
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of Mg and Ti doping in O3 type NaNi1/2Mn1/2O2 on reversibility and phase transition during electrochemical Na intercalation†
Kei
Kubota
 ab,
Naoya
Fujitani
a,
Yusuke
Yoda
a,
Kazutoshi
Kuroki
a,
Yusuke
Tokita
a and
Shinichi
Komaba
*ab
ab,
Naoya
Fujitani
a,
Yusuke
Yoda
a,
Kazutoshi
Kuroki
a,
Yusuke
Tokita
a and
Shinichi
Komaba
*ab
aDepartment of Applied Chemistry, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: komaba@rs.tus.ac.jp
bElements Strategy Initiative for Catalysts and Batteries (ESICB), Kyoto University, 1-30 Goryo-Ohara, Nishikyo-ku, Kyoto 615-8245, Japan
First published on 21st May 2021
Abstract
O3 type layered sodium nickel manganese oxide, NaNi1/2Mn1/2O2, which is isostructural with α-NaFeO2, has attracted attention as a promising positive electrode material for sodium-ion batteries owing to its large reversible capacity of ca. 200 mA h g−1. To improve the cycle stability for practical use, O3 type NaNi1/2Mn1/2O2 materials with Mg or/and Ti substitution are synthesized. The materials with Mg or Ti substitution exhibit better capacity capability, and Mg and Ti co-substituted material demonstrates even better capacity capability, with an initial discharge capacity of 200 mA h g−1 without any capacity loss due to substitution. Substitution of Mg2+ and Ti4+, which are larger ions than Ni2+ or Mn4+, results in a larger in-plane lattice of the O3 type structure, in contrast to the shrinkage during charging, and this has the potential to delay the phase transition during charging. In contrast to the non-substituted NaNi1/2Mn1/2O2, the Mg and Ti co-substituted material demonstrates more continuous phase transitions and lattice parameter changes, and no significant shrinkage of the interslab spacing in the layered structure, as evidenced by ex situ and operando X-ray diffraction. The coexistence of Mg and Ti enhances not only the reversibility of the structural change but also the structural stability at the surface, resulting in the excellent sodium battery performance.
Introduction
Since acceptable charge–discharge cycling performance of a sodium-ion battery full cell at room temperature was first demonstrated by using hard carbon as a negative electrode and Na[Ni1/2Mn1/2]O2 as a positive electrode material,1 much effort has been devoted by many researchers in developing further high-energy electrode materials.2 We recently developed an extremely high-capacity hard carbon, and the negative electrode achieved a large reversible capacity of more than 450 mA h g−1 in a non-aqueous Na cell.3 On the other hand, a positive electrode material with outstanding Na battery performance like LiCoO2 in lithium-ion batteries has been not found yet.As Ni-rich Ni–Mn–Co-based layered oxides have been extensively studied in lithium-ion batteries, Ni–Mn-based layered oxides have attracted attention as high capacity positive electrode materials for sodium-ion batteries.4 In particular, O3 type (α-NaFeO2 type) layered oxides are promising candidates as practically feasible high-capacity positive electrode materials because of the high capacity obtainable in the moderately high voltage range to avoid severe anodic electrolyte decomposition5 and almost the stoichiometric Na amount in the formula to deliver a balanced initial discharge capacity with a charge capacity.6
Among the O3 type layered oxides, O3 type Na[Ni1/2Mn1/2]O2 is very attractive owing to the large reversible capacity of ca. 200 mA h g−1.7 However, the available capacity was limited to 120 mA h g−1 in the full cell1 because of the narrower voltage range of approximately 2.0–3.8 V vs. Na+/Na to avoid the severe capacity decay by charging beyond 4.0 V. The capacity decay is known to be due to the significant change in interslab spacing by extraction of almost all sodium ions from the structure8 and surface deterioration.9 To improve the cycle stability, coating with MgO was carried out by Hwang et al., and the performance is partly enhanced.9 As we found in P2 type Na2/3Ni1/3Mn2/3O2,10 Ti-substitution is effective to enhance the performance, but the obtained reversible capacity in the O3 type was limited to less than 150 mA h g−1.11,12 Recently, Tarascon's group reported significantly improved performance by the co-substitution of Ti and Cu in Na[Ni1/2Mn1/2]O2 as Na[Ni1/2−xCuxMn1/2−yTiy]O2 (ref. 13) and by that of Ti and Zn as Na[Ni1/2−xZnxMn1/2−yTiy]O2.14
Here, we have synthesized O3 type Na[Ni1/2Mn1/2]O2 with Mg and/or Ti substitution as Na[Ni1/2−xMn1/2−yMgxTiy]O2 (x = 0 and 1/18, y = 0 and 1/6) and investigated the influence of the titanium and magnesium dopants on the crystal structure and electrochemical Na intercalation properties. To elucidate the relationship between electrochemical and structural properties, structural changes in the bulk and surface of the layered oxides during electrochemical Na (de)intercalation are systematically studied by using operando and ex situ X-ray diffraction and X-ray absorption spectroscopy as well as TEM observation. Based on the results, the influence of the dopants titanium and magnesium is discussed in order to understand the suitable dopants for layered oxide positive electrode materials for Na-ion batteries.
Experimental
Synthesis procedures
Na[Ni1/2−xMn1/2−yMgxTiy]O2 (x = 0 and 1/18, y = 0 and 1/6), i.e. Na[Ni1/2Mn1/2]O2, Na[Ni4/9Mn1/2Mg1/18]O2, Na[Ni1/2Mn1/3Ti1/6]O2, and Na[Ni4/9Mn1/3Mg1/18Ti1/6]O2, was prepared by a solid–state reaction. Reagent grade Na2CO3 (Nacalai Tesque, Inc., 99.8%), Ni(OH)2 (Wako Co., Inc., >95.0%), and Mn2O3 prepared by calcining MnCO3 (Kishida Chem. Co., Ltd, 44% as Mn content) at 700 °C were mixed at stoichiometric ratio by planetary ball-milling with acetone at 600 rpm for 12 h by using a planetary ball mill (PULVERISETTE 7 classic line, FRITSCH GmbH). The mixture was dried and pressed into pellets, followed by heating for 24 h in air at 800 °C. After the calcination, the samples were taken out from the furnace without the cooling process, and then immediately transferred into an argon-filled glove box. The samples were cooled to room temperature in the glove box and were kept inside to avoid the exposure to moisture in air. The synthesized samples of Na[Ni1/2Mn1/2]O2, Na[Ni4/9Mn1/2Mg1/18]O2, Na[Ni1/2Mn1/3Ti1/6]O2, and Na[Ni4/9Mn1/3Mg1/18Ti1/6]O2 are, hereinafter, referred to as “Non-sub”, “Mg-sub”, “Ti-sub”, and “Mg–Ti-sub”, respectively.Structural and morphological characterization
Structures of the Na[Ni1/2−xMn1/2−yMgxTiy]O2 samples were identified by using an X-ray diffractometer (SmartLab, Rigaku Corporation) equipped with a high-speed position sensitive detector (D/teX Ultra, Rigaku Corporation) and Ni-filtered Cu Kα radiation as an X-ray source. A homemade air-tight sample holder was used for the XRD measurements to avoid the sample exposure to air. Detailed structures of the samples were determined with a synchrotron X-ray diffractometer at BL02B2 (ref. 15) and BL19B2 (ref. 16) of SPring-8 in Japan, equipped with a large Debye–Scherrer camera. For ex situ measurements, samples were electrochemically prepared in the coin type cells, the constitution of which is described in the next section. The composite electrodes, taken out from the coin cells, were rinsed with dimethyl carbonate (DMC) to remove an electrolyte salt and then dried at room temperature in an Ar-filled glovebox. The oxide composites were separated from the aluminum current collectors and crushed into powders. As-prepared and electrochemically tested samples were put into glass capillaries with 0.3 and 0.5 mm in diameter, respectively. The glass capillaries were sealed with a resin in the glovebox or by heating and melting the end of glass capillaries to eliminate sample exposure to air. To minimize the effect of X-ray absorption by the samples, the wavelength of the incident X-ray beam was set to 0.5 Å using a silicon monochromator and was calibrated with NIST standard reference material 674b CeO2 as an external standard reference. Structural parameters were refined by the Rietveld method with the program RIETAN-FP.17 The observed pattern was compared to the simulated patterns using the FAULTS program18 based on the structural models having stacking faults. Schematic illustrations of the crystal structures of the sample were drawn using the program VESTA.19 Structural changes during an initial charging process were examined for O3 type Na[Ni1/2Mn1/2]O2 and Na[Ni4/9Mn1/3Mg1/18Ti1/6]O2 as representatives of the samples and by an operando XRD technique using a battery-cell attachment (Rigaku Corporation)20 and the data were collected using an X-ray diffractometer (MultiFlex, Rigaku Corporation) equipped with a high-speed position sensitive detector (D/teX Ultra, Rigaku Corporation) and non-monochromatized but Ni-filtered Cu Kα radiation as an X-ray source. Particle morphology of the samples was confirmed with a scanning electron microscope (SEM, JCM-6000, JEOL Ltd.). The outermost surface structure and morphology of the samples were observed with a JEM-2100F transmission electron microscope (JEOL Ltd.) at an accelerating voltage of 200 kV. The composition of the samples was confirmed by inductively coupled plasma atomic emission spectrometry (ICP-AES) using SPS3500 (Hitachi High-Tech Corporation).Electrochemical characterization
Positive electrodes were prepared by the following procedures. A slurry with a mixture of 80 wt% Na[Ni1/2−xMn1/2−yMgxTiy]O2, 10 wt% acetylene black (AB, Strem Chemicals, Inc.) and 10 wt% poly(vinylidene fluoride) (PVdF, Polysciences, Inc.) with N-methyl pyrrolidone (Kanto Chemical Co., Ltd.) as a solvent was prepared and pasted on aluminum foil and then dried at 80 °C under vacuum. Single side coated electrodes were punched into disks of 10 mm or 15 mm in diameter and evaluated as a positive electrode for the R2032 type coin cells with an Al-clad type of stainless-steel cap for the positive electrode side (Hohsen Corp.). The active material loading was ∼2.5 mg cm−2. The separator used was a glass fiber filter (GB-100R, ADVANTEC, Co.), the electrolyte solution was 1.0 mol dm−3 NaPF6 dissolved in the solvent mixture of ethylene carbonate (EC) and propylene carbonate (PC) at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol/vol (Kishida Chemical Co., Ltd.), and the negative electrode was sodium metal (purity > 99%, Kanto Chemical Co., Ltd). The electrode preparation and fabrication of the coin cells were carried out in an Ar-filled glove box. Galvanostatic charge–discharge tests were conducted with a charge/discharge measurement system (TOSCAT-3100, TOYO System Co., Ltd.) in the voltage range of 2.2–4.5 V at C/20 (ca. 12.0 mA g−1) at 25 °C.
:1 vol/vol (Kishida Chemical Co., Ltd.), and the negative electrode was sodium metal (purity > 99%, Kanto Chemical Co., Ltd). The electrode preparation and fabrication of the coin cells were carried out in an Ar-filled glove box. Galvanostatic charge–discharge tests were conducted with a charge/discharge measurement system (TOSCAT-3100, TOYO System Co., Ltd.) in the voltage range of 2.2–4.5 V at C/20 (ca. 12.0 mA g−1) at 25 °C.
