
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bottom-up assembly of bimetallic nanocluster catalysts from oxide-supported single-atom precursors†
Bidyut B.
Sarma‡
 a,
Giovanni
Agostini
b,
Marcos G.
Farpón
c,
Carlo
Marini
b,
Norbert
Pfänder
d and
Gonzalo
Prieto
*ac
a,
Giovanni
Agostini
b,
Marcos G.
Farpón
c,
Carlo
Marini
b,
Norbert
Pfänder
d and
Gonzalo
Prieto
*ac
aMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany. E-mail: prieto@mpi-muelheim.mpg.de; prieto@itq.upv.es
bALBA Synchrotron Light Source, Carrer de la Llum 2-26, Cerdanyola del Vallès, 08290, Barcelona, Spain
cITQ Instituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas (UPV-CSIC), Av. Los Naranjos s/n, 46022 Valencia, Spain
dMax-Planck-Institut für Chemische Energiekonversion, Stiftstraße 34-36, 45470 Mülheim an der Ruhr, Germany
First published on 5th March 2021
Abstract
The precise synthesis and stabilization of oxide-supported bimetallic clusters in the low-to-sub nanometer size regime is highly relevant in various fields, from optics and sensing to electrochemistry and catalysis. In surface-driven phenomena such as catalysis, avoiding metal segregation and agglomeration is essential for performance and stability under relevant operation conditions. Here we show how high-temperature oxidative crystal redispersion and oxide atom-trapping phenomena provide a widely valid route towards atomically dispersed bimetallic precursors which, upon reductive metal agglomeration, result in very small, uniformly sized and remarkably stable bimetallic clusters. For a PdPt/MgO system, oxidative redispersion leads to isolated Pd and Pt atoms stabilized by the MgO support up to overall surface metal contents of ca. 1.0 Mat nm−2, beyond which loading, atom-trapping oxide sites become exhausted and metal aggregation sets in. On the contrary, more conventional, milder-temperature activation protocols lead to significant metal segregation and markedly bimodal particle size populations already from comparatively lower metal contents. As proven by in situ X-ray absorption spectroscopy and atomic-resolution STEM microscopy, the stabilization of isolated Pd and Pt cations within nanometer distances on the common MgO support is essential for the synthesis of ca. 1 nm bimetallic PdPt clusters by reductive agglomeration. The uniformly sized PdPt aggregates developed on MgO from single-atom precursors display a notably higher activity for the oxidative activation of methane with carbon dioxide (dry reforming) at 923 K compared to analogue materials synthesized via milder calcination/reduction protocols. Moreover, despite their high surface-to-volume ratio, the small bimetallic clusters in the former display an outstanding stability against metal agglomeration under the demanding reaction conditions, likely as a result of a lower driving force for Ostwald ripening growth processes. The synthesis concept, which is amenable to other combinations of 4d and 5d transition metals, contributes to the rationalization of the possibilities and bounds of oxidative metal redispersion phenomena and provides a technically simple and potentially general route to high-temperature stable, (sub)nanometer bimetallic clusters for catalysis and other applications where bimetallic effects and thermal stability are of importance.
Introduction
Supported bimetallic nanoclusters are important functional materials in areas spanning from photoluminescence, to sensing, electrochemistry and catalysis.1–7 As solid catalysts, synergistic effects ascribed to the concomitant presence of two metals within nanosized aggregates, nanoclusters or nanoparticles, have been shown to unlock favourable, in some instances unique, catalytic performances in a broad range of conversion processes.7–10 Pioneering work in this area by Sinfelt et al. in alkane dehydrogenation and hydrogenolysis reactions11,12 was followed by a surge of studies which demonstrated the potential of bimetallic catalysts in a wide array of reactions, from oxidation,13–15 to hydrogenation,16–20 reforming,21 biomass conversion,5 C–C coupling,22 or acetoxylation catalysis.23 The distinctive catalytic properties of bimetallics compared to their monometallic counterparts has been associated to factors such as differences in lattice strain and surface topology, intermetallic charge transfer phenomena, or atomic-scale surface dilution phenomena of one metal within a matrix of second metal (also referred to as single-atom alloys).9,24,25 Most of these effects are more prominent at small particle sizes, e.g. in the low- and sub-nm regime, where a higher fraction of highly reactive, coordinatively unsaturated, surface sites (cus) often dominates catalytic performance.With the aforementioned encouraging application background, the interest in developing effective, precise and scalable synthesis strategies for supported bimetallic nanoclusters and nanoparticles has witnessed a notable thrust over the last decades. Colloidal methods rooted in the co-reduction of precursor salts in solution, in the presence of organic surface capping agents, is a widely extended synthesis approach.10,26,27 However, despite their capacity to induce monodispersity, colloidal methods show limitations to produce particles of sizes approaching the sub-nm regime e.g. <2 nm. In order to produce very small bimetallic clusters on solid surfaces, methods based on the adsorption of molecular metal complexes from solution, followed by their reductive decomposition, have also been intensively explored.28 These methods face limitations connected to the need for suitable molecular precursors showing adequate adsorption energies on a common carrier as well as comparable thermal stability to prevent step-wise decompositions and thus metal segregation. Scalable synthesis routes which utilize less complex and more widely available organic and inorganic metal precursors are preferred for catalytic applications.
The synthesis of ultrasmall (∼1–2 nm) bimetallic nanoparticles was demonstrated by Regalbuto and coworkers by strong electrostatic adsorption from amine metal precursors by careful adjustment of the electrostatic charges of the support's surface and the metal precursors in solution, respectively.29 In a similar approach, Ding et al. adsorbed double complex metal salts, created via electrostatic interaction of oppositely charged inorganic salts, on a silica carrier.30 Subsequent thermal decomposition led to bimetallic nanoparticles of ca. 3 nm in size. Methods based on electrostatic adsorption are thus promising for the synthesis of very small bimetallic aggregates, although the overall metal loading is typically limited by the adsorption capacity of the oxide support.29 Other approaches, including radiolysis,31 sonochemical synthesis32 and step-wise ion-exchange and framework demetallation of zeotype solid precursors33 have also been demonstrated for the synthesis of supported bimetallic nanoparticles in the <2 nm size range.
Recently, the interest in oxide-supported atomically dispersed metals in catalysis, often referred to as single-atom catalysts,34,35 has spurred research on synthesis routes leading to atomic-level dispersion of metals on oxide carriers. Processes of oxidative crystal disruption and re-dispersion, coupled by atom trapping on oxide surfaces, offer a promising strategy to atomically disperse transition metals – which form highly mobile (sub-)oxides at high temperatures – on suitable carriers.36–38 Here we show that two metals can be oxidatively co-dispersed, down to the atomic level, on a common oxide support. The atomic-level structure and reduction behaviour of the resulting bimetallic materials is investigated with Scanning-Transmission Electron Microscopy (STEM), and in situ X-ray absorption spectroscopy. Metal reduction with hydrogen leads to very small bimetallic clusters, stabilized on a MgO carrier, which can only be achieved from atomically dispersed precursors and show remarkable stability under harsh reaction conditions in the dry reforming of methane with CO2. Moreover, the synthesis route is amenable to different combinations of 4d and 5d transition metals.
