
Enabling highly reversible sodium metal cycling across a wide temperature range with dual-salt electrolytes†
Akila C.
Thenuwara
 a,
Pralav P.
Shetty
a,
Neha
Kondekar
b,
Chuanlong
Wang
c,
Weiyang
Li
c and
Matthew T.
McDowell
*ab
a,
Pralav P.
Shetty
a,
Neha
Kondekar
b,
Chuanlong
Wang
c,
Weiyang
Li
c and
Matthew T.
McDowell
*ab
aG.W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: mattmcdowell@gatech.edu
bSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA
cThayer School of Engineering, Dartmouth College, Hanover, NH 03755, USA
First published on 20th April 2021
Abstract
New rechargeable battery chemistries that can operate at lower temperatures than Li-ion batteries are necessary for emerging applications such as electric flight. Sodium metal anodes have shown high coulombic efficiency and encouraging cycling behavior for high-energy batteries at room temperature, but electrochemical behavior of sodium at low temperatures has rarely been explored. Here, we show that sodium metal anodes can be cycled with high coulombic efficiency down to −50 °C when using diglyme-based electrolytes with dual salts, which surpasses the low-temperature performance of graphite anodes in Li-ion batteries. This novel electrolyte shows low overpotential and an average coulombic efficiency of 99.0% over 50 cycles when tested at −40 °C, as well as promising performance over a range of higher temperatures. The high coulombic efficiency of sodium cycling at low temperatures is attributed to the formation of a uniform SEI that is rich in inorganic species, as revealed by cryogenic transmission electron microscopy and X-ray photoelectron spectroscopy. These results are superior to lithium metal, which has exhibited relatively low coulombic efficiency at low temperatures in a variety of electrolytes. Our findings thus demonstrate the particular promise of sodium-metal-based batteries for use in cold environments.
1. Introduction
Rechargeable batteries that can operate efficiently at low temperatures (<0 °C) present a significant research and development challenge. Reduced temperatures have a marked effect on ion migration, electrolyte viscosity, and interface dynamics within batteries and can cause increased cell polarization and severe capacity fade.1–6 For instance, conventional Li-ion batteries do not behave well below about −20 °C due to sluggish solid-state diffusion in graphite, electrolyte freezing, unfavorable desolvation energetics, and lithium plating during charge.2–4,7–11 The graphite anode is a key limiting factor at low temperatures, which has motivated the investigation of lithium metal anodes (as well as other alternatives) for energy storage under challenging low-temperature conditions.1,12–15 The electrolyte plays an important role in the performance of lithium metal anodes at low temperatures. Meng and coworkers developed a pressurized gas-based electrolyte,15,16 and Xia and coworkers developed an ester-based electrolyte coupled with an organic cathode,14 with both showing cycling capabilities below −50 °C. Work on carbonate electrolytes with ionic liquid additives has shown promise for operation of lithium titanate electrodes down to −30 °C.17 Very recently, Wang and coworkers utilized fluorinated non-polar ether electrolytes for low-temperature energy storage.13 Our group has shown that low operating temperatures have a distinct effect on the morphology of deposited lithium and the structure and composition of the solid-electrolyte interphase (SEI); furthermore, many electrolyte combinations exhibit extremely low coulombic efficiency (CE) for lithium cycling at low temperatures.12 A recent study has shown that the CE for lithium cycling can be improved at low temperatures by adding small amounts of carbonates to ether-based electrolytes, but higher CEs are required for long-term stable cycling at low temperatures.18The sodium metal anode also shows promise for batteries because of its high theoretical specific capacity (1165 mA h g−1) and greater abundance than lithium.19–25 Despite fundamental issues such as dendrite formation, dead sodium accumulation, and continuous SEI growth, recent advances in electrolyte engineering and cathode material development have enabled sodium metal batteries to exhibit high CE (>99%) at room temperature.26–29 Cui and coworkers showed that sodium metal in a diglyme-based electrolyte with sodium hexafluorophosphate (NaPF6) salt exhibits excellent reversibly at room temperature owing to the uniform SEI containing sodium oxide and sodium fluoride.29 This inorganic-rich SEI was able to inhibit dendritic growth of sodium and limit the extent of SEI formation, leading to efficient cycling at room temperature.29 Recently, Zhang and coworkers showed that concentrated sodium salts in ether electrolytes can enable highly reversible sodium metal batteries,30 although the high concentration of salts decreases ionic conductivity and increases viscosity, leading to cathode wetting issues.31 Despite these advances in room-temperature sodium metal-based batteries, there is a fundamental knowledge gap related to how temperature affects sodium metal anodes. Specifically, the electrochemical behavior, morphological evolution, and the nature of the electrolyte interface at temperatures < −20 °C has not been sufficiently explored.32
Here, a dual-salt electrolyte formulation was designed that enables cycling of sodium metal with high CE at temperatures below −40 °C. This novel electrolyte (0.8 M sodium triflate and 0.2 M NaBF4 in diglyme) shows relatively high ionic conductivity at low temperature (0.8 mS cm−1 at −40 °C), and it enables sodium nucleation on stainless steel substrates with low overpotential. The electrolyte allows for high capacities (>4.0 mA h cm−2) of sodium metal to be deposited as highly uniform and compact films at low temperature, which is in contrast to the high surface area and small particle sizes of deposited lithium metal under such conditions.12,18 SEI formed at different temperatures was investigated with X-ray photoelectron spectroscopy (XPS) and cryogenic transmission electron microscopy (cryo-TEM), revealing the chemical and structural effects of the salt combination on the SEI. These findings indicate the promise of sodium metal as a low-temperature battery anode, and they are an important step toward enabling cost-effective, high energy sodium metal batteries for a wide range of temperatures.