Hard X-ray absorption spectroscopy
Hard X-ray absorption spectroscopy was performed at beam line BL-12C in the Photon Factory, High Energy Accelerator Research Organization (KEK), Japan. The spectra were collected with a silicon monochromator in the transmission mode. The intensities of incident and transmitted X-ray were measured using ionization chambers at room temperature. The energy was calibrated by setting the first inflection point in the spectrum of a Cu foil to 8979 eV. Electrode samples were electrochemically prepared with the coin cells. The positive electrodes were taken out from the coin cells, rinsed with DMC solvent, and then dried at room temperature in an Ar-filled glovebox. The several electrodes with Al-foil current collectors were stacked and sealed in an oxygen and moisture barrier polymer film in the Ar-filled glovebox. Absorption spectroscopy data were normalized and treated using the ATHENA program in the Demeter software package.21 Fourier transform of χ(k)k3–k plots, where χ and k are oscillatory components of normalized absorption and angular wavenumber, respectively, was performed using the ATHENA program.Results and discussion
Characterization of as-prepared Na[Ni1/2−xMn1/2−yMgxTiy]O2
Crystal structures of the synthesized samples were first identified by an XRD method. Fig. 1a displays powder synchrotron XRD patterns of as-prepared Non-sub, Mg-sub, Ti-sub, and Mg–Ti-sub samples. Observed Bragg diffraction peaks can be indexed as an O3 type layered phase with a space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, which is isostructural to α-NaFeO2.22 The formation of a rock salt type NiO impurity is often a concern in the synthesis of Na[Ni1/2Mn1/2]O2-related materials.23–25 When the diffraction angle region around 13.7° (λ = 0.5 Å) is magnified, one can see NiO and MgO like peaks with quite small intensities (see Fig. S1 in the ESI†). Considering the rock salt type impurities, Rietveld refinements were carried out on the synchrotron XRD patterns, and the refinement results are shown in Fig. S2–S5 and Table S1–S4.† The refinement results for all the samples represent small differences between the observed and calculated intensities and low R values, indicating good agreement between the observed and calculated patterns. The refined lattice parameters a = 2.96277(4) Å and c = 15.9132(2) Å for Non-sub are identical with those in previous reports.7,26,27 In our Non-sub sample, the concerned mass fraction of the NiO phase is quite small (0.2 mass%), indicating that an almost NiO-free O3 type phase of Na[Ni1/2Mn1/2]O2 is synthesized in this study.
m, which is isostructural to α-NaFeO2.22 The formation of a rock salt type NiO impurity is often a concern in the synthesis of Na[Ni1/2Mn1/2]O2-related materials.23–25 When the diffraction angle region around 13.7° (λ = 0.5 Å) is magnified, one can see NiO and MgO like peaks with quite small intensities (see Fig. S1 in the ESI†). Considering the rock salt type impurities, Rietveld refinements were carried out on the synchrotron XRD patterns, and the refinement results are shown in Fig. S2–S5 and Table S1–S4.† The refinement results for all the samples represent small differences between the observed and calculated intensities and low R values, indicating good agreement between the observed and calculated patterns. The refined lattice parameters a = 2.96277(4) Å and c = 15.9132(2) Å for Non-sub are identical with those in previous reports.7,26,27 In our Non-sub sample, the concerned mass fraction of the NiO phase is quite small (0.2 mass%), indicating that an almost NiO-free O3 type phase of Na[Ni1/2Mn1/2]O2 is synthesized in this study.
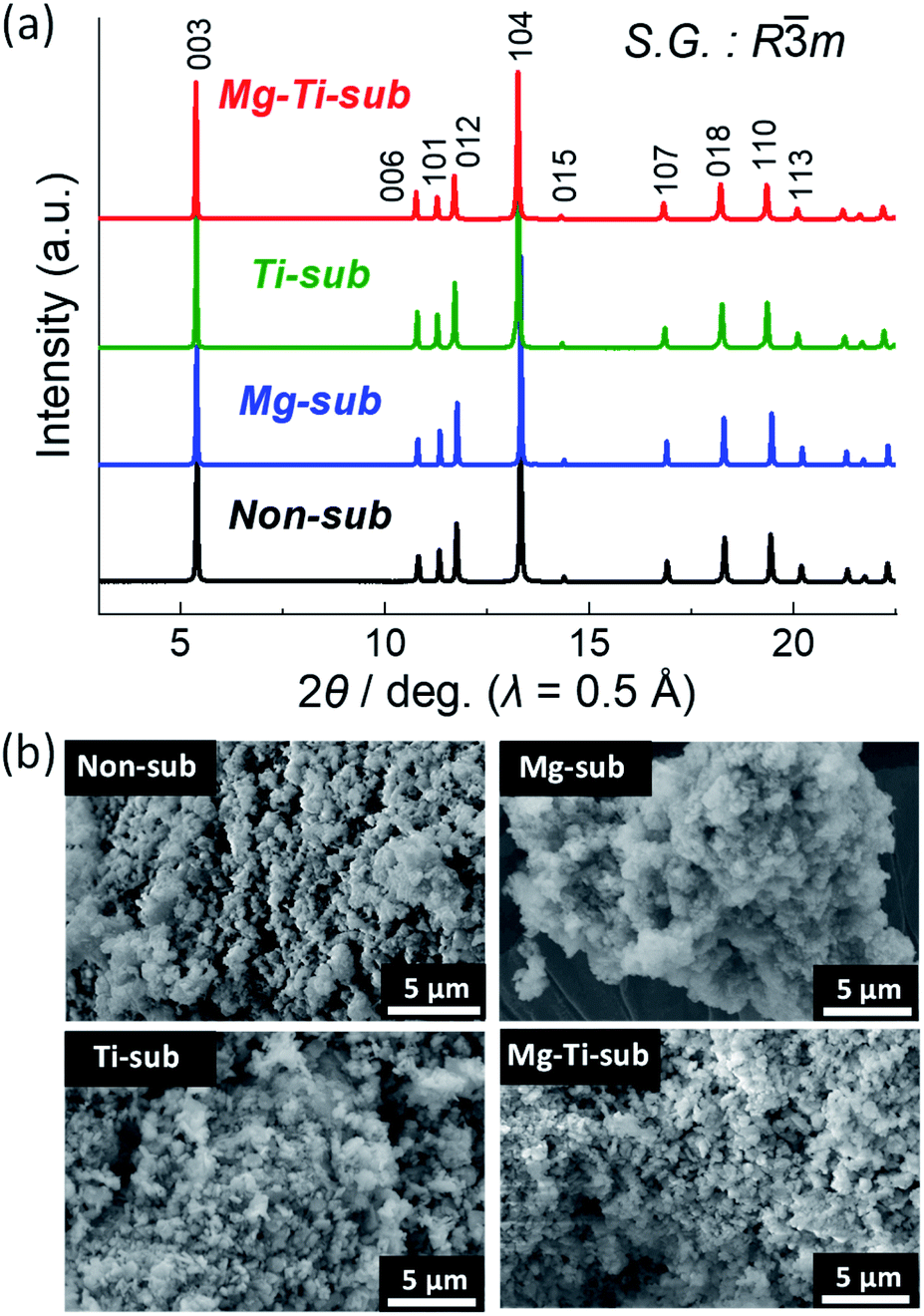 | ||
| Fig. 1 (a) Synchrotron powder XRD patterns and (b) SEM images of pristine Na[Ni1/2Mn1/2]O2 (Non-sub), Na[Ni4/9Mn1/2Mg1/18]O2 (Mg-sub), Na[Ni1/2Mn1/3Ti1/6]O2 (Ti-sub), and Na[Ni4/9Mn1/3Mg1/18Ti1/6]O2 (Mg–Ti-sub). | ||
Similar to the Non-sub sample, two rock-salt type phases are detected as MgO and MgO–NiO solid-solution phases in the XRD patterns of Mg-sub and Mg–Ti-sub samples (see Fig. S1†). For the Rietveld refinements, three phases of O3 type NaMeO2 (Me = Ni, Mn, Mg, Ti), (Mg, Ni)O, and MgO were used and the results are shown in Fig. S3 and S5.† The refined phase fractions of NaMeO2, (Mg, Ni)O, and MgO for the Mg-sub sample are 98.77 mass% (97.04 mol%), 0.78 mass% (2.03 mol%), and 0.45 mass% (0.93 mol%), respectively, and those for the Mg–Ti-sub sample are 99.77 mass% (99.45 mol%), 0.17 mass% (0.45 mol%), and 0.06 mass% (0.10 mol%), respectively. The trace amounts of rock-salt type MgO and Mg1−xNixO are indeed detected in the Mg-doped samples as previously reported in the literature,9,28 but the amounts in our Mg-substituted samples are negligibly small compared to the reported values.
In contrast to the three samples, no rock-salt type phase is confirmed in the Ti-sub sample (see Fig. S4†). Even in the Mg and Ti co-substitution, the phase fractions of MgO and Mg1−xNixO phases in Mg–Ti-sub are slightly lower than those in Mg-sub. Ti-substitution might disturb the formation of rock-salt type phases under our synthesis conditions although a NiO impurity phase was found in the reported patterns of Na[Ni1/2Mn1/2−xTix]O2 (0 ≤ x ≤ 0.5).12 Furthermore, Ti-containing samples of Ti-sub and Mg–Ti-sub exhibit larger values of lattice volume than those in Non-sub and Mg-sub, which agrees with a significant increase in ionic radii by replacing 1/6 of Mn4+ (0.53 Å) with Ti4+ (0.605 Å) than that by replacing 1/18 of Ni2+ (0.69 Å) with Mg2+ (0.72 Å) in six-folded coordination.29 All the results prove that Mg or Ti substitution and Mg and Ti co-substitution are successful in Na[Ni1/2Mn1/2]O2 to form an isostructural O3 type phase. ICP-AES data in Table S5† also confirm that metal ratios of the Na[Ni1/2−xMn1/2−yMgxTiy]O2 samples are almost identical to the targeted ones.
Particle morphology of the as-prepared samples was observed by using SEM as shown in Fig. 1b. All the four samples show almost the same morphology. Particles of several hundred nanometers in diameter are agglomerated homogeneously.
Electrochemical properties
Electrochemical properties of the Na[Ni1/2−xMn1/2−yMgxTiy]O2 electrodes were examined by using coin-type aprotic Na cells. The mixture of EC:PC (1:1 v/v) was used as an electrolyte solvent because the Non-sub electrode in a Na cell filled with 1.0 mol dm−3 NaPF6 dissolved in EC:PC (1:1 v/v) demonstrates a higher initial Coulombic efficiency than those in the cases of PC or EC:DEC (1:1 v/v) solvent (see Fig. S6†). Fig. 2a displays galvanostatic charge/discharge curves in the wide voltage range of 2.2–4.5 V at a current density of 12 mA g−1 for the initial 20 cycles. Mg-sub and Ti-sub electrodes deliver slightly smaller discharge capacities of ca. 185 mA h g−1 than 202 mA h g−1 for Non-sub at the initial cycle. Mg–Ti-sub delivers a large reversible capacity of 198 mA h g−1, which is almost identical to that of Non-sub. The initial Coulombic efficiency of Non-sub and Mg-sub electrodes is almost the same value of 80% while Ti-sub electrode exhibits slightly lower efficiency of 76%. Mg–Ti-sub electrode demonstrates the highest efficiency of 86%.
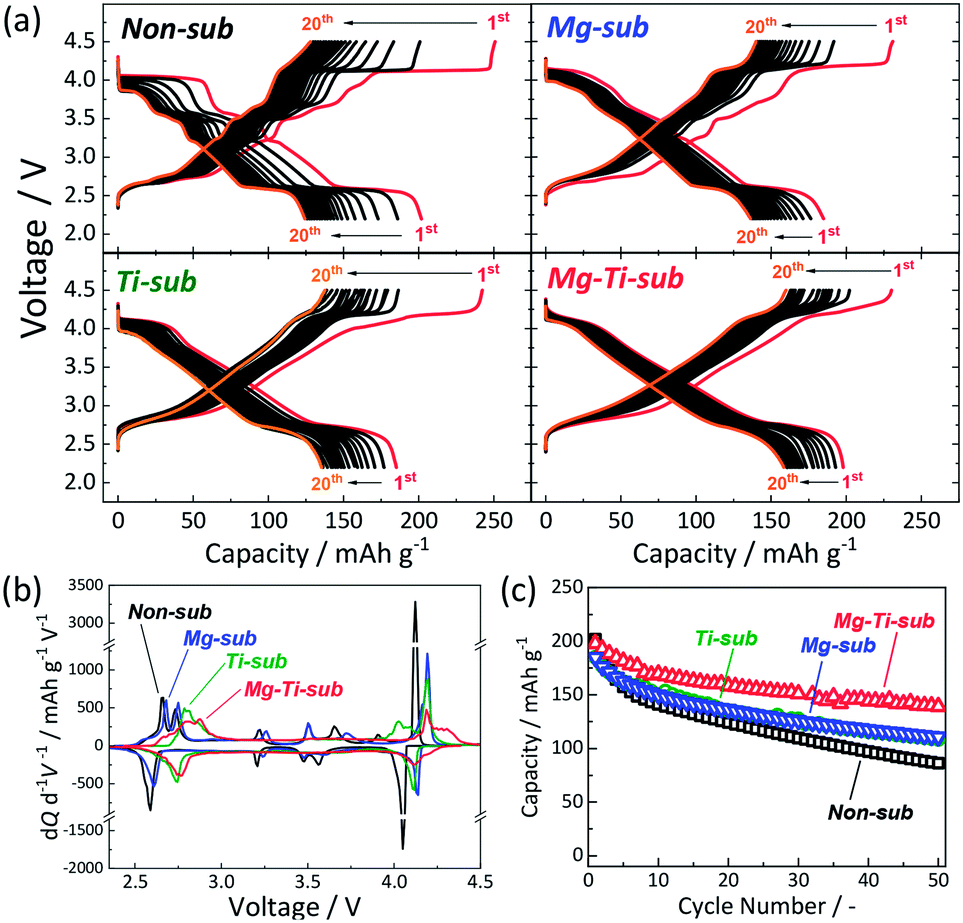 | ||
| Fig. 2 (a) Charge–discharge curves, (b) dQ/dV curves, and (c) cycle stability of Non-sub, Mg-sub, Ti-sub, and Mg–Ti-sub electrodes in Na cells. Galvanostatic charge–discharge tests were carried out at a current density of 12 mA g−1. | ||
More obvious differences are found in the initial charge/discharge voltage profiles. Ti-containing samples of Ti-sub and Mg–Ti-sub represent sloping voltage profiles while Non-sub and Mg-sub samples show stepwise voltage profiles which are obviously observed as redox peaks in the differential curves in Fig. 2b. Compared to Non-sub and Mg-sub, Ti-sub and Mg–Ti-sub exhibit smaller numbers of redox peaks in the differential profiles. Such sloping voltage profiles were consistent with those reported for O3 type Na[Ni1/2Mn1/2−xTix]O2 (0 ≤ x ≤ 0.5),12,30 and P2 type Na2/3[Ni1/3Mn2/3−xTix]O2,10 and suppressed phase evolution is expected for Ti-sub and Mg–Ti-sub. Furthermore, the Ti-substitution raises the redox voltages of the plateau region in 2.5–3.0 V in Fig. 2a (peak shift towards higher voltage in the differential curves in Fig. 2b), which are consistent with those reported for Na0.9[Ni0.45MnxTi0.55−x]O2 (ref. 30) and Na2/3[Ni1/3Mn2/3−xTix]O2.10
In contrast to the clear difference between Mg-containing and Ti-containing electrodes in the voltage profile, in the cycle performance (Fig. 2c), Mg-sub and Ti-sub electrodes similarly exhibit improved cycle stability for 50 cycles with ca. 60% of the initial discharge capacities at the 50th cycle compared to 43% of that of Non-sub. The co-substitution demonstrates further enhanced cycle performance. The Mg–Ti-sub electrode delivers 70% of the initial discharge capacity at the 50th cycle. Similar significant improvement is observed for Cu and Ti co-substituted13,24,31 and Zn and Ti co-substituted Na[Ni1/2Mn1/2]O2.14 Thus, dual-substitution with a tetravalent titanium ion and a divalent metal ion, which includes not only Cu2+ and Zn2+ ions but also Mg2+ ion, is effective in improving the electrochemical properties of Na[Ni1/2Mn1/2]O2 and significantly enhances cycle stability even in the wide voltage range including the high potential region above 4.0 V vs. Na+/Na.