Experimental
Materials
Metal precursors platinum(II) acetylacetonate (97%), palladium(II) acetylacetonate (99%), iridium(III) acetylacetonate (97%), as well as solvent acetone (>99.9%) were purchased from Sigma-Aldrich (Merck) and used as received. For the synthesis of MgO, Mg(OCH3)2 (7–8 wt% solution in methanol) was purchased from Alfa-Aesar and technical-grade toluene was in-house distilled. Bulk PdO (99.97%) and PtO2 hydrate (≥99.9%) were purchased from Sigma-Aldrich (Merck).Synthesis of MgO support material
A MgO xerogel was synthesized employing magnesium(II) methoxide (7–8 wt% in methanol) as precursor.39 100 mL of the Mg(OCH3)2 stock solution was further diluted in 1 L toluene. Then Milli-Q water was dropwise added to the solution until complete precipitation and the resulting suspension was stirred overnight. The resulting gel was collected by centrifugation (9000 rpm), dried at 298 K in air overnight and finally at 353 K for 12 hours. The solid obtained was ground, sieved (200–300 μm) and calcined under synthetic air flow (50 mL min−1) at 773 K in a packed-bed reactor for 5 hours (heating ramp from room temperature of 2 K min−1).Synthesis of bimetallic atomically dispersed precursors
Metals were dispersed on the surface of the MgO support via oxidative redispersion. Equimolar amounts of the corresponding metal acetylacetonate precursors required to achieve a pre-set total metal content (0.2–4.0 Mat nm−2) after air-annealing at 1073 K, were dissolved in 40 mL of acetone. These metal contents corresponded to overall weight loadings in the range of 0.3–5.4 wt% (Table S1 in the ESI†). Then the MgO support material (previously ground to a fine powder) was suspended in the solution and the slurry sonicated. Solvent was then evaporated to dryness in a rotary evaporator under vacuum (in the range of 430–100 mbar) at 313 K and the solid was dried at 353 K overnight. The dried impregnate was then transferred to an uncapped porcelain crucible and heated to 1073 K under a stagnant air atmosphere in a muffle oven for 10 hours, using a temperature ramp of 2 K min−1. Materials were denoted as xM1M2/MgO, where x indicates the surface-specific metal content corresponding to the state after air-annealing at 1073 K, and M1 and M2 represent the two metals incorporated, in an equimolar content, on MgO.Synthesis of MgO-supported bimetallic nanoclusters
Bimetallic nanoclusters were generated on the surface of the MgO carrier by reduction of the corresponding oxide precursors under flow of 20% H2/N2 gas mixture. Reduction was performed in a vertically aligned packed-bed reactor using a gas space velocity of 100 mL g−1 min−1. The temperature was increased from RT to 673 K for Pt–Pd and 773 K for Pt–Ir at a constant heating rate of 2 K min−1. In order to elucidate the significance of the atomic dispersion of the oxidic precursors for the synthesis of the bimetallic nanoclusters, additional materials were also synthesized from precursors which had not been submitted to the high-temperature oxidative crystal redispersion treatment. In one instance, the acetylacetonate metal precursors were directly decomposed on the surface of the MgO support, followed by metal reduction, by treating the dried impregnate under flow of 20% H2/N2 gas mixture as described above. Alternatively, the impregnated acetylacetonate precursors were decomposed by calcination in air flow at temperatures of 623 K or 773 K, followed by reduction of the corresponding oxidic precursors in flow of 20% H2/N2 as detailed previously.N2 physisorption
Nitrogen physisorption isotherms were recorded at 77 K using a Micromeritics 3Flex v4.04 instrument. Prior to the measurement, samples were dried at 423 K under vacuum for 5 h. Specific surface areas were derived using the BET method in the relative pressure (P/P0) range from 0.05–0.30.Powder X-ray diffraction (XRD)
Powder X-ray diffraction patterns were collected on a Stoe STADI P transmission diffractometer equipped with a primary Ge (111) monochromator and a position-sensitive detector. Data were collected using MoKα1 radiation (λ = 0.7093 Å) in the 2θ range between 5 and 50° with a step width of 0.015° and a measuring time per step of 20 s. Samples were filled into glass capillaries (Ø 0.5 mm) and 8 consecutive full scans were collected and summed-up. Acquired patterns were evaluated qualitatively by comparison with entries from the ICCD PDF-2 powder pattern database.CO chemisorption
CO chemisorption was used to quantify metal surface atoms after exposure of selected catalysts to dry reforming reaction conditions for 70 h (vide infra) and thus determine metal-surface specific reaction rates (TOF). Experiments were performed in a Quantachrome Autosorb-1C equipment using the double isotherm method. Prior to chemisorption, catalyst samples (150 mg, pellet size of 200–400 μm) were treated in situ in H2 flow (25 mL min−1) at 673 K for 5 h to reverse any surface metal reoxidation due to handling of the catalyst in air after reactor off-loading. Then, samples were degassed at 1.3 Pa for 2 h at the reduction temperature, and the temperature was lowered to 308 K. Pure CO was admitted and the first chemisorption isotherm (total CO uptake) was recorded at 308 K. After evacuation at 308 K, a second isotherm (reversible CO uptake) was registered. The amount of strongly chemisorbed CO was then obtained by subtracting the two isotherms. The CO pressure range studied was 0.5–11.0 × 104 Pa. A CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ms surface chemisorption stoichiometry of 1:1 was considered.
:Ms surface chemisorption stoichiometry of 1:1 was considered.
Scanning-transmission electron microscopy (STEM) and energy-dispersive X-ray (EDS) spectroscopy
High-angle annular dark-field scanning-transmission microscopy (HAADF-STEM) experiments were performed using a Cs-probe-corrected Hitachi HD-2700 dedicated Scanning-Transmission Electron Microscope equipped with a cold field-emission gun and two EDAX Octane T Ultra W EDS detectors and operated at 200 kV. Prior to observation, finely ground powder samples were dry-cast on Cu grids coated with a holey carbon film. Metal nanoparticle size distributions were generated from populations of at least 100 particles coming from different micrographs. The following eqn (1) was used to estimate the average number of atoms from the number-averaged metal cluster size, assuming a spherical cluster shape: | (1) |
X-ray absorption spectroscopy (XAS)
X-Ray absorption spectra were recorded at the Pd K-edge (24.350 keV) and Pt L3-edge (11.564 keV), respectively, at the CLÆSS beamline station (BL22) of the ALBA synchrotron light source, Barcelona (Spain).40 The X-ray beam was monochromatized using a (311) double crystal monochromator. Ex situ measurements on oxidic catalyst precursors were performed at room temperature either in transmission or fluorescence modes, depending on the weight metal loading on the sample. For transmission measurements, powder samples were pressed into pellets (Ø = 31 mm) with optimized thickness after dilution, when required, with boron nitride. Absorption spectra were registered with ion chambers filled with appropriate gases to absorb 15% and 80% in the I0 and I1 at the different photon energies, respectively. For fluorescence measurements, sample pellets were mounted in a 45° angle with respect to the incoming beam and the spectra were registered using a fluorescence solid-state Silicon Drift Detector placed with an angle of 90° respect to the incoming beam. High-purity bulk metal oxide reference materials were measured in transmission mode as described above.In situ X-ray Absorption Near Edge Structure (XANES) experiments were performed to follow the reduction behaviour of selected bimetallic oxide precursors. The powder samples were hand-pressed into a pellet sample holder and mounted in a multipurpose gas–solid cell, previously described by Guilera et al.41 and equipped with a gas dosing system with Bronkhorst mass flow controllers and temperature control. The sample pellet was oriented in a 45° angle with respect to the incoming beam and the measurements were performed in fluorescence mode. The cell was purged with 20% H2/He at room temperature and the total gas flow was then set to 100 mL min−1. Next, the temperature was increased from 298 K to 773 K at a heating rate of 5 K min−1. XANES spectra were collected at the corresponding metal edge energy with an energy resolution of 1 eV and a temperature resolution of one full spectrum every 6 K of temperature change during heating ramps. EXAFS spectra were collected at RT both prior to and after the reduction treatment. For all bimetallic samples, the procedure was repeated at the corresponding X-ray absorption energy edge of each of the two metals.
