
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bottom-up wet-chemical synthesis of a two-dimensional porous carbon material with high supercapacitance using a cascade coupling/cyclization route†
Yongjie
Xu
abc,
Reiner Sebastian
Sprick
 b,
Nick J.
Brownbill
bd,
Frédéric
Blanc
bd,
Qingyin
Li
a,
John W.
Ward
*bc,
Shijie
Ren
*a and
Andrew I.
Cooper
*bc
b,
Nick J.
Brownbill
bd,
Frédéric
Blanc
bd,
Qingyin
Li
a,
John W.
Ward
*bc,
Shijie
Ren
*a and
Andrew I.
Cooper
*bc
aCollege of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, P. R. China. E-mail: rensj@scu.edu.cn
bDepartment of Chemistry, Materials Innovation Factory, University of Liverpool, Liverpool L69 7ZD, UK. E-mail: aicooper@liverpool.ac.uk; john.ward@liverpool.ac.uk
cLeverhulme Research Centre for Functional Materials Design, University of Liverpool, Liverpool, UK
dStephenson Institute for Renewable Energy, University of Liverpool, Liverpool L69 7ZD, UK
First published on 8th February 2021
Abstract
Wet-chemical bottom-up synthesis methods for two-dimensional (2D) layered materials are less explored than the top-down exfoliation of bulk materials. Here, we set out to synthesize a graphyne-type material by a wet-chemical synthesis method using Sonogashira–Hagihara cross-coupling polycondensation of a multifunctional monomer, 2, bearing alkyne and vinyl bromide functionalities. Spectroscopic and chemical analysis revealed that upon C–C bond formation, an unanticipated Bergman cyclization occurred to give an aromatic 2D porous carbon material (2D-PCM). 2D-PCM is a black material with graphene-like layers and a bulk structure that is similar to irregular graphite. It is porous with a hierarchical pore structure and an apparent Brunauer–Emmett–Teller surface area of 575 m2 g−1. The material has excellent electrochemical performance as an electrode in supercapacitors with a specific capacitance of 378 F g−1 at the current density of 0.1 A g−1, which surpasses state-of-the-art carbon materials, suggesting that wet-chemical methods might give functional benefits over top-down processing routes.
Carbon materials such as diamond, graphite and amorphous carbon have long been exploited for their specific properties, such has hardness (diamond) or electric conductivity (graphite, amorphous carbon). Recently, new carbon allotropes such as graphene, buckminsterfullerene, carbon nanotubes, and schwarzites have been discovered and these have received much interest for their potential applications.1 Graphene possesses high electrical conductivity, mechanical flexibility, and high chemical stability, and has been used in many advanced applications.2,3 The relatively low surface area of graphene, however, limits its efficiency in energy storage devices.4 To overcome this drawback, significant attention has been focused on the preparation of high surface area 2D porous carbon materials that exhibit electrical conductivity.5 2D carbon materials can be