X-ray absorption spectroscopy
To confirm the oxidation state of nickel and manganese ions as well as titanium ions in pristine and electrochemically tested Na[Ni1/2−xMn1/2−yMgxTiy]O2, hard X-ray absorption spectroscopy was carried out for Non-sub and Mg–Ti-sub sample electrodes as representatives of the synthesized samples. Fig. 3a and b display ex situ Ni K-edge X-ray absorption near-edge structure (XANES) spectra of Non-sub and Mg–Ti-sub electrodes, respectively. Ni K-edge for both pristine samples is located at a similar energy to that of Ni2+O, suggesting that nickel ions have a divalent state in both the pristine Non-sub and Mg–Ti-sub electrodes. The Ni K-edge gradually shifts towards higher energy through a charging process, and the K-edge energies after fully charging to 4.5 V are almost the same as that for the O2 type [Ni1/34+Mn2/3]O2.32 Upon discharging to 2.2 V, the Ni K-edge shifts back to the initial energy position. The results suggest a reversible redox reaction with the Ni2+/4+ redox couple for both the Non-sub and Mg–Ti-sub electrodes during charging and discharging processes in Na cells. Corresponding to the reversible change in Ni K-edge energy, a reversible change in the Ni–O bond distance, decreasing during charging and increasing during discharging, is confirmed in the Fourier-transform Ni K-edge extended X-ray absorption fine structure (EXAFS) spectra in Fig. S7a and S7b.†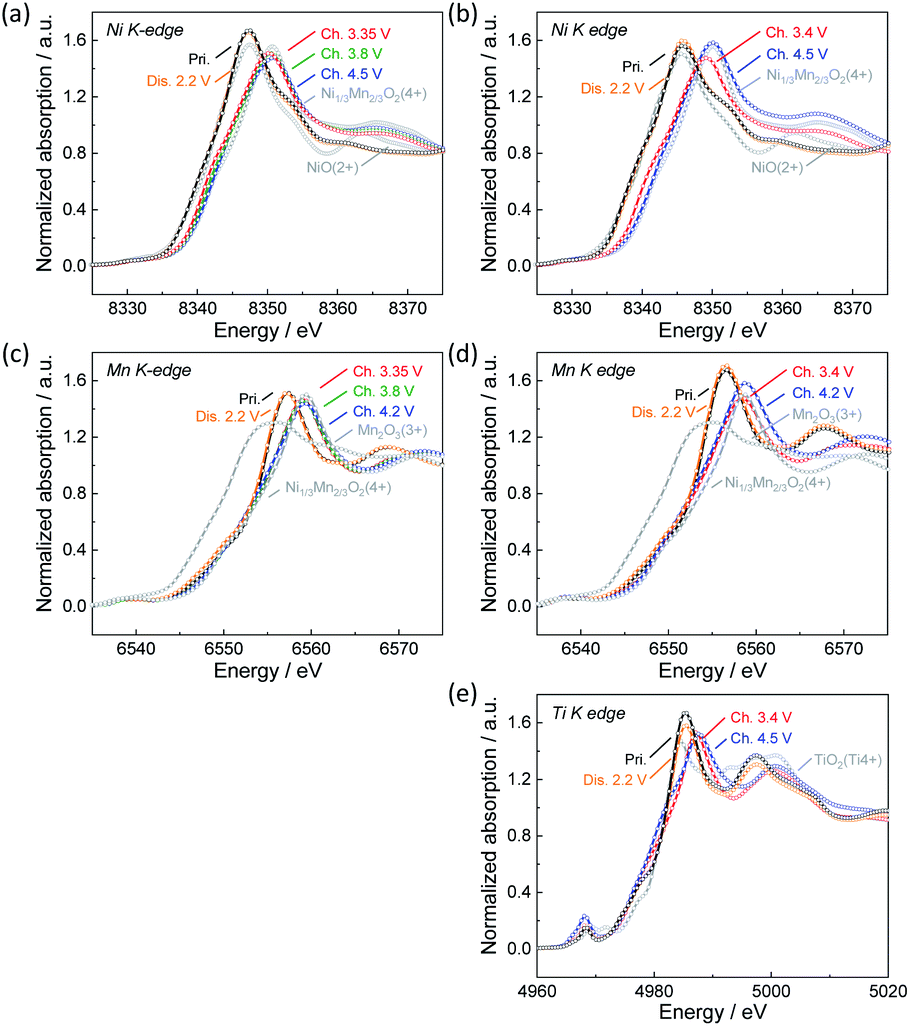 | ||
| Fig. 3 Ex situ XANES spectra of (a) and (c) Non-sub and (b), (d) and (e) Mg–Ti-sub electrodes at (a) and (b) Ni K-edge, (c) and (d) Mn K-edge, and (e) Ti K-edge. The tested electrodes were prepared by charging and discharging in Na cells followed by taking the electrodes from the cells. | ||
In contrast to the Ni K-edge, no significant changes in Mn K-edge energy are observed in the ex situ Mn K-edge spectra during charge and discharge for both the Non-sub and Mg–Ti-sub electrodes as shown in Fig. 3c and d, respectively. The Mn K-edge energies of both samples are almost the same as that for [Ni1/3Mn2/34+]O2,32 indicating that manganese ions are tetravalent and electrochemically inactive during charge and discharge. Fourier-transform EXAFS spectra in Fig. S7c and S7d† clearly confirm no significant changes in Mn–O bond distances. On the other hand, the maximum absorption peak, the so-called white line, located at ca. 6557 eV, is slightly shifted to ca. 2 eV higher energy side by charging and reversibly returns to its original position by discharging. This indicates reversible changes in the local structures of manganese coordination as previously reported for Na[Ni1/2Mn1/2]O2.7 We expected the change in Mn-to-Me distance (Me = Ni, Mn, Mg, and Ti) to be different from the change in Ni-to-Me distance, but there is no significant difference, resulting in almost the same degree of contraction of Mn–Me and Ni–Me distances on charge and elongation on discharge. The energy change of the white line at the Mn K-edge is probably due to the change in the local structure in response to the redox of nickel ion. Between Non-sub and Mg–Ti-sub electrodes, the redox behavior of electrochemically active nickel and inactive manganese ions is found to be very similar, consistent with that previously reported for Na[Ni1/2Mn1/2]O2 (ref. 7) and Na[Ni1/2Mn1/2−xTix]O2 (0 ≤ x ≤ 0.5).12 It should be noted that Ti K-edge XANES spectra in Fig. 3e and Fourier-transform EXAFS spectra in Fig. S7e† exhibit slight but certain changes in K-edge energy and Ti–O bonds, respectively, during charge and discharge. Tetravalent titanium ions at octahedral sites have an electronic 3d0 configuration. In the octahedrally coordinated d0 transition metal, a pseudo (or second-order) Jahn–Teller effect (PJTE) occurs when the empty d-orbitals of the metal mix with the filled p-orbitals of the ligands (oxide ions in our case).33 The PJTE produces a distorted structure with a low-symmetry configuration like the Jahn–Teller effect. Thus, as proposed for O3–Na[Ti1/4Fe1/4Co1/4Ni1/4]O2,34 the PJTE may be related to the local structural changes during charge and discharge in the present study. Since the Mg–Ti-sub electrode delivers almost the same discharge capacity as the Non-sub electrode, almost the same amount of Na should be extracted by charging, even thouth the redox-inactive Mg is subsituted. This implies that ligands of oxide ions partially participate in the redox reaction of Mg–Ti-sub.
Operando and ex situ XRD
 | ||
| Fig. 4 Contour plots of operando XRD patterns for (a) Non-sub and (b) Mg–Ti-sub electrodes during 1st charging to 4.5 V. | ||
Fig. 4a shows that the pristine O3 type Na[Ni1/2Mn1/2]O2 phase changes in rhombohedral O3 → monoclinic O′3 → monoclinic P′3 and hexagonal P3 → monoclinic O′3 → rhombohedral O3 during the initial charging process. The first half sequence until P3 phase formation is almost the same as that previously reported with ex situ XRD patterns.7Operando XRD successfully reveals the complicated multiple structural changes without the formation of a hydrated phase, which is often observed in ex situ XRD patterns due to slight leakage of the sealed sample holder and moisture exposure of the charged materials despite the conventional argon atmosphere measurement.7,35,36
As we reported previously, by Na extraction upon charging, the O3 type phase first transforms into the O′3 type one as a two-phase reaction in the approximate compositional range of 0.94 > x > 0.85 in Nax[Ni1/2Mn1/2]O2.7 According to Delmas' notation, the O′3 type structure refers to a distorted O3 type structure, generally having a monoclinic lattice.37 The reflections of the O3 type phase, such as 101, 104, and 018, disappear with reduced peak intensity, and new peaks corresponding to the O′3 type phase, such as 110, 20![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , 11, 20
, 11, 20![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) , 111, and 11 peaks, appear with increasing peak intensity during the O3–O′3 transition (see magnified XRD patterns in Fig. S9† with the peak intensities attributed to an Al-coated Be window and an Al current collector subtracted). The 110/20, 20/111, and 11/202 doublets in the O′3 type phase correspond to 101, 104, and 018 singlet reflections in the O3 type phase, respectively, based on the crystallographic relationship between the monoclinic cell and the rhombohedral one with hexagonal setting as follows:38
, 111, and 11 peaks, appear with increasing peak intensity during the O3–O′3 transition (see magnified XRD patterns in Fig. S9† with the peak intensities attributed to an Al-coated Be window and an Al current collector subtracted). The 110/20, 20/111, and 11/202 doublets in the O′3 type phase correspond to 101, 104, and 018 singlet reflections in the O3 type phase, respectively, based on the crystallographic relationship between the monoclinic cell and the rhombohedral one with hexagonal setting as follows:38
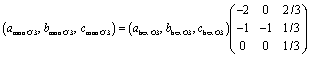 | (1) |
However, the peak splitting is not continuous, suggesting a two-phase reaction for the O3–O′3 transition. The corresponding flat voltage plateau is confirmed at 2.65 V on charge in Fig. 2a.