2. Results and discussion
2.1. Temperature-dependent electrochemical investigation
To understand how choice of electrolyte salt affects sodium electrochemistry, we first investigated CE at both ambient (20 °C) and low-temperature (−40 °C) conditions in half-cells containing a sodium metal counter electrode and a stainless-steel working electrode. The salts investigated were 1.0 M NaTFSI (sodium bis(trifluoromethylsulfonyl)imide), NaOTf (sodium triflate), NaBF4, NaClO4, and NaPF6. Diglyme (bis(2-methoxyethyl) ether) was selected as the solvent due to its low freezing point (−64 °C). Fig. 1a shows the CE of sodium cycling in these five electrolytes at 20 °C, where 0.5 mA h cm−2 of sodium was electrodeposited and stripped for 50 cycles. The results demonstrate that the CE strongly depends on choice of salt. The electrolyte with 1.0 M NaPF6 showed the highest CE at 20 °C, with an average CE of 99.5% over 50 cycles (Fig. 1a). The electrolyte with 1.0 M NaBF4 exhibited an average CE of 98.5% over 50 cycles, and the electrolyte with 1.0 M NaOTf showed an average CE of 96.5% over 50 cycles (Fig. 1a). The other two salts (1.0 M NaTFSI or NaClO4) showed extremely low CE (<25%). At −40 °C, all electrolytes showed poor CE except for the electrolyte with 1.0 M NaOTf (Fig. 1b). Nucleation of sodium on the working electrode was difficult in electrolytes other than that with NaOTf at −40 °C, demonstrating the challenges of designing electrolytes for sufficient low-temperature performance. NaPF6 and NaClO4 electrolytes are not included in Fig. 1b because they did not allow for nucleation of sodium at −40 °C. The electrolyte with 1.0 M NaOTf exhibited an average CE of 90.3% at −40 °C, which is lower than that at 20 °C.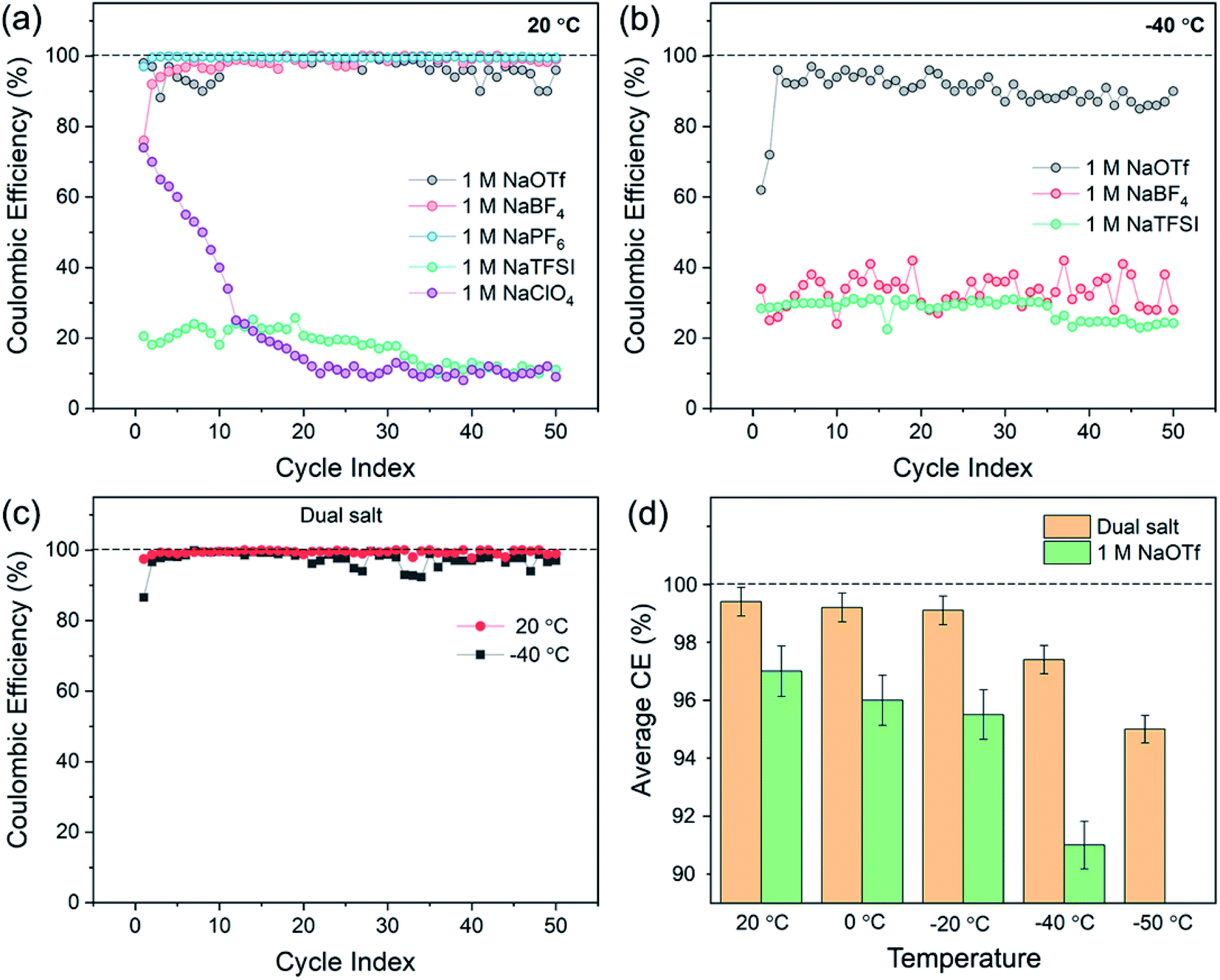 | ||
| Fig. 1 Coulombic efficiency (CE) of sodium deposition/stripping in half cells using electrolytes containing various single salts in diglyme solvent at (a) 20 °C and (b) −40 °C. (c) CE measurements carried out at 20 °C (red) and −40 °C (black) using 0.8 M NaOTf and 0.2 M NaBF4 salts in diglyme (the “dual-salt” electrolyte). For (a–c), the areal capacity for each half cycle was 0.5 mA h cm−2 and the current density was 0.5 mA cm−2. (d) The average CE over 50 cycles from 20 °C to −50 °C using two electrolytes: the dual-salt combination in diglyme and 1.0 M NaOTf in diglyme. The error bars show standard deviation. | ||
Based on these results, we investigated combinations of NaOTf with other sodium salts. This effort was motivated by prior studies on lithium metal electrodes, where dual-salt electrolytes have been shown to increase CE33–37 and have also enabled “anode-free” Li metal batteries that can sustain 90 charge/discharge cycles at 40 °C.37 The effects of multiple salts on sodium metal electrochemistry has not yet been sufficiently explored, however. Through systematic experimentation, we found that combining 0.8 M NaOTf and 0.2 M NaBF4 leads to much-improved sodium reversibility over a wide range of temperatures (Fig. 1c and d). At 20 °C, this dual-salt electrolyte showed a remarkable average CE of 99.4% (Fig. 1c), which is similar to the highest-performing single-salt electrolyte in Fig. 1a (1.0 M NaPF6). At −40 °C, the dual-salt electrolyte showed better CE than all the single-salt electrolytes, with an average CE of 97.4% over 50 cycles (Fig. 1c). As shown in Fig. 1d, the dual-salt electrolyte also showed a CE of >95% across the range of temperatures from 20 °C to −50 °C, which is higher than all other single salts across this temperature range. Thus, this dual-salt electrolyte shows encouraging sodium deposition/stripping behavior down to −50 °C. Furthermore, these CE values at low temperatures are substantially higher than previous results for lithium metal in ether-based electrolytes.12,18
Additional CE experiments were carried out to determine how deposition of different capacities (from 0.5 mA h cm−2 to 4 mA h cm−2) at −40 °C affects cycling reversibility. Fig. 2a shows that with higher plating capacities, the CE increases to >99% after the first few cycles at −40 °C. This important result suggests that the dual-salt electrolyte causes sodium to deposit in a uniform and dense fashion without excessive exposed surface area that would serve to increase the extent of SEI growth in thicker films. Fig. S1† shows that the higher CE with higher deposited capacity is sustained over at least 50 cycles. In addition to these experiments, we also investigated sodium plating and stripping in Na/Na symmetric cells as a function of temperature (Fig. 2b). The dual-salt electrolyte showed stable sodium plating/stripping with low overpotentials from 20 °C to −60 °C. For comparison, Fig. 2b shows data using the 1.0 M NaOTf electrolyte. While low overpotential was also observed, substantial overpotential fluctuations are evident at −60 °C, indicating unstable sodium plating/stripping and the formation of inactive metal at this temperature. Other single-salt electrolytes showed similar fluctuations (Fig. S2†). Conductivity measurements (Fig. S3†) showed that both electrolytes exhibited very similar low-temperature conductivities, suggesting that the observed unstable sodium plating is not a result of anomalous decreases in conductivity. Instead, it is likely a result of the complex interplay among the properties of the SEI, Na+ solvation/desolvation phenomena, and charge transfer kinetics. Finally, the dual-salt electrolyte enabled excellent long-term cycling of symmetric cells at −40 °C (Fig. S4†), with low and steady overpotential (∼50 mV) over 300 h of cycling, while a similar experiment with only NaOTf showed a steady increase of overpotential and irregular voltage fluctuations (Fig. S4†).