synthesized by either top-down or bottom-up approaches.6 Top-down methods extract 2D carbon from bulk materials, using methods such as exfoliation, while bottom-up methods commonly use chemical vapor deposition, template methods, or carbonization.7 Bottom-up wet-chemical synthesis approaches are less explored in this context, and carbon allotrope graphdiyne-type materials have only very recently been reported using such methods.8,9 In principle, wet-chemical synthesis methods allow for a degree of structural diversity and fine tuning of properties such as porosity. Also, new high carbon-content materials have the potential to exhibit high charge-carrier mobilities and good physicochemical stability, which are desirable properties applications such as gas storage and separation,10 photocatalysis,11 photodetectors,12 and energy storage and generation,13 for instance in Li-ion batteries,14 supercapacitors,15 fuel cells,16 and perovskite solar cells.17
In this work, our initial goal was to synthesize a graphyne-type material using a Sonogashira–Hagihara cross-coupling reaction using a A2B2-type monomer, 2, bearing both bromide and alkyne functionalities (Fig. 1a). The TMS-protected precursor, 1, was synthesized using a previously reported method18 and deprotected to afford 2 which was used directly in a Pd(0)-catalysed Sonogashira–Hagihara cross-coupling polycondensation reaction at 150 °C. A black powder (2D-PCM) was formed, which after Soxhlet extraction to remove any unreacted monomers or oligomers, was found to be insoluble in THF, toluene, N,N-dimethylformamide, and N-methyl-2-pyrrolidone. 2D-PCM was stable under a variety of conditions, including in 1 M aqueous H2SO4, 6 M aqueous KOH, and 1 M Na2SO4 aqueous solution. Analysis by solid-state Magic Angle Spinning (MAS) Nuclear Magnetic Resonance (NMR), X-Ray Photoelectron Spectroscopy (XPS), Fourier Transformation-InfraRed (FT-IR) and chemical functional group tests revealed that very few sp-hybridized carbons were present in the material, which consisted mainly of sp2-hybridized carbons. We tentatively ascribe this to an in situ Bergman cyclization that occurs after the initial Sonogashira–Hagihara cross-coupling reaction to give a polyaromatic 2D porous carbon material. Bergman cyclization19 has been used previously in the polycondensation of aromatic diynes and multi-ethynyl aromatic compounds at high temperatures to yield layered polyphenylenes20 and amorphous conjugated microporous polymers, respectively.21 A reaction temperature of 150 °C was required for this Sonogashira–Hagihara cross-coupling/Bergman cyclization cascade reaction, but this temperature is much lower than normally required for Bergman cyclization reactions or the bottom-up synthesis of 2D carbon materials from organic precursors.22 Sonogashira–Hagihara coupling of the alkyne with the vinyl bromide results in an intermediate enediyne that is poised for Bergman cyclisation. Unactivated