu–RGO samples are given in Fig. 2a. The obtained XRD pattern of Cs2AgBiBr6 (DP) shows its characteristic diffraction peaks at 12.8, 15.1, 22.2, 25.1, 26.0, and 31.8° (2θ), corresponding to the 111, 200, 022, 311, 222, 400 and 044 planes, respectively (JSPCDS #01-084-8699). This indicates that Cs2AgBiBr6 consists of a cubic structure with a = 11.2711 Å and space group symmetry Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, which accounts for the Bi/Ag ordering in a rock-salt-like superstructure. The calculated coherent diffraction domain size is 107 (±5) nm, similar to their physical thickness (120–170 nm). Both DP–RGO and DP–Cu–RGO composites show the same cubic Cs2AgBiBr6 DP crystal phase in the XRD patterns, indicating that the type of phase is not affected by the addition of RGO or Cu–RGO under the chosen synthesis conditions. However, the calculated coherent diffraction domain size of Cs2AgBiBr6 is reduced to 52 (±5) nm in DP–Cu–RGO.
m, which accounts for the Bi/Ag ordering in a rock-salt-like superstructure. The calculated coherent diffraction domain size is 107 (±5) nm, similar to their physical thickness (120–170 nm). Both DP–RGO and DP–Cu–RGO composites show the same cubic Cs2AgBiBr6 DP crystal phase in the XRD patterns, indicating that the type of phase is not affected by the addition of RGO or Cu–RGO under the chosen synthesis conditions. However, the calculated coherent diffraction domain size of Cs2AgBiBr6 is reduced to 52 (±5) nm in DP–Cu–RGO.
 | ||
| Fig. 2 (a) XRD patterns and (b) UV-Vis DRS spectra of Cs2AgBiBr6 (DP) and DP–Cu–RGO nanocomposite among others for comparison. Inset in (b) shows photographs of DP, Cu–RGO and DP–Cu–RGO from left to right. | ||
UV-Vis DRS spectra of Cs2AgBiBr6, RGO, and DP–Cu–RGO nanocomposites are shown in Fig. 2b. The spectrum of Cs2AgBiBr6 shows a broad absorption starting around 600 nm in the visible region, corresponding to an indirect band gap of 2.0 eV (Fig. S4a†).34 The DP–Cu–RGO nanocomposite shows a similar absorption onset attributed to the Cs2AgBiBr6, but an upward shift in the absorption baseline due to the presence of Cu–RGO (Fig. S4b†), since this absorbs broadly in the UV-Vis region (Fig. 2b).
XPS was used to probe the chemical oxidation state of Cu, Cs, Ag, Bi and Br and their chemical environment (Fig. 3a–d and S5†). Compared to DP, the Cs 3d, Ag 3d, Bi 4f and Br 3d peaks of DP–Cu–RGO show lower intensity, suggesting the DP in DP–Cu–RGO is partially wrapped by RGO (Fig. 3a, b, S5c and d†).35 Further, Ag 3d spectra of DP–Cu–RGO exhibit two extra spin–orbit doublets at 368.3 eV and 374.4 eV related to Ag–O bond (Fig. 3a).36 Similarly, Bi 4f spectra of DP–Cu–RGO show two extra peaks at binding energies 163.0 eV and 157.5 eV (Fig. 3b).37 These results indicate that the perovskite is at least in part anchored to RGO via bonds between the metals (Ag and Bi) in the perovskite octahedra and residual O-groups on RGO. The O 1s spectrum of DP–Cu–RGO is deconvoluted into three peaks centered at 532.0, 530.3, and 529.3 eV (Fig. 3c). These are attributed to residual C–OH in RGO, residual C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or OC–O in RGO, and Ag–O/Bi–O bonds, respectively.36,38 Cu-LLM spectra shows a peak just below 570 eV in DP–Cu–RGO composites, which deconvolutes into three peaks with binding energy of 568.3, 568.8, and 570.2 eV, corresponding to Cu, CuO, and Cu2O (Fig. 3d).39