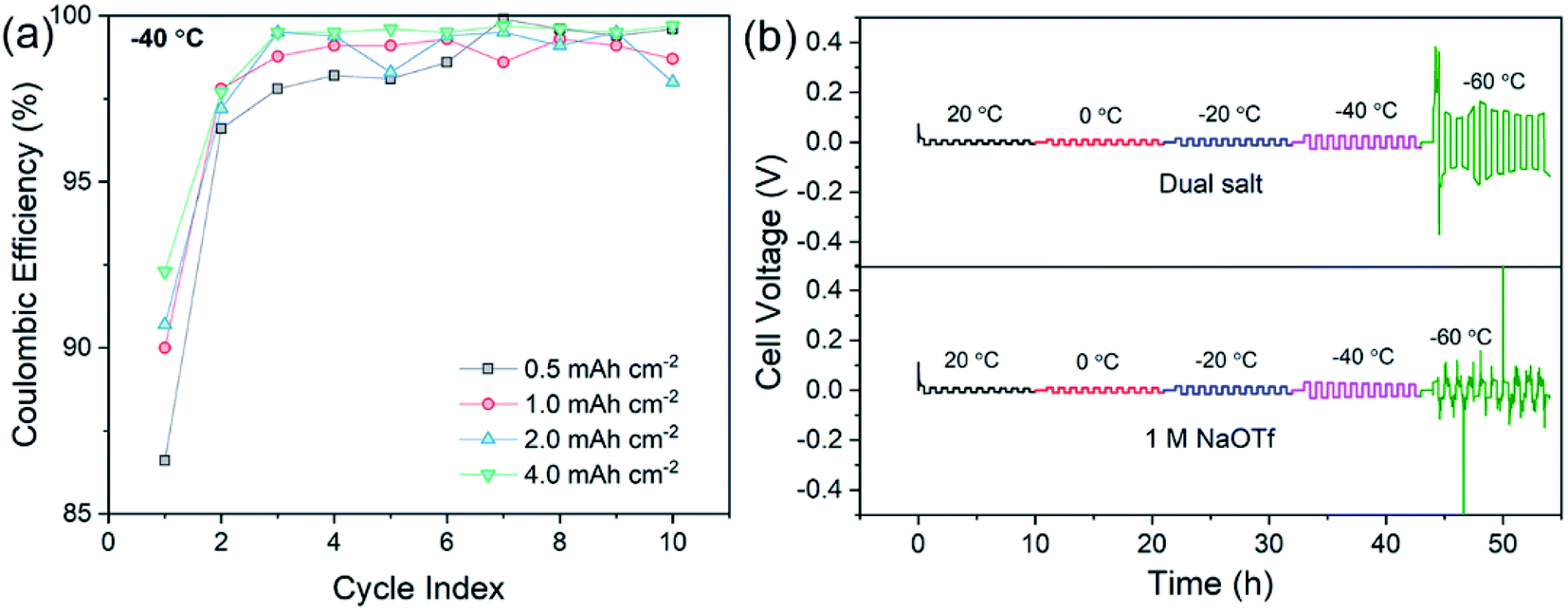 | ||
| Fig. 2 (a) Capacity-dependent CE measurements carried out at −40 °C using the dual-salt electrolyte. The current density was fixed at 0.5 mA cm−2. (b) Temperature-dependent galvanostatic cycling of Na/Na symmetric cells using two electrolytes: the dual salt in diglyme and 1.0 M NaOTf in diglyme. A current of 0.5 mA cm−2 was used with 30 min half-cycles. | ||
2.2. Morphological analysis
Since the morphology of electrodeposited alkali metals is often directly correlated to CE,38–42 we examined the morphology of sodium electrodeposited from different electrolytes as a function of temperature using scanning electron microscopy (SEM). As expected, our dual-salt electrolyte resulted in the deposition of highly uniform, compact, and non-dendritic films at both 20 °C and −40 °C (Fig. 3a and d), which gives rise to the high CE measured under these conditions. Fig. S5† shows cross-sectional SEM images of similar samples. At 20 °C, the electrolytes containing 1.0 M NaPF6, 1.0 M NaOTf, and 1.0 M NaBF4 also showed uniform, non-dendritic sodium morphology (Fig. 3b and c and S6†), while electrolytes containing the other two salts (1.0 M NaTFSI and 1.0 M NaClO4, see Fig. S6†) showed nodular sodium deposits and increased film porosity, consistent with prior studies.28,29 The increased film porosity can accelerate SEI formation, which likely contributes to the much lower CE values during cycling of the 1.0 M NaTFSI and 1.0 M NaClO4 electrolytes (Fig. 1a). At −40 °C, most of the single-salt electrolytes exhibited non-uniform and porous sodium morphology (Fig. 3f and S7†), consistent with the decreased CE. Among the single-salt electrolytes, the electrolyte with 1.0 M NaOTF showed deposits with the greatest uniformity at −40 °C (Fig. 3e), which correlates with its relatively high CE. Overall, the dual-salt electrolyte produced sodium films that appear to have the smoothest and least porous morphology at −40 °C. The uniform sodium films have a low surface area that is exposed to the electrolyte, which reduces parasitic SEI growth and dead sodium formation, leading to enhanced sodium reversibility.38 We note that this uniform and compact sodium morphology at low temperatures is distinct from prior studies on lithium metal in a variety of different electrolytes.12,18,43–45 These studies have shown that low temperatures (<−20 °C) cause the formation of smaller Li particles, which have high surface area and therefore substantial SEI growth and lower CE. Our findings regarding sodium in this dual-salt electrolyte thus indicate the significant promise of sodium metal for enabling low-temperature battery systems. | ||
| Fig. 3 SEM images after electrodeposition of 0.25 mA h cm−2 of sodium onto stainless steel electrodes at different temperatures in three different electrolytes using a current density of 0.5 mA cm−2. (a and d) Dual-salt electrolyte at 20 °C and −40 °C; (b and e) 1.0 M NaOTf electrolyte at 20 °C and −40 °C; (c and f) 1.0 M NaBF4 at 20 °C and −40 °C. | ||
Based on these promising results, we investigated the temperature-dependent electrochemical behavior of dual-salt-electrolyte-based full cells with Na3V2(PO4)3 (NVP) cathode material and sodium metal anodes. Fig. 4a shows the charge/discharge curves for a cell that was sequentially cooled to lower temperatures for each cycle. These data show that from 0 °C to −60 °C, the Na/NVP cell exhibited good reversibility, and at −40 °C about ∼50% of the specific capacity attained at 0 °C was retained. Fig. S8† shows data from the first cycle of a different cell that was operated only at −40 °C, demonstrating similar results as in Fig. 4a. Fig. 4b shows that the dual-salt electrolyte enabled stable charging/discharging for 80 cycles at −40 °C with relatively high CE. While the areal capacity of this cell is low, these data demonstrate that this dual-salt electrolyte enables the operation of sodium metal batteries at a much lower temperature than is possible for Li-ion batteries.