enediynes require high temperatures to undergo Bergman cyclisation, but the temperature can be reduced significantly in the presence of metals via chelation-controlled cyclisation.23 We propose that the palladium catalyst used for the Sonogashira–Hagihara reaction also promotes the Bergman cyclisation through chelation of the enediyne and enables the polymerisation to occur at relatively low temperature in high yield. We investigated the polymerisation reaction at various temperatures ranging from 20 °C to 200 °C and found 150 °C to be optimal for both yield and materials properties. No reaction was observed in the absence of palladium catalyst, further supporting the proposed cascade reaction sequence of palladium-catalysed cross-coupling followed by Bergman cyclization.
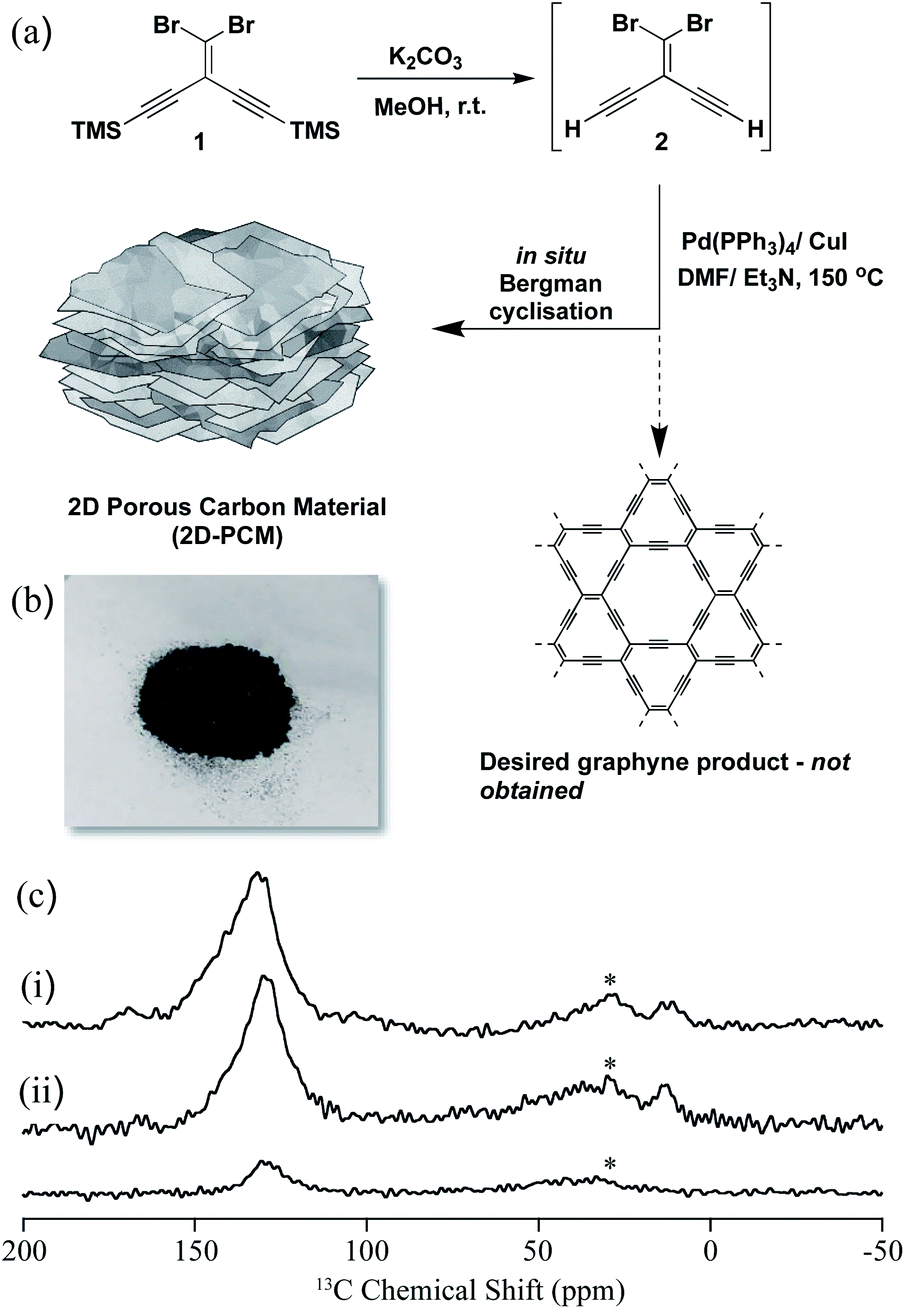 | ||
| Fig. 1 (a) Synthesis of 2D-PCM; (b) photograph of 2D-PCM; (c) stack plot of the 13C NMR spectra of 2D-PCM recorded at 9.4 T and under magic angle spinning (MAS) at 10 kHz. (i) Directly excited 13C NMR spectrum, (ii) 13C cross polarisation (CP) NMR spectra obtained at contact times of 2 ms (top) and 50 μs (bottom). Further experimental details are given in the ESI.† The asterisks (*) at 30 ppm denote spinning sidebands from the sp2-hybridized carbons at 130 ppm. | ||
The 13C MAS NMR spectrum of 2D-PCM obtained under direct 13C excitation (Fig. 1c(i)) showed a dominant signal centred around 130 ppm that showed that the material was composed almost entirely of sp2-hybridized carbons.24 This was supported by the 13C spectra obtained under cross polarisation (CP) from 1H (Fig. 1c(ii)), which indicated that this 13C resonance arises from carbons that are strongly coupled to 1H; that is, most likely protonated carbons. The signal is fairly broad with a full width at half maximum of around 25 ppm, strongly indicating inhomogeneous broadening and distribution of 13C chemical shifts that supports some significant structural disorder. Two very small 13C signals were also observed around 170 and 20 ppm (Fig. 1c(i)), which were tentatively assigned to some residual N,N-dimethylformamide from the synthesis of 2D-PCM. Within the signal-to-noise of the directly excited 13C NMR spectrum obtained (see Fig. S3† for an additional spectrum obtained with a longer recycle delay that would favour the detection of unprotonated carbons), no sp-hybridized carbons were observed in the 60–85 ppm region that would be expected for highly conjugated systems such as polyynes25 and polyenynes (e.g., poly(diacetylene)26 and poly(triacetylene)),27 thus ruling out a graphyne-type material. To further probe the absence of alkyne groups and sp-hybridized carbons, a number of chemical functional group tests were performed including bromination, azide–alkyne cycloaddition reaction with an azo-dye, and oxidation with potassium permanganate (Fig. S6–S11†): all of these tests proved negative for alkyne groups.
