
A new metric to control nucleation and grain size distribution in hybrid organic–inorganic perovskites by tuning the dielectric constant of the antisolvent†
Blaire A.
Sorenson‡
 *a,
Lucy U.
Yoon‡
b,
Eric
Holmgren
b,
Joshua J.
Choi
b and
Paulette
Clancy
a
*a,
Lucy U.
Yoon‡
b,
Eric
Holmgren
b,
Joshua J.
Choi
b and
Paulette
Clancy
a
aJohns Hopkins University, Department of Chemical and Biomolecular Engineering, 3400 North Charles Street, Baltimore, MD 21218, USA. E-mail: bsorens4@jhu.edu; Tel: +1-410-16-7170
bUniversity of Virginia, Department of Chemical Engineering, Thornton Hall, P.O. Box 400259, Charlottesville, VA 22904-4259, USA. E-mail: jjc6z@virginia.edu; Tel: +1-434-924-3072
First published on 16th January 2021
Abstract
In perovskite research, there is a widely exploited but poorly explained phenomenon in which the addition of “antisolvents (ATS)” to precursor solutions results in higher-quality films. We explain the mechanism and driving force underlying an antisolvent-driven solvent extraction process. Density functional theory calculations uncover the defining effects of antisolvent choice on the extent of complexation between a lead salt and a methylammonium cation in solution. We experimentally validate the computational results using ultraviolet-visible spectroscopy and 207Pb nuclear magnetic spectroscopy of methylammonium lead iodide solutions, containing both a processing solvent and an antisolvent. Furthermore, we uncover, and subsequently identify, the appearance of new species in solution as a result of the addition of the antisolvent. We observe that the choice of antisolvent has a substantial effect on the nature of the complexation of the methylammonium lead iodide (MAPbI3) precursor species, whose origin we explain at an atomic level; specifically, the lower the dielectric of the antisolvent, the stronger the intermolecular binding energy between methylammonium cation (MA+) cation and PbI3− plumbate, independent of the solvent or antisolvent interaction with the lead salt. Thin films were characterized using scanning electron microscopy; images of the films show how the addition of an antisolvent influences and, importantly, can be used to alter thin-film grain size. Grain size and distribution in thin films is reflected by the choice of antisolvent, promoting slower nucleation rates, a lower nucleation density, and hence larger final grain size.
1 Introduction
Hybrid organic–inorganic perovskites (HOIPs) have shown significant and growing effectiveness as prospective solar cell materials since their emergence in 2009.1 With their current certified solar cell efficiency of over 25.0%,2 HOIPs are now a competitive solar cell material with mature techniques established for thin film fabrication.3,4 Multiple studies have emphasized the importance of understanding how HOIP film formation is governed by solution processing to achieve uniform film morphology, large grain size and high crystallinity.5,6 All of these desirable characteristics are directly correlated to processing choices in the search for improved device performance.7–15 Unfortunately, the focus on increasing power conversion efficiency (PCE) has outstripped our understanding, and hence control, of the fundamentals of solution chemistry that gives rise to the nucleation and growth of HOIP crystallization pathways and hence film quality.Solvent “engineering” for perovskites was first proposed in 2014 to expedite nucleation and achieve smooth, full-coverage perovskite films by the addition of antisolvents during spin-casting.16 This has been one of the most important contributions to the field, as it has proven to achieve uniform coverage, pinhole-free surface morphology and better device performance while inducing fast nucleation.16–19 Zhou et al. later demonstrated an alternative method incorporating antisolvents using an “antisolvent bath method”,20,21 in which the perovskite thin film is immersed in a bath of antisolvent. This eliminated the difficulties of precise timing and uniform deposition needed for antisolvent treatment during spin-casting and it reduced film damage. Currently, the criteria used to constitute an antisolvent in the context of perovskite solar cells is that the antisolvent is miscible with the processing solvent used to dissolve HOIP precursor species such as the lead salts and methylammonium iodide (MAI), and that it does not dissolve the perovskite or perovskite species.5
The wide range of antisolvents used to date for perovskite films has produced variable results for film quality, dependent on the chemistry of the antisolvent.16,22–24 Xiao et al.18 studied the effect of different antisolvents on methylammonium lead iodide (MAPbI3) thin films deposited from dimethylformamide (DMF) to show that films exposed to acetonitrile, benzonitrile and tetrohydrofuran produced transparent films, for reasons that are still not fully understood. Alcohol-containing antisolvents such as methanol, ethanol, and ethylene glycol produced a large amount of PbI2 in the resulting thin film due to the high solubility of the perovskite in these solvents. Toluene, chlorobenzene and xylene, however, produced good coverage and large grain sizes.25
To date, the induced nucleation observed as a result of antisolvent addition is thought to occur through a solvent extraction process, induced by the antisolvent.20,21 But the mechanism by which this occurs is still largely undefined. Further, no thorough study has been conducted on the interactions between the processing solvent and antisolvents, nor the interaction of antisolvents with HOIP precursor species. To shed light on this, we have conducted an in-depth examination on the effect of using four commonly used antisolvents, dichloromethane (DCM), chlorobenzene (CB), anisole (ANS) and toluene (TOL), on an archetypal perovskite precursor solution containing MAPbI3 in DMF.
The focus of this work is to uncover the mechanism and driving force underlying antisolvent–solvent extraction and explain how antisolvents promote fast HOIP nucleation. To do so, we employ a combination of computational and experimental approaches. Computationally, we use density functional theory (DFT) to probe interactions at an atomic-level. We include experimental validation of these calculations using Ultraviolet Visible spectroscopy (UV-Vis), and 207Pb Nuclear Magnetic Spectroscopy (207Pb-NMR) of the perovskite precursor solutions containing DMF and antisolvents. We use scanning electron microscopy (SEM) to characterize thin films deposited from different solution choices.