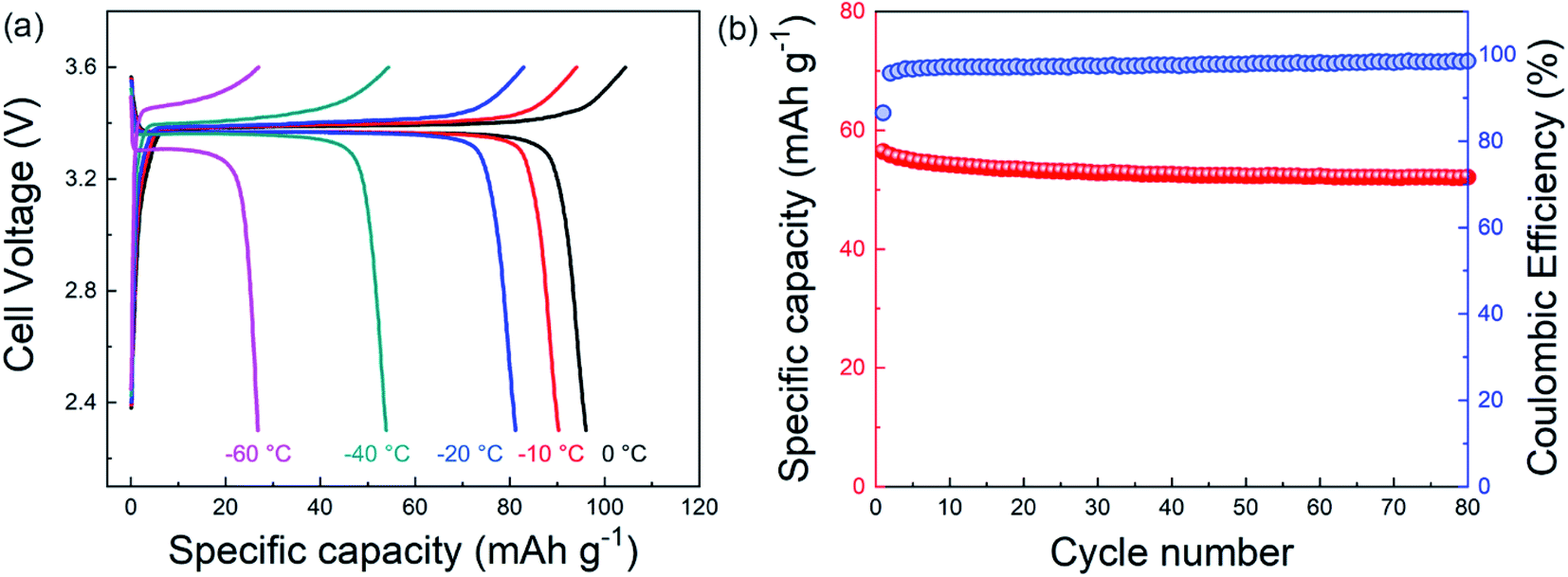 | ||
| Fig. 4 (a) Charge–discharge profiles of a Na/Na3V2(PO4)3 (NVP) cell using the dual-salt electrolyte as a function of temperature. The charging/discharging rate was 0.2C (1C = 110 mA g−1). Data for 20 °C was not gathered likely due to reactions between NVP and the electrolyte at this temperature which precluded cycling. (b) Specific capacity (based on the active mass in the cathode) and CE as a function of cycle number for a Na/NVP cell operated at −40 °C. The areal capacity for this Na/NVP cell was ∼0.1 mA h cm−2. | ||
2.3. Chemistry and structure of the solid-electrolyte interphase
Since the properties of the SEI can determine electrochemical behavior of alkali metals, we next investigated the chemical and structural characteristics of the SEI as a function of temperature with X-ray photoelectron spectroscopy (XPS) and cryogenic transmission electron microscopy (cryo-TEM). XPS can provide spatially-averaged information regarding the chemical properties of the SEI, and cryo-TEM can reveal the nanoscale structure of the SEI.46 For XPS, we electrodeposited a fixed amount of sodium onto copper foil, disassembled and washed with diglyme in a glove box, and transferred to the XPS using a vacuum transfer holder (see Experimental section). Fig. 5a–f show the C 1s, F 1s, and Na 1s core-level spectra from SEI formed at different temperatures (20 °C and −40 °C). These data compare two different electrolytes: 1.0 M NaOTf in diglyme (Fig. 5a–c) and the dual-salt mixture in diglyme (Fig. 5d–f). The C 1s spectra for the single and dual-salt electrolytes (Fig. 5a and d) feature four peaks at 20 °C with binding energies of 293.2 eV (C–Fx), 289.5 eV (C–Ox), 286.7 eV (O–C–O, C–SOx), and 285.0 eV (C–C, C–H).28,29 For the dual-salt electrolyte (Fig. 5d), the SEI formed at −40 °C shows a lower-intensity C 1s signal compared to that at room temperature. This indicates that the SEI formed at lower temperature in the dual-salt electrolyte features less carbon content, which may be due to kinetically limited decomposition of the solvent, consistent with our previous studies of lithium systems.12,18 The F 1s spectra in Fig. 5b and e show two peaks at 683.1 eV (F–Na) and 688.0 eV (F–C) for both electrolytes. An additional shoulder at about 686.2 eV (F–B) is present in the dual-salt electrolyte due to decomposition of NaBF4 (Fig. 5e). While the F–C peak is dominant in these plots, the peak at 683.1 eV was clearly dominant for both electrolytes at both temperatures after sputtering thin layers of material to expose the SEI interior (Fig. S9†). This suggests that the interior of the SEI is rich in NaF. The Na 1s spectra in Fig. 5c and f show single peaks at ∼1071–1072 eV, which can be attributed to overlapping contributions of Na–O and Na–F.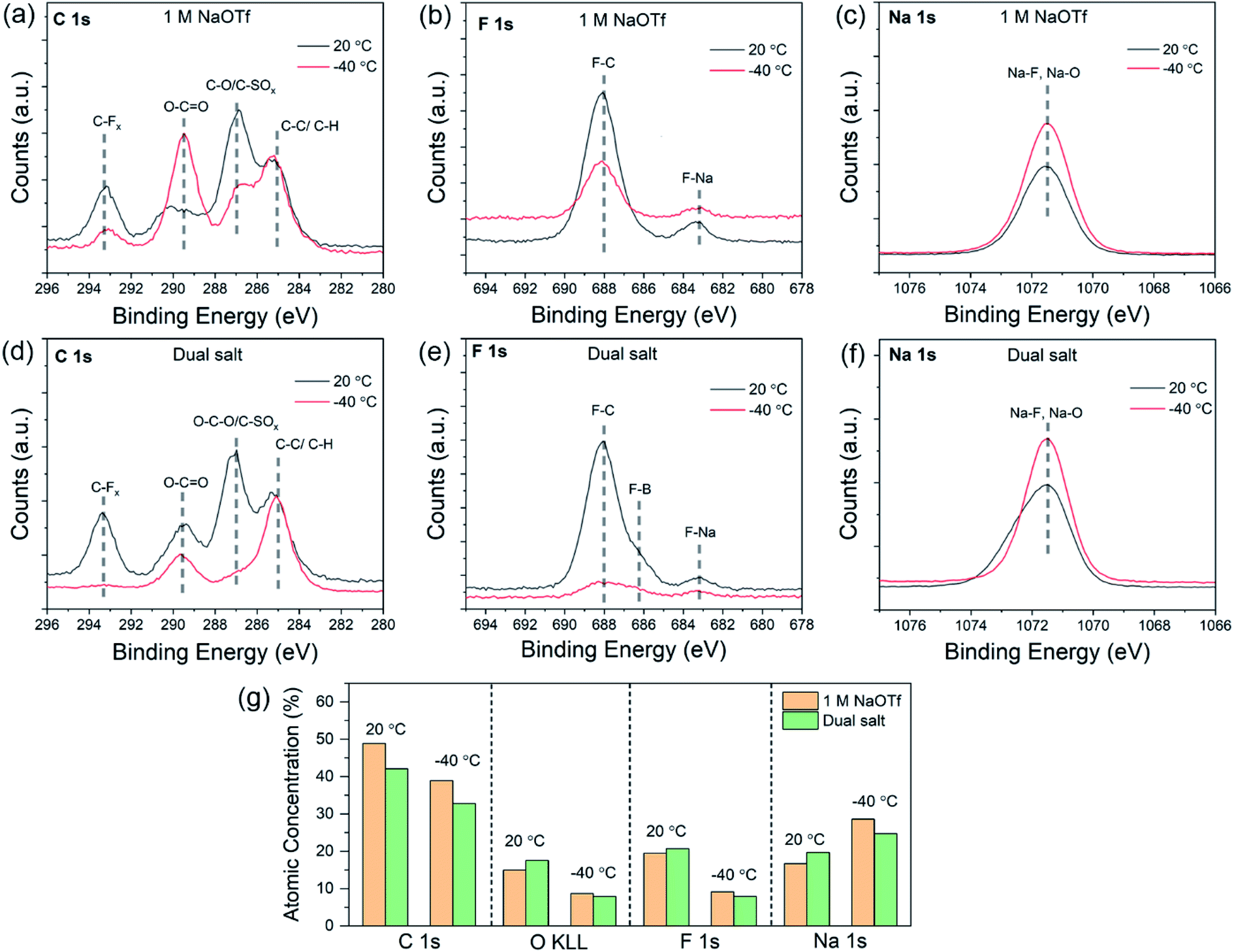 | ||
| Fig. 5 Temperature-dependent SEI chemistry probed by vacuum-transfer XPS. Sodium was electrodeposited at 0.5 mA cm−2 and vacuum-transferred to the XPS instrument. (a–c) High-resolution C 1s, F 1s, and Na 1s XPS spectra for SEI formed on sodium with the 1.0 M NaOTf electrolyte at 20 °C and −40 °C. (d–f) High-resolution C 1s, F 1s, and Na 1s XPS spectra for SEI formed on sodium with the dual-salt electrolyte at 20 °C and −40 °C. (g) Atomic concentrations of various species measured by integrating core level spectra for SEI samples generated at 20 °C and −40 °C. Since the O 2p core level overlapped with the Na KL23L23 Auger peak, the O KL23L23 core level was used to quantify oxygen content. | ||
