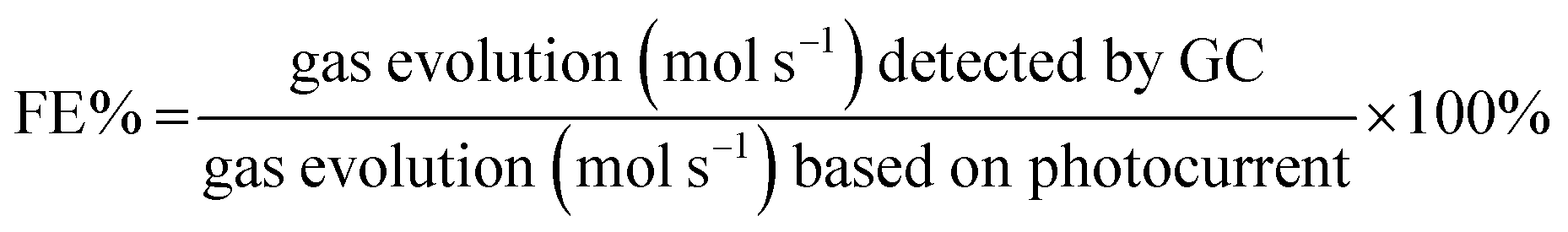