Data reduction and extraction of the χ(k) function has been performed using the Athena code (version 0.9.26),42 while EXAFS data analysis was performed using the Artemis (Demeter software package). For data fitting a k-range 4–10.8 Å−1 was chosen with a Rbkg value of 1. Temperature-resolved in situ XANES spectra have been analysed using the MCR-ALS (Multivariate Curved Regression with Alternative Least Squares) chemometric method43,44 applying the algorithm developed by Jaumot, Tauler and co-workers.45
Catalysis
Catalysis experiments have been performed in a PIDEng microactivity setup, equipped with a tubular 316 stainless-steel reactor (ID = 7.8 mm) placed inside an annular oven. Gas feeds i.e. Ar (99.99%, as dilutant) and a certified CH4/CO2 mixture (60/40 v/v%, Linde) were fed using calibrated mass flow controllers. About 200 mg of catalyst (200–400 μm) were diluted with SiC granules (600–800 μm) to achieve an isothermal packed bed with an overall volume of 1.9 cm3. The catalyst was reduced prior to reaction at 673 K (2 h, heating rate of 2 K min−1) under flow of 20% H2/Ar (100 mL min−1). After in situ activation, reaction tests were performed at a constant temperature of 923 K and atmospheric pressure, feeding a reactant gas mixture with a molar composition Ar/CH4/CO2: 4.4/1.5/1.0 at a weight hourly space velocity (WHSV) of 6.9 h−1. The gas reactor outlet was analysed periodically in a SCION 456 gas chromatograph, equipped with a TCD detector and Scionbond-Q and Molsieve 5A capillary columns using H2 as a carrier gas. Hydrogen was quantified offline in a Bruker 450GC Rapid Refinery Gas Analyzer (RRGA), equipped with two TCD channels and Molsieve 5A/Hayesep-Q and Molsieve 13X/Hayesep-Q columns and N2 as carrier gas. Gas samples had been collected at the reactor outlet using sealed gas transfer bags after 30 h on-stream. Forward methane conversion rates have been corrected by an approach-to-equilibrium term (η) defined as:46 | (2) |
 | (3) |
Results and discussion
Synthesis of supported bimetallic nanoparticles
Palladium and platinum were dispersed on a high-surface MgO support material, previously obtained via a sol–gel route. The nanostructure of this material was predominantly composed by rod-like nanocrystals, which exposed a specific surface area of 250 m2 g−1 (Fig. S1 in the ESI†). Bimetallic catalysts were synthesized by dispersion of the corresponding acetylacetonate metal precursors on the as-synthesized MgO carrier, in a 1:1 atomic ratio, followed by different thermal activation treatments to decompose the organometallic precursors and reduce metals to their elemental state. Fig. 1 summarizes those thermal activation treatments applied to produce different materials starting from a common dry impregnate.
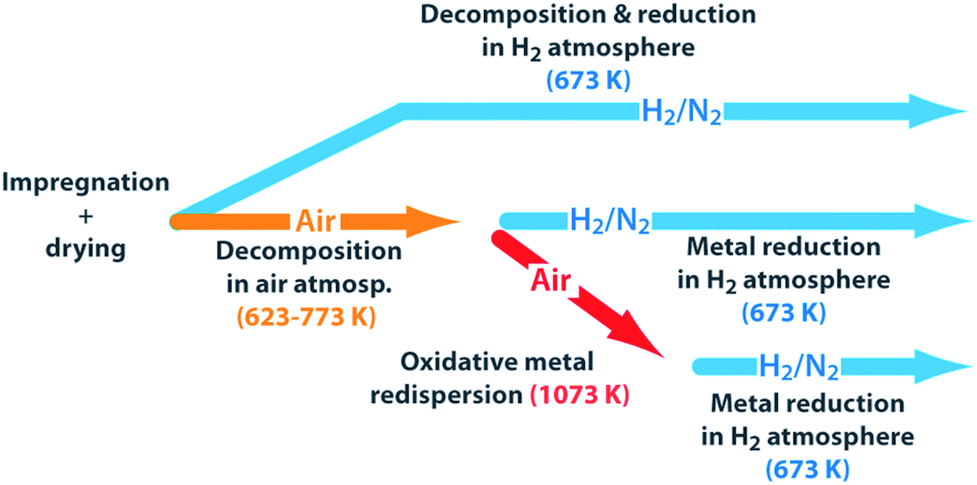 | ||
| Fig. 1 Schematics of the different activation treatments applied from a single dry (Pd(acac)2+Pt(acac)2)/MgO impregnate to produce bimetallic catalysts. acac: acetyl acetonate. | ||
Powder X-ray diffraction was applied to assess the presence of crystalline metal phases on materials with different surface-specific metal content and thermal activation history. Fig. 2 shows the diffraction patterns recorded for 1.0PdPt/MgO catalysts derived from a single impregnate precursor through different thermal activation routes. The dry impregnate showed diffractions corresponding to the MgO carrier (Fd3m), indicating the amorphous character of the acetylacetonate metal precursors deposited on the support surface. Following the decomposition of the supported metal precursors and metal reduction, in a single activation step under a reductive (20% H2/N2) atmosphere, additional diffractions emerged which could be indexed to a fcc metallic phase (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), indicating metal reduction. Close examination of the emerging diffraction lines enabled their assignment to metallic Pt (Fig. S2†). Even though the asymmetric skew of the diffraction lines towards lower d-spacings does not enable to rule out contributions from PdPt alloy or Pd crystallites, this result clearly points to metal segregation.
m), indicating metal reduction. Close examination of the emerging diffraction lines enabled their assignment to metallic Pt (Fig. S2†). Even though the asymmetric skew of the diffraction lines towards lower d-spacings does not enable to rule out contributions from PdPt alloy or Pd crystallites, this result clearly points to metal segregation.
 | ||
| Fig. 2 X-ray diffraction patterns for PdPt/MgO materials synthesized from a common dry impregnate (bottom trace) following different activation routes. Selected diffraction patterns from the ICCD PDF-2 database are also included for reference. | ||
Conventional activation routes based on air-calcination of the supported organometallic precursors, followed by a H2-reduction treatment, were also examined. Decomposition of the metal precursors in air at a temperature of either 633 K or 773 K resulted in weak, though comparatively sharper diffraction signals for Pt (Fig. 2). Again, no obvious signs for metallic Pd crystalline phases were detected by XRD. The observation of metallic species after calcination under an oxidative atmosphere indicates that the decomposition of the acetylacetonate moieties in the molecular precursors suffices to drive (partial) metal reduction to a zero-valent state. As judged from the narrower diffraction line-shape, larger Pt crystallites were obtained in this case compared to the direct thermal decomposition of the dry impregnate under a reducing 20%H2/N2 atmosphere. Subsequent reduction led to a slight increase in relative intensity for the Pt diffractions, likely as a result of the expected increase in the degree of metal reduction. In marked contrast, annealing the air-calcined catalyst precursor to a higher temperature of 1073 K in air led, on the one hand, to a noticeable sharpening of the MgO diffractions indicating the sintering of the oxide support, but also to the complete vanishment of those diffractions corresponding to crystalline phases of the supported metals. This observation points to an oxidative disruption of metal(oxide) crystallites and redispersion of metallic phases into structures lacking long-range order. Interestingly, as shown in Fig. 2, neither after the subsequent H2-reduction treatment could diffraction signals for metallic phases be detected in this case, despite H2-temperature programmed reduction showing a noticeable cumulative H2 consumption of 0.08 mmol H2 g−1, associated to metal reduction, at temperatures <673 K (Fig. S3†). In conjunction, these results suggest that, in contrast to any of the more conventional catalyst activation routes discussed above, oxidative metal redispersion results, after reduction, in metallic structures which are either amorphous in nature or highly dispersed, thus with very small, XRD-silent (<2 nm) crystalline domains.