2 Computational and experimental Methods
2.1 DFT computational details
Solvent molecules and perovskite precursor species were built using the molecule visualization tool, Avogadro.26 Their geometries were optimized using the quantum mechanical software package, Orca,27 which we have employed extensively in the past to study perovskite moieties.9,28,29 DFT calculations of the molecular geometries were converged using a pure generalized gradient approximation (GGA) B97 (030 and 31) and a polarized basis set, TZVPP32 with dispersion corrections, as recommended by Grimme.31,33 All the molecular species and complexes were further optimized using PW6B95, the well-ranked hybrid functional in Grimme et al.'s thorough benchmark.34 The Geometrical Counterpoise Correction (gCP)35 was added to remove artificial over-binding effects from basis set superposition errors (BSSE). All systems were given “Tight SCF” and slow convergence criteria with a grid size of 7.The processing solvent chosen for this study was DMF as it is one of the most widely employed solvents for HOIPs and has been shown to produce films with superior morphology compared to other processing solvents.16,36 The antisolvents included in this study are DCM, CB, ANS, and TOL (acronyms described in Section 1). For each set of simulations, we used a single explicit solvent molecule in conjunction with the COSMO implicit solvation model37,38 to incorporate the dielectric of the solvent, or solvent mixture, into the simulations.
2.2 Absorbance characterization
To examine the effect of MAI concentration in MAPbI3 precursor solution, we prepared a solution of 2.5 mM lead iodide (PbI2) (TCI, 99.999%) and varying concentrations (2.5 mM to 0.75 M) of methylammonium iodide (MAI) (Dyesol, 99.99%) dissolved in DMF (Sigma-Aldrich, 99.8%). To study the effect of the dielectric constant of the antisolvent in a MAPbI3 precursor solution, we maintained constant concentrations of PbI2 and MAI as we varied the molar ratio of DMF to the antisolvent (DCM, CB, ANS, and TOL) from 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, to 2:1, to 1:1. The solutions were stirred until the precursors were completely dissolved and the solution became clear. The solutions were kept in a N2-purged glovebox and transferred into airtight capped cuvettes until immediately before the absorbance measurement in order to minimize ambient exposure. Absorbance measurements were taken using a PerkinElmer Lambda 950S spectrophotometer. The corresponding DMF or DMF–antisolvent solvent mixture was used as a blank reference for calibration. It should be noted that absorbance spectra of excess MAI with PbI2 concentrations above 2.5 mM could not be measured due to the saturation of measured signal.
:1, to 2:1, to 1:1. The solutions were stirred until the precursors were completely dissolved and the solution became clear. The solutions were kept in a N2-purged glovebox and transferred into airtight capped cuvettes until immediately before the absorbance measurement in order to minimize ambient exposure. Absorbance measurements were taken using a PerkinElmer Lambda 950S spectrophotometer. The corresponding DMF or DMF–antisolvent solvent mixture was used as a blank reference for calibration. It should be noted that absorbance spectra of excess MAI with PbI2 concentrations above 2.5 mM could not be measured due to the saturation of measured signal.
2.3 207Pb-NMR characterization
We performed 207Pb-NMR spectroscopic measurements of DMF and DMF–antisolvent solution samples using a Varian NMRS 600 at a frequency of 125.7 MHz. The delay time, acquisition time, and pulse were set at values of 10 ms, 42.6 ms, and 3.125 μs, respectively. The chemical shifts were referenced to Pb(NO3)2 in D2O. We prepared the samples using an identical procedure described in the Experimental absorbance characterization section. Solutions were stored and transferred into airtight NMR tubes in the N2 glovebox prior to measurement.2.4 Thin film fabrication
We prepared a precursor solution of 1 M MAPbI3 in DMF and stirred overnight in a N2 glovebox to ensure a complete precursor dissolution. For film fabrication, we cleaned glass substrates by sonicating in a Hellmanex soap solution and then in deionized H2O for 10 min. The substrates were further sonicated in isopropanol and acetone for 10 min each. The cleaned substrates were UV–ozone treated for 10 min and brought into the N2 glovebox for spin-coating and an annealing process. On the substrate, we statically dispensed 45 μL of precursor solution, immediately initiated spin-coating at 4000 rpm. Six seconds after the start of the spin-coating process, we rapidly dispensed 200 μL of antisolvent onto the substrate. After the spin-coating was finished, we annealed the thin film at 110 °C for 10 min. The samples were stored in a glovebox prior to SEM characterization. We performed planar SEM imaging with a FEI Quanta 650 SEM operating at a 2 kV accelerating voltage to examine the morphologies and grain sizes of films prepared via extraction with different antisolvents.3 Results and discussion
3.1 Solvent–antisolvent interactions
DFT calculations provided information on the pre-nucleation moieties and the interactions of the precursor species, solvent and antisolvents studied in this work. To understand the molecular-scale interactions that occur between DMF and the different antisolvents, we calculated the intermolecular binding energy between solvent molecules for both the solvent interacting with itself, e.g., two DMF molecules, which we refer to as “same-solvent” pairs and for mixtures of DMF with antisolvent molecules e.g., one DMF and one DCM molecule, denoted as “DMF–ATS” in Fig. 1 (see Fig. S1 in the ESI† for values of solvent–solvent intermolecular binding energies). | ||
Fig. 1 Intermolecular binding energies (in kcal mol−1) of 2:1 binary solvent mixtures used in perovskite precursor solution processing and arranged in decreasing value of the mixture's dielectric constant: the binding energy of DMF to an antisolvent is shown as green circles ( ). The binding energy of “same-solvent” pairs is shown as blue triangles ( ). The binding energy of “same-solvent” pairs is shown as blue triangles ( ). ). | ||
Sorenson et al.29 previously showed that a small system containing as few as one to three explicitly modeled solvent molecules can provide accurate trends in intermolecular binding energies for perovskite precursor species in solution. For the mixed DMF–antisolvent simulations, we calculated the static dielectric of each DMF–antisolvent solution using a 2:1 ratio of DMF:antisolvent for each antisolvent in this study. The dielectric constants of pure DMF, DCM, CB, ANS and TOL are 36.7, 8.98, 5.62, 4.33, 2.38, respectively.39 Using the dielectric mixing rules based on the Kirkwood model,40,41 and a 2:1 ratio of DMF to antisolvent, this resulted in dielectric values of the precursor solutions being 28.54, 24.38, 23.73, and 22.66 for DMF:DCM, DMF:CB, DMF:ANS and DMF:TOL, respectively.