In the subsequent O′3–P′3 transition in the approximate compositional range of 0.85 > x > 0.74 in Nax[Ni1/2Mn1/2]O2, the reflections of the O′3 type phase, such as 110, 20, 11, 20, 111, and 11, disappear with reduced peak intensity, and new peaks corresponding to the P′3 type phase, such as 110, 20, 11, 11, and 11 peaks, appear at the diffraction angles different from those for the O′3 type one as shown in Fig. 4a, S8a and S9a,† indicating a two-phase reaction for the O′3–P′3 transition. The corresponding flat charging voltage plateau is confirmed at 2.75 V in Fig. 2a. P′3 structure generally has a low-symmetry monoclinic unit cell, unlike the high-symmetry rhombohedral P3 type one.39 A vectorial relationship between the rhombohedral P3 type cell in hexagonal setting and the monoclinic P′3 type one is formulated as follows:40
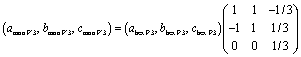 | (2) |
In the early P′3 phase region of 0.74 > x > 0.70 in Nax[Ni1/2Mn1/2]O2 just after the O′3–P′3 transition, except for the 00l reflections, diffraction peaks such as 201 and 11 and 202 and 11 reflection pairs are very low in intensity, and the peak positions of the pairs gradually become close to each other, like broad and anisotropic reflections of 015 and 018, respectively, in the rhombohedral P3 phase (Fig. 4a and S9a†). Similar anisotropic peak-broadening was reported for O′3–P′3 transition in NaxVO2, which is related to the formation of stacking faults.41 The O′3–P′3 transition in this study might induce stacking faults and/or OP2-like stacking formation, where an octahedral type and a prismatic type Na layers are alternatively stacked with sandwiching MeO2 slabs.42 The formation of stacking faults and residual O′3 domains41 under the non-equilibrium operando condition might affect the anisotropic peak broadening. In our previous data, ex situ XRD patterns also displayed relatively broad peaks of 10l and 01l reflections compared to 00l ones for the P3 type phases in 0.7 > x > 0.45 in Nax[Ni1/2Mn1/2]O2,7 implying the formation of stacking faults and/or slightly different a and b lattice parameters as a P′3 type phase. In the previous ex situ data, clear differences in the a and b lattice parameters were confirmed at x = 0.5.7
Similar to the previous report,7 slight but certain peak separation is confirmed in the range of 0.6 > x > 0.5 in Nax[Ni1/2Mn1/2]O2 (Fig. 4a and S9a†). For example, 202 and 11 reflections are observed at different diffraction angles of 56.8° and 57.5°, respectively. At Na content x = 0.5 in NaxMeO2, Na-vacancy ordering is expected as Didier et al. reported for P′3 type Na0.5VO2 (ref. 41) and our group for P′3 type Na0.5[Fe1/2Co1/2]O2 (ref. 43) and Na0.5CrO2.44 Thus, we carried out an ex situ synchrotron XRD measurement for the electrochemically prepared Nax[Ni1/2Mn1/2]O2 electrode by charging to 3.35 V. For the Rietveld refinement, the amon. × 2bmon. × cmon. superlattice model with a space group of P21/m similar to P′3 type Na0.5[Fe1/2Co1/2]O2 (ref. 43) was used because 1½0 and 1½ superlattice peaks, corresponding to 110 and 11 reflections, respectively, in the refined lattice, are observed as shown in Fig. 5a. Rietveld refinement results in Fig. 5a and Table S6† display small R-factors and good agreement between the calculated and observed patterns. This suggests that P′3 type Na∼0.5[Ni1/2Mn1/2]O2 has in-plane Na ordering in the structure as shown in the schematic illustration of Fig. 5b. The Na-vacancy ordering generally stabilizes the structure, resulting in a voltage jump at the specific Na composition41,43–45 as confirmed in the voltage curves in the narrow region of 0.575 > x > 0.548 in Nax[Ni1/2Mn1/2]O2 (Fig. 4a).
 | ||
| Fig. 5 (a) Rietveld refinement results for the ex situ synchrotron XRD pattern of the Non-sub electrode prepared by charging to 3.35 V and floating for 12 h. (b) Schematic illustration of the refined crystal structure projected along the c-axis. | ||
Furthermore, sandwiching this compositional region, flat charging voltage plateaus are observed at 3.2 V and 3.5 V in the ranges of 0.560 > x > 0.575 and 0.548 > x > 0.500, respectively. This indicates two-phase reactions accompanied by Na-vacancy ordering. Despite the two-phase regions, two P′3 phases were hardly distinguished in the laboratory-scale XRD patterns.
At the end of the later two-phase region at x = ca. 0.50 in Nax[Ni1/2Mn1/2]O2, 201 and 11 reflections are overlapped at 45.3° in 2θ and become a singlet broad peak as 015 reflection of a non-distorted P3 type phase (see Fig. S9a†).
After the single-phase P3 region of 0.50 > x > 0.44, a two-phase region corresponding to P3–O′3 transition is found in 0.44 > x > 0. New peaks corresponding to an O′3 type phase, such as 110, 11, 20, 111, and 11 reflections, appear with increasing peak intensity upon charge (Fig. 4a and S9a†). A corresponding flat charging voltage plateau at 3.7 V in Fig. 2a and 4a (top) agrees with the two-phase reaction. The O′3 type phase was not observed in the previous ex situ XRD patterns of NaxNi1/2Mn1/2O2 electrodes.7 Instead, a hydrated phase was observed due to significant moisture sensitivity of the O′3 type phase as described above.
The calculated interslab distance of the O′3 type phase is 5.67 Å at x = 0.37 in Nax[Ni1/2Mn1/2]O2 which slightly smaller than 5.69 Å for the last P3 type phase but much larger than 5.30 Å for the as-prepared O3 type phase. Generally, by Na extraction on charge, most O3 type NaMeO2 (Me = 3d transition metals) materials transform into a P3 type (including P′3 type) phase and then into O3 type NaxMeO2 (0 < x < 0.3) having a smaller interslab distance (<5.0 Å) through a two-phase reaction of P3–O3 transition and/or formation of a P3–O3 intergrowth phase without formation of the O3 or O′3 type NaxMeO2 phases having such large interslab spacing.2,46,47
Such large interslab-spacing O3 and O′3 type phases are commonly reported for the charged Ni-rich O3 type layered oxides such as O′3–NaNiO2,48 O3–Na[Ni0.5Co0.5]O2,49 O3– and O′3–Na[Ni0.8Co0.15Al0.05]O2,50 O3–Na[Ni0.60Fe0.25Mn0.15]O2,51 O3–Na[Ni0.5Mn0.4Ti0.1]O2,13 and O3–Na0.98Ca0.01[Ni0.5Mn0.5]O2.52 The fact suggests that increasing the amount of Jahn–Teller active low-spin Ni3+ ion (t2g6eg1) would be the driving force of the P3–O′3 transition. Similar to our study, Yu et al. recently studied structural changes of O3 type Na[Ni1/2Mn1/2]O2 by an in situ XRD measurement during the initial charging to 4.0 V, and P3–O3 transition above 3.6 V and formation of the large interslab spacing of the O3 phase was confirmed,27 which is consistent with our results in this study.
Further charging through the single-phase O′3 region in 0.38 > x > 0.31 in Nax[Ni1/2Mn1/2]O2 (Fig. 4a), a two-phase reaction of O′3–O3 transition proceeds in 0.31 > x > 0, and new peaks corresponding to the O3 type phase, such as 101 and 104 appear at 37° and 46° in 2θ, respectively, with increasing peak intensity (see Fig. S9a†). A corresponding flat charging voltage plateau is confirmed at 4.13 V in a coin cell (Fig. 2a) and 4.24 V in an operando cell (Fig. 4a). 003 reflection of the O3 phase is located at 20.1° in 2θ (d = 4.42 Å) without variation of the angle position, and the O3 type phase should have almost no sodium in the structure according to the current passing through the whole charging process as well as the same order of magnitude of the measured interslab distance (4.42 Å) with those of O3–Li0.02Ni1.02O2 (4.45 Å),53 O1–Ni1.02O2 (4.33 Å),53 and O′1–NiO2 (4.53 Å).54 The oxidation states of nickel and manganese ions in the fully charged state, which are confirmed to be close to Ni4+ and Mn4+ by XANES spectra (Fig. 3a and c), also support almost complete Na extraction upon charging. Consequently, a significant reduction in interslab distances is observed from 5.67 Å of O′3 to 4.42 Å of the O3 type phase. Variations of the interslab distances are discussed in detail in a later section.
Compared to Non-sub, the primary O3 type pristine phase in Mg–Ti-sub has a wider Na content range of 1.0 ≥ x > 0.92 in NaxMeO2 (Me = Ni4/9Mn1/3Mg1/18Ti1/6) as a single phase. The reflections of the O3 type phase, such as 003, 006, 101, 012, 104, and 018, continuously shift during charging. By further charging to x < 0.92, peak intensities of the reflections gradually reduce, and new peaks corresponding to the O′3 type phase, such as 110, 20, 11, 20, 111, and 11 peaks, appear with increasing peak intensity as a two-phase reaction of O3–O′3 transition in 0.92 > x > 0.82 in NaxMeO2 (Fig. 4b and S9b†). However, in contrast to the typical two-phase reaction of O3–O′3 transition in Non-sub (Fig. 4a), the O3–O′3 transition in Mg–Ti-sub has a slightly narrower range in Na content with lower diffraction intensities of the O′3 type phase. Furthermore, the prior rhombohedral O3 type phase coexists even in the subsequent O′3–P3 transition region in 0.82 > x > 0.73. The high peak-intensities and continuous peak-shift for the prior O3 type phase look like O3–P3 transition without formation of the O′3 type phase. One can indeed see a relatively sloping voltage plateau corresponding to the O3–O′3 transition (Fig. 2a) and a corresponding broad oxidation peak at 2.79 V in the dQ/dV curve of Fig. 2b. However, a distinct oxidation peak is observed at 2.87 V in Fig. 2b, indicating a two-phase reaction of the O′3–P3 transition. Thus, the dQ/dV curves prove the two two-phase reactions: the O3–O′3 transition at 2.79 and the O′3–P3 transition at 2.87 V.
During the O′3–P3 transition in 0.82 > x > 0.73, the reflections of the P3 type phase, such as 003, 006, 101, 012, 015, and 018, appear with increasing intensity as a two-phase reaction (Fig. 4b and S9b†). After complete transition into the P3 type phase, the peak position continuously changes, and a long Na content range of the single-phase P3 region is observed in 0.73 > x > 0.22 in NaxMeO2. No clear peak separation is found in the P3 region in Mg–Ti-sub (Fig. 4b and S9b†) unlike the Non-sub case (Fig. 4a and S9a†). Even in the ex situ synchrotron XRD pattern of the P3 phase at 3.40 V in Fig. 6a, no superlattice peaks are observed unlike the P′3–Na∼0.5[Ni1/2Mn1/2]O2 phase (Fig. 5a). As no sharp redox peaks are also confirmed in the P3 phase region of the dQ/dV curve (Fig. 2b), Mg and Ti co-substitution successfully disturbs Na-vacancy ordering and enlarges a Na content range of single-phase regions.
 | ||
| Fig. 6 (a) Ex situ synchrotron XRD patterns of Mg–Ti-sub electrodes in the pristine and charged state at 3.40, 4.20, and 4.50 V through floating at each voltage for 12 h, 2 h, and 2 h, respectively. (b) and (c) Simulated XRD patterns in comparison to the observed ones in the charged state of (b) 4.20 V and (c) 4.50 V. Schematic illustrations of the plausible crystal structures are drawn. | ||
By further charging, the reflections of the P3 type phase, such as 003, 006, 101, 012, 015, and 018, disappear with reducing intensity, and new peaks appear as a two-phase reaction in 0.22 > x > 0.21 in NaxMeO2 (Fig. 4b and S9b†) with a corresponding charging voltage plateau at 4.20 V (Fig. 2a). The new diffraction peaks can be indexed as an OP2 type phase with a space group of P3m1 (ref. 42) as shown in the ex situ synchrotron XRD pattern in Fig. 6a. After the single-phase region of the OP2-like phase in 0.21 > x > 0.09 (Fig. 4b), new diffraction peaks appear as a two-phase reaction (Fig. 4b) and can be indexed as an O3 type phase as shown in the ex situ synchrotron XRD pattern in Fig. 6a.