To summarize these findings in combination with XPS depth profiling results (Fig. S9 and S10†), Fig. 5g shows the atomic concentrations of different elements found in the SEI for both electrolytes at 20 °C and −40 °C. Both electrolytes at −40 °C contain fewer organic species and are chemically distinct from the SEIs formed at 20 °C. These changes in SEI composition at low temperatures can be attributed to altered decomposition kinetics of the electrolyte. Furthermore, since the NaBF4 salt can readily be decomposed to form NaF,28 the dual salt combination likely leads to SEI films that are rich in inorganic species at both 20 °C and −40 °C.
Cryo-TEM was used to investigate the nanoscale structure of the SEI formed in the dual-salt electrolyte, as has been performed in previous work on other electrolyte systems.47–49 Cryo-TEM samples were prepared in a similar fashion to our previous work,18 in which sodium was directly electrodeposited onto a Cu TEM grid within a coin cell, followed by electrochemical stripping of the sodium so that the SEI remains. Fig. 6a shows a high-resolution image of SEI formed at −40 °C. This image is overlaid with false-colored lattice fringes of different crystallites found in this region (the original image is shown in Fig. S11†). This SEI film clearly exhibits the “mosaic” structural motif, with inorganic crystallites embedded in an amorphous matrix. While it is difficult to ascribe these individual sets of lattice fringes to full crystal structures, the various lattice spacings are consistent with Na2CO3, Na2O, and Na2SO4 crystallites, which are expected to be found in the SEI. Cryo-TEM experiments on SEI grown at 20 °C also showed a similar mosaic structure (Fig. S12†). Thus, the SEI at both temperatures is rich in sodium-based inorganic compounds, which is consistent with the XPS results in Fig. 5. One difference between the SEI formed at different temperatures was the average crystallite size: the crystallite size was smaller at −40 °C (191 ± 24 nm2) compared to the SEI formed at 20 °C (306 ± 43 nm2), as measured across multiple recorded images. Selected area electron diffraction (SAED) patterns from SEI formed at −40 °C and 20 °C (Fig. 6b and c, respectively) are consistent with the likely existence of Na2CO3, Na2O, Na2SO4 and NaF phases, and the more diffuse diffraction rings at −40 °C again indicate smaller crystallites. Inorganic-rich SEI is generally considered to be beneficial for plating/stripping of alkali metals,28,29 and the high inorganic content in the SEI at both temperatures here likely enables the high CE observed with the dual-salt electrolyte across a wide range of temperatures. This is in contrast to prior work on lithium metal cycling at low temperature,12,18 which has shown that in certain electrolytes the SEI can exhibit low crystallinity, which is correlated with low CE during cycling.