In order to further explore the bounds of the crystallite oxidative redispersion phenomenon for the synthesis of PdPt/MgO bimetallic catalysts, a set of materials with various surface-specific metal contents in the range of 0.2–4.0 Mat nm−2 (M = Pd + Pt, Pd:Pt = 1) were synthesized by impregnation with the acetyl acetonate metal precursors, followed by air calcination and further annealing at 1073 K. The X-ray diffractograms for this series of materials, both after air-annealing and following the subsequent reduction treatment under hydrogen, are compiled in Fig. S4.† For reference purposes, also the diffractogram for the metal-free MgO carrier, submitted to the same air annealing treatment, is shown. Only diffractions corresponding to MgO are detected for catalysts with metal contents ≤1 Mat nm−2 indicating that highly dispersed metal species, with crystalline domains below the detection limit of XRD are obtained in this range of metal contents. In contrast, further increasing metal loading to ≥2 Mat nm−2 led to observable diffraction signals corresponding to PdO and fcc metallic crystals. In this case, the latter diffractions could be indexed to a PdPt alloy phase given the d-spacing correspondence with an equimolar, bulk PdPt alloy (Fig. S2†). Particularly the alloy phase grew in relative intensity upon increasing metal content from 2.0 Mat nm−2 to 4.0 Mat nm−2. Following the successive H2 reduction treatment, no signs of crystalline phases could be detected for catalysts with metal contents ≤1 Mat nm−2. In the case of materials with metal loadings ≥2 Mat nm−2, those diffractions for PdO vanished completely, indicating the reduction of palladium oxide. Simultaneously, signals for PdPt grew in intensity, signifying further development of alloy crystals via reduction of metal oxidic species.
The results indicate that metal (oxide) crystallite disaggregation into highly dispersed species can be achieved via the high-temperature annealing (1073 K) in an air atmosphere, up to surface metal contents of ca. 1 Mat nm−2. Surplus in metal content beyond this threshold lead to aggregation into metal(oxide) crystals. In the case of palladium, separate PdO crystals are clearly observed, whereas in the case of Pt only metallic phases are detected by XRD. This is in keeping with the known instability of platinum oxides at temperatures in excess to ca. 600 K, which typically results in the release of lattice oxygen even under oxidative gas environments.47
Analysis of the evolution of the specific surface area after air annealing with the metal content provided further insights into the metal redispersion phenomenon. As shown in Fig. S5,† the as-synthesized MgO support showed a BET specific surface area of 250 m2 g−1. Annealing to 1073 K led to a very significant sintering, reflected in a ca. seven-fold drop in surface area to 34 m2 g−1, indicating that the applied annealing temperature promotes surface mobility and agglomeration on MgO. After metal deposition, however, increasingly higher specific surface areas in the range of 37–57 m2 g−1 were stabilized after annealing for PdPt/MgO materials with increasing metal contents from 0.2 to 1 Mat nm−2. This stabilization levels off at metal contents ≥1 Mat nm−2. At even higher loadings (>2 Mat nm−2) the BET surface area decreases again. This behaviour is reminiscent of that observed previously for series of monometallic supported catalysts synthesized by dispersing different 4d and 5d transition metals on CeO2via air-annealing at 1073 K,48 for which a comparable stabilization of the oxide carrier against sintering was observed also up to surface-specific metal contents of ca. 1 Mat nm−2.
Electron microscopy insights into metal dispersion
Spherical aberration-corrected high-angle annular dark-field scanning-transmission electron microscopy (Cs-HAADF-STEM) was applied to study the dispersion of metal species at various stages throughout catalyst synthesis. Fig. 3 shows representative micrographs for 1.0PdPt/MgO catalysts obtained via the activation of a common dry impregnate precursor through either decomposition of the metal precursors by air-calcination at 623 K or 773 K, followed by reduction in 20%H2/N2 at 673 K, or via the combination of both decomposition and reduction steps in a single treatment under 20%H2/N2 at 673 K. As observed, in all cases, very small metal nanoparticles (<5 nm) coexist with larger metal agglomerates (up to >50 nm in size). These results are in line with those retrieved with XRD, indicating that the latter population of large metal crystals are by and large responsible for the observed diffraction signals. Compositional analysis of the latter population by energy-dispersive X-ray spectroscopy (EDS) additionally revealed differences in composition among particles, with a significant fraction of the metal agglomerates being notably enriched in platinum with respect to the equimolar overall composition, indicating severe metal segregation. Both metal segregation and the markedly bimodal metal dispersion are undesired features for catalysis, given that a significant fraction of Pt remains unmixed and inaccessible to reactants, in the bulk of large crystals. | ||
| Fig. 3 HAADF-STEM micrographs of PdPt/MgO catalysts (Pd:Pt = 1) obtained from an identical dry impregnate precursor via different precursor decomposition routes, i.e. by treating the dry impregnate directly in 20%H2/N2 at 673 K (a, b); by decomposing the metal precursor in air at 623 K followed by metal reduction in 20%H2/N2 at 673 K (c, d); and by decomposing the metal precursor in air at 623 K, followed by air-calcination at 773 K and metal reduction in 20%H2/N2 at 673 K (e, f). Panels g–i show close-up details, alongside line EDS compositional scans for representative large (>20 nm) metal nanoparticles in panel (a). | ||
A notably different picture aroused when the catalyst precursor underwent air annealing at 1073 K following the air decomposition of the organometallic precursors. As shown in Fig. 4, no metal aggregates could be observed after air-annealing, in contrast to any of the counterpart materials activated through more conventional procedures (Fig. 3). Higher-magnification imaging revealed isolated metal atoms, which can be identified owing to their considerably higher Z-contrast, on the MgO surface (Fig. 4a and b). An appealing feature of HAADF-STEM is that the measured contrast intensity (I) follows a simple relationship with the atomic number (Z) for sufficiently thin samples, i.e. I = k·Zα, where k is a constant under a given set of experimental imaging conditions. The exponent α would take a value of 2 for a genuine Rutherford electron scattering phenomenon.49 In practice, values in the range of 1.7 ± 0.3 are typically observed due to screening effects associated to atom core electrons.50,51Fig. 4c and d show the result of a local contrast analysis for those areas framed in Fig. 4a, each populated with five isolated metal atoms. In both areas, atom-sized motifs can be grouped into two major levels of contrast intensity, which are marked with diamonds and stars, respectively, on the corresponding 3D contour plots. Analysis of the average intensity differences between these two levels, corrected for the contrast contribution from the MgO support, and considering the difference in atomic number between Pd (Z = 46) and Pt (Z = 78), translates into an experimental value for the exponent α of 1.4 in the HAADF intensity relation, which falls well within the range of expected values. This analysis supports the assignment of the atom-sized motifs to Pd and Pt atoms, respectively, isolated, within nanometer distances, on the MgO surface.
 | ||
| Fig. 4 HAADF-STEM analysis of 1.0PdPt/MgO after air calcination and annealing at 1073 K (a–d) and subsequent H2-reduction at 673 K (e–h). Panels (b) and (e) provide representative micrographs of the two materials at identical magnification, illustrating the reductive agglomeration of atomically dispersed metals into bimetallic nanoclusters following H2-reduction. Panel (a) shows a close-up view of a region in panel (b) where the isolated metal atoms can be discerned. Panels (c) and (d) display 3D Z-contrast maps of the corresponding regions framed in panel (a). Panel f shows the EDS compositional profiles determined along the line scans depicted on panel e. The inset to panel (h) shows the nanoparticle size distribution determined after H2-reduction. | ||
Following the reduction treatment, the surface of the MgO crystals became populated with metal nanoclusters, as a result of the reductive agglomeration of the atomically dispersed species (Fig. 4e–h). In contrast to previously discussed catalyst activation procedures, a monomodal and remarkably narrow size distribution arises, which is centred at around 1.2 nm (Fig. 4h), i.e. corresponding to ca. 60 metal atoms per cluster. Moreover, as shown in Fig. 4f, EDS analysis revealed the bimetallic character of these clusters. The EDS linescans, collected while scanning the sample with the Cs-corrected e-beam probe (ca. 0.1 nm diameter), did not indicate any substantial metal segregation, or a core–shell metal distribution which have previously been computationally predicted for sub-nm PdPt nanocluster models,52,53 suggesting mixing of the two metals directly after reduction activation. No larger metal agglomerates could be observed by STEM, which is in agreement with the absence of contributions from metallic crystals in the corresponding X-ray diffractograms, both after air annealing and following subsequent reduction. These results might now be further rationalized in view of the complementary electron microscopy insights. On the one hand, after high-temperature air annealing, metal species appear to be atomically trapped on MgO, therefore lacking long range order. On the other hand, after H2-reduction, the very small size of those bimetallic aggregates produced via reductive agglomeration from the atomically dispersed precursor, falls below the minimum crystalline domain size detectable by XRD.