O or OC–O in RGO, and Ag–O/Bi–O bonds, respectively.36,38 Cu-LLM spectra shows a peak just below 570 eV in DP–Cu–RGO composites, which deconvolutes into three peaks with binding energy of 568.3, 568.8, and 570.2 eV, corresponding to Cu, CuO, and Cu2O (Fig. 3d).39
 | ||
| Fig. 3 (a) Ag 3d, (b) Bi 4f, (c) O 1s and (d) Cu XPS spectra. Raw data is presented in grey circles. (e) Valence band XPS spectrum. (f) UPS spectrum of nanoplates prepared for 60 min. (g) Band structure of Cs2AgBiBr6, where Eg, Ef, Φ, EVB, and ECB represent the band gap energy, Fermi level, electronic workfunction, valence band edge and conduction band edge, respectively. | ||
Valence-band (VB) XPS was used to determine the energy gap between the VB edge potential and the Fermi level (Ef) (Fig. 3e). The calculated value from the intercept between the tangent of the onset and the baseline of the spectra is approximately 1.4 eV, which is consistent with reported VB energy values.32 Furthermore, UPS served to measure the electronic workfunction, i.e. the difference between Ef and the vacuum level, found to be 4.3 eV (Fig. 3f). With this information and the band gap value from UV-Vis DRS (2.0 eV), we constructed the electronic band diagram of Cs2AgBiBr6, as shown in Fig. 3g. The resulting CB edge potential is at −3.7 eV from the vacuum level (−0.6 VNHE at pH = 7). This indicates that Cs2AgBiBr6 possesses a suitable CB edge potential for multielectron CO2 reduction to low carbon products such as CO (CO2/CO, −0.53 VNHE at pH = 7) and CH4 (CO2/CH4, −0.24 VNHE at pH = 7).
PL emission spectra were recorded for DP and DP–Cu–RGO to understand the charge carrier dynamics between phases (Fig. 4a). DP exhibits a wide typical PL peak around 600 nm (2.0 eV) and an additional peak around 670 (1.85 eV), corresponding to indirect band gap transitions of Cs2AgBiBr6.34,40 The normalized PL intensity of the DP–Cu–RGO nanocomposite is much lower than that of DP, which can be attributed to suppressed electron–hole recombination and photoinduced charge carrier separation between Cs2AgBiBr6 and Cu–RGO. Furthermore, time-resolved PL measurements were carried out to confirm the charge transport behavior on DP–Cu–RGO nanocomposites (Fig. 4b). Average charge carrier lifetimes (τav) were determined from fitting the resulting decay curves with a three-exponential function (Table S1†), consistent with three decay processes, such as nonradiative and radiative relaxations originating from the direct formation of free charge carriers and the indirect formation of self-trapped excitons.13 DP–Cu–RGO showed lower τav values than DP (0.2 vs. 1.5 ns), which indicates the presence of a nonradiative pathway that suppresses electron–hole pair recombination and thereby improves charge carrier separation. This electronic transport behavior further confirms an excellent interface at the heterostructure between the DP and Cu–RGO. Further, this is supported by photocurrent measurements on samples mounted on electrodes. The DP and DP–Cu–RGO electrodes exhibit a photocurrent response to on–off cycles under light irradiation with a bias of 0 V vs. Ag/AgCl. Interestingly, the DP–Cu–RGO based electrode exhibits much higher photocurrent response than that of DP–RGO and DP (Fig. 4c), consistent with the better charge separation observed by PL.