The morphology of 2D-PCM was studied by scanning electron microscope (SEM) and transition electron microscope (TEM). This bulk material has macroscopic order and layers that are clearly visible in the SEM and TEM. Such well-defined layers are not typically observed for carbon materials made by wet-chemical synthesis. SEM images of the material showed that the material was composed of micrometre-sized, smooth-surfaced sheets that were stacked together (Fig. S12a†). Several sheets were found to be irregularly aggregated at the region near the edge of this bulk material (Fig. 2a and S12b†). Another twisted edge of one of the aggregates gave a clearer cross-sectional view of the sheet-by-sheet aggregated structure (Fig. 2b). It appears that slits between the sheets of 2D-PCM provide extra pore structures, which might contribute to the total specific surface area and, hence, energy storage applications.22 TEM images showed that the aggregated sheets are formed by layer-by-layer stacking of thin, flexible and wrinkled graphene-like 2D layers (Fig. 2c, d and S13†).
 | ||
| Fig. 2 (a and b) Scanning electron microscope of 2D-PCM at different magnifications; (c and d) transmission electron microscope images of 2D-PCM at different magnifications. | ||
Powder X-ray diffraction (PXRD) of 2D-PCM shows two broad peaks at 22° and 40°, which can be assigned to the (002) and (100) planes in the structure28 (Fig. 3a). This indicates an overall 3D amorphous structure of bulk material was formed by the irregular stacking of layers that are formed during the wet chemical synthesis. Raman spectroscopy of 2D-PCM reveals three prominent peaks at 1365 cm−1, 1575 cm−1 and 2800 cm−1 that are assigned to D band, G band and 2D band characteristics based on comparison with other 2D carbon materials (Fig. 3b).8,29 The D band corresponds to the breathing vibration of sp2 carbon domains and the intensity is strongly associated with structural defects, while G band corresponds to the first-order scattering of the E2g mode for in-phase stretching vibration of sp2 carbon domains.30 The intensity ratio of D band to G band is 0.83, indicating a relatively ordered structure in the bulk material. A broad 2D peak at 2800 cm−1 indicates that a multi-layered structure exists.31 These results are consistent with the layered structure observed by SEM and TEM. X-ray photoelectron spectroscopy (XPS) shows that 2D-PCM is mainly composed of carbon. A small amount of oxygen could be observed, which was also observed by energy dispersive X-ray spectroscopy (Fig. 3c and S4†). The existence of oxygen can be attributed to absorbed atmospheric O2 and/or oxidization on the surface when the sample is exposed to air.15 The C 1s curve-fit spectrum (Fig. 3d) shows two sub-peaks, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C at 284.9 eV and C–O at 287.0 eV.32 IR spectra revealed absorption bands around 1436 cm−1 and 1622 cm−1 that were assigned to the skeletal vibrations of the aromatic rings and stretching vibrations of CC bonds (Fig. S5†). A very weak absorption band located at 2170 cm−1 is assigned to C
C at 284.9 eV and C–O at 287.0 eV.32 IR spectra revealed absorption bands around 1436 cm−1 and 1622 cm−1 that were assigned to the skeletal vibrations of the aromatic rings and stretching vibrations of CC bonds (Fig. S5†). A very weak absorption band located at 2170 cm−1 is assigned to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond stretching vibrations indicating the presence of some sp-hybridized carbons—most likely terminal alkyne end groups—at a concentration that could likely not be detected by solid-state NMR.
C bond stretching vibrations indicating the presence of some sp-hybridized carbons—most likely terminal alkyne end groups—at a concentration that could likely not be detected by solid-state NMR.
 | ||
| Fig. 3 (a) PXRD pattern of 2D-PCM; (b) Raman spectrum of 2D-PCM; (c) full XPS spectrum shown from 0 up to 1100 eV and (d) in the C 1s region 2D-PCM with fits. | ||
The porosity of 2D-PCM was investigated by nitrogen adsorption/desorption experiments at 77 K. An apparent Brunauer–Emmett–Teller surface area (SABET) of 575 m2 g−1 was determined from the nitrogen sorption isotherm (Fig. 4a) with a total pore volume of 0.4 cm3 g−1 at P/P0 = 0.99. The nitrogen adsorption isotherm shows a steep uptake in the low relative pressure range (P/P0 < 0.01), indicative of micropores.33 At higher relative pressure, the isotherm shows a continuous rise with a H4-hysteresis loop indicating the presence of slit-like mesopores.34 We propose that these slit-like pores stem from the irregular aggregation of the layers, as observed in the SEM and TEM images. Pore size distributions were calculated from the nitrogen sorption isotherms using non-local density functional theory (NL-DFT) carbon slit pore model. 2D-PCM exhibits a considerable fraction of micropores with a pore width range of 0.9 to 2 nm, and a proportion of mesopores with pore width between 2 to 6 nm (Fig. 4b). Besides the micropores and mesopores, which are formed by irregular aggregation, we postulate that 2D-PCM might also have intrinsic ultramicropores within its molecular network. Nitrogen adsorption experiments are not suitable for the pore size analysis of these ultramicropores, and we therefore carried out CO2 adsorption measurements (Fig. 4c and S16†) to probe this intrinsic ultramicroporosity.35 The pore size distribution curve derived from the CO2 adsorption isotherm shows ultramicropores with pore widths of 0.36, 0.55 and 0.82 nm (Fig. 4d).