In situ X-ray absorption spectroscopy
X-ray absorption spectroscopy was applied to assess the dispersion, reduction kinetics and local coordination with metal specificity. Fig. 5 shows the extended X-ray absorption fine-structure (EXAFS) spectra recorded at the PdK and PtL3 absorption edges, respectively, for 1.0PdPt/MgO after oxidative metal redispersion at 1073 K. The spectra showed no evidence for neither 1st-shell M–M nor 2nd-shell M–O–M coordination, compatible with the absence of neither metallic nor metal oxide agglomerates. For both metals, the spectra could be fitted to a structural model where isolated metal atoms isomorphically substitute Mg2+ in direct sub-surface crystallographic positions underneath MgO step-edges, in agreement with previous results for monometallic catalysts synthesized similarly.54 Alternatively, structural models with metal cations sitting at more generic sub-surface MgO positions with a vicinal vacancy, provide also an acceptable description of the experimental spectra (Fig. S6†). As reported recently by our group,54 alternative site models, such as those proposed earlier, based on metal atoms adsorbed onto F-centers or divacancy sites on the MgO surface,55,56 with lower first- and second-shell coordination numbers for the metal adatom, did not provide a sensible fit of the experimental EXAFS data. These results indicate that both metals become atomically dispersed and stabilized by the MgO lattice following the high temperature oxidative redispersion treatment.
vacancy, provide also an acceptable description of the experimental spectra (Fig. S6†). As reported recently by our group,54 alternative site models, such as those proposed earlier, based on metal atoms adsorbed onto F-centers or divacancy sites on the MgO surface,55,56 with lower first- and second-shell coordination numbers for the metal adatom, did not provide a sensible fit of the experimental EXAFS data. These results indicate that both metals become atomically dispersed and stabilized by the MgO lattice following the high temperature oxidative redispersion treatment.
 | ||
| Fig. 5 X-ray absorption spectroscopy characterization of atomically dispersed bimetallic PdPt/MgO catalysts. (a–b) Fourier-transform of the phase-uncorrected k3-weighted EXAFS function for spectra recorded at the Pd–K (a) and Pt-L3 (b) XAS edges for 1.0PdPt/MgO synthesized via oxidative metal redispersion at 1073 K. The corresponding spectra for bulk metal oxides and metallic foils reference materials are also included. Scatter traces correspond to experimental data (closed symbols: real components; open symbols: imaginary components). Full lines correspond to the fits of the experimental data to structural models corresponding to single metal atoms sitting under a MgO step-edge, depicted on panel (c). See Tables S2 and S3 in the ESI† for fitting results. | ||
The reduction behaviour of the atomically dispersed bimetallic 1.0PdPt/MgO was studied with in situ X-ray absorption near-edge structure (XANES) spectroscopy. Fig. 6 shows the temperature-resolved XANES spectra recorded at the PdK and PtL3 edges, respectively, during the in situ reduction of the catalyst under flow of 20%H2/N2 in the temperature range of 298–773 K. For comparison, results for monometallic 0.5Pd/MgO and 0.5Pt/MgO analogues, synthesized identically, are also included. In both cases, progressive dampening of the white line intensity indicated metal reduction. Metal-specific information on the reduction kinetics was extracted via a MCR-ALS chemometric method to track the evolution of the concentration of the oxidized and reduced components as a function of the temperature. As shown in Fig. 6c and f, a decline of the oxidic contribution to the convoluted spectra sets in from milder temperatures for Pd, and drops faster as the temperature increases in the range <523 K, indicating that Pd reduction proceeds at lower temperatures than that of Pt. Moreover, comparison of the concentration profiles determined by MCR-ALS for mono- and bimetallic counterparts reveals that the concomitant presence of both metals on MgO shifts Pt reduction to lower temperatures, suggesting that hydrogen activation and spillover from nascent Pd clusters assists the reduction of more stable oxidic Pt species.
 | ||
| Fig. 6 Temperature-resolved X-ray Absorption Near-Edge Structure (XANES) analysis of metal reduction in a bimetallic 1.0PdPt/MgO catalyst (a–c) and the corresponding monometallic 0.5Pd/MgO and 0.5 Pt/MgO monometallic counterparts (d–f). Temperature-resolved XANES spectra at the PdK-edge (a, d) and the PtL3-edge (b, e) during the exposure of the atomically dispersed catalyst precursors to a flow of 20%H2/N2 in the temperature range of 298–773 K. The panels include the corresponding XANES spectra for reference metal foils and metal oxides (PdO and PtO2, respectively). Temperature-resolved evolution of the relative abundance of the oxidic metal state and the as-reduced metal states in the temperature range screened. For the sake of direct comparison, dotted lines in panel (c) show the reduction behaviour for Pd and Pt, respectively, in the corresponding monometallic materials. | ||
Following in situ reduction in the XAS cell, EXAFS spectra were recorded to investigate the average local coordination of the two metals. Fig. 7 shows the corresponding spectra for 1.0PdPt/MgO after reduction of the atomically dispersed bimetallic precursor. Spectra for the oxidic precursor, metallic foils and the corresponding atomically dispersed monometallic 0.5Pd/MgO and 0.5Pt/MgO after reduction, are also shown for comparison. Wavelet transforms of the PdK-EXAFS spectra for the ex-single-atom 1.0PdPt/MgO after reduction show essentially only Pd–M (M = Pd, Pt) scattering contributions, indicating the quantitative reductive agglomeration of palladium. At the PtL3 edge, however, next to the 1st-shell metal–metal component, a second scattering contribution for Pt–O is observed, indicating that a fraction of the atomically dispersed platinum cations, likely those stabilized in higher-confinement sub-surface positions, did not undergo reduction at the applied activation temperature and thus did not engage in metal agglomeration. This is in contrast to the results obtained for the corresponding 1.0PdPt/MgO bimetallic catalyst which had not undergone oxidative redispersion, for which no significant M–O scattering contributions for either metal were observed following reduction under identical conditions (Fig. S7†), indicating that the lower metal reducibility in the former is associated to the oxidative redispersion of metal crystallites and the trapping and stabilization of isolated metal cations by the lattice of the MgO support.