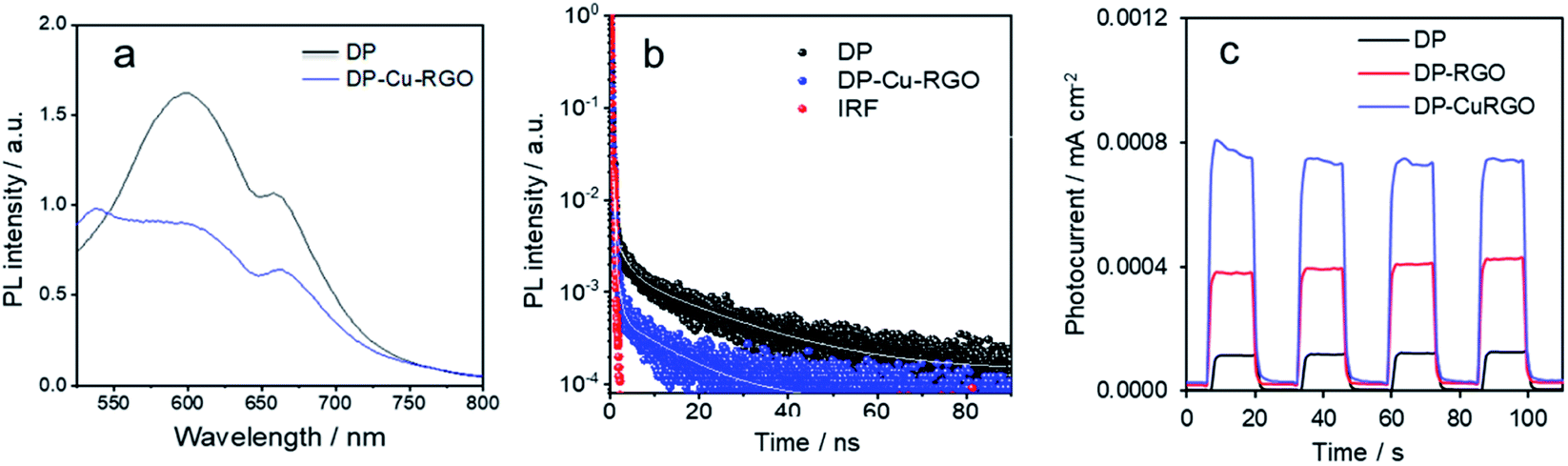 | ||
| Fig. 4 (a) Steady-state (λex = 470 nm), and (b) time resolved (λex = 470 nm; λem = 600 nm) PL spectra of Cs2AgBiBr6 (DP) and DP–Cu–RGO nanocomposite. ‘IRF’ in (b) stands for instrument response function. (c) Photocurrent response measured at 0 V against Ag/AgCl electrode. | ||
N2 adsorption–desorption on Cs2AgBiBr6 (DP), DP–RGO, DP–Cu–RGO, and a physical mixture of DP and Cu–RGO was carried out to study their surface area (Fig. 5a). All the samples show typical type II isotherms with type III hysteresis which are characteristic of non-porous materials and slit-like pores, which might be due to interparticle porosity.41 The DP–Cu–RGO nanocomposite shows relatively higher BET surface area (82 m2 g−1) than Cs2AgBiBr6 nanoplates (62 m2 g−1), due to the presence of RGO, a two-dimensional material, and observed smaller DP particles in the composite (Fig. 1c–e). The BET surface area of DP–RGO is similar to that of the DP–Cu–RGO nanocomposite (77 vs. 82 m2 g−1). It is worth noting that the BET surface area of DP–Cu–RGO is larger than the physical mixture of DP and Cu–RGO (72 m2 g−1), indicating that the Cu–RGO presence during the mechanochemical synthesis has a templating effect increasing the surface area of the composites.28,42–44 In addition, CO2 physisorption was also carried out, as CO2 adsorption is the first and a key step in photocatalytic CO2 reduction (Fig. 5b). The CO2 physisorption of DP–Cu–RGO (5.2 cm3 g−1) is relatively higher than that of DP (4.2 cm3 g−1), physical mixture of DP and Cu–RGO (4.7 cm3 g−1), and DP–RGO (3.8 cm3 g−1), in agreement with the trends observed in BET surface area.
 | ||
| Fig. 5 (a) N2-adsorption–desorption isotherms (• ads, △ des) and (b) CO2 adsorption–desorption isotherms at 298 K. | ||
To test their thermal stability, 100 mg of the as-prepared Cs2AgBiBr6 DP nanoplates were placed in an alumina crucible and heated to 100 and 300 °C for 1 h in an oven under ambient conditions (air with relative humidity 60–70%, Fig. S6†). Upon heating, no additional diffraction related to the formation of secondary phases such as Cs3Bi2Br9 or AgBr is observed, just narrowing of the XRD peaks due to sintering.45 These results show the high thermal stability of Cs2AgBiBr6 DP nanoplates, like CsPbBr3 and unlike organic–inorganic halide perovskites.46 Regarding long-term ambient stability at room temperature and ambient conditions with relative humidity 60–70%, pure Cs2AgBiBr6 nanoplates are stable for at least two days, but decompose to AgBr, Cs(OH)2 and Cs3Bi2Br9 after 8 days (Fig. S7†). Interestingly, the DP–Cu–RGO composite showed relatively higher stability under the same environment and no phase change even after 8 days (Fig. S8†), with only nanoparticle sintering observed over time. The high stability of the DP–Cu–RGO composite under 60–70% relative humidity ambient conditions indicates that the RGO, of more hydrophobic character, acts as a protection layer for the Cs2AgBiBr6 DP.