Even in the ex situ synchrotron XRD patterns of the Mg–Ti-sub electrodes prepared by charging and floating for 2 h (Fig. 6a), broad peaks are observed not only for the fully charged O3 but also the prior OP2-like phase. In the latter case, the ideal P3-derived OP2 type structure consists of two MeO2 slabs in the trigonal unit cell, which stack along the c-axis with the ABCAAB oxygen stacking sequence as shown in Fig. S10a.† In the OP2 type structure, two triangular prismatic Na sites exist in the same P type interlayer space and are in an upside-down symmetric relationshipt. One side of the Na prism in the P type Na layer shares the edge with MeO6 octahedra, and the other side shares the face. In the other O type interlayer space, Na or vacancy occupies octahedral sites. The Na or vacancy octahedra share only edge with MeO6 octahedra. Alternative stacking of the P type Na layer and the edge-shared O type layer (hereinafter referred to as the Oe type layer) with the MeO2 slabs forms an OP2 type structure and theoretically provides sharp diffraction peaks as shown in Fig. 6b. However, the observed 00l reflections are very broad, indicating a disordered stacking sequence of P and Oe layers as stacking faults. According to previous reports,6,28,32 we simulated XRD patterns of the stacking-faulted layered structures using the FAULTS program18 as shown in Fig. 6b.
The broad 00l peaks are successfully simulated with the stacking fault model in which P-type and Oe-type layers are randomly stacked at the P:Oe ratio of 50:50 in Fig. 6b. However, 013 and 015 reflections still have a sharper peak profile than the observed ones. Thus, we further employed stacking-faults including an O1-type Of layer (Fig. S10a†) for the XRD simulations. An O1 type MeO2 phase is ideally isostructural to Cdl2.53,55 The O1 type structure consists of a single MeO2 slab in a trigonal unit cell with an ABAB oxygen stacking sequence along the c-axis as shown in Fig. S10a.† The interslab Na or vacancy site has an octahedral coordination, and the octahedron shares face with the MeO6 octahedra in both upper and lower sides. Thus, the interslab layer is, henceforth, denoted as the Of type layer. The simulated pattern at P:Oe:Of ratio of 50:40:10 is in good agreement with the observed one in Fig. 6b. From the results, the continuous peak shift of 00l reflections for the OP2-like phase (Fig. 4b) is explainable by the continuous formation of the P–Oe intergrowth phase, that is, solid-solution-like P3–O3 transition and by partial formation of Of layer as stacking faults. Although quantitative analysis is difficult in a further complicated model, an ideal OP2 domain might be included in the observed P–Oe–Of phase because 011 and 013 reflection peaks are slightly sharper in the observed pattern (Fig. 6b), and local P Oe P Oe P stacking was confirmed with high resolution-scanning TEM (HR-STEM) for Nax[Mn1/4Fe1/4Co1/4Ni1/4]O2 (0.31 < x < 0.18).34 Furthermore, in this study, the OP2-like region is isolated from the prior P3 phase and the subsequent O3 phase through the two two-phase regions of P3–OP2 and OP2–O3 transitions (Fig. 4b). All the results indicate formation of a stable P–Oe intergrowth phase as an intermediate phase between P3 and O3 phases.
On the other hand, the fully charged electrode prepared by floating at 4.50 V for 2 h represents an asymmetric peak profile of 003 reflection at 5.79° in 2θ (wavelength λ = 0.5 Å). The peak is tailing on the high angle side. A peak shoulder is also observed at 13.33° between 012 and 104 reflections. These peaks are assigned to an O1 type phase.53,55 Even in the O3-like main phase, 10l and 01l reflections are slightly broader than 110 reflection, indicating stacking faults including the O1 like Of layer, that is, O3–O1 (Oe–Of) intergrowth phase as reported by Croguennec et al. for NiO2.53 Thus, we first considered the stacking-faulted phase consisting of the Oe and Of layers and simulated XRD patterns with different Oe–Of ratios as shown in Fig. S11.† Similar to the simulated patterns in the literature, the broadened 10l and 01l reflections of the simulated patterns are consistent with those in the observed synchrotron XRD pattern. However, 113O3/111O1 reflections located at 21.06° in λ = 0.5 Å (68.6° in CuKα X-ray) are broader and have lower intensities than those of the simulations (Fig. S11†). The O1 type phase formed at the high voltage is often reported to have migrated transition metal ions in the interslab space.14 Thus, we applied the stacking-faults model with a Me-migrated Of layer, which is, henceforth, described as the Of(Me) type Me0.5Me0.5O2 layer (Me = transition metals and Mg) as shown in Fig. S10b.† Consequently, the two patterns at the Oe/Of(Me) ratio of 95:5 and 40:60 are simulated and summed up at 0.67:0.33 ratio. The simulations produce a good agreement between the calculated and observed patterns (Fig. 6c). As irreversible structural changes were reported for LixNiO2 during the floating process in a high voltage region,56 the Me-migration to form the Of(Me)–Me0.5Me0.5O2 unit might occur during the floating period at 4.50 V for 2 h and not during the prior galvanostatic charging process because a reversible structural change into a pristine O3 type phase is confirmed by ex situ synchrotron XRD for the discharged electrode without the floating charging process as shown in a later section. Thus, migration of a transition metal into the Na layer would be negligibly small during the galvanostatic charge and discharge. Since the Oe/Of(Me) ratio of the main phase is 95:5, an almost pure O3 type MeO2 phase consisting of Oe type stacking is confirmed to be mainly formed by fully charging Mg–Ti-sub like the Non-sub case.
The ex situ XRD pattern of the fully charged Mg–Ti-sub electrode is compared to those of Non-sub, Mg-sub, and Ti-sub ones in Fig. S12.† Only the Mg-sub electrode represents distinct reflections of the O1 type phase (or Of-layer rich phase) in addition to the O3 type phase containing Na, even though the Mg content is very low, 1/18 (see Fig. S12b†). Consequently, the Mg–Ti-sub electrode relatively retains sharp diffraction peaks and suppresses irreversible migration of transition metal ions. Furthermore, the 003 reflection of Mg–Ti-sub is located at a lower diffraction angle compared to Non-sub, indicating a wider interslab spacing in the fully charged state of Mg–Ti-sub.
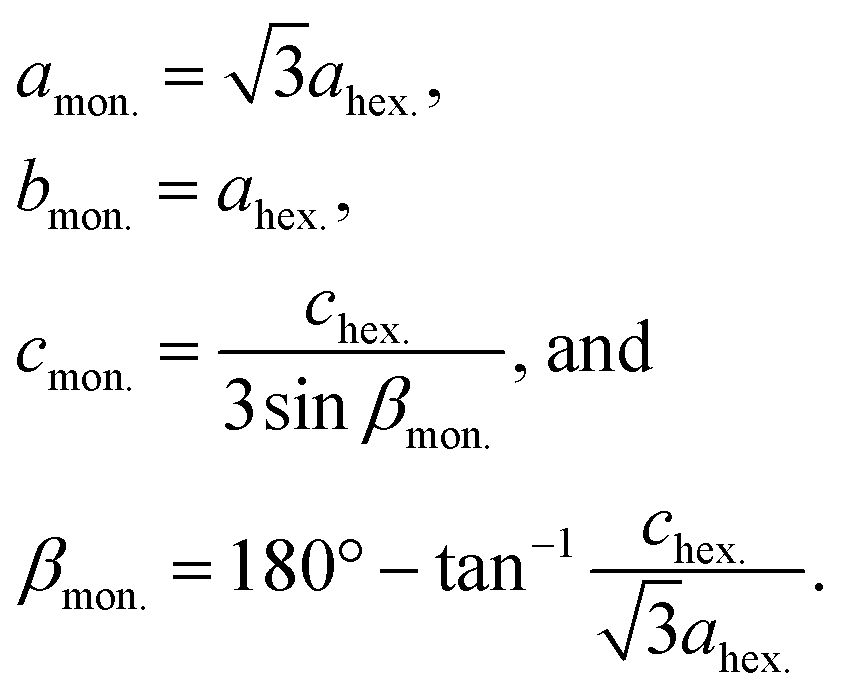 | (3) |
Furthermore, averaged interslab distance dinterslab and in-plane Me–Me distance dMe–Me were calculated using the following equations:
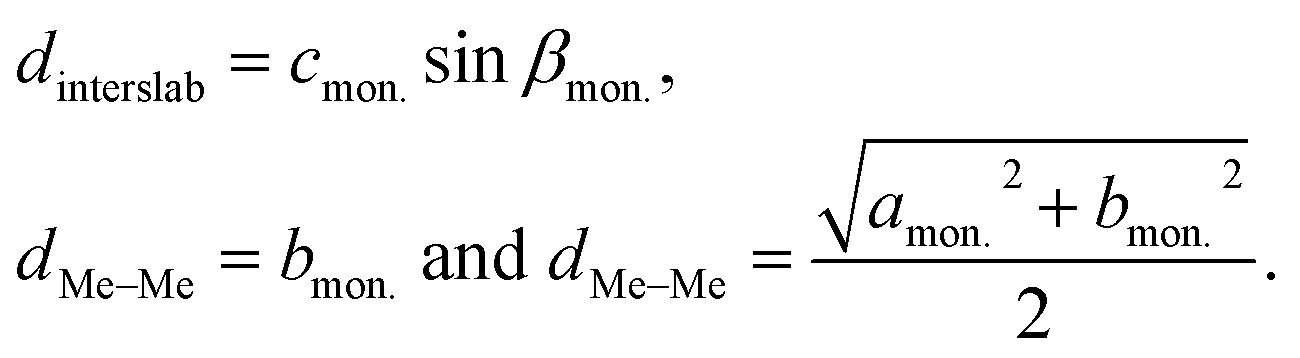 | (4) |
These distances are depicted in the schematic illustrations of the crystal structures in Fig. S13.† The calculated dinterslab and dMe–Meversus Na content plots are shown and compared in Fig. 7.