 | ||
| Fig. 7 X-ray absorption study of the structure of bimetallic clusters in PdPt/MgO. (a, d) Modulus and (b, e) imaginary Fourier-Transform (FT) terms of the EXAFS function, and wavelet transform of the EXAFS function (c, f) at the Pd–K (a–c) and Pt-L3 (d–f) absorption edges for a 1.0PdPt/MgO single-atom precursor, as-synthesized via oxidative redispersion at 1073 K, and the corresponding catalyst after in situ reduction in 20%H2/N2. In panels (a and b) and (d and e), the corresponding spectra for monometallic 0.5Pd/MgO and 0.5 Pt/MgO single-atom analogues in their oxidic state as well as the corresponding metallic foils are also included for reference purposes. The inset to panel (b) shows the imaginary FT component of simulated EXAFS spectra for a pure Pd crystal as well as a PdPt equimolar alloy. In panels (c) and (f), M stands for either Pd or Pt. | ||
Owing to the incomplete reduction of Pt species, and thus the convolution of different 1st- and 2nd-shell contributions to the corresponding PtL3-EXAFS spectra, further analyses on the bimetallic character of the resulting metal nanoclusters focused on spectral features at the PdK edge. After reduction activation, all FT EXAFS spectra show a scattering contribution centred at a radial distance of 2.5 Å (without phase correction), corresponding to M–M coordination in metallic clusters (Fig. 7a). Careful evaluation of the imaginary part (Fig. 7b), however, reveals significant differences in the relative intensity for those three function maxima in the radial distance region of 2.0–3.0 Å between mono- and bimetallic catalysts produced by reductive activation of atomically dispersed precursors. Whilst for 0.5Pd/MgO, the amplitude of the three maxima increases progressively on increasing radial distance, for 1.0PdPt/MgO the amplitude levelled off after the second maximum. The corresponding PdK-EXAFS spectra were simulated for Pd–Pd and Pd–Pt paths in mono- and bimetallic clusters, respectively (see inset to Fig. 7b). For the Pd–Pd a systematic increase in the amplitude for the three maxima in the mentioned radial distance range of the imaginary FT component is observed, well in line with the experimental results for the monometallic 0.5Pd/MgO catalyst. In contrast, the amplitude order reversed for the second and third maxima for the simulated spectra for the Pd–Pt path. Moreover, the signals of the simulated EXAFS patterns are partially in anti-phase. These observations suggest strongly that the change in the monotony for the EXAFS FT-imaginary component for the bimetallic 1.0PdPt/MgO, compared to the monometallic 0.5Pd/MgO counterpart, arises from a convolution of two contributions associated to Pd–Pd and Pd–Pt scattering paths in the former. These results tie in well with those from EDS nanospectroscopy and indicate the bimetallic nature of the small clusters developed on MgO upon reductive agglomeration from the atomically dispersed precursors. Fitting of the EXAFS spectra (Fig. S8†) resulted in average Pd–Pd and Pd–Pt coordination numbers (CN) of 2.5 ± 0.8 and 2.1 ± 0.9, indicative for the average very small size of the bimetallic nanoclusters (Table S4†). Taken together, XAS results demonstrate the atomically dispersed state of the catalyst precursors after the oxidative crystal redispersion treatment, the existence of bimetallic effects on the metal reduction kinetics and the development of bimetallic clusters following reductive agglomeration, with sizes at the lowest frontier of the nanometer regime.
Catalysis
To assess the implications of the thermal history-dependent metal aggregate size on catalytic applications under severe reaction conditions, 1.0PtPd/MgO catalysts were evaluated in the dry reforming of methane (DRM) with CO2 at 923 K. DRM is a reaction of eminent interest in the context of the valorisation of abundant (unconventional) natural gas resources with a simultaneous chemical recycling of waste CO2. Bimetallic nanoparticles,57–60 including PdPt,57 have shown significant potential with high reaction rates and limited carbon build-up at moderate temperatures. Catalysts synthesized by either air calcination at 623 K or oxidative redispersion at 1073 K, followed, in both cases, by reduction at 673 K with hydrogen, were selected for the catalysis investigation. Reaction settings were adjusted to achieve approach-to-equilibrium factors <0.01 for this reversible reaction,46 thus ensuring kinetic control.Under the applied reaction conditions, the neat MgO support showed a mass-specific methane conversion rate between sevenfold and more than one order of magnitude lower than any of the PdPt/MgO catalysts, evidencing that the magnesia support and the reactor walls are rather inert at the applied process conditions and thus the supported metal clusters/nanoparticles account for the activity observed. With the bimetallic catalysts, in both cases, a transient reaction regime was observed for times on stream <30 h, during which the CH4 conversion rate increased (Fig. S9†). Similar “induction” regimes have been previously observed for bimetallic catalysts exposed to methane reforming conditions and have been ascribed to an initial reduction/surface restructuring of the active metallic phases.61 Following this transient period, the ex-single-atom PdPt/MgO catalyst afforded a more than threefold higher conversion TOF (Fig. 8a). This observation agrees with the higher abundance of coordinatively unsaturated surface sites (cus) on the small bimetallic clusters developed on the latter catalyst, which are known to be more effective for the kinetically relevant C–H bond cleavage in methane reforming catalysis.46,62,63 The metal-specific reaction rate achieved with the bimetallic clusters (9.7 × 10−2 mol min−1 gmetal−1) is higher than those reported for Ni-based catalysts at higher reaction temperatures in the range of 1023–1073 K.61,64 A H2/CO molar ratio of 0.65 was determined for the syngas product following the transient period, which is slightly higher than the value of 0.55 predicted under the assumption of thermodynamic equilibrium considering the occurrence of both DRM and the reverse water-gas-shift (RWGS) reactions at the applied operation temperature.
 | ||
| Fig. 8 Catalytic results for methane conversion on bimetallic PdPt/MgO catalysts. (a) Forward methane conversion rate per unit metal surface atom (Turnover Frequency, TOF); (b) evolution of the normalized activity parameter, i.e. a = rCH4(t)/rCH4(t0) with the time-on-stream, following the initial induction period observed after the initiation of the catalytic test (t0 = 30 h); and (c) representative HAADF-STEM micrographs prior to catalysis and after ca. 70 hours on stream under methane dry reforming conditions, for 1.0PdPt/MgO catalysts activated from the same dry impregnate precursor via either air-calcination at 623 K or the same followed by air-annealing at 1073 K (oxidative metal redispersion into single-atom species), followed by reduction in 20%H2/Ar at 673 K. Reaction conditions: T = 923 K, P = 1 bar, feed Ar/CH4/CO2 = 4.4/1.5/1.0 (molar), WHSV = 6.9 h−1. | ||
Next to activity, analysis of the reaction performance for ca. 40 h on-stream, following the initial induction period, revealed remarkable differences in stability. Fig. 8b depicts the evolution of the normalized activity factor with the reaction time. No relevant decay in activity was observed for the most active 1.0PdPt/MgO catalyst generated from the corresponding atomically dispersed precursor. In marked contrast, a significant activity decay, corresponding to a zero-order deactivation rate constant of 3.6 × 10−3 h−1, was obvious for the catalyst generated from metal oxide agglomerates on the MgO carrier. To shed light into these different stabilities under reaction conditions, HAADF-STEM was applied to study metal dispersion on the catalysts recovered after the catalytic test. As shown in Fig. 8c, the (sub)nanometer metal clusters were found to persist on the surface of the catalyst developed from an atomically dispersed precursor, where only few slightly larger, albeit <5 nm, metal nanoparticles could be detected. In contrast, the catalyst for which activation did not involve oxidative atomic redispersion, and thus it displayed a bimodal starting metal particle size distribution prior to catalysis, showed an evident depletion in the population of the smallest metal clusters after the prolonged exposure to reaction conditions. Essentially only those comparatively larger (>5 nm) particles could be observed after catalysis. These results point to a significant metal particle growth in this case, which accounts for the observed catalyst deactivation. Reinforcing these microscopy observations, XRD provided also evidence for the limited growth experienced by the small metal clusters derived from single-atom precursors during exposure to reaction conditions (Fig. S10†). These results are reminiscent of the high stability observed previously with Ni–MgO catalysts derived from solid solutions,61,64,65 where the active metal exsolves from lattice sites within the support under reaction conditions. It can therefore be inferred from these results that the narrow nanocluster size distribution achieved via reductive agglomeration from isolated metal atoms dampens metal growth processes even under high-temperature reaction conditions, in spite of their higher surface-to-volume ratio and thus intrinsic lower stability. This effect is thus likely associated to a minimization of the driving force for Ostwald ripening mechanisms by the achievement of low-polydispersity nanoclusters. On the contrary, such ripening processes are favoured with the markedly bimodal metal nanoparticle size distributions obtained via more conventional catalyst synthesis and activation protocols.