 | ||
| Fig. 6 Cryo-TEM investigation of the structure of the SEI formed at −40 °C and 20 °C with the dual-salt electrolyte. (a) High resolution cryo-TEM image of the SEI formed at −40 °C with colored overlays of lattice fringes extracted with fast Fourier transform (FFT) analysis. The FFT of the image is shown in the inset with colored circles corresponding to the lattice fringes. The 3.2 Å lattice fringes could arise from Na2O (111) or Na2CO3 (201), the 3.0 Å fringes likely arise from Na2CO3 (002), and the 2.8 Å fringes could arise from crystal planes in Na2O, Na2CO3, or Na2SO4. (b) SAED pattern of SEI formed at −40 °C and (c) SAED pattern of SEI formed at 20 °C; both images feature the common diffraction rings from sodium-containing crystalline compounds labeled. | ||
To link these investigations to transport properties at the sodium interface, electrochemical impedance spectroscopy (EIS) was performed on sodium electrodes in both the dual-salt and the 1.0 M NaOTf-based electrolytes. For these experiments, sodium symmetric cells underwent one galvanostatic cycle at either 20 °C or −40 °C to form the SEI, and then EIS was subsequently carried out over a range of temperatures (Fig. S13†). The charge transfer resistance (RCT) was extracted from the Nyquist plots using the equivalent circuit model shown in the inset of Fig. S13a.† For SEI formed at 20 °C, the RCT increases from ∼25 Ω at 20 °C to ∼250–300 Ω at −60 °C. Interestingly, SEI formed at −40 °C exhibits lower RCT values across the entire temperature range, including ∼100–125 Ω at −60 °C. The lower impedance for SEI formed at low temperatures is likely associated with the thinner nature of this SEI observed with XPS. Finally, the dual-salt electrolyte has slightly higher RCT than the single-salt electrolyte across the temperature range.
3. Conclusion
In summary, we have developed a new electrolyte that enables electrochemical cycling of sodium metal anodes at low temperatures with high coulombic efficiency. The synergy between NaOTf and NaBF4 salts in diglyme solvent leads to highly efficient sodium deposition/stripping down to −50 °C, as well as relatively high reversible capacity in sodium metal batteries with the intercalation cathode Na3V2(PO4)3 at −40 °C. The promising electrochemical behavior is associated with the uniform and compact morphology of the deposited sodium, as well as the high inorganic content in the SEI across the examined temperature range. Our findings suggest that the development of sodium metal-based batteries with excellent rechargeability at low temperatures is possible by designing electrolytes to control sodium morphology and SEI composition. Furthermore, the high CE observed for sodium cycling herein exceeds prior results on lithium metal, indicating that sodium metal is particularly promising for low-temperature batteries. Since conventional Li-ion batteries are unable to reversibly deliver power below approximately −20 °C, these findings could open up new avenues for rechargeable energy storage under extreme temperature conditions, with important implications for electric flight, deep-sea vehicles, and space exploration.4. Experimental section
4.1. Materials
All the chemicals used in this study were purchased from commercial vendors and used without further purification unless otherwise stated. The chemicals used for electrolyte preparation were purchased from Sigma-Aldrich, and they include the following: bis(2-methoxyethyl) ether (99.5%, anhydrous), sodium hexafluorophosphate (NaPF6, 98%), sodium tetrafluoroborate (NaBF4, 98%), sodium trifluoromethanesulfonate (NaOTf, 98%), sodium perchlorate (NaClO4, ACS reagent, ≥98.0%), and sodium trifluoromethanesulfonimide (NaTFSI, 97%).4.2. Electrolyte preparation
Bis(2-methoxyethyl) ether (diglyme) solvent was dried with molecular sieves (3 Å) so that the water content was <15 ppm (Karl Fischer titration). The diglyme-based electrolytes were prepared by dissolving sodium salts in the solvent inside an Ar-filled glove box (Vigor), where the moisture and oxygen content were <0.3 ppm. The following electrolyte compositions were used in this study:(1) 1.0 M NaPF6 in diglyme
(2) 1.0 M NaBF4 in diglyme
(3) 1.0 M NaOTf in diglyme
(4) 1.0 M NaClO4 in diglyme
(5) 1.0 M NaTFSI in diglyme
(6) 0.8 M NaOTf + 0.2 M NaBF4 in diglyme (referred to as “dual salt”)
4.3. Cell fabrication and electrochemistry
2032-type coin cells were used for all electrochemical measurements except for electrolyte ionic conductivity measurements, where a custom Swagelok cell was used. The coin cells and Swagelok cells were assembled inside an Ar-filled glove box. The ionic conductivity values of the electrolytes as a function of temperature were determined by carrying out electrochemical impedance spectroscopy (EIS) in Swagelok cells. For these measurements, an electrolyte-soaked fiberglass membrane was sandwiched between two stainless-steel electrodes. The applied frequency range was 1 MHz to 0.1 Hz with an AC amplitude of 5 mV. EIS measurements were carried out after holding at the specified temperature for at least 2 h.Sodium metal (Sigma-Aldrich, 99.8%) was used as the sodium source for all experiments, and 0.5-in diameter disks (0.8 mm thick) were used in coin cells. Na/Na symmetric cells were fabricated in coin cells with freshly cut Na metal and 40 μL of electrolyte. For coulombic efficiency (CE) measurements, coin cells were assembled with stainless steel as the working electrode and the freshly cut Na metal foil as the counter electrode with 40 μL of electrolyte. CE measurements were performed by plating and stripping with a current density of 0.5 mA cm−2 over 1 h (0.5 mA h cm−2 deposited or stripped per half cycle), with an upper voltage cutoff of 1.0 V. To investigate the impedance of the SEI formed at different temperatures, Na/Na symmetric cells were assembled with various electrolytes and cycled once (0.5 mA cm−2 for 30 min each of plating and stripping) at the desired temperature before carrying out EIS. All electrochemical measurements were performed with a Bio-Logic VMP3 potentiostat, and cells were tested within an ESPEC MC-812R environmental chamber to control temperature between 20 °C and −60 °C. Coin cells were held at a specified temperature for at least 1 h before testing to ensure thermal equilibrium.
4.4. Cathode material synthesis, electrode fabrication, and full-cell electrochemical testing
Na3V2(PO4)3 was produced via a solution-based synthesis using an optimized recipe reported previously.50,51 The phase purity of the Na3V2(PO4)3 was verified with X-ray diffraction (Fig. S14†). The synthesized Na3V2(PO4)3 powder was mixed with carbon black (C-NERGY) and polyvinylidene fluoride (Arkema) in a weight ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1 in 1-methyl-2-pyrrolidone (Sigma-Aldrich) to form a slurry. The prepared slurry was cast onto an Al foil, which was fully dried in a vacuum oven. The active material mass loading was typically 1 mg cm−2. For the electrochemical testing of full cells, a current density of 0.2C (1C = 110 mA g−1) was employed with voltage cutoffs between 2.3 V and 3.6 V using a standard battery tester (CT2001A, LAND Electronics Co., Ltd).
:3:1 in 1-methyl-2-pyrrolidone (Sigma-Aldrich) to form a slurry. The prepared slurry was cast onto an Al foil, which was fully dried in a vacuum oven. The active material mass loading was typically 1 mg cm−2. For the electrochemical testing of full cells, a current density of 0.2C (1C = 110 mA g−1) was employed with voltage cutoffs between 2.3 V and 3.6 V using a standard battery tester (CT2001A, LAND Electronics Co., Ltd).