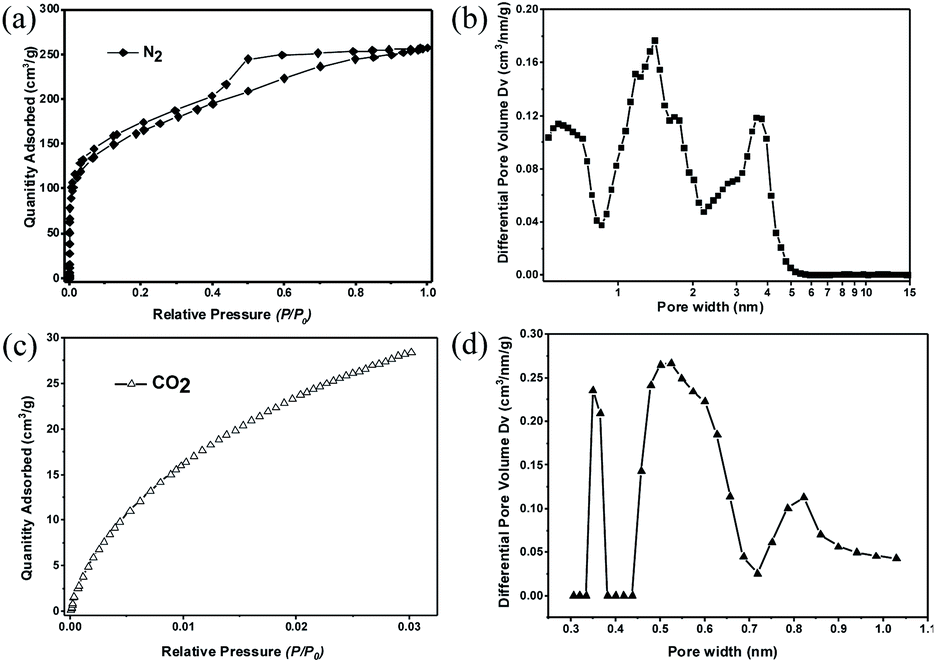 | ||
| Fig. 4 (a) Nitrogen sorption isotherm of 2D-PCM measured at 77 K; (b) pore size distribution of 2D-PCM calculated via a NL-DFT model based on the N2 sorption isotherm; (c) CO2 adsorption of 2D-PCM measured at 273 K; (d) pore size distribution curve of 2D-PCM calculated via a NL-DFT model based on the CO2 adsorption isotherm. | ||
For supercapacitance measurements, a conventional three-electrode set-up with 2D-PCM as the working electrode was fabricated.36 Cyclic voltammetry (CV) curves at different scan rates (5 mV s−1 to 200 mV s−1) showed a quasi-rectangular shape suggesting electric double-layer capacitive characteristics (Fig. 5a). Weak redox peaks superposed in the CV curves indicate faradaic pseudocapacitance during the charge–discharge process, which may be a result of the small amount of oxygen in the structure. Galvanostatic charge–discharge tests were carried out to further verify this observation. Fig. 5b shows the typical charge–discharge profiles at the current density from 0.1 A g−1 to 10 A g−1. At the large current density, the curves show nearly symmetrical triangular shapes, suggesting fast ion and electron transport within the 2D-PCM electrodes. The specific capacitance calculated by the slope of discharge curve at the current density of 0.1 A g−1 is approximately 378 F g−1, which is higher than many state-of-the-art 2D carbon-based materials including reduced graphene oxide (42 F g−1), 2D porous carbon nanosheets (244–304 F g−1 at 0.1 A g−1),37 nitrogen-doped holey graphitic carbon (206 F g−1 at 0.1 A g−1),38 2D conductive MOFs and 2D COFs (256–396 F g−1 at 0.2 A g−1)39–41 (for more details see Fig. S17† and for a comprehensive compilation of literature data see Table S1†). The specific capacitance has a value of 175 F g−1 even when the current density is increased to 10 A g−1, indicating a good rate performance (Fig. 5c).42 The maximum power (4.3 kW kg−1) and energy density (69.4 kW h kg−1) were calculated based on the Ragone plot (Fig. S18†). We believe that this high specific capacitance can be attributed, in part, to the high surface area and hierarchical pore structure, which provides a large electroactive electrode/electrolyte interface and enhanced electron and ion transport within the material. High surface areas have been shown to be beneficial for electrode materials in Li-ion batteries14,43 and supercapacitors.15 We attribute the weak alkyne stretching vibration band in the FT-IR spectra to incomplete Bergman cyclization at the edges of the material. It is conceivable that these residual alkyne linkages may be lowering the atom density and increasing the theoretical capacity,43 when compared with purely sp2-hybridized carbon materials such as graphene. The inherent 2D layered structure is likely to improve the conductivity, promoting fast charge transmission.