Jointly, the results underscore how adjusting both the surface metal content and the thermal activation route is essential for the development and stabilization of very small supported bimetallic nanoparticles. Fig. 9 summarizes graphically the different explored paths from a common starting dry impregnate and the consequences for metal mixing and dispersion. Following the simultaneous deposition of acetyl acetonate compounds of the two selected metals on the MgO support, direct decomposition of these metal precursors and metal reduction to zero-valent state under a reductive atmosphere (20%H2/N2) leads to a markedly bimodal metal particle size distribution, combining very small (<3 nm) nanoparticles with larger (>10 nm) agglomerates, as well as a noticeable size-dependent metal segregation, and thus suboptimal metal mixing. Decomposition of the molecular metal precursors via air calcination (623–773 K) results in highly dispersed metal oxide species, however, accompanied by bulkier metallic agglomerates. Further thermal treatment in the presence of H2 leads to an essentially complete metal reduction which, however, does not revert the bimodal size distribution and metal segregation occurred during air calcination. However, when higher temperature air annealing follows decomposition of the metal precursors, instability of metal (oxide) crystals drives oxidative disruption into mobile monoatomic species of each metal which, up to an overall surface content of ca. 1 Mat nm−2, can be trapped and stabilized by the oxide carrier at near-surface lattice positions. Further reductive metal agglomeration from isolated (single) atoms of the two metals, initially spaced within nanometer distances, leads to the development of uniformly sized, down to sub-nanometric, bimetallic clusters with particularly high stability to sintering even under harsh reaction conditions. While this synthesis concept is illustrated in greater detail for the showcase of PdPt nanoclusters, the methodology holds broader generality. Indeed, an identical synthesis route has been demonstrated to also develop (sub)nanometer PtIr bimetallic clusters on MgO (Fig. S11 in the ESI†).
 | ||
| Fig. 9 Schematic representation of the nanostructure of PdPt/MgO materials obtained from a common dry impregnate via the decomposition of metal acetylacetonate precursors through various activation procedures. The route in the dotted frame corresponds to the oxidative crystal redispersion and atom trapping into atomically dispersed metals on MgO, followed by reductive agglomeration into ∼1 nm monodispersed bimetallic clusters. | ||
Conclusions
The combination of oxidative disruption of metal (oxide) crystallites at high temperatures with atom-trapping phenomena on nanoparticulate oxides enables single-atom species for two different metals to be stabilized, within nanometer distances, on a common oxide support material. For supported bimetallic PdPt/MgO catalysts, atomic resolution HAADF-STEM, X-ray diffraction and X-ray absorption spectroscopy show that this atomic co-dispersion for the two metals is effective up to an overall surface-specific metal content of about 1 Mat nm−2, beyond which the metal atom-trapping capacity of the MgO support appears to exhaust and metal clustering into crystallites sets in. Subsequent reductive metal agglomeration using hydrogen as reducing agent shows the reduction kinetics to differ for each metal, and reveals bimetallic effects, likely due to H2 dissociation and spillover effects from early nascent metallic nanoclusters. Unlike more conventional activation routes based on air-calcination of the metal precursors at milder temperatures (623–773 K) followed by reduction, which result in bimodal metal particle size distributions and noticeable metal segregation, the reductive activation from single-atom dispersed bimetallic precursors delivers very small (ca. 1 nm) and bimetallic clusters on the oxide carrier. The latter display higher catalytic activity and a remarkable stability under relevant conditions for the high-temperature (923 K) dry reforming of methane with CO2 into syngas, likely as a result of a minimized driving force for Ostwald ripening particle growth mechanisms associated to the high cluster monodispersity. The synthesis route, which is herein shown to be applicable to different combinations of 4d and 5d metals, provides access to very small, (sub)nanometer sized bimetallic clusters with high stability for applications in catalysis and other technologies where combining bimetallic effects, highly reactive metal surfaces and high thermal stability is important.Conflicts of interest
There are no conflicts to declare.Acknowledgements
XAS experiments were performed at the CLAESS BL22 beamline of the ALBA Light Source, Cerdanyola del Vallés, Spain: experiment 2019023278. The authors are grateful to beamline staff members, as well as to E. Andrés, M. E. Martínez-Monje and I. López-Luque (ITQ) for assistance with XAS measurements. J. Ternieden (MPI-KOFO) is acknowledged for XRD measurements. M. Mayer (MPI-KOFO) is thanked for his assistance with the synthesis of the MgO support. P. N. Plessow and F. Studt (KIT) are gratefully acknowledged for the development of the DFT-optimized structural models used in EXAFS data fitting. Authors are grateful to Prof. Ferdi Schüth for providing lab facilities and supportive access to analytical methods at the MPI-KOFO. This research received funding from the Max Planck Society and the Spanish Ministry of Science and Innovation (RTI2018-096399-A-100 and SEV-2016-0683). B. B. S. acknowledges the Alexander von Humboldt Foundation for a postdoctoral grant. M. G. F. acknowledges the Spanish Ministry of Science, Innovation and Universities for a FPU predoctoral scholarship (FPU17/04701). Open Access funding provided by the Max Planck Society.References
- M. Zhou, C. Li and J. Fang, Chem. Rev., 2021, 121, 736–795 CrossRef CAS PubMed.
- J. Rick, M.-C. Tsai and B. Hwang, Nanomaterials, 2015, 6, 5 CrossRef PubMed.
- Q. Shao, P. Wang and X. Huang, Adv. Funct. Mater., 2019, 29, 1806419 CrossRef.
- B.-W. Zhang, H.-L. Yang, Y.-X. Wang, S.-X. Dou and H.-K. Liu, Adv. Energy Mater., 2018, 8, 1703597 CrossRef.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075 RSC.
- K. Loza, M. Heggen and M. Epple, Adv. Funct. Mater., 2020, 30, 1909260 CrossRef CAS.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099 RSC.
- K. D. Gilroy, A. Ruditskiy, H.-C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS.
- X. Peng, Q. Pan and G. L. Rempel, Chem. Soc. Rev., 2008, 37, 1619–1628 RSC.
- J. H. Sinfelt, J. L. Carter and D. J. C. Yates, J. Catal., 1972, 24, 283–296 CrossRef CAS.
- J. Schwank, AIChE J., 1985, 31, 1405 CrossRef.
- S. I. Murahashi, T. Naota and N. Hirai, J. Org. Chem., 1993, 58, 7318–7319 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS.
- N. Agarwal, S. J. Freakley, R. U. McVicker, S. M. Althahban, N. Dimitratos, Q. He, D. J. Morgan, R. L. Jenkins, D. J. Willock, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Science, 2017, 358, 223–227 CrossRef CAS.
- M. Zaarour, J. Cazemier and J. Ruiz-Martínez, Catal. Sci. Technol., 2020, 10, 8140–8172 RSC.
- P. Lu, T. Teranishi, K. Asakura, M. Miyake and N. Toshima, J. Phys. Chem. B, 1999, 103, 9673–9682 CrossRef CAS.
- S. Hermans, R. Raja, J. M. Thomas, B. F. G. Johnson, G. Sankar and D. Gleeson, Angew. Chem., Int. Ed., 2001, 40, 1211–1215 CrossRef CAS PubMed.