The photocatalytic CO2 reduction with water vapor as a proton source on all the samples was carried out under simulated solar light (1 sun, Fig. 6a–d). The photocatalytic activity of Cs2AgBiBr6 nanoplates (DP) is 1.49 (±0.09) μmol CH4 g−1 h−1, 0.54 (±0.03) μmol CO g−1 h−1 and 0.19 (±0.01) μmol H2 g−1 h−1. This is relatively lower than conventional all-inorganic CsPbBr3 halide perovskite nanocrystals prepared by mechanochemical synthesis and tested in the same photocatalytic reactor (Fig. 6a and b). This can be attributed to the indirect band gap of Cs2AgBiBr6, compared with the direct band gap of CsPbBr3, despite being smaller (Fig. S9†). Interestingly, DP–RGO shows twice the activity of DP, something that was not achieved previously in CsPbBr3–RGO (Fig. 6c and d). We anticipate that this could be due to the presence of Ag–O and Bi–O between Cs2AgBiBr6 and RGO which can help in fast transfer of holes and thereby water oxidation.18,32 More importantly, the composite DP–Cu–RGO shows remarkable photocatalytic activity of 10.7 (±0.6) μmol CH4 g−1 h−1, 1.9 (±0.3) μmol CO g−1 h−1 and 1.0 (±0.2) μmol H2 g−1 h−1, much higher than that of pure Cs2AgBiBr6 nanoplates, DP–RGO and Cu–RGO (Fig. 6c and d). RGO was approximately 1 wt% in all the Cs2AgBiBr6–RGO composites, which is known to be optimal in RGO-photocatalyst composites.29 The optimum DP–Cu–RGO composite contains 1 wt% Cu–RGO content with an optimal 5 wt% of Cu content in Cu–RGO (Fig. S10a†). A further increase of Cu content in Cu–RGO suppressed the photocatalytic activity for CO2 reduction, assigned to an increased Cu particle size and poorer dispersion. The CH4 selectivity also increases for DP–Cu–RGO compared with DP, from 46.0 (±0.5) to 93.0 (±0.5)%. Overall, the photocatalytic activity of DP–Cu–RGO, on a total electron (e−) consumption basis is 93 μmol e− g−1 h−1 with 1 sun, which is almost 13 times higher than that of pure DP (7 μmol e− g−1 h−1). Further, a 32% enhancement in photocatalytic activity is observed on electron consumption basis when light intensity is doubled (2 sun). However, selectivity of CH4 decreased (18%) as more H2 evolution is observed (Fig. 7a). We attribute this to the heat generated under concentrated light, changing the adsorption of H2O and CO2 on the photocatalyst surface (the reactor temperature was 22 and 39 °C under 1 and 2 sun, respectively).