4.5. Scanning electron microscopy (SEM)
A Hitachi SU8030 microscope with an accelerating voltage of 3 kV was used to examine the morphology of electrodeposited sodium. For ex situ SEM analysis, the sodium samples were prepared by electrodepositing sodium on stainless-steel foil at a current density of 0.5 mA cm−2 for 0.5 h, yielding an areal capacity of 0.25 mA h cm−2. The cells were then disassembled in an Ar-filled glove box and washed with 0.5 mL of diglyme, and the sample was dried under vacuum before imaging. During loading into the SEM, the samples were exposed to atmosphere for ∼5 s, and evacuation of the loading chamber took ∼30 s.4.6. Cryogenic transmission electron microscopy
The cryo-TEM measurements were carried out using an FEI Tecnai F30 TEM operated at 300 kV. TEM samples were prepared by carrying out a single sodium plating/stripping cycle (0.5 mA cm−2 for 2 h) using a copper TEM grid as the working electrode in a coin cell. After the sodium plating/stripping cycle, the coin cell was disassembled in an Ar-filled glove box, and the TEM grid was washed with 0.5 mL of diglyme and dried in vacuum before loading it into a Gatan cryogenic cooling holder. The holder was exposed to atmosphere for ∼10 s as the holder was transferred from the argon environment into the TEM load lock, where it was brought to vacuum. Liquid nitrogen was then added to the holder's external dewar, and the temperature of the sample was maintained at −175 °C for imaging. After temperature stabilization, imaging was conducted while recording with a Gatan OneView camera. Under these conditions, beam-induced sample degradation was minimized at electron dose levels below 5 × 102 electrons per Å2 per s. Thus, to optimize the tradeoff between image resolution and sample degradation, all images were recorded at a dose rate between 1 and 5 × 102 electrons per Å2 per s.4.7. X-ray photoelectron spectroscopy
For XPS analysis, Na/Cu half cells were constructed and galvanostatic sodium plating (0.5 mA cm−2 for 2 h) was carried out at two different temperatures: 20 °C and −40 °C. Once the sodium plating was completed, these cells were disassembled in an Ar-filled glove box, washed with 1 mL of diglyme, and dried in vacuum before being placed into a vacuum transfer holder for transfer to the XPS instrument without exposure to atmosphere. XPS analysis was performed using a Thermo K-Alpha XPS with an Al Kα source, a 400 μm spot size, and 15 W X-ray gun power. The base pressure was 5 × 10−8 torr, and the analyzer pass energy was 50 eV with a resolution of 0.05 eV and a dwell time of 100 ms. Surface charging was compensated with a flood gun with slow electrons and Ar+ ions. Charge referencing for the peaks was performed using C 1s at 284.8 eV. For XPS depth profiles, a series of three etches of 60 s each was performed.Author contributions
AT: conceptualization, investigation, data curation, formal analysis, writing, visualization; PP: investigation, formal analysis, visualization; NK: investigation; CW: investigation, formal analysis, writing; WL: writing, supervision, funding acquisition; MTM: conceptualization, formal analysis, writing, supervision, project administration, funding acquisition.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by Early Career Faculty grants from NASA's Space Technology Research Grants Program under awards 80NSSC18K1507 and 80NSSC18K1514. This work was performed in part at the Georgia Tech Institute for Electronics and Nanotechnology, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (ECCS-2025462). M. T. M. acknowledges support from a Sloan Research Fellowship in Chemistry from the Alfred P. Sloan Foundation.References
- A. Gupta and A. Manthiram, Adv. Energy Mater., 2020, 10, 2001972 CrossRef CAS PubMed.
- M. C. Smart, B. V. Ratnakumar and S. Surampudi, J. Electrochem. Soc., 1999, 146, 486–492 CrossRef CAS.
- R. V. Bugga and M. C. Smart, ECS Trans., 2010, 25, 241–252 CrossRef.
- Q. Li, D. Lu, J. Zheng, S. Jiao, L. Luo, C.-M. Wang, K. Xu, J.-G. Zhang and W. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 42761–42768 CrossRef CAS PubMed.
- S. S. Zhang, K. Xu and T. R. Jow, Electrochim. Acta, 2004, 49, 1057–1061 CrossRef CAS.
- S. S. Zhang, K. Xu and T. R. Jow, J. Power Sources, 2003, 115, 137–140 CrossRef CAS.
- Q. Li, S. Jiao, L. Luo, M. S. Ding, J. Zheng, S. S. Cartmell, C.-M. Wang, K. Xu, J.-G. Zhang and W. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 18826–18835 CrossRef CAS PubMed.
- M. C. Smart, J. F. Whitacre, B. V. Ratnakumar and K. Amine, J. Power Sources, 2007, 168, 501–508 CrossRef CAS.
- H. P. Lin, D. Chua, M. Salomon, H. C. Shiao, M. Hendrickson, E. Plichta and S. Slane, ECS Solid State Lett., 2001, 4, A71 CrossRef CAS.
- C. K. Huang, J. S. Sakamoto, J. Wolfenstine and S. Surampudi, J. Electrochem. Soc., 2000, 147, 2893 CrossRef CAS.
- Y. Ein-Eli, S. R. Thomas, R. Chadha, T. J. Blakley and V. R. Koch, J. Electrochem. Soc., 1997, 144, 823–829 CrossRef CAS.
- A. C. Thenuwara, P. P. Shetty and M. T. McDowell, Nano Lett., 2019, 19, 8664–8672 CrossRef CAS PubMed.
- X. Fan, X. Ji, L. Chen, J. Chen, T. Deng, F. Han, J. Yue, N. Piao, R. Wang, X. Zhou, X. Xiao, L. Chen and C. Wang, Nat. Energy, 2019, 4, 882–890 CrossRef CAS.
- X. Dong, Y. Lin, P. Li, Y. Ma, J. Huang, D. Bin, Y. Wang, Y. Qi and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 5623–5627 CrossRef CAS PubMed.
- C. S. Rustomji, Y. Yang, T. K. Kim, J. Mac, Y. J. Kim, E. Caldwell, H. Chung and Y. S. Meng, Science, 2017, 356, eaal4263 CrossRef PubMed.
- Y. Yang, Y. Yin, D. M. Davies, M. Zhang, M. Mayer, Y. Zhang, E. S. Sablina, S. Wang, J. Z. Lee, O. Borodin, C. S. Rustomji and Y. S. Meng, Energy Environ. Sci., 2017, 13, 2209–2219 RSC.
- M. Uhlemann, M. Madian, R. Leones, S. Oswald, S. Maletti, A. Eychmüller and D. Mikhailova, ACS Appl. Mater. Interfaces, 2020, 12, 37227–37238 CrossRef CAS PubMed.
- A. C. Thenuwara, P. P. Shetty, N. Kondekar, S. E. Sandoval, K. Cavallaro, R. May, C.-T. Yang, L. E. Marbella, Y. Qi and M. T. McDowell, ACS Energy Lett., 2020, 5, 2411–2420 CrossRef CAS.