More negative values of the intermolecular binding energy correspond to stronger interactions between each species or solvents. Fig. 1 shows that DMF has a stronger affinity for all the antisolvents we tested than it does with itself. We observe that DMF has the largest attraction to TOL, followed – in order – by ANS, CB and DCM. Antisolvents containing aromatic groups (TOL, ANS and CB) show increased affinity to DMF (ranging between −3.45 and −3.96 kcal mol−1) compared to DCM (−2.86 kcal mol−1), which has no aromatic groups, and the intermolecular binding energy between two DMF molecules (−1.95 kcal mol−1). Since aromatic rings involve regions of both positive and negative charge on either side of the conjugated ring, the polar oxygen atom on DMF, with its lone pair of electrons, would be more attracted to this region of positive charge. The two electronegative chlorine atoms on the small dichloromethane molecule exhibit more repulsion when mixed with DMF than the other antisolvents in this study. Given the results in Fig. 1, the addition of an antisolvent to the DMF precursor solution will clearly promote strong DMF–antisolvent interactions.
3.2 Antisolvent–HOIP precursor interactions
To understand the nature of the solvent–antisolvent extraction mechanism, we first probed molecular-scale interactions between an MAPbI3 precursor species, DMF and each of the antisolvents; then we probed the interaction of the MA+ cation to the DMF or antisolvent-coordinated PbI3− plumbate. The resulting intermolecular binding energies are reported in Fig. 2 (see Fig. S2 in ESI† for calculated intermolecular binding energy values). The strength of these interactions provides information about the effects of antisolvent on the complexation between the PbI3− and the MA+ cation, and how each solvent complexes with an MAPbI3 precursor species. The MAPbI3 species was chosen based on the idea by Sorenson et al.29 that this was the smallest and most energetically favorable perovskite “building block” that contained both the methylammonium cation and an iodoplumbate.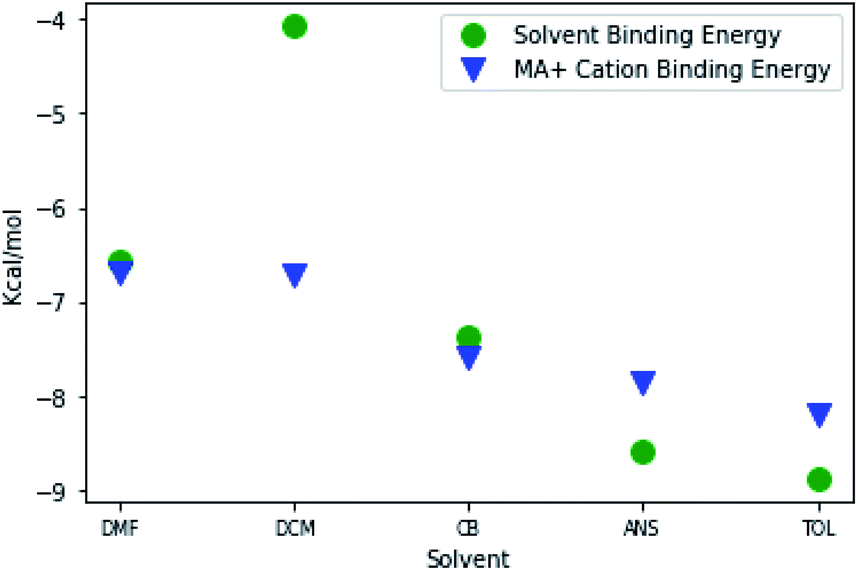 | ||
Fig. 2 Intermolecular binding energies of DMF and antisolvents to MAPbI3 species ( ). The binding energies of a MA+ cation to a complex formed between Pb and a single molecule of either DMF or antisolvent ( ). The binding energies of a MA+ cation to a complex formed between Pb and a single molecule of either DMF or antisolvent ( ). All binding energies are given in kcal mol−1. ). All binding energies are given in kcal mol−1. | ||
Most antisolvents, with the exception of DCM, have a considerably stronger attraction to the MAPbI3 precursor species than the processing solvent DMF. Fig. 2 shows the strength of the antisolvents to the MAPbI3 precursor species denoted by green circles. It shows that CB (−7.38 kcal mol−1), ANS (−8.59 kcal mol−1) and TOL (−8.88 kcal mol−1) each have a stronger affinity for the MAPbI3 precursor species by 0.8 to 2.3 kcal mol−1 compared to DMF (−6.66 kcal mol−1). The strength of the intermolecular binding energy between the antisolvent and an MAPbI3 precursor species increases with decreasing dielectric constant of the solution. We postulate that antisolvent–solvent extraction mechanism is partially driven by the stronger attraction of the antisolvent to the plumbate, thereby displacing DMF solvent molecules that surround the precursor species.
Stevenson et al. showed that solvents commonly used in perovskite thin film processing, such as DMF, form dative bonds with the Pb atom in the PbXn plumbate.28 We adopt that study's use of the Mayer Bond Order (MBO), a quantum mechanical measure of the electron density between atoms, to measure the interaction and coordination between antisolvents and the MAPbI3 precursor species. We found that DMF is the only solvent in this study that possessed a detectable MBO between the solvent and MAPbI3, specifically between the oxygen atom, and the lead atom (MBO of 0.176), indicating electron donation from DMF to the lead, characteristic of a dative bond. All the antisolvents we studied revealed no detectable MBO between any atom in the MAPbI3 species, to any atom in the antisolvent molecules, indicating a lack of dative bond formation.
Looking at the interaction of the antisolvents with the MAPbI3 precursor species, we found that TOL, ANS and CB all interact with the lead atom in MAPbI3 through non-covalent π interactions, and not the electronegative chlorine or oxygen atom on CB or ANS, respectively (as illustrated in Fig. 3). The lack of detection of an MBO between pairs of atoms here indicates that the region of charge that the antisolvent shares with the lead atom extends over most, if not all, of the carbon atoms in the conjugated ring, and not an isolated atom in the antisolvent molecule. Individually, each carbon atom does not donate enough electron density to form a measurable MBO value.