 | ||
| Fig. 6 (a and c) CO, CH4, H2 and (b and d) O2 production rates from photocatalytic CO2 reduction by using water vapor as a proton source on different photocatalysts under simulated solar light for 4 h. | ||
 | ||
| Fig. 7 (a) CO, CH4 and H2 production on DP–Cu–RGO with 1 sun and 2 suns of simulated sunlight. (b) Reusability experiments for three successive runs of on DP–Cu–RGO. (c) Schematic diagram for the charge separation and transfer mechanism of CO2 reduction on DP–Cu–RGO nanocomposites under simulated sunlight. | ||
Based on our findings, this remarkable improvement in photocatalytic activity can be attributed to the enhanced charge separation and suppressed recombination of electron–hole pairs in the closely interfaced DP–Cu–RGO composites, along with higher light absorption and CO2 adsorption. Furthermore, the relatively very low photocatalytic activity of a physical mixture of Cs2AgBiBr6 nanoplates and Cu–RGO (manually mixed powder samples) confirms our argument (Fig. 6c). The total O2 evolved on both pure composite photocatalysts was consistent with a CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2 and (CO + H2):O2 theoretical stoichiometry of 1:2 and 2:1, respectively, and with the actual involvement of 4, 8, 2 and 2 e− to form O2, CH4, CO and H2, respectively. Hence, photocatalytic CO2 conversion to CH4 (and minor CO and H2) by using water vapor as a proton source was achieved without a hole scavenger such as an alcohol. Furthermore, the apparent quantum efficiency (AQE) for CH4 production on DP–Cu–RGO is found to be 0.89 (±0.21)% at 590 nm, which is interestingly very close to the AQE of CsPbBr3–Cu–RGO (1.1% at 523 nm)18 and much higher than other efficiencies reported for similar heterogeneous photocatalysts (Table S2†). Therefore, Cs2AgBiBr6 could be a potential safer alternative to CsPbBr3 containing toxic Pb2+. We also carried out various control experiments on pure Cs2AgBiBr6 nanoplates and DP–Cu–RGO composite in the absence of CO2, H2O, photocatalyst or light irradiation, and confirm that the carbon source is CO2 (Fig. S10b and c†). This was further supported by 13CO2 experiments using GCMS (Fig. S11†). Hence, we confirm that there was no involvement of a secondary carbon source from organic contamination in the photocatalytic production of CO and CH4 in this work.47,48 Finally, we examine the recyclability of the pure Cs2AgBiBr6 nanoplates and DP–Cu–RGO composite by evacuation and refilling of the reactor with a fresh CO2 and water vapor mixture for three successive times. The photocatalytic activity of the Cs2AgBiBr6 nanoplates drops drastically during the three cycles (Fig. S12†), whereas the DP–Cu–RGO composite continued to produce similar activity even on the third cycle (Fig. 7b). This is attributed to the improved chemical stability of DP–Cu–RGO due to its RGO content (see previous stability tests in Fig. S7 and S8†). As shown in Fig. S13 and S14,† there is no change in the crystal structure and morphology of DP–Cu–RGO after the three photocatalytic cycles, while DP undergoes phase change as well as decomposition to AgBr and Cs(OH)2.
:O2 and (CO + H2):O2 theoretical stoichiometry of 1:2 and 2:1, respectively, and with the actual involvement of 4, 8, 2 and 2 e− to form O2, CH4, CO and H2, respectively. Hence, photocatalytic CO2 conversion to CH4 (and minor CO and H2) by using water vapor as a proton source was achieved without a hole scavenger such as an alcohol. Furthermore, the apparent quantum efficiency (AQE) for CH4 production on DP–Cu–RGO is found to be 0.89 (±0.21)% at 590 nm, which is interestingly very close to the AQE of CsPbBr3–Cu–RGO (1.1% at 523 nm)18 and much higher than other efficiencies reported for similar heterogeneous photocatalysts (Table S2†). Therefore, Cs2AgBiBr6 could be a potential safer alternative to CsPbBr3 containing toxic Pb2+. We also carried out various control experiments on pure Cs2AgBiBr6 nanoplates and DP–Cu–RGO composite in the absence of CO2, H2O, photocatalyst or light irradiation, and confirm that the carbon source is CO2 (Fig. S10b and c†). This was further supported by 13CO2 experiments using GCMS (Fig. S11†). Hence, we confirm that there was no involvement of a secondary carbon source from organic contamination in the photocatalytic production of CO and CH4 in this work.47,48 Finally, we examine the recyclability of the pure Cs2AgBiBr6 nanoplates and DP–Cu–RGO composite by evacuation and refilling of the reactor with a fresh CO2 and water vapor mixture for three successive times. The photocatalytic activity of the Cs2AgBiBr6 nanoplates drops drastically during the three cycles (Fig. S12†), whereas the DP–Cu–RGO composite continued to produce similar activity even on the third cycle (Fig. 7b). This is attributed to the improved chemical stability of DP–Cu–RGO due to its RGO content (see previous stability tests in Fig. S7 and S8†). As shown in Fig. S13 and S14,† there is no change in the crystal structure and morphology of DP–Cu–RGO after the three photocatalytic cycles, while DP undergoes phase change as well as decomposition to AgBr and Cs(OH)2.