 | ||
| Fig. 5 (a) Cyclic voltammograms; (b) galvanostatic charge–discharge curves; (c) specific capacitance at various current densities; (d) Nyquist plots with the inset showing the enlarged part of the high-frequency region; (e) imaginary part capacitance–frequency curves of 2D-PCM electrode; (f) capacitance of 2D-PCM for a 2500-cycle charge–discharge stability tested at a current density of 1 A g−1. | ||
The kinetic behaviour of 2D-PCM was studied with electrochemical impedance spectroscopy (EIS) in the frequency range 100 kHz to 0.01 Hz. Nyquist plots show a small quasi-semicircle in the high-frequency range, followed by a straight sloped line in the low-frequency region (Fig. 5d). The intercept with the Z′ axis is around 0.45 Ω, representing internal resistance that derives from the resistance of the electrolyte. The diameter of the high-frequency semi-circle corresponds to the charge transfer resistance (RCT) of the electrode kinetics. 2D-PCM possesses a RCT of 0.30 Ω, which is significantly lower than that of other 2D porous materials, including graphene coupled porous polymers (0.69 Ω),44 Aza-CMP (1.40 Ω)45 and graphdiyne (14.7 Ω).46 In the low-frequency region, 2D-PCM displays linear plots with a relatively large slope, indicating a low diffusive resistance of electrolyte ion within the hierarchical pores. The imaginary part capacitance–frequency curves derived from Electrochemical Impedance Spectroscopy (EIS) profiles can be used to evaluate the charge–discharge performance of electrode materials (Fig. 5e). The location of the peak in the curves represents the relaxation frequency (fR), at which purely resistive behaviour is transformed into purely capacitive behaviour. Calculated from the reciprocal of 2πfR, the relaxation time τR of 2D-PCM is 0.51 s, indicating a fast charging and discharging capability and thus a high-power density. We also assembled a two-electrode system supercapacitor device and tested the stability by cycling experiments. 2D-PCM exhibited excellent performance stability without loss in capacitance after 2500 charge–discharge cycles at current density of 1 A g−1 (Fig. 5f). Overall, 2D-PCM exhibited good energy storage performance with excellent cycle life.