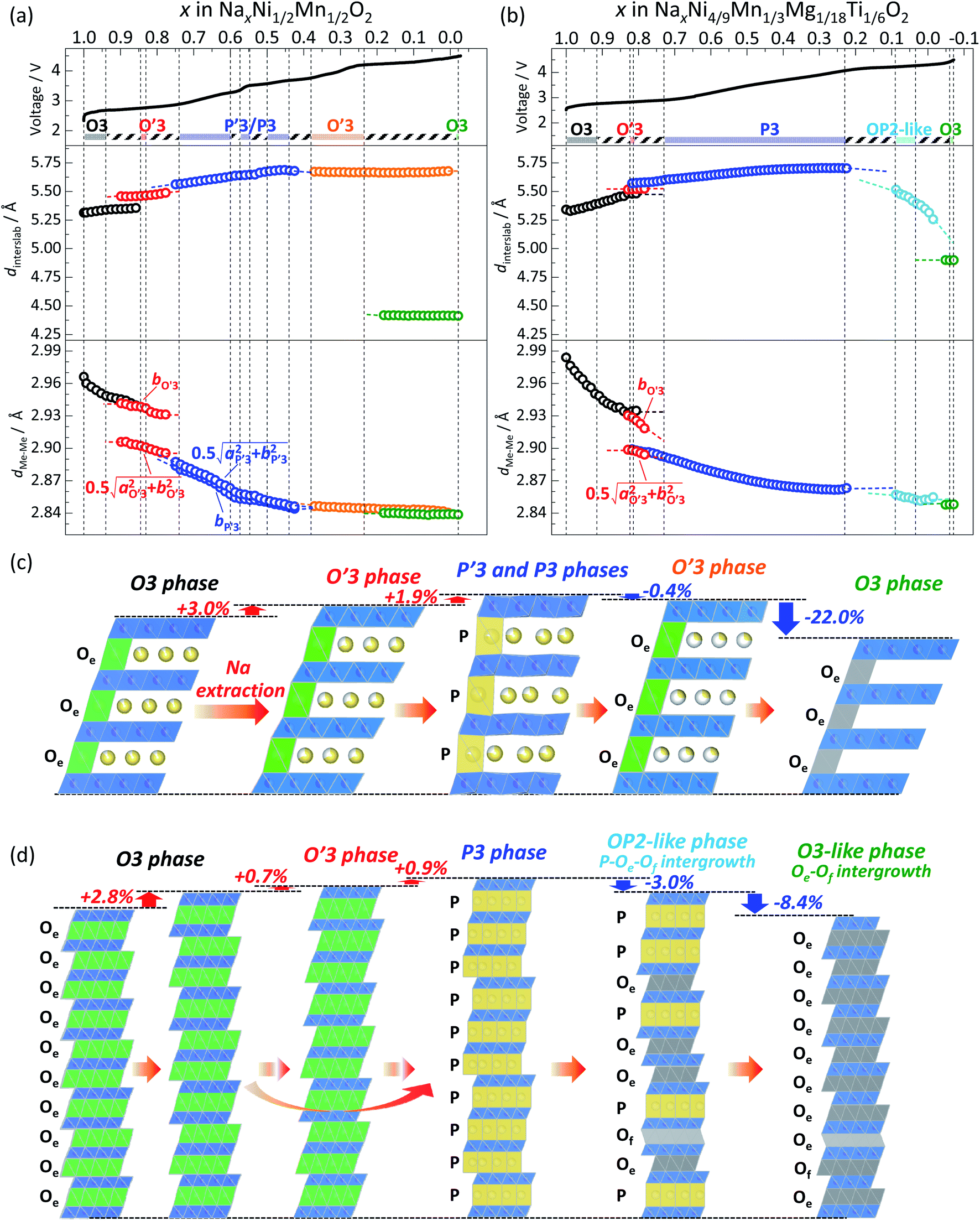 | ||
| Fig. 7 Variations of interslab distance dinterslab and Me–Me distance dMe–Me (Me includes Ni, Mn, Mg, and Ti) for (a) Non-sub and (b) Mg–Ti-sub electrodes during charging to 4.5 V at the 1st cycle. Schematic illustrations of structural changes of layered oxides for (c) Non-sub and (d) Mg–Ti-sub during Na extraction on charging. | ||
In the Non-sub sample, discrete dinterslab values of 5.30–5.35 and 5.46 Å are observed for the O3 and O′3 phases, respectively, and the dMe–Me value of the O′3 phase corresponding to bmon.O′3 is obviously different and quite smaller (2.91 Å) than that of the O3 type one (2.95–2.96 Å). The fact agrees with the two-phase reaction of O3–O′3 transition as mentioned above. Another dMe–Me value derived from 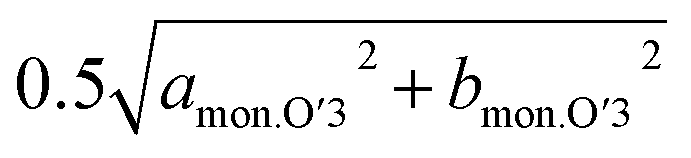 is, however, almost the same as that of O3, and continuously reduces from 2.94 Å to 2.93 Å during the O3–O′3 transition. As Saadoune et al. referred to the O′3 phase as an intermediate stage of O3–P3 transition in Nax[Ni0.6Co0.4]O2,58 our results also indicate that the O′3 phase acts as an intermediate phase between the prior O3 type phase and the following P′3 phase. Despite the distinct and larger values of dinterslab for the P′3 phase (5.57 Å) than that for the O′3 type one (5.46 Å), dMe–Me of the P′3 phase corresponding to bmon.P′3 is almost the same (2.89 Å) as that corresponding to
is, however, almost the same as that of O3, and continuously reduces from 2.94 Å to 2.93 Å during the O3–O′3 transition. As Saadoune et al. referred to the O′3 phase as an intermediate stage of O3–P3 transition in Nax[Ni0.6Co0.4]O2,58 our results also indicate that the O′3 phase acts as an intermediate phase between the prior O3 type phase and the following P′3 phase. Despite the distinct and larger values of dinterslab for the P′3 phase (5.57 Å) than that for the O′3 type one (5.46 Å), dMe–Me of the P′3 phase corresponding to bmon.P′3 is almost the same (2.89 Å) as that corresponding to  of the O′3 one (2.91 Å), which is similarly found in the relationship between O3 and O′3 as mentioned above. Consequently, dMe–Me values are significantly different between O3 and P′3 type phases, but each value is close to the two different dMe–Me values of the O′3 type one. These results support the hypothesis that the O′3 type phase is formed as an intermediate phase to compensate the obvious difference in dMe–Me values between O3 and P′3 type phases. In addition, oxidation of Ni2+ ion, formation of Jahn–Teller active Ni3+ ion ((t2g)6(eg)1 in low spin state), and possible Na/vacancy ordering associated by Ni2+/Ni3+ ordering like the Mn3+/Mn4+ one in NaxMnO2 (ref. 59) might stabilize the monoclinic O′3 structure and facilitate O′3 phase formation as only O′3 type phases are formed in NaxMnO2 by Na extraction.60
of the O′3 one (2.91 Å), which is similarly found in the relationship between O3 and O′3 as mentioned above. Consequently, dMe–Me values are significantly different between O3 and P′3 type phases, but each value is close to the two different dMe–Me values of the O′3 type one. These results support the hypothesis that the O′3 type phase is formed as an intermediate phase to compensate the obvious difference in dMe–Me values between O3 and P′3 type phases. In addition, oxidation of Ni2+ ion, formation of Jahn–Teller active Ni3+ ion ((t2g)6(eg)1 in low spin state), and possible Na/vacancy ordering associated by Ni2+/Ni3+ ordering like the Mn3+/Mn4+ one in NaxMnO2 (ref. 59) might stabilize the monoclinic O′3 structure and facilitate O′3 phase formation as only O′3 type phases are formed in NaxMnO2 by Na extraction.60
By further charging from x = 0.74 to x = 0.6 in Nax[Ni1/2Mn1/2]O2, two different dMe–Me values of the P′3 type phase, corresponding to bmon.P′3 and  , gradually become close to each other although the initial values are very similar (2.89 and 2.91 Å) at x = 0.74. The P′3 phase which is observed in 0.7 > x > 0.6 in Nax[Ni1/2Mn1/2]O2 might be a non-distorted P3 type as mentioned above for the contour plots (Fig. 4a). In the dinterslab and dMe–Me plots in Fig. 7a, the dinterslab and dMe–Me values were calculated as a monoclinic P′3 lattice even in the P3-like region. Two different dMe–Me values are observed again in the range of 0.6 > x > 0.5 in Nax[Ni1/2Mn1/2]O2 (Fig. 7a) due to the formation of the Na-vacancy ordered phase, which is confirmed in Fig. 5. Note that lattice parameters for the P′3 phases were calculated as a single phase even in the two-phase regions of 0.560 > x > 0.575 and 0.548 > x > 0.500 in Nax[Ni1/2Mn1/2]O2 sandwiching the single-phase Na-ordered P′3 region of 0.575 > x > 0.548. This is because that the diffraction peaks are overlapped, and the lattice parameters of the two phases are quite similar to each other. Nevertheless, almost constant values of dinterslab (5.640–5.647 Å) and dMe–Me (2.853–2.852 Å and 2.858–2.856 Å) in 0.6 > x > 0.5 indicate two-phase reaction regions surrounding the narrow single-phase region (Fig. 7a).
, gradually become close to each other although the initial values are very similar (2.89 and 2.91 Å) at x = 0.74. The P′3 phase which is observed in 0.7 > x > 0.6 in Nax[Ni1/2Mn1/2]O2 might be a non-distorted P3 type as mentioned above for the contour plots (Fig. 4a). In the dinterslab and dMe–Me plots in Fig. 7a, the dinterslab and dMe–Me values were calculated as a monoclinic P′3 lattice even in the P3-like region. Two different dMe–Me values are observed again in the range of 0.6 > x > 0.5 in Nax[Ni1/2Mn1/2]O2 (Fig. 7a) due to the formation of the Na-vacancy ordered phase, which is confirmed in Fig. 5. Note that lattice parameters for the P′3 phases were calculated as a single phase even in the two-phase regions of 0.560 > x > 0.575 and 0.548 > x > 0.500 in Nax[Ni1/2Mn1/2]O2 sandwiching the single-phase Na-ordered P′3 region of 0.575 > x > 0.548. This is because that the diffraction peaks are overlapped, and the lattice parameters of the two phases are quite similar to each other. Nevertheless, almost constant values of dinterslab (5.640–5.647 Å) and dMe–Me (2.853–2.852 Å and 2.858–2.856 Å) in 0.6 > x > 0.5 indicate two-phase reaction regions surrounding the narrow single-phase region (Fig. 7a).
At the end of the latter two-phase region at x = ca. 0.50 in Nax[Ni1/2Mn1/2]O2, the two dMe–Me values corresponding to 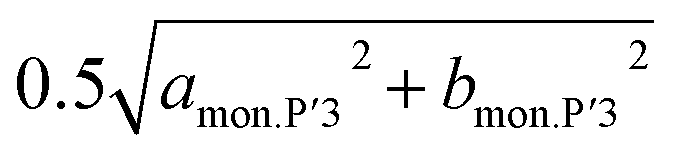 and bmon.P′3 are almost the same (2.85 Å), indicating a non-distorted P3 phase. The dMe–Me slightly decreases from 2.850 Å to 2.845 Å, while dinterslab gradually increases from 5.66 Å to 5.69 Å, implying a single-phase reaction in 0.5 > x > 0.44 in Nax[Ni1/2Mn1/2]O2. In the subsequent two-phase region of P3–O′3 transition in 0.44 > x > 0.38, slight reduction of dinterslab value from 5.69 Å for the last P3 type phase to 5.67 Å for the O′3 type phase is confirmed. In contrast, the two dMe–Me values for the O′3 type phase corresponding to
and bmon.P′3 are almost the same (2.85 Å), indicating a non-distorted P3 phase. The dMe–Me slightly decreases from 2.850 Å to 2.845 Å, while dinterslab gradually increases from 5.66 Å to 5.69 Å, implying a single-phase reaction in 0.5 > x > 0.44 in Nax[Ni1/2Mn1/2]O2. In the subsequent two-phase region of P3–O′3 transition in 0.44 > x > 0.38, slight reduction of dinterslab value from 5.69 Å for the last P3 type phase to 5.67 Å for the O′3 type phase is confirmed. In contrast, the two dMe–Me values for the O′3 type phase corresponding to 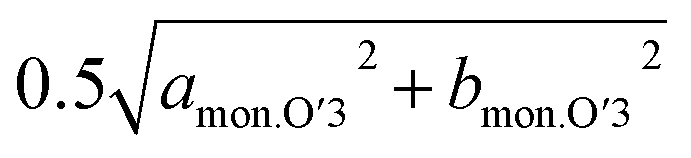 and bmon.O′3, which are almost the same (2.845 Å), are close to those for the prior P3 type phase, which is consistent with the results reported by Yu et al. in the literature.27
and bmon.O′3, which are almost the same (2.845 Å), are close to those for the prior P3 type phase, which is consistent with the results reported by Yu et al. in the literature.27
The most significant reduction of dinterslab value is visible in the O′3–O3 transition in 0.31 > x > 0 in Nax[Ni1/2Mn1/2]O2 (Fig. 7a). The dinterslab value obviously reduces from 5.67 Å of O′3 to 4.42 Å of the O3 type phase. In contrast, dMe–Me values for O′3 and O3 phases are 2.85 Å and 2.84 Å, respectively, and almost same.
Consequently, the Non-sub electrode demonstrates the phase transitions from O3 → O′3 → P′3/P3 → O′3 → O3 phase by Na extraction during the charging process (Fig. 7c). The relative changes of the dinterslab values in percentages are as follows: +3.0% at the two-phase O3–O′3 transition, +1.9% at the two-phase O′3–P′3 transition, +2.2% in the single-phase P′3–P3 region, −0.4% at the two-phase P3–O′3 transition, and −22.0% at the two-phase O′3–O3 transition. In contrast, the relative changes of the dMe–Me values in percentages are much smaller and −4.2% in total and ca. −1.8% at maximum between the smaller and larger dMe–Me at the two-phase O′3–P′3 transition (Fig. 7a). These values are consistent with those reported for Nax[Ni1/2Mn1/2]O2.27 The shrinkage of the interslab distance is known to generate microcracks between primary submicron particles, leading to electrical isolation of the primary particles.27,52 In fact, as shown in Fig. 8a, the ex situ XRD of the Non-sub electrode after 10 cycles proves a residual P3 (or P′3) type phase, although the reversibly reverted O3 phase is the main phase. This partial irreversible structural change is similarly reported by Yu et al. with observation of a trace amount of an O′3 phase after 10 and 20 cycles.52
 | ||
| Fig. 8 (a) and (b) Ex situ synchrotron XRD patterns of (a) Non-sub and (b) Mg–Ti-sub electrodes in pristine and discharged states at the 10th cycle. TEM images of (c)–(f) Non-sub and (g)–(j) Mg–Ti-sub particles taken from the (c, d, g and h) pristine and (e, f, i and j) discharged electrodes at the 10th cycle. Blue squares in ‘c’, ‘e’, ‘g’, and ‘i’ panels represent the observed area of the magnified images of ‘d’, ‘f’, ‘h’, and ‘j’ panels. | ||
From the detailed identification of the structural changes in Non-sub Nax[Ni1/2Mn1/2]O2 upon charging (Fig. 4a, 5 and 7c) and variations of dinterslab and dMe–Me values calculated from lattice parameters (Fig. 7a) as well as reported results in the literature, characteristic features are found as follows: (i) formation of the O′3 type intermediate phase to compensate a big difference in dMe–Me values between pristine O3 and P3 type phases, (ii) Na-vacancy ordering at specific Na content of x ∼ 0.5 in NaxMeO2 (Me = Cr, Mn, Fe, Co, Ni) having a narrow single-phase region surrounded by two two-phase reaction regions, (iii) formation of the O′3 type phase in x < 0.5 in NaxMeO2 having a large interslab spacing as commonly observed in Ni-rich NaxMeO2, (iv) slight shrinkage of dinterslab by P3–O′3 transition, and (v) no significant change in dMe–Me values in x < 0.5 in NaxMeO2.