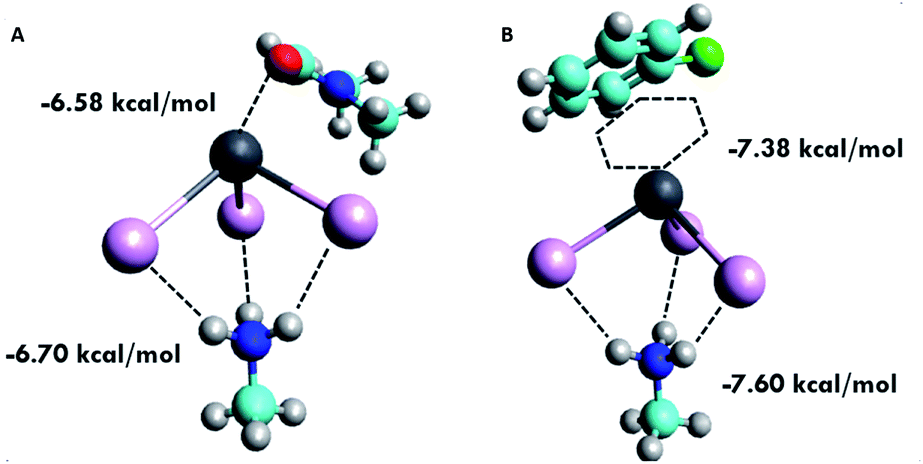 | ||
Fig. 3 Visualization of the intermolecular binding of (A) DMF solvent coordinated through a dative bond with MAPbI3 species and (B) the chlorobenzene antisolvent showing a π-cation interaction between the conjugated ring and the Pb atom in the MAPbI3 species. (Color code: oxygen ( ), hydrogen ( ), hydrogen ( ), nitrogen ( ), nitrogen ( ), carbon ( ), carbon ( ), lead ( ), lead ( ), iodine ( ), iodine ( ).) ).) | ||
The concept of the antisolvent–solvent extraction mechanism, as we show, is largely driven by, first, the affinity of DMF to the antisolvents that we have calculated above and, secondly, by the antisolvent having stronger, non-covalent interaction with the lead atom, facilitating the exchange of a processing solvent molecule for an antisolvent.5
The correlation we observed between decreasing solution dielectric and increasing intermolecular binding energy, also extends to the strength of MA+ cation complexation to the iodoplumbate to form MAPbI3 precursor species, as seen in Fig. 2. As a result, this increase in the strength of complexation between MA+ cations and plumbates in solution is a direct view into the earliest stages of nucleation.
 | (1) |
Eqn (1) 42 shows how the total free energy change, ΔG, is a function of the saturation ratio S = C/Cs, where C is the concentration of the solute in the perovskite precursor solution, and Cs is the equilibrium solubility limit (r is the nuclei radius, Vm is the molar volume of the nucleus, R is the gas constant, T is temperature, γCL is the interfacial energy between the solvent and the nucleus).
Supersaturation is a driving force of nucleation and growth and will ultimately dictate the final crystal size distribution. As we have shown, antisolvents lower the overall dielectric constant of the perovskite solution and promote complexation between the MA+ cation and PbI3− plumbate, leading to an increase in the formation of new MAPbI3 precursor species, and thus reducing the amount of MA cations and lead salts in solution. Unlike DMF, or other processing solvents, the antisolvent's inability to dissolve lead salts and perovskites43 implies that it also cannot dissolve the smallest perovskite building block, e.g., MAPbI3, or clusters of these building blocks. Supersaturation is achieved by increasing the saturation ratio, S in eqn (1), by simultaneously reducing Cs, the solubility limit of MA+ cation and lead salts, and increasing C, the concentration of newly generated insoluble (MAPbI3) species. In general, the supersaturation intervals are decreased with increasing dielectric constant of the solvent or the solution and is evident when you consider that the dielectric constant is a measure of the solvents polarity.44
In our work here, the addition of each tested antisolvent to different samples of the precursor solution reduces the dielectric constant of the system. All our systems contained the processing solvent DMF, giving rise to dielectric constants of 28.54, 24.38, 23.73, and 22.66 for DMF:DCM, DMF:CB, DMF:ANS and DMF:TOL, respectively. It is therefore palpable that the lower the dielectric of the antisolvent, the smaller the supersaturation interval will be, and therefore the smaller the grain size in the resulting thin films.
3.3 UV-Vis absorbance results analysis
Previous studies have assigned the absorbance peaks responsible for iodoplumbate species: PbI2 at 330 nm, PbI3− at 370 nm, andPbI42− at 422 nm.45 In good agreement with the reported literature,46 we observed that the addition of excess MAI in increasing concentrations promotes the formation of higher-ordered iodoplumbate species, represented by absorbance increases in the 370 and 422 nm peaks (Fig. 4A). The presence of an isosbestic point at 388 nm indicates that species transition from PbI3− to PbI42− states upon the addition of excess MAI. To probe the role of the antisolvent in an environment where the concentrations of PbI2 and MAI are relatively low and at similar degree, we prepared solutions of equimolar 2.5 mM PbI2 and MAI in DMF (control) and in 1:1 molar ratios of DMF to various antisolvents. As shown in Fig. 4B, we observed that the addition of antisolvent shows an increase in higher iodoplumbate species formation in low precursor concentration environment. We also discovered that the extent of PbI3− formation is inversely correlated with the dielectric constant trend of an antisolvent incorporated in the solution. Specifically, samples that contain a lower dielectric constant antisolvent yielded a higher degree of PbI3− formation. This suggests that, in a low concentration of PbI2 and MAI precursor solution, addition of a lower dielectric constant antisolvent shows an analogous effect to that of the addition of excess MAI.