- A. P. Cohn, N. Muralidharan, R. Carter, K. Share and C. L. Pint, Nano Lett., 2017, 17, 1296–1301 CrossRef CAS PubMed.
- R. Cao, K. Mishra, X. Li, J. Qian, M. H. Engelhard, M. E. Bowden, K. S. Han, K. T. Mueller, W. A. Henderson and J.-G. Zhang, Nano Energy, 2016, 30, 825–830 CrossRef CAS.
- B. Lee, E. Paek, D. Mitlin and S. W. Lee, Chem. Rev., 2019, 119, 5416–5460 CrossRef CAS PubMed.
- L. Fan and X. Li, Nano Energy, 2018, 53, 630–642 CrossRef CAS.
- X. Zheng, C. Bommier, W. Luo, L. Jiang, Y. Hao and Y. Huang, Energy Storage Mater., 2019, 16, 6–23 CrossRef.
- Y. Wang, Y. Wang, Y.-X. Wang, X. Feng, W. Chen, X. Ai, H. Yang and Y. Cao, Chem, 2019, 5, 2547–2570 CAS.
- T. Liu, Y. Zhang, Z. Jiang, X. Zeng, J. Ji, Z. Li, X. Gao, M. Sun, Z. Lin, M. Ling, J. Zheng and C. Liang, Energy Environ. Sci., 2019, 12, 1512–1533 RSC.
- H. Che, S. Chen, Y. Xie, H. Wang, K. Amine, X.-Z. Liao and Z.-F. Ma, Energy Environ. Sci., 2017, 10, 1075–1101 RSC.
- G. G. Eshetu, G. A. Elia, M. Armand, M. Forsyth, S. Komaba, T. Rojo and S. Passerini, Adv. Energy Mater., 2020, 10, 2000093 CrossRef CAS.
- P. M. L. Le, T. D. Vo, H. Pan, Y. Jin, Y. He, X. Cao, H. V. Nguyen, M. H. Engelhard, C. Wang, J. Xiao and J.-G. Zhang, Adv. Funct. Mater., 2020, 30, 2001151 CrossRef CAS.
- Z. W. Seh, J. Sun, Y. Sun and Y. Cui, ACS Cent. Sci., 2015, 1, 449–455 CrossRef CAS PubMed.
- J. Zheng, S. Chen, W. Zhao, J. Song, M. H. Engelhard and J.-G. Zhang, ACS Energy Lett., 2018, 3, 315–321 CrossRef CAS.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- Y. You, H.-R. Yao, S. Xin, Y.-X. Yin, T.-T. Zuo, C.-P. Yang, Y.-G. Guo, Y. Cui, L.-J. Wan and J. B. Goodenough, Adv. Mater., 2016, 28, 7243–7248 CrossRef CAS PubMed.
- S. Liu, Q. Zhang, X. Wang, M. Xu, W. Li and B. L. Lucht, ACS Appl. Mater. Interfaces, 2020, 12, 33719–33728 CrossRef CAS PubMed.
- S. Jiao, X. Ren, R. Cao, M. H. Engelhard, Y. Liu, D. Hu, D. Mei, J. Zheng, W. Zhao, Q. Li, N. Liu, B. D. Adams, C. Ma, J. Liu, J.-G. Zhang and W. Xu, Nat. Energy, 2018, 3, 739–746 CrossRef CAS.
- X. Shangguan, G. Xu, Z. Cui, Q. Wang, X. Du, K. Chen, S. Huang, G. Jia, F. Li, X. Wang, D. Lu, S. Dong and G. Cui, Small, 2019, 15, 1900269 CrossRef PubMed.
- T. T. Beyene, H. K. Bezabh, M. A. Weret, T. M. Hagos, C.-J. Huang, C.-H. Wang, W.-N. Su, H. Dai and B.-J. Hwang, J. Electrochem. Soc., 2019, 166, A1501–A1509 CrossRef CAS.
- R. Weber, M. Genovese, A. J. Louli, S. Hames, C. Martin, I. G. Hill and J. R. Dahn, Nat. Energy, 2019, 4, 683–689 CrossRef CAS.
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M.-H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed.
- S. Tang, Z. Qiu, X.-Y. Wang, Y. Gu, X.-G. Zhang, W.-W. Wang, J.-W. Yan, M.-S. Zheng, Q.-F. Dong and B.-W. Mao, Nano Energy, 2018, 48, 101–106 CrossRef CAS.
- T.-S. Wang, Y. Liu, Y.-X. Lu, Y.-S. Hu and L.-Z. Fan, Energy Storage Mater., 2018, 15, 274–281 CrossRef.
- B. Sun, P. Li, J. Zhang, D. Wang, P. Munroe, C. Wang, P. H. L. Notten and G. Wang, Adv. Mater., 2018, 30, 1801334 CrossRef PubMed.
- Q. Zhang, Y. Lu, L. Miao, Q. Zhao, K. Xia, J. Liang, S.-L. Chou and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 14796–14800 CrossRef CAS PubMed.
- J. Wang, W. Huang, A. Pei, Y. Li, F. Shi, X. Yu and Y. Cui, Nat. Energy, 2019, 4, 664–670 CrossRef CAS.
- K. Yan, J. Wang, S. Zhao, D. Zhou, B. Sun, Y. Cui and G. Wang, Angew. Chem., Int. Ed., 2019, 58, 11364–11368 CrossRef CAS PubMed.
- M. T. McDowell, F. J. Q. Cortes, A. C. Thenuwara and J. A. Lewis, Chem. Mater., 2020, 32, 8755–8771 CrossRef CAS.
- W. Huang, H. Wang, D. T. Boyle, Y. Li and Y. Cui, ACS Energy Lett., 2020, 5, 1128–1135 CrossRef CAS.
- Y. Li, Y. Li, A. Pei, K. Yan, Y. Sun, C.-L. Wu, L.-M. Joubert, R. Chin, A. L. Koh, Y. Yu, J. Perrino, B. Butz, S. Chu and Y. Cui, Science, 2017, 358, 506 CrossRef CAS PubMed.
- X. Wang, M. Zhang, J. Alvarado, S. Wang, M. Sina, B. Lu, J. Bouwer, W. Xu, J. Xiao, J.-G. Zhang, J. Liu and Y. S. Meng, Nano Lett., 2017, 17, 7606–7612 CrossRef CAS PubMed.
- J. Huang, X. Guo, X. Du, X. Lin, J.-Q. Huang, H. Tan, Y. Zhu and B. Zhang, Energy Environ. Sci., 2019, 12, 1550–1557 RSC.
- Z. Jian, W. Han, X. Lu, H. Yang, Y.-S. Hu, J. Zhou, Z. Zhou, J. Li, W. Chen, D. Chen and L. Chen, Adv. Energy Mater., 2013, 3, 156–160 CrossRef CAS.
- Q. An, F. Xiong, Q. Wei, J. Sheng, L. He, D. Ma, Y. Yao and L. Mai, Adv. Energy Mater., 2015, 5, 1401963 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta00842k |
| This journal is © The Royal Society of Chemistry 2021 |