Conclusions
In summary, a bulk polyaromatic 2D porous carbon material (2D-PCM) was synthesized using a Sonogashira–Hagihara cross-coupling polycondensation reaction with a A2B2-type monomer containing both bromide and alkyne functionalities. Spectroscopic and chemical analysis of the material determined that the expected graphyne-like material was not obtained, but rather a polyaromatic material containing sp2-hybridized carbons. We propose that this is the result of a Bergman-type cyclization after the Sonogashira–Hagihara coupling reaction. SEM and TEM images reveal well-defined stacked layers that are similar to irregular graphite.472D-PCM is highly porous with a hierarchical pore structure and a Brunauer–Emmett–Teller surface area of 575 m2 g−1, and has excellent electrochemical performance as an electrode in supercapacitors with a specific capacitance of 378 F g−1 at the current density of 0.1 A g−1, which surpasses many state-of-the-art carbon materials.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (51973128, 21574087), Science and Technology Department of Sichuan Province (2019YJ0128), the Leverhulme Trust via the Leverhulme Research Centre for Functional Materials Design, and the Engineering and Physical Sciences Research Council (EPSRC) (Doctoral Training Studentship to N. J. B. and grant EP/N004884/1). We thank B. Layla Mehdi for helpful discussions.Notes and references
- J. Zhang, M. Terrones, C. R. Park, R. Mukherjee, M. Monthioux, N. Koratkar, Y. S. Kim, R. Hurt, E. Frackowiak, T. Enoki, Y. Chen, Y. S. Chen and A. Bianco, Carbon, 2016, 98, 708–732 CrossRef CAS.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS.
- M. Inagaki and F. Kang, J. Mater. Chem. A, 2014, 2, 13193–13206 RSC.
- P. Yao, P. Chen, L. Jiang, H. Zhao, H. Zhu, D. Zhou, W. Hu, B.-H. Han and M. Liu, Adv. Mater., 2010, 22, 5008–5012 CrossRef CAS.
- K. Gao, Q. Niu, Q. Tang, Y. Guo and L. Wang, J. Electron. Mater., 2018, 47, 337–346 CrossRef CAS.
- J. M. Tour, Chem. Mater., 2014, 26, 163–171 CrossRef CAS.
- K. Dimos, F. Arcudi, A. Kouloumpis, I. B. Koutselas, P. Rudolf, D. Gournis and M. Prato, Nanoscale, 2017, 9, 10256–10262 RSC.
- G. Li, Y. Li, H. Liu, Y. Guo, Y. Li and D. Zhu, Chem. Commun., 2010, 46, 3256–3258 RSC.
- Z. Jia, Z. Zuo, Y. Yi, H. Liu, D. Li, Y. Li and Y. Li, Nano Energy, 2017, 33, 343–349 CrossRef CAS.
- T. D. Daff, S. P. Collins, H. Dureckova, E. Perim, M. S. Skaf, D. S. Galvao and T. K. Woo, Carbon, 2016, 101, 218–225 CrossRef CAS.
- N. Yang, Y. Liu, H. Wen, Z. Tang, H. Zhao, Y. Li and D. Wang, ACS Nano, 2013, 7, 1504–1512 CrossRef CAS.
- Z. W. Jin, Q. Zhou, Y. H. Chen, P. Mao, H. Li, H. B. Liu, J. Z. Wang and Y. L. Li, Adv. Mater., 2016, 28, 3697–3702 CrossRef CAS.
- Q. Peng, A. K. Dearden, J. Crean, L. Han, S. Liu, X. Wen and S. De, Nanotechnol., Sci. Appl., 2014, 7, 1–29 CAS.
- S. Zhang, H. Liu, C. Huang, G. Cui and Y. Li, Chem. Commun., 2015, 51, 1834–1837 RSC.
- H. Du, H. Yang, C. Huang, J. He, H. Liu and Y. Li, Nano Energy, 2016, 22, 615–622 CrossRef CAS.
- B. Kang and J. Y. Lee, J. Phys. Chem. C, 2014, 118, 12035–12040 CrossRef CAS.
- C. Kuang, G. Tang, T. Jiu, H. Yang, H. Liu, B. Li, W. Luo, X. Li, W. Zhang, F. Lu, J. Fang and Y. Li, Nano Lett., 2015, 15, 2756–2762 CrossRef CAS.
- N. P. Bowling and R. J. McMahon, J. Org. Chem., 2006, 71, 5841–5847 CrossRef CAS.
- Y. Xiao and A. Hu, Macromol. Rapid Commun., 2011, 32, 1688–1698 CrossRef CAS.
- Q. Sun, C. Zhang, Z. Li, H. Kong, Q. Tan, A. Hu and W. Xu, J. Am. Chem. Soc., 2013, 135, 8448–8451 CrossRef CAS.
- X.-M. Zhang, X. Ding, A. Hu and B.-H. Han, Polym. Chem., 2015, 6, 4734–4741 RSC.