 | ||
| Fig. 4 Absorbance spectra of (A) 2.5 mM PbI2 solution with varying concentrations of MAI (2.5 mM, 0.1 M, 0.25 M, 0.5 M, and 0.75 M). As the MAI concentration increases, higher-ordered iodoplumbates are observed, indicated by an increase in the absorbance peak at 422 nm. (B) Equimolar 2.5 mM PbI2 and MAI solutions dissolved in pure DMF and in 1:1 molar ratios of DMF and various antisolvents. | ||
To investigate the effect of adding antisolvents with different dielectric constants into perovskite precursor environments in which higher-order iodoplumbates species are dominant, we performed UV-Vis absorbance measurements on 1:1 molar ratios of DMF to various antisolvents that incorporated 2.5 mM PbI2 and 0.75 M MAI solutions (using a procedure provided in the Methods section). As shown in Fig. 5, the absorbance spectrum of a perovskite precursor in pure DMF (control) yielded a predominant peak at around 422 nm. As these various antisolvents were introduced into the precursor solutions, the absorbance peaks at 422 nm broadened significantly, and showed an increasing blue-shift towards lower wavelengths (Fig. 5). The extent of blue-shift also depended on the molar ratio of DMF to antisolvent incorporated in each solution. As we decreased the molar ratio of DMF to antisolvent from 3:1 to 1:1, we clearly observed a more pronounced and distinctive peak formation between 422 nm and 372 nm (see Fig. 5 A–C). With the exception of DCM, the increment in the blue-shift in absorbance showed a strong correlation with the dielectric constant of the choice of antisolvents incorporated into the solution. For instance, the addition of TOL, which has the lowest dielectric constant, resulted in the largest blue-shift of the 422 nm absorption peak. A similar trend was followed by ANS and CB.
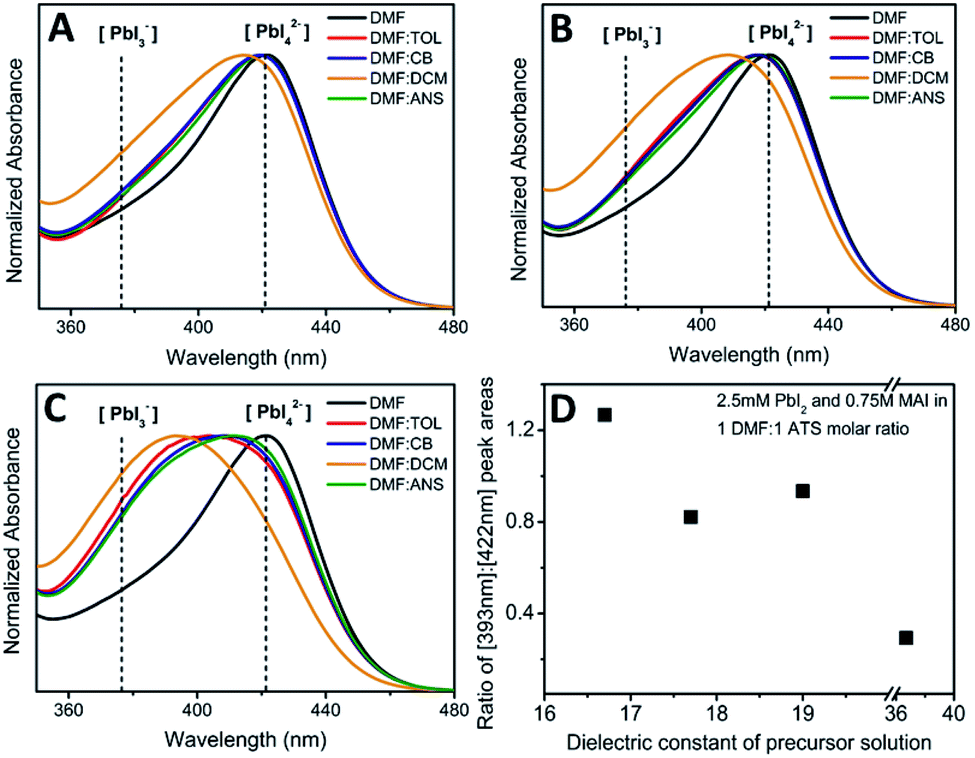 | ||
| Fig. 5 Normalized absorbance spectra of 2.5 mM PbI2 and 0.75 M MAI dissolved in pure DMF and with a (A) 3:1 (B) 2:1 and (C) 1:1 molar ratio of DMF to a set of antisolvents with varying dielectric constant, as listed in the inset color key. The new absorbance peak positions of PbI3− (372 nm) and PbI42− (422 nm) are marked with dotted vertical lines. (D) The ratio between the areas under the new peak, appearing at 393 nm, and at 422 nm plotted as a function of dielectric constant of precursor solution. The trend shows a higher ratio of 393 nm and 422 nm peaks for lower dielectric constant precursor solutions. | ||
The blue-shifted peak does not correspond to the positions of either the PbI3− or PbI42− peaks, suggesting the appearance of another, as yet unidentified, precursor species that have a characteristic absorption between 372 nm and 422 nm. Deconvolution of the absorbance peak into two peaks shows that the contribution is composed mostly from peaks at 393 nm (the new peak that we postulate to be a species of the form MAxPbI4 in the presence of an antisolvent) and at 422 nm (the PbI42− peak). To more clearly compare the degree of formation of the new precursor species responsible for the absorption at 393 nm, the ratio of the areas under the 393 nm and 422 nm peaks in 1:1 molar ratio was plotted with respect to the dielectric constant of the precursor solution. The DCM plot was not included, as the 393 nm peak could not be fitted. The resulting data (shown in Fig. 5D) shows a general trend in which more of the new species (postulated to be MAxPbI4) forms as a result of incorporating an antisolvent with increasingly lower dielectric constant into the precursor solution.