- J. K. Edwards, B. Solsona, E. Ntainjua N, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS PubMed.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sorensen, C. H. Christensen and J. K. Norskov, Science, 2008, 320, 1320–1322 CrossRef CAS PubMed.
- J. M. Parera and J. N. Beltramini, J. Catal., 1988, 112, 357–365 CrossRef CAS.
- H. Miura, Y. Tanaka, K. Nakahara, Y. Hachiya, K. Endo and T. Shishido, Angew. Chem., Int. Ed., 2018, 57, 6136–6140 CrossRef CAS PubMed.
- M. Chen, D. Kumar, C. W. Yi and D. W. Goodman, Science, 2005, 310, 291–293 CrossRef CAS PubMed.
- F. Gao and D. W. Goodman, Chem. Soc. Rev., 2012, 41, 8009 RSC.
- F. (Feng) Tao, Chem. Soc. Rev., 2012, 41, 7977 RSC.
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179–1201 RSC.
- P.-C. Chen, X. Liu, J. L. Hedrick, Z. Xie, S. Wang, Q.-Y. Lin, M. C. Hersam, V. P. Dravid and C. A. Mirkin, Science, 2016, 352, 1565–1569 CrossRef CAS PubMed.
- O. S. Alexeev and B. C. Gates, Ind. Eng. Chem. Res., 2003, 42, 1571–1587 CrossRef CAS.
- A. Wong, Q. Liu, S. Griffin, A. Nicholls and J. R. Regalbuto, Science, 2017, 358, 1427–1430 CrossRef CAS PubMed.
- K. Ding, D. A. Cullen, L. Zhang, Z. Cao, A. D. Roy, I. N. Ivanov and D. Cao, Science, 2018, 362, 560–564 CrossRef CAS PubMed.
- M. Treguer, C. de Cointet, H. Remita, J. Khatouri, M. Mostafavi, J. Amblard, J. Belloni and R. de Keyzer, J. Phys. Chem. B, 1998, 102, 4310–4321 CrossRef CAS.
- Y. Mizukoshi, K. Okitsu, Y. Maeda, T. A. Yamamoto, R. Oshima and Y. Nagata, J. Phys. Chem. B, 1997, 101, 7033–7037 CrossRef CAS.
- T. Iida, D. Zanchet, K. Ohara, T. Wakihara and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2018, 57, 6454–6458 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- M. Flytzani-Stephanopoulos and B. C. Gates, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 545–574 CrossRef CAS PubMed.
- A. Datye and Y. Wang, Natl. Sci. Rev., 2018, 5, 630–632 CrossRef CAS.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. Pereira Hernandez, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- Z. Tang, P. Liu, H. Cao, S. Bals, H. J. Heeres and P. P. Pescarmona, ACS Catal., 2019, 9, 9953–9963 CrossRef CAS PubMed.
- S. Utamapanya, K. J. Klabunde and J. R. Schlup, Chem. Mater., 1991, 3, 175–181 CrossRef CAS.
- L. Simonelli, C. Marini, W. Olszewski, M. Ávila Pérez, N. Ramanan, G. Guilera, V. Cuartero and K. Klementiev, Cogent Phys., 2016, 3, 1231987 Search PubMed.
- G. Guilera, F. Rey, J. Hernández-Fenollosa and J. J. Cortés-Vergaz, J. Phys.: Conf. Ser., 2013, 430, 12057 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- W. H. Cassinelli, L. Martins, A. R. Passos, S. H. Pulcinelli, C. V Santilli, A. Rochet and V. Briois, Catal. Today, 2014, 229, 114–122 CrossRef CAS.
- A. Voronov, A. Urakawa, W. van Beek, N. E. Tsakoumis, H. Emerich and M. Rønning, Anal. Chim. Acta, 2014, 840, 20–27 CrossRef CAS PubMed.
- J. Jaumot, R. Gargallo, A. de Juan and R. Tauler, Chemom. Intell. Lab. Syst., 2005, 76, 101–110 CrossRef CAS.
- J. Wei and E. Iglesia, J. Phys. Chem. B, 2004, 108, 4094–4103 CrossRef CAS.
- L. K. Ono, B. Yuan, H. Heinrich and B. R. Cuenya, J. Phys. Chem. C, 2010, 114, 22119–22133 CrossRef CAS.
- B. B. Sarma, J. Kim, J. Amsler, G. Agostini, C. Weidenthaler, N. Pfänder, R. Arenal, P. Concepción, P. Plessow, F. Studt and G. Prieto, Angew. Chem., 2020, 132, 5855–5864 CrossRef.
- M. M. J. Treacy, Microsc. Microanal., 2011, 17, 847–858 CrossRef CAS PubMed.
- P. Hartel, H. Rose and C. Dinges, Ultramicroscopy, 1996, 63, 93–114 CrossRef CAS.
- Z. W. Wang, Z. Y. Li, S. J. Park, A. Abdela, D. Tang and R. E. Palmer, Phys. Rev. B Condens. Matter, 2011, 84, 73408 CrossRef.
- L. O. Paz-Borbón, R. L. Johnston, G. Barcaro and A. Fortunelli, J. Phys. Chem. C, 2007, 111, 2936–2941 CrossRef.
- L. O. Paz-Borbón, R. L. Johnston, G. Barcaro and A. Fortunelli, J. Chem. Phys., 2008, 128, 134517 CrossRef PubMed.
- B. B. Sarma, P. N. Plessow, G. Agostini, P. Concepción, N. Pfänder, L. Kang, F. R. Wang, F. Studt and G. Prieto, J. Am. Chem. Soc., 2020, 142, 14890–14902 CrossRef CAS PubMed.
- A. Del Vitto, L. Giordano, G. Pacchioni and U. Heiz, J. Phys. Chem. B, 2005, 109, 3416–3422 CrossRef CAS PubMed.
- H. Xu, C.-Q. Xu, D. Cheng and J. Li, Catal. Sci. Technol., 2017, 7, 5860–5871 RSC.
- Y. Zhu, K. Chen, C. Yi, S. Mitra and R. Barat, Chem. Eng. Commun., 2018, 205, 888–896 CrossRef CAS PubMed.
- J. Zhang, H. Wang and A. Dalai, J. Catal., 2007, 249, 300–310 CrossRef CAS.
- C. Dai, S. Zhang, A. Zhang, C. Song, C. Shi and X. Guo, J. Mater. Chem. A, 2015, 3, 16461–16468 RSC.
- R. K. Singha, A. Shukla, A. Sandupatla, G. Deo and R. Bal, J. Mater. Chem. A, 2017, 5, 15688–15699 RSC.
- Y. Song, E. Ozdemir, S. Ramesh, A. Adishev, S. Subramanian, A. Harale, M. Albuali, B. A. Fadhel, A. Jamal, D. Moon, S. H. Choi and C. T. Yavuz, Science, 2020, 367, 777 CrossRef CAS PubMed.
- J. Wei and E. Iglesia, Angew. Chem., Int. Ed., 2004, 43, 3685–3688 CrossRef CAS PubMed.
- K. Klier, J. S. Hess and R. G. Herman, J. Chem. Phys., 1997, 107, 4033–4043 CrossRef CAS.
- Z. Zuo, S. Liu, Z. Wang, C. Liu, W. Huang, J. Huang and P. Liu, ACS Catal., 2018, 8, 9821–9835 CrossRef CAS.
- Y. H. Hu and E. Ruckenstein, Catal. Lett., 1997, 43, 71–77 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta00421b |
| ‡ Present address: Karlsruhe Institute of Technology, Institute of Catalysis Research and Technology (IFKT), Hermann-von-Helmholtz-Platz 1, Eggenstein-Leopoldshafen, 76344, Germany. |
| This journal is © The Royal Society of Chemistry 2021 |