Photocatalytic CO2 reduction over DP–Cu–RGO is proposed as shown in Fig. 7c. Briefly, upon light irradiation, photoinduced electrons are promoted to the conduction band of the Cs2AgBiBr6 with band gap of 2.0 eV and then migrate through the DP–Cu–RGO heterostructure, eventually reaching the Cu nanoparticles.49 There, CO2, either weakly chemisorbed directly at the Cu surface or spilled over onto Cu facets in contact with the RGO support, undergoes reduction, likely via the activated CO2 in a multi proton-coupled process (Fig. 7c).50,51 In parallel, photogenerated holes in the valence band of Cs2AgBiBr6 oxidize surface-bound water to O2 and protons on the perovskite surface or on the RGO surface upon migration, possibly via Ag–O bonding.52 Protons then need to migrate across the interface at the DP–Cu–RGO heterostructure to Cu and the activated CO2 to produce CH4 (and minor CO) and H2. Here Cu plays an important role facilitating the use of photoinduced electrons to activate CO2 and form CH4.51,53,54 Along with the extended light absorption and CO2 adsorption with the addition of Cu–RGO, the direct growth of Cs2AgBiBr6 on the Cu–RGO ensures a good interface and a large contact area, which improves charge separation to facilitate the challenging eight-electron reduction of CO2 to CH4.
Conclusions
We have successfully demonstrated a fast, surfactant/ligand-free and scalable mechanochemical synthesis of Cs2AgBiBr6 double perovskite (DP) nanoplates and their nanocomposites with Cu-decorated reduced graphene oxide (Cu–RGO). The resulting Cs2AgBiBr6–Cu–RGO nanocomposite shows significantly improved photocatalytic activity for CO2 reduction to CH4 (minor CO and H2) using water vapor as proton and electron source, with 93.0 (±0.5)% CH4 selectivity and an apparent quantum efficiency of 0.89 (±0.21)%, in the absence of expensive hole scavengers such as alcohols but just simply water vapor. The optimum photocatalytic activity of 93 μmol e− h−1 g−1 (at 1 sun) on total electron consumption basis is obtained for Cs2AgBiBr6–Cu–RGO, which is 13 times higher than Cs2AgBiBr6 double perovskite nanoplates. The activity increases to 122 μmol e− h−1 g−1 at 2 suns. The high performance of the Cs2AgBiBr6–Cu–RGO nanocomposite for photocatalytic reduction of CO2 reflects the formation of an excellent interface between the Cs2AgBiBr6 and Cu–RGO, which promotes efficient photoinduced charge separation and lower recombination of electron–hole pairs. This is a result of electron transfer from the Cs2AgBiBr6 to Cu via RGO and simultaneous hole migration from Cs2AgBiBr6 to its surface or to the RGO, along with extended light absorption, larger surface area and improved CO2 adsorption. In addition, improved reusability thanks to the hydrophobic character of RGO makes this Cs2AgBiBr6–Cu–RGO photocatalyst a potential Pb-free halide perovskite photocatalyst candidate for long-term application in solar fuel production. This strategy could be readily extended to prepare diverse Pb-free halide double perovskites for solar fuel applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SK and SE thank the EPSRC grants EP/S030727/1, EP/S017615/1, EP/R035407/1, and EP/R035407/2 for financial support. The X-ray photoelectron (XPS) data collection was performed at the EPSRC National Facility for XPS (HarwellXPS), operated by Cardiff University and UCL, under Contract No. PR16195.References
- X. Yang, S. Zhang, P. Li, S. Gao and R. Cao, J. Mater. Chem. A, 2020, 8, 20897–20924 RSC.
- D. Zhang, G. Li and J. C. Yu, J. Mater. Chem., 2010, 20, 4529–4536 RSC.
- Y. Shiraishi, M. Hashimoto, K. Chishiro, K. Moriyama, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2020, 142, 7574–7583 CrossRef CAS PubMed.
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- J. Ran, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- P. Usubharatana, D. McMartin, A. Veawab and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2006, 45, 2558–2568 CrossRef CAS.
- C. Bozal-Ginesta and J. R. Durrant, Faraday Discuss., 2019, 215, 439–451 RSC.
- T. Faunce, S. Styring, M. R. Wasielewski, G. W. Brudvig, A. W. Rutherford, J. Messinger, A. F. Lee, C. L. Hill, H. deGroot, M. Fontecave, D. R. MacFarlane, B. Hankamer, D. G. Nocera, D. M. Tiede, H. Dau, W. Hillier, L. Wang and R. Amal, Energy Environ. Sci., 2013, 6, 1074–1076 RSC.