The absorption of energy observed in UV-Vis spectra has its origin in the excitation of electrons from their ground state to a higher-energy (excited) molecular orbital. This links the absorption peaks in the UV-Vis spectrum to the difference in energy between the Highest Occupied Molecular Orbital (HOMO) and the LUMO (Lowest Unoccupied Molecular Orbital). To test this connection and help identify the precursor species giving rise to the new absorption peak, we performed DFT calculations to obtain the HOMO–LUMO gap energies. The species studied here were built in a so-called “Jacob's ladder” approach, starting with calculation of ΔHOMO–LUMO gaps for different lead salts (PbI2, PbI3−, or PbI42−). This allowed us to identify precursor species as candidates based on their ΔHOMO–LUMO energy gap, looking for candidates that lie between the ΔHOMO–LUMO energy gaps for PbI3− and PbI42−, and reported in Fig. 6. The HOMO–LUMO gaps were calculated for each precursor species in DMF as well as the 2:1 molar ratio of DMF to antisolvent.
 | ||
| Fig. 6 Differences in HOMO–LUMO energy levels (eV) in different DMF/antisolvent solutions for perovskite precursor species. Horizontal dashed lines indicate the characteristic ΔHOMO–LUMO energy gap for PbI3− and PbI42−, which are used as upper and lower bounds to identify perovskite precursor species in Fig. 5A. | ||
DFT calculations of the value of the ΔHOMO–LUMO gap are typically an underestimate by about a factor of two; the quality of this approximation to the true energy difference depends sensitively on the nature of the exchange–correlation functional chosen and the amount of Hartree–Fock exchange that is incorporated.47 However, for our purposes, we only need to observe trends in the DFT-generated gaps.48,49 Our DFT results suggest that the new absorbance peak that arises when an antisolvent is added is likely to be due to an increasing population of MAxPbI4 species. There is a very clear trend that the lower the dielectric constant of the solution, the more abundant the MAxPbI4 species that exist compared to PbI42−. These results are thus consistent with our computational findings that a lower dielectric constant environment enhances the incorporation of MA+ cations with iodoplumbate species.
3.4 207Pb-NMR results analysis
207Pb-NMR was employed to probe the change in electron density around the lead atoms in different solvent–antisolvent combinations. First, we studied the effect of changing concentrations of PbI2 and MAI, where we found that increasing PbI2 concentration alone in DMF resulted in an upfield shift (see Fig. S1 in ESI†). It should be noted that the appearance of a single peak, despite the presence of multiple iodoplumbate species in solution, is likely due to chemical exchange occurring on the same order of magnitude time scale as that of the detection period.50 This is consistent with a previous report on solution-state 207Pb-NMR study of lead halide perovskites.51 Therefore, a single peak represents the averaged frequency of the relative populations of iodoplumbate species present in the solution sample. A single peak also indicates that all the iodoplumbate species in the solution are in dynamic equilibrium. Increasing MAI concentration with a fixed PbI2 concentration initially caused a downfield shift until a considerable excess of MAI was introduced (20 MAI:1 Pb molar ratio), at which point an upfield shift was observed (see Fig. S2 in ESI†). To understand the effect of varying the dielectric constant of the antisolvent on the electron density around the lead atoms, we performed 207Pb-NMR studies on solution samples of 0.075 M PbI2 and 0.2 M MAI in a 2:1 molar ratio of DMF to antisolvent; the results are shown in Fig. 7. Similarly, a set of samples prepared with identical PbI2 and MAI concentrations, but in 3:1 ratios of DMF to antisolvent was also measured and analyzed. The results showed an identical trend of chemical shift as a function of dielectric constant of the precursor solution to the results for the 2:1 ratio (see Fig. S3 in ESI†). Only a single peak was observed from each sample during the 207Pb-NMR measurements, which suggests that multiple species are in dynamic equilibrium, with exchange rates faster than the time scale of the NMR measurements. As a result, the chemical shift represents the average change in relative amounts of multiple species in solution, as well as an overall change in electronic shielding around the lead atoms.
 | ||
| Fig. 7 (A) Solution-state 207Pb-NMR spectra of 0.075 M PbI2 and 0.2 M MAI in pure DMF and in a 2:1 molar ratio of DMF to antisolvents with varying dielectric constants. Pb(NO3)2 was used as a standard. (B) 207Pb-NMR chemical shift as a function of the dielectric constant of the precursor solution. The result shows a clear trend in downfield chemical shift with incorporation of a lower dielectric constant antisolvent. | ||
The 207Pb-NMR results in Fig. 7A and B show a clear trend of downfield shift caused by the incorporation of an antisolvent with lower dielectric constant, which, in turn, lowers the dielectric of the precursor solution. Fig. S4 and S5 in the ESI† also show the effect of increasing the MAI concentration on both 3:1 and 2:1 binary solvent mixtures of DMF and antisolvent. These results suggest that the electronic shielding around the lead atoms decreases with the addition of the antisolvents. This trend contradicts the general effect of dielectric constant on chemical shift in NMR.52 Instead, the trend we observed should be viewed as a result of reduced electron density around the lead atoms caused by increased binding of MA+ cations to iodoplumbate species. Using this interpretation, our 207Pb-NMR results also support the computational findings that the π interactions between the Pb atom in precursor species and the antisolvent do indeed lack electron donation from the antisolvent to the Pb.