As in the case of Non-sub (Fig. 7a), variations of dinterslab and dMe–Me values for Mg–Ti-sub were also calculated from the lattice parameters and are plotted in Fig. 7b. Both the initial dinterslab and dMe–Me values of the O3 type phase at the starting points (Fig. 7b) are larger for Mg–Ti-sub than for Non-sub (Fig. 7a), which are consistent with those of pristine powder (Table S1 and S4†). In contrast to the almost constant dinterslab and dMe–Me values of the O3 phase in Non-sub (Fig. 7a), Mg–Ti-sub represents distinct increase in dinterslab from 5.33 to 5.48 Å and reduction in dMe–Me from 2.98 to 2.94 Å in the single-phase O3 region in 1.0 > x > 0.84 in NaxMeO2 (Me = Ni4/9Mn1/3Mg1/8Ti1/6) upon charge (Fig. 7b). By further charging, the dinterslab slightly increases from 5.48 Å for O3 to 5.52 Å for O′3 in the subsequent O3–O′3 transition in 0.92 > x > 0.82 and further but slightly increases to 5.57 Å for P3 in the O′3–P3 transition in 0.82 > x > 0.73. The overall increase in dinterslab in the O3–O′3–P3 transition of Mg–Ti-sub is 1.6% (0.7% in the O3–O′3 transition and 0.9% in the O′3–P3 transition), which is much lower than 2.8% in the prior single-phase O3 region of Mg–Ti-sub (Fig. 7b) and 4.9% in the O3–O′3–P′3 transition of Non-sub (Fig. 7a). Furthermore, dMe–Me slightly reduces from 2.94 Å for O3 to 2.90 Å for P3 in the O3–O′3–P3 transition of Mg–Ti-sub, and the reduction rate is 1.5% (Fig. 7b), which is lower than 2.0% in the O3–O′3–P′3 transition of Non-sub (Fig. 7a). In Mg–Ti-sub, significant variations in both dinterslab and dMe–Me within the O3 region result in the small differences between O3 and P3 type phases on the transition.
In principle, when Na+ ions are extracted from the layered transition metal oxides during the charging process, electrostatic repulsion between oxide-ion layers sandwiching the Na layer is pronounced. Transition metal ions are simultaneously oxidized, and Me–O distances shorten. The shortening of the in-plane Me–Me distance in the MeO2 slab competes with electrostatic Na+–Na+ repulsion in the Na layer. Further electrochemical charging and oxidation of transition metal ions lead to shorter Me–Me distance and stronger Na+–Na+ repulsion despite the reduced Na content, resulting in the formation of a P type Na layer having a large interslab space by gliding the MeO2 slabs. Such a stabilization mechanism of the P type phase was proposed for potassium-containing layered 3d transition metal oxides,61 which generally crystallize into P2 or P3 type phases.62,63 Therefore, the longer Me–Me distance of Mg–Ti-sub in the pristine state delays O3–P3 transition and enlarges the Na content range of the O3 phase region, resulting in the significant variation in dMe–Me in the O3 region. In spite of the large initial dMe–Me (2.98 Å), the minimum dMe–Me value of the O3 type phase just before the O3–O′3 transition is ca. 2.94 Å (Fig. 7b) and almost the same with ca. 2.95 Å in Non-sub (Fig. 7a). Furthermore, the dMe–Me value of the initial P3 phase (2.90 Å) is larger and closer to that of the prior O3 phase in Mg–Ti-sub compared to that of the initial P′3 phase in Non-sub (2.89 Å) due to the substitution of Ni2+ and Mn4+ for larger ions of Mg2+ and Ti4+, respectively. The smaller differences in the dMe–Me values between O3 and P3 phases are expected to result in the facile P3 phase formation partially skipping O′3 phase formation in Mg–Ti-sub (Fig. 4b and 7b) compared to Non-sub (Fig. 4a and 7a).
By further charging and in the single-phase P3 region in 0.73 > x > 0.22 in NaxMeO2, the dinterslab value continuously increases from 5.57 to 5.72 Å, while the dMe–Me value gradually decreases from 2.90 to 2.86 Å (Fig. 7b). In the subsequent two-phase P3–OP2-like transition in 0.22 > x > 0.21, the dinterslab value reduces from 5.72 Å for P3 to 5.55 Å for the OP2-like phase. In the OP2-like P–Oe–Of intergrowth phase region and the following transition region into the almost O3-like Oe–Of intergrowth phase, dinterslab of the OP2-like phase further reduces to 5.35 Å gradually. Finally, the dinterslab value reduces from 5.35 Å for the OP2-like phase to 4.90 Å for the O3-like phase by Na extraction on charging to 4.5 V. Compared to the Non-sub case (ca. 4.42 Å) in Fig. 7a, the O3 type phase has a larger dinterslab value of ca. 4.90 Å. Substitution with electrochemically inactive Mg2+ ion would disturb complete Na extraction even by fully charging to 4.50 V, resulting in the larger dinterslab value in Mg–Ti-sub.
Consequently, the Mg–Ti-sub electrode demonstrates the phase transitions from O3 → (O′3→) P3 → OP2-like P–Oe intergrowth → O3 phase by Na extraction during the charging process (Fig. 7d). The change in dMe–Me values is −4.3%, which is almost the same as −4.2% in Non-sub (Fig. 7a). However, the maximum change is just −1.5% between the initial and end phases of the P3 type, which is attributed to the certain reduction within the primary O3 type single-phase region (−1.2%, which is double of −0.6% for the O3 region in Non-sub). The large Na content range of the O3 type phase and the smooth O3–P3 transition almost skipping an O′3 intermediate phase significantly contribute to the gradual structural changes without specific Na-ordering phases. The relative changes of the dinterslab values in percentages are as follows: +2.8% in the primary O3 single-phase region, +1.6% at the two-phase O3–P3 transition (+0.7% for O3–O′3 and +0.9% for O′3–P3 transition), +2.0% in the single-phase P3 region, −3.0% at the two-phase reaction of P3–OP2-like transition, −3.6% in the single-phase OP2-like P–Oe intergrowth, and −8.4% at the two-phase reaction from OP2-like to O3-like phase (Fig. 7d). The dinterslab values gradually increase and decrease in Mg–Ti-sub, resulting in the smaller dinterslab change (−8.4%) at the final OP2–O3 transition than −22.0% at the final O′3–O3 transition in Non-sub (Fig. 7c). No significant difference in the XRD patterns and no residual intermediate phase are confirmed for the electrode after 10 cycles compared to the pristine elctrode as shown in Fig. 8b. The gradual change in dinterslab values would disturb electrical isolation of primary particles in Mg–Ti-sub. Furthermore, Mg and Ti co-substitution stabilizes the O3 type phase in the fully charged state as discussed above with Fig. 6 and S12.†
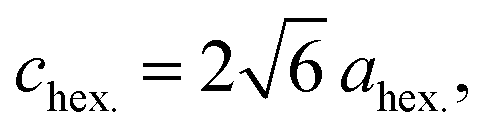 the O3 type trigonal lattice in the hexagonal setting can be converted into a cubic lattice, such as a cubic rock-salt type structure.64 The lattice constant ratio chex./ahex. of the second phases is close to
the O3 type trigonal lattice in the hexagonal setting can be converted into a cubic lattice, such as a cubic rock-salt type structure.64 The lattice constant ratio chex./ahex. of the second phases is close to 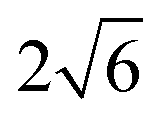 (Fig. S12†). The fact indicates a gradual and irreversible structural change into a cation-disordered rock-salt type cubic phase during charging to 4.50 V and floating processes. Even without the floating process, surface deterioration and formation of the disordered rock-salt type cubic phase were proved by TEM images for the cycled Na[Ni1/2Mn1/2]O2.27 Herein, we conducted ex situ TEM observation for cycled Non-sub and Mg–Ti-sub electrodes.
(Fig. S12†). The fact indicates a gradual and irreversible structural change into a cation-disordered rock-salt type cubic phase during charging to 4.50 V and floating processes. Even without the floating process, surface deterioration and formation of the disordered rock-salt type cubic phase were proved by TEM images for the cycled Na[Ni1/2Mn1/2]O2.27 Herein, we conducted ex situ TEM observation for cycled Non-sub and Mg–Ti-sub electrodes.
Fig. 8c–j display ex situ TEM images of the two samples in the pristine and discharged state at the 10th cycle. Before charge–discharge tests, both Non-sub (Fig. 8c and d) and Mg–Ti-sub (Fig. 8g and h) pristine samples exhibit fine lattice fringes and smooth surface morphology without any cracks. After galvanostatic charge–discharge for 10 cycles, Non-sub (Fig. 8e and f) represents corrosion-like pits in the outer edge of 5–10 nm in depth (Fig. 8f). Such surface deterioration is similar to that reported by Yu et al. and probably attributed to a side reaction with the electrolyte and dissolution of transition metals into the electrolyte.27 In contrast, the TEM images of the cycled Mg–Ti-sub (Fig. 8i and j) display no significant change in the outermost surface (Fig. 8j). The fact agrees with a smaller change of structures in bulk compared to Non-sub (Fig. S12†).
These results suggest that Mg and Ti co-substitution stabilizes layered structures in both bulk and surface. Mg-substitution would be effective as a HF scavenger, protecting the particle surface against the electrolyte attack.9 The substitution only of Mg2+ results in slight improvement of cycle stability (Fig. 2c) and formation of a huge amount of a Me-migrated O1-like phase after floating at 4.50 V for 2 h (Fig. S12†). Substitution only with Ti4+ also presents similar electrochemical properties and a highly stacking-faulted phase at 4.50 V.
Elemental combination of the dopants and the parent transition metals as well as substitution amount would be important for enhancing the electrochemical properties of layered oxides.4 Consequently, co-substitution of large and electrochemically inactive Mg2+ and Ti4+ ions is found to be highly effective to improve the electrochemical performance and structural stability of O3 type Na[Ni1/2Mn1/2]O2 during charge–discharge cycles. Our findings from the systematic studies on the influencing substitution metals are believed to accelerate further development of layered oxide positive electrode materials for practical Na-ion batteries.
Conclusions
Mg or Ti substituted, and Mg and Ti co-substituted O3 type NaNi1/2Mn1/2O2 are synthesized as positive electrode materials for Na-ion batteries. Mg- or Ti-substituted materials deliver slightly smaller reversible capacities of 185 mA h g−1 and exhibit better capacity capability in non-aqueous Na cells than non-substituted ones. The Mg and Ti co-substituted material demonstrates a large initial discharge capacity of 200 mA h g−1 almost same as the non-substituted one and much better capacity capability. Operando XRD reveals that co-substitution of large Mg2+ and Ti4+ ions delays O3–O′3 transition, enlarges the Na content range of the primary O3 type single-phase region, and reduces lattice mismatch between the charged O3 and the subsequent P3 type phases, leading to gradual structural changes during Na extraction on charge. Furthermore, Mg and Ti co-substitution produces an OP2-like P–Oe–Of intergrowth phase as an intermediate buffer phase between the P3 and the O3 phases, resulting in suppression of the significant shrinkage in interslab spacing which is observed in non-substituted Nax[Ni1/2Mn1/2]O2. Ex situ synchrotron XRD confirms the P′3 type Na-ordered phase at x ∼ 0.5 in the non-substituted Nax[Ni1/2Mn1/2]O2 phase, which exhibits the characteristic features of Na-vacancy ordering, such as voltage jump at the specific Na content and two-phase reaction regions surrounding the Na-ordered phase. However, the Na-ordering behaviors are not observed in the Mg and Ti co-substituted material. Furthermore, at the outermost surface, in the case of Mg and Ti co-substitution, the particle surface after cycling is as smooth as pristine, which is confirmed by ex situ TEM observation. On the other hand, non-substituted Na[Ni1/2Mn1/2]O2 represents deteriorated particle surface after cycling. Mg and Ti co-substitution enhances the structural stability in terms of not only the reversibility of structural changes but also surface protection against the electrolyte at high voltage. The comprehensive modification of the layered structures is generated by Mg and Ti co-substitution leading to the superior sodium battery performance.Author contributions
N. Fujitani, Y. Yoda, and K. Kuroki conducted the material synthesis and the structural and electrochemical characterization. K. Kubota and N. Fujitani analyzed the experimental data and prepared the manuscript. Y. Tokita conducted the XRD simulations of the faulted structures. S. Komaba supervised the project and co-wrote the manuscript. All the authors discussed the results and contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Dr Toshinari Ichihashi and Prof. Yasushi Idemoto in the Tokyo University of Science for TEM measurements. This study was in part supported by the Japan Science and Technology Agency (JST) through the Adaptable and Seamless Technology Transfer Program through Target-driven R&D (A-STEP), Project No. AS2614056L and CONCERT-Japan program of the Strategic International Research Cooperative Program (SICORP), Grant No. JPMJSC17C1 and by the Element Strategy Initiative of the Ministry of Education Culture, Sports, Science and Technology, Japan (MEXT), Grant Number JPMXP0112101003. The synchrotron X-ray diffraction experiments were performed at the BL02B2 and BL19B2 of SPring-8 with the approval of JASRI (Proposal No. 2015B1532, 2017B1312, 2018A1630, and 2018B1776). The synchrotron hard X-ray absorption experiments were performed at the BL-12C of the Photon Factory under the approval of the Photon Factory Program Advisory Committee (Proposal No. 2018G673).References
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- A. Kamiyama, K. Kubota, D. Igarashi, Y. Youn, Y. Tateyama, H. Ando, K. Gotoh and S. Komaba, Angew. Chem., Int. Ed., 2021, 60, 5114–5120 CrossRef CAS PubMed.
- K. Kubota, S. Kumakura, Y. Yoda, K. Kuroki and S. Komaba, Adv. Energy Mater., 2018, 8, 1703415 CrossRef.
- K. Kubota, M. Dahbi, T. Hosaka, S. Kumakura and S. Komaba, Chem. Rec., 2018, 18, 459–479 CrossRef CAS PubMed.
- Y. Yoda, K. Kubota, K. Kuroki, S. Suzuki, K. Yamanaka, T. Yaji, S. Amagasa, Y. Yamada, T. Ohta and S. Komaba, Small, 2020, 16(9), 2006483 CrossRef CAS PubMed.
- S. Komaba, N. Yabuuchi, T. Nakayama, A. Ogata, T. Ishikawa and I. Nakai, Inorg. Chem., 2012, 51, 6211–6220 CrossRef CAS PubMed.
- S. Mariyappan, Q. Wang and J. M. Tarascon, J. Electrochem. Soc., 2018, 165, A3714–A3722 CrossRef CAS.
- J. Y. Hwang, T. Y. Yu and Y. K. Sun, J. Mater. Chem. A, 2018, 6, 16854–16862 RSC.
- H. Yoshida, N. Yabuuchi, K. Kubota, I. Ikeuchi, A. Garsuch, M. Schulz-Dobrick and S. Komaba, Chem. Commun., 2014, 50, 3677–3680 RSC.
- X. Qi, Y. Wang, L. Jiang, L. Mu, C. Zhao, L. Liu, Y.-S. Hu, L. Chen and X. Huang, Part. Part. Syst. Charact., 2016, 33, 538–544 CrossRef CAS.
- P. F. Wang, H. R. Yao, X. Y. Liu, J. N. Zhang, L. Gu, X. Q. Yu, Y. X. Yin and Y. G. Guo, Adv. Mater., 2017, 29, 1700210 CrossRef PubMed.
- Q. Wang, S. Mariyappan, J. Vergnet, A. M. Abakumov, G. Rousse, F. Rabuel, M. Chakir and J. M. Tarascon, Adv. Energy Mater., 2019, 9, 1901785 CrossRef.
- S. Mariyappan, T. Marchandier, F. Rabuel, A. Iadecola, G. Rousse, A. V. Morozov, A. M. Abakumov and J. M. Tarascon, Chem. Mater., 2020, 32, 1657–1666 CrossRef CAS.
- S. Kawaguchi, M. Takemoto, K. Osaka, E. Nishibori, C. Moriyoshi, Y. Kubota, Y. Kuroiwa and K. Sugimoto, Rev. Sci. Instrum., 2017, 88(9), 085111 CrossRef CAS PubMed.
- K. Osaka, Y. Yokozawa, Y. Torizuka, Y. Yamada, M. Manota, N. Harada, Y. Chou, H. Sasaki, A. Bergamaschi and M. Sato, AIP Conf. Proc., 2019, 2054, 050008 CrossRef.
- F. Izumi and K. Momma, Solid State Phenom., 2007, 130, 15–20 CAS.
- M. Casas-Cabanas, M. Reynaud, J. Rikarte, P. Horbach and J. Rodriguez-Carvajal, J. Appl. Crystallogr., 2016, 49, 2259–2269 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- J. Rigaku, 2011, 27, 32–35, https://www.rigaku.com/journal/summer-2011-volume-27-no-2/32-35.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- C. Delmas, C. Fouassier and P. Hagenmuller, Physica B+C, 1980, 99, 81–85 CrossRef CAS.
- M. Sathiya, Q. Jacquet, M.-L. Doublet, O. M. Karakulina, J. Hadermann and J.-M. Tarascon, Adv. Energy Mater., 2018, 8, 1702599 CrossRef.
- H. R. Yao, P. F. Wang, Y. Gong, J. N. Zhang, X. Q. Yu, L. Gu, C. Y. OuYang, Y. X. Yin, E. Y. Hu, X. Q. Yang, E. Stavitski, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2017, 139, 8440–8443 CrossRef CAS PubMed.
- J. Breger, K. Kang, J. Cabana, G. Ceder and C. P. Grey, J. Mater. Chem., 2007, 17, 3167–3174 RSC.
- K. S. Kang, Y. S. Meng, J. Breger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS PubMed.
- T. Y. Yu, H. H. Ryu, G. Han and Y. K. Sun, Adv. Energy Mater., 2020, 10(8), 2001609 CrossRef CAS.
- A. Mullaliu, K. Kuroki, M. Keller, K. Kubota, D. Buchholz, S. Komaba and S. Passerini, Inorg. Chem., 2020, 59, 7408–7414 CrossRef CAS PubMed.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- L. T. Zheng and M. N. Obrovac, Electrochim. Acta, 2017, 233, 284–291 CrossRef CAS.
- J. Wang, Z. F. Zhou, Y. S. Li, M. Li, F. Wang, Q. R. Yao, Z. M. Wang, H. Y. Zhou and J. Q. Deng, J. Alloys Compd., 2019, 792, 1054–1060 CrossRef CAS.
- K. Kubota, Y. Yoda and S. Komaba, J. Electrochem. Soc., 2017, 164, A2368–A2373 CrossRef CAS.
- P. S. Halasyamani, Chem. Mater., 2004, 16, 3586–3592 CrossRef CAS.
- J. C. Kim, D. H. Kwon, J. H. Yang, H. Kim, S. H. Bo, L. J. Wu, H. Kim, D. H. Seo, T. Shi, J. Y. Wang, Y. M. Zhu and G. Ceder, Adv. Energy Mater., 2020, 10(13), 2001151 CrossRef CAS.
- Z. H. Lu and J. R. Dahn, Chem. Mater., 2001, 13, 1252–1257 CrossRef CAS.
- D. Buchholz, L. G. Chagas, C. Vaalma, L. M. Wu and S. Passerini, J. Mater. Chem. A, 2014, 2, 13415–13421 RSC.
- C. Delmas, J. J. Braconnier, C. Fouassier and P. Hagenmuller, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1981, 36, 1368–1374 Search PubMed.
- Y. Shao-Horn, S. Levasseur, F. Weill and C. Delmas, J. Electrochem. Soc., 2003, 150, A366–A373 CrossRef CAS.
- C. Fouassier, G. Matejka, J.-M. Reau and P. Hagenmuller, J. Solid State Chem., 1973, 6, 532–537 CrossRef CAS.
- M. Blangero, D. Carlier, M. Pollet, J. Darriet, C. Delmas and J.-P. Doumerc, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 184116 CrossRef.
- C. Didier, M. Guignard, M. R. Suchomel, D. Carlier, J. Darriet and C. Delmas, Chem. Mater., 2016, 28, 1462–1471 CrossRef CAS.
- N. Yabuuchi, M. Kajiyama, J. Iwatate, H. Nishikawa, S. Hitomi, R. Okuyama, R. Usui, Y. Yamada and S. Komaba, Nat. Mater., 2012, 11, 512–517 CrossRef CAS PubMed.
- K. Kubota, T. Asari, H. Yoshida, N. Yabuuchi, H. Shiiba, M. Nakayama and S. Komaba, Adv. Funct. Mater., 2016, 26, 6047–6059 CrossRef CAS.
- N. Yabuuchi, I. Ikeuchi, K. Kubota and S. Komaba, ACS Appl. Mater. Interfaces, 2016, 8, 32292–32299 CrossRef CAS PubMed.
- J. L. Kaufman and A. Van der Ven, Phys. Rev. Mater., 2019, 3, 015402 CrossRef CAS.
- J.-J. Braconnier, C. Delmas, C. Fouassier and P. Hagenmuller, Mater. Res. Bull., 1980, 15, 1797–1804 CrossRef CAS.
- X. Li, Y. Wang, D. Wu, L. Liu, S. H. Bo and G. Ceder, Chem. Mater., 2016, 28, 6575–6583 CrossRef CAS.
- M. H. Han, E. Gonzalo, M. Casas-Cabanas and T. Rojo, J. Power Sources, 2014, 258, 266–271 CrossRef CAS.
- P. Vassilaras, D. H. Kwon, S. T. Dacek, T. Shi, D. H. Seo, G. Ceder and J. C. Kim, J. Mater. Chem. A, 2017, 5, 4596–4606 RSC.
- L. T. Zheng, R. Fielden, J. C. Bennett and M. N. Obrovac, J. Power Sources, 2019, 433, 226698 CrossRef CAS.
- F. Ding, C. Zhao, D. Zhou, Q. Meng, D. Xiao, Q. Zhang, Y. Niu, Y. Li, X. Rong, Y. Lu, L. Chen and Y.-S. Hu, Energy Storage Mater., 2020, 30, 420–430 CrossRef.
- T.-Y. Yu, J. Kim, J.-Y. Hwang, H. Kim, G. Han, H.-G. Jung and Y.-K. Sun, J. Mater. Chem. A, 2020, 8, 13776–13786 RSC.
- L. Croguennec, C. Pouillerie, A. N. Mansour and C. Delmas, J. Mater. Chem., 2001, 11, 131–141 RSC.
- J. M. Tarascon, G. Vaughan, Y. Chabre, L. Seguin, M. Anne, P. Strobel and G. Amatucci, J. Solid State Chem., 1999, 147, 410–420 CrossRef CAS.
- G. G. Amatucci, J. M. Tarascon and L. C. Klein, J. Electrochem. Soc., 1996, 143, 1114–1123 CrossRef CAS.
- L. Croguennec, C. Pouillerie and C. Delmas, J. Electrochem. Soc., 2000, 147, 1314–1321 CrossRef CAS.
- K. Mukai, H. Nozaki, Y. Ikedo, J. Sugiyama, K. Ariyoshi and T. Ohzuku, J. Power Sources, 2009, 192, 684–688 CrossRef CAS.
- I. Saadoune, A. Maazaz, M. Menetrier and C. Delmas, J. Solid State Chem., 1996, 122, 111–117 CrossRef CAS.
- X. Li, X. H. Ma, D. Su, L. Liu, R. Chisnell, S. P. Ong, H. L. Chen, A. Toumar, J. C. Idrobo, Y. C. Lei, J. M. Bai, F. Wang, J. W. Lynn, Y. S. Lee and G. Ceder, Nat. Mater., 2014, 13, 586–592 CrossRef CAS PubMed.
- X. Chen, Y. C. Wang, K. Wiaderek, X. H. Sang, O. Borkiewicz, K. Chapman, J. LeBeau, J. Lynn and X. Li, Adv. Funct. Mater., 2018, 28(50), 1805105 CrossRef.
- H. Kim, D. H. Seo, A. Urban, J. Lee, D. H. Kwon, S. H. Bo, T. Shi, J. K. Papp, B. D. McCloskey and G. Ceder, Chem. Mater., 2018, 30, 6532–6539 CrossRef CAS.
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Chem. Rev., 2020, 120, 6358–6466 CrossRef CAS PubMed.
- K. Kubota, Electrochemistry, 2020, 88, 507–514 CrossRef CAS.
- J. Kikkawa, T. Akita, M. Tabuchi, M. Shikano, K. Tatsumi and M. Kohyama, J. Appl. Phys., 2008, 103, 104911 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta01164b |
| This journal is © The Royal Society of Chemistry 2021 |