- P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834–2860 CrossRef CAS.
- Y. Dong, Y. Zhao, S. Zhang, Y. Dai, L. Liu, Y. Li and Q. Chen, J. Mater. Chem. A, 2018, 6, 21729–21746 RSC.
- H. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS.
- J. Hou, S. Cao, Y. Wu, Z. Gao, F. Liang, Y. Sun, Z. Lin and L. Sun, Chem.–Eur. J., 2017, 23, 9481–9485 CrossRef CAS PubMed.
- Y.-F. Xu, M.-Z. Yang, B.-X. Chen, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed.
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed., 2018, 57, 13570–13574 CrossRef CAS PubMed.
- S. Wan, M. Ou, Q. Zhong and X. Wang, Chem. Eng. J., 2019, 358, 1287–1295 CrossRef CAS.
- L.-Y. Wu, Y.-F. Mu, X.-X. Guo, W. Zhang, Z.-M. Zhang, M. Zhang and T.-B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed.
- Z.-C. Kong, J.-F. Liao, Y.-J. Dong, Y.-F. Xu, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS.
- S. Kumar, M. Regue, M. A. Isaacs, E. Freeman and S. Eslava, ACS Appl. Energy Mater., 2020, 3, 4509–4522 CrossRef CAS.
- W. Ke and M. G. Kanatzidis, Nat. Commun., 2019, 10, 965 CrossRef PubMed.
- T. C. Jellicoe, J. M. Richter, H. F. J. Glass, M. Tabachnyk, R. Brady, S. E. Dutton, A. Rao, R. H. Friend, D. Credgington, N. C. Greenham and M. L. Böhm, J. Am. Chem. Soc., 2016, 138, 2941–2944 CrossRef CAS PubMed.
- X. Wang, T. Zhang, Y. Lou and Y. Zhao, Mater. Chem. Front., 2019, 3, 365–375 RSC.
- G. Volonakis and F. Giustino, Appl. Phys. Lett., 2018, 112, 243901 CrossRef.
- M. R. Filip, S. Hillman, A. A. Haghighirad, H. J. Snaith and F. Giustino, J. Phys. Chem. Lett., 2016, 7, 2579–2585 CrossRef CAS PubMed.
- T. Wang, D. Yue, X. Li and Y. Zhao, Appl. Catal., B, 2020, 268, 118399 CrossRef CAS.
- L. Zhou, Y.-F. Xu, B.-X. Chen, D.-B. Kuang and C.-Y. Su, Small, 2018, 14, 1703762 CrossRef PubMed.
- Y. Wang, H. Huang, Z. Zhang, C. Wang, Y. Yang, Q. Li and D. Xu, Appl. Catal., B, 2021, 282, 119570 CrossRef CAS.
- Z. Zhang, Y. Liang, H. Huang, X. Liu, Q. Li, L. Chen and D. Xu, Angew. Chem., Int. Ed., 2019, 58, 7263–7267 CrossRef CAS PubMed.
- S. Eslava, A. Reynal, V. G. Rocha, S. Barg and E. Saiz, J. Mater. Chem. A, 2016, 4, 7200–7206 RSC.
- B. S. Gonçalves, H. G. Palhares, T. C. C. d. Souza, V. G. d. Castro, G. G. Silva, B. C. Silva, K. Krambrock, R. B. Soares, V. F. C. Lins, M. Houmard and E. H. M. Nunes, J. Mater. Res. Technol., 2019, 8, 6262–6274 CrossRef.
- R. López and R. Gómez, J. Sol-Gel Sci. Technol., 2012, 61, 1–7 CrossRef.
- A. Li, Q. Cao, G. Zhou, B. V. K. J. Schmidt, W. Zhu, X. Yuan, H. Huo, J. Gong and M. Antonietti, Angew. Chem., Int. Ed., 2019, 58, 14549–14555 CrossRef CAS PubMed.
- N. Pai, J. Lu, M. Wang, A. S. R. Chesman, A. Seeber, P. V. Cherepanov, D. C. Senevirathna, T. R. Gengenbach, N. V. Medhekar, P. C. Andrews, U. Bach and A. N. Simonov, J. Mater. Chem. A, 2020, 8, 2008–2020 RSC.