3.5 Effect of antisolvent on grain sizes of MAPbI3 film
Finally, we extended these atomic/molecular-scale studies by looking at the ultimate effect of changing the antisolvent at a more macroscopic level. To do so, we tested the computational findings by characterizing the grain sizes of MAPbI3 thin films prepared from solutions of each antisolvent in this study. Based on classical nucleation theory,53 we hypothesized that a lower overall dielectric constant of the solution will promote interactions between the MA+ cation and iodoplumbate species, and that this would result in higher nucleation rates, higher nucleation density, and smaller final grain size. In contrast, a higher dielectric constant solvent would result in fewer nuclei and thus larger grains. Films were prepared using an antisolvent dripping method, in which an abrupt supersaturation of a MAPbI3 precursor solution, followed by nucleation and growth of MAPbI3 crystallites, is induced by dripping the antisolvent during the spin-coating process. A detailed description of this process is provided in the “Thin film fabrication” section in Experimental methods. Images of film samples prepared with 1 M MAPbI3 in DMF and incorporating various dielectric constant antisolvents in this dripping process are shown in Fig. S6 of the ESI.†Numerous studies in the literature24,54–56 have reported the significance of antisolvent-induced perovskite crystallization to yield smooth, high-quality films that are ideal for device application. Similar to previous studies,57,58 we observed that the films fabricated with 1 M MAPbI3 in DMF precursor solutions using a one-step method yielded a needle-like morphology (Fig. 7A and S7 in ESI†), possibly indicating the formation of perovskite–DMF adducts. Similarly, introducing antisolvent directly to the precursor solution prior to spin-coating also produced an impure phase of MAPbI3, represented by pale yellow discoloration and needle-like structure. On the other hand, films fabricated using an antisolvent dripping method yielded smoother, pinhole-free morphologies with rounder grains, which allowed us to observe a more coherent comparison of grain sizes between films. As shown in Fig. 8A and S8,† the SEM images of samples prepared using antisolvents of varying dielectric constant show a consistent trend of decreased grain sizes with lower dielectric constant antisolvents. The grain size distribution analysis (Fig. 8B) revealed that antisolvents with a lower dielectric constant yield smaller average grain sizes, but also a narrower distribution of the grain sizes. The only exception to this trend was the film made with DCM; the morphology of the sample was quite similar to that of the DMF sample in which no antisolvent dripping was employed, with rougher, irregular grains and a less uniform film coverage with many pinholes. The surface of the film resembled that of the DMF sample, with a significantly duller appearance compared to other antisolvent-incorporated films (see Fig. S6 in the ESI†). Based on this result, we suspect that DCM with its (relatively) high dielectric constant has similar effects to DMF on the crystallization and growth behavior of MAPbI3. These results for grain size add further supporting evidence to our hypothesis that the dielectric of the precursor solution, tuned by the judicious addition of a suitable choice of antisolvent can, indeed, promote slower nucleation rates, a lower nucleation density, and hence larger final grain size.
 | ||
| Fig. 8 (A–E) SEM images showing grain sizes of MAPbI3 thin films prepared by the antisolvent dripping method. (A) With no antisolvent dripping process, MAPbI3 in pure DMF yields grains with needle-like morphology. (B)–(E) MAPbI3 films made with various antisolvents using a dripping process results in a smoother, more continuous grain morphology. (F) Histogram of grain size population analyzed from MAPbI3 films prepared with various antisolvents. As the dielectric constant of the antisolvent incorporated in the thin film increases, the larger and more poly-dispersed grains were formed. | ||
4 Conclusions
This work has provided significant evidence to support the widely held, but largely unexplained, hypothesis that antisolvents “extract” processing solvents when applied during HOIP processing through an unknown atomic-scale mechanism. Using complementary approaches of DFT calculations, UV-Vis and 207Pb NMR experiments, we provide details on the complexation between the precursor species and explain how the dielectric of the precursor solution can be used to control nucleation by the addition of an antisolvent. We demonstrate the ability of antisolvents to be tuned (using its dielectric constant) to promote the formation of a newly identified MAxPbI4 species, which decreases the solute concentration of MA+ and PbI3− in solution, thus decreasing the saturation ratio S in eqn (1). Importantly, our findings suggest that control over grain size and distribution in the final thin film can be gained by making a rational choice of antisolvents based on its dielectric constant and the nature of the interaction between the antisolvent and precursor plumbates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Cornell Center for Materials Research with funding from the NSF MRSEC program (DMR-1719875). BAS thanks the Ford Foundation for support of her doctoral research. The intense amount of computing resources required for this study was provided by the Cornell Institute for Computational Science and Engineering (ICSE) and by the Maryland Advanced Research Computing Center (MARCC), which is partially funded by the state of Maryland. JJC acknowledges the U.S Department of Energy, Office of Basic Energy Sciences under award no. DE-SC0016144.References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS.
- https://www.nrel.gov/pv/cell-efficiency.html-01-24-2019.pdf, The National Renewable Energy Laboratory, accessed October 19 2018.
- N. J. Jeon, H. Na, E. H. Jung, T.-Y. Yang, Y. G. Lee, G. Kim, H.-W. Shin, S. I. Seok, J. Lee and J. Seo, Nat. Energy, 2018, 3, 682–689 CrossRef CAS.
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim and J. H. Noh, et al. , Science, 2017, 356, 1376–1379 CrossRef CAS.
- W. A. Dunlap-Shohl, Y. Zhou, N. P. Padture and D. B. Mitzi, Chem. Rev., 2018, 119, 3193–3295 CrossRef.
- H.-S. Kim, A. Hagfeldt and N.-G. Park, Chem. Commun., 2019, 55, 1192–1200 RSC.
- A. Sharenko and M. F. Toney, J. Am. Chem. Soc., 2016, 138, 463–470 CrossRef CAS.
- B. J. Foley, J. Girard, B. A. Sorenson, A. Z. Chen, J. S. Niezgoda, M. R. Alpert, A. F. Harper, D.-M. Smilgies, P. Clancy and W. A. Saidi, et al. , J. Mater. Chem. A, 2017, 5, 113–123 RSC.
- A. G. Ortoll-Bloch, H. C. Herbol, B. A. Sorenson, M. Poloczek, L. A. Estroff and P. Clancy, Crystal Growth & Design, 2019 Search PubMed.
- L. Li, Y. Chen, Z. Liu, Q. Chen, X. Wang and H. Zhou, Adv. Mater., 2016, 28, 9862–9868 CrossRef CAS.
- Y. Zhou, M. Yang, A. L. Vasiliev, H. F. Garces, Y. Zhao, D. Wang, S. Pang, K. Zhu and N. P. Padture, J. Mater. Chem. A, 2015, 3, 9249–9256 RSC.
- G. E. Eperon, V. M. Burlakov, P. Docampo, A. Goriely and H. J. Snaith, Adv. Funct. Mater., 2014, 24, 151–157 CrossRef CAS.
- D. Wang, Z. Liu, Z. Zhou, H. Zhu, Y. Zhou, C. Huang, Z. Wang, H. Xu, Y. Jin and B. Fan, et al. , Chem. Mater., 2014, 26, 7145–7150 CrossRef CAS.
- T. Salim, S. Sun, Y. Abe, A. Krishna, A. C. Grimsdale and Y. M. Lam, J. Mater. Chem. A, 2015, 3, 8943–8969 RSC.