- S. Han, D. Q. Wu, S. Li, F. Zhang and X. L. Feng, Adv. Mater., 2014, 26, 849–864 CrossRef CAS.
- A. Basak, S. Mandal and S. S. Bag, Chem. Rev., 2003, 103, 4077–4094 CrossRef CAS.
- R. M. Silverstein, F. X. Webster, D. J. Kiemle and D. L. Bryce, Spectrometric Identification of Organic Compounds, 2014, vol. 8 Search PubMed.
- W. A. Chalifoux, R. McDonald, M. J. Ferguson and R. R. Tykwinski, Angew. Chem., 2009, 121, 8056–8060 CrossRef.
- L. Luo, C. Wilhelm, A. Sun, C. P. Grey, J. W. Lauher and N. S. Goroff, J. Am. Chem. Soc., 2008, 130, 7702–7709 CrossRef CAS.
- A. M. Boldi, J. Anthony, V. Gramlich, C. B. Knobler, C. Boudon, J. P. Gisselbrecht, M. Gross and F. Diederich, Helv. Chim. Acta, 1995, 78, 779–796 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz and M. Thommes, Science, 2011, 332, 1537–1541 CrossRef CAS.
- Z. Luo, C. Cong, J. Zhang, Q. Xiong and T. Yu, Carbon, 2012, 50, 4252–4258 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- A. Ferrari, J. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. Novoselov and S. Roth, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- H. Estrade-Szwarckopf, Carbon, 2004, 42, 1713–1721 CrossRef CAS.
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Adv. Mater., 2012, 24, 2357–2361 CrossRef CAS.
- K. S. Sing, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- X. Huang, Y. C. Zhao and B. H. Han, Chem. Commun., 2016, 52, 6597–6600 RSC.
- L. Hao, B. Luo, X. Li, M. Jin, Y. Fang, Z. Tang, Y. Jia, M. Liang, A. Thomas, J. Yang and L. Zhi, Energy Environ. Sci., 2012, 5, 9747 RSC.
- X. D. Zhuang, F. Zhang, D. Q. Wu, N. Forler, H. W. Liang, M. Wagner, D. Gehrig, M. R. Hansen, F. Laquai and X. L. Feng, Angew. Chem., Int. Ed., 2013, 52, 9668–9672 CrossRef CAS.
- Z. Xiang, D. Cao, L. Huang, J. Shui, M. Wang and L. Dai, Adv. Mater., 2014, 26, 3315–3320 CrossRef CAS.
- J. Liu, X. Song, T. Zhang, S. Liu, H. Wen and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 2–15 CrossRef.
- J. Liu, X. Song, Y. Zhou, Z. Xie, Y. Li, Y. Liu, J. Sun, Y. Ma, O. Terasaki and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 1081–1086 CrossRef CAS.
- M. Li, J. Liu, Y. Li, G. Xing, X. Yu, C. Peng and L. Chen, CCS Chem., 2020, 2, 696–706 CrossRef.
- S. P. Pang, Y. Hernandez, X. L. Feng and K. Mullen, Adv. Mater., 2011, 23, 2779–2795 CrossRef CAS.
- C. Huang, S. Zhang, H. Liu, Y. Li, G. Cui and Y. Li, Nano Energy, 2015, 11, 481–489 CrossRef CAS.
- X. D. Zhuang, F. Zhang, D. Q. Wu and X. L. Feng, Adv. Mater., 2014, 26, 3081–3086 CrossRef CAS.
- K. Yan, X. Yanhong, G. Zhaoqi and J. Donglin, Angew. Chem., Int. Ed., 2011, 50, 8753–8757 CrossRef.
- K. Krishnamoorthy, S. Thangavel, J. C. Veetil, N. Raju, G. Venugopal and S. J. Kim, Int. J. Hydrogen Energy, 2016, 41, 1672–1678 CrossRef CAS.
- Z. Zhang, M. Dou, H. Liu, L. Dai and F. Wang, Small, 2016, 12, 4193–4199 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional MAS NMR spectra, energy dispersive X-ray spectra, FT-IR spectra, chemical functional group tests, galvanostatic charge–discharge curves, and Ragone plots. See DOI: 10.1039/d0ta11649a |
| This journal is © The Royal Society of Chemistry 2021 |