- C. Xu, S. De, A. M. Balu, M. Ojeda and R. Luque, Chem. Commun., 2015, 51, 6698–6713 RSC.
- A. H. Slavney, T. Hu, A. M. Lindenberg and H. I. Karunadasa, J. Am. Chem. Soc., 2016, 138, 2138–2141 CrossRef CAS PubMed.
- W. Zhu, L. Zhang, P. Yang, C. Hu, H. Dong, Z.-J. Zhao, R. Mu and J. Gong, ACS Energy Lett., 2018, 3, 2144–2149 CrossRef CAS.
- G. B. Hoflund, Z. F. Hazos and G. N. Salaita, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 11126–11133 CrossRef CAS.
- G. Yan, B. Jiang, Y. Yuan, M. Kuang, X. Liu, Z. Zeng, C. Zhao, J.-H. He and W. Mai, ACS Appl. Mater. Interfaces, 2020, 12, 6064–6073 CrossRef CAS PubMed.
- H. Yu, P. Xu, D. W. Lee and X. Li, J. Mater. Chem. A, 2013, 1, 4444–4450 RSC.
- A. I. Aria, P. R. Kidambi, R. S. Weatherup, L. Xiao, J. A. Williams and S. Hofmann, J. Phys. Chem. C, 2016, 120, 2215–2224 CrossRef CAS PubMed.
- W. Gao, C. Ran, J. Xi, B. Jiao, W. Zhang, M. Wu, X. Hou and Z. Wu, ChemPhysChem, 2018, 19, 1696–1700 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol, T. Siemieniewska, Handbook of Heterogeneous Catalysis, 2008, pp. 1217–1230, DOI:10.1002/9783527610044.hetcat0065.
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- W.-J. Ong, L.-L. Tan, S.-P. Chai and S.-T. Yong, Chem. Commun., 2015, 51, 858–861 RSC.
- S. C. Rood, H. B. Ahmet, A. Gomez-Ramon, L. Torrente-Murciano, T. R. Reina and S. Eslava, Appl. Catal., B, 2019, 242, 358–368 CrossRef CAS.
- Z. Xiao, W. Meng, J. Wang and Y. Yan, ChemSusChem, 2016, 9, 2628–2633 CrossRef CAS PubMed.
- F. Igbari, Z.-K. Wang and L.-S. Liao, Adv. Energy Mater., 2019, 9, 1803150 CrossRef.
- C.-C. Yang, Y.-H. Yu, B. van der Linden, J. C. S. Wu and G. Mul, J. Am. Chem. Soc., 2010, 132, 8398–8406 CrossRef CAS PubMed.
- S. Kumar, M. A. Isaacs, R. Trofimovaite, L. Durndell, C. M. A. Parlett, R. E. Douthwaite, B. Coulson, M. C. R. Cockett, K. Wilson and A. F. Lee, Appl. Catal., B, 2017, 209, 394–404 CrossRef CAS.
- X.-J. Lv, S.-X. Zhou, C. Zhang, H.-X. Chang, Y. Chen and W.-F. Fu, J. Mater. Chem., 2012, 22, 18542–18549 RSC.
- M. Regue, S. Kumar and S. Eslava, in Heterogeneous Catalysis for Energy Applications, The Royal Society of Chemistry, 2020, pp. 80–115, 10.1039/9781788019576-00080.
- D. Raciti and C. Wang, ACS Energy Lett., 2018, 3, 1545–1556 CrossRef CAS.
- F. Sordello, M. Ghibaudo and C. Minero, ACS Appl. Mater. Interfaces, 2017, 9, 23800–23809 CrossRef CAS PubMed.
- N. Li, B. Wang, Y. Si, F. Xue, J. Zhou, Y. Lu and M. Liu, ACS Catal., 2019, 9, 5590–5602 CrossRef CAS.
- N. Sharma, T. Das, S. Kumar, R. Bhosale, M. Kabir and S. Ogale, ACS Appl. Energy Mater., 2019, 2, 5677–5685 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available in DOI: 10.1039/d1ta01281a. All data generated during this research are openly available from the Imperial Research Data Repository. See DOI: 10.14469/hpc/8188 |
| This journal is © The Royal Society of Chemistry 2021 |