- G. Yang, C. Wang, H. Lei, X. Zheng, P. Qin, L. Xiong, X. Zhao, Y. Yan and G. Fang, J. Mater. Chem. A, 2017, 5, 1658–1666 RSC.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897 CrossRef CAS.
- A. J. Pearson, J. Mater. Res., 2017, 32, 1798–1824 CrossRef CAS.
- M. Xiao, F. Huang, W. Huang, Y. Dkhissi, Y. Zhu, J. Etheridge, A. Gray-Weale, U. Bach, Y.-B. Cheng and L. Spiccia, Angew. Chem., Int. Ed., 2014, 53, 9898–9903 CrossRef CAS.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate and A. Hagfeldt, et al. , Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- Y. Zhou, M. Yang, W. Wu, A. L. Vasiliev, K. Zhu and N. P. Padture, J. Mater. Chem. A, 2015, 3, 8178–8184 RSC.
- Y. Zhou, M. Yang, O. S. Game, W. Wu, J. Kwun, M. A. Strauss, Y. Yan, J. Huang, K. Zhu and N. P. Padture, ACS Appl. Mater. Interfaces, 2016, 8, 2232–2237 CrossRef CAS.
- D. Bi, W. Tress, M. I. Dar, P. Gao, J. Luo, C. Renevier, K. Schenk, A. Abate, F. Giordano and J.-P. C. Baena, et al. , Sci. Adv., 2016, 2, e1501170 CrossRef.
- M. Yavari, M. Mazloum-Ardakani, S. Gholipour, M. M. Tavakoli, S.-H. Turren-Cruz, N. Taghavinia, M. Grätzel, A. Hagfeldt and M. Saliba, Adv. Energy Mater., 2018, 1800177 CrossRef.
- S. Paek, P. Schouwink, E. N. Athanasopoulou, K. Cho, G. Grancini, Y. Lee, Y. Zhang, F. Stellacci, M. K. Nazeeruddin and P. Gao, Chem. Mater., 2017, 29, 3490–3498 CrossRef CAS.
- T. Bu, L. Wu, X. Liu, X. Yang, P. Zhou, X. Yu, T. Qin, J. Shi, S. Wang and S. Li, et al. , Adv. Energy Mater., 2017, 7, 1700576 CrossRef.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- J. Stevenson, B. Sorenson, V. H. Subramaniam, J. Raiford, P. P. Khlyabich, Y.-L. Loo and P. Clancy, Chem. Mater., 2016, 29, 2435–2444 CrossRef.
- B. A. Sorenson, S. S. Hong, H. C. Herbol and P. Clancy, Comput. Mater. Sci., 2019, 170, 109138 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2005, 109, 5656–5667 CrossRef CAS.
- H. Kruse and S. Grimme, J. Chem. Phys., 2012, 136, 04B613 CrossRef.
- G. Eperon and V. Burlakov, Adv. Funct. Mater., 2014, 24, 151–157 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- D. M. York and M. Karplus, J. Phys. Chem. A, 1999, 103, 11060–11079 CrossRef CAS.
- https://depts.washington.edu/eooptic/linkfiles/dielectric_chart[1].pdf, The University of Washington, accessed June 1 2020.
- J. G. Kirkwood, J. Chem. Phys., 1939, 7, 911–919 CrossRef CAS.
- P. Wang and A. Anderko, Fluid Phase Equilib., 2001, 186, 103–122 CrossRef CAS.
- A. E. Nielsen, Kinetics of precipitation, Pergamon, 1964, vol. 18 Search PubMed.
- M. Jung, S.-G. Ji, G. Kim and S. I. Seok, Chem. Soc. Rev., 2019, 48, 2011–2038 RSC.
- T. D. Storm, R. A. Hazleton and L. E. Lahti, J. Cryst. Growth, 1970, 7, 55–60 CrossRef CAS.
- K. G. Stamplecoskie, J. S. Manser and P. V. Kamat, Energy Environ. Sci., 2015, 8, 208–215 RSC.
- A. Sharenko, C. Mackeen, L. Jewell, F. Bridges and M. F. Toney, Chem. Mater., 2017, 29, 1315–1320 CrossRef CAS.
- G. Zhang and C. B. Musgrave, J. Phys. Chem. A, 2007, 111, 1554–1561 CrossRef CAS.
- J.-L. Bredas, Mater. Horiz., 2014, 1, 17–19 RSC.
- J. P. Perdew, Int. J. Quantum Chem., 1985, 28, 497–523 CrossRef.
- I. R. Kleckner and M. P. Foster, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 942–968 CrossRef CAS.
- F. Coleman, G. Feng, R. W. Murphy, P. Nockemann, K. R. Seddon and M. Swadźba-Kwaśny, Dalton Trans., 2013, 42, 5025–5035 RSC.
- I. G. Shenderovich, A. P. Burtsev, G. S. Denisov, N. S. Golubev and H.-H. Limbach, Magn. Reson. Chem., 2001, 39, S91–S99 CrossRef CAS.
- J. De Yoreo, Nat. Mater., 2013, 12, 284 CrossRef CAS.
- D. Gedamu, I. M. Asuo, D. Benetti, M. Basti, I. Ka, S. G. Cloutier, F. Rosei and R. Nechache, Sci. Rep., 2018, 8, 1–11 CrossRef CAS.
- B.-E. Cohen, S. Aharon, A. Dymshits and L. Etgar, J. Phys. Chem. C, 2016, 120, 142–147 CrossRef CAS.
- Y. Yu, S. Yang, L. Lei, Q. Cao, J. Shao, S. Zhang and Y. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 3667–3676 CrossRef CAS.
- K. Yan, M. Long, T. Zhang, Z. Wei, H. Chen, S. Yang and J. Xu, J. Am. Chem. Soc., 2015, 137, 4460–4468 CrossRef CAS.
- A. A. Petrov, I. P. Sokolova, N. A. Belich, G. S. Peters, P. V. Dorovatovskii, Y. V. Zubavichus, V. N. Khrustalev, A. V. Petrov, M. Gratzel and E. A. Goodilin, et al. , J. Phys. Chem. C, 2017, 121, 20739–20743 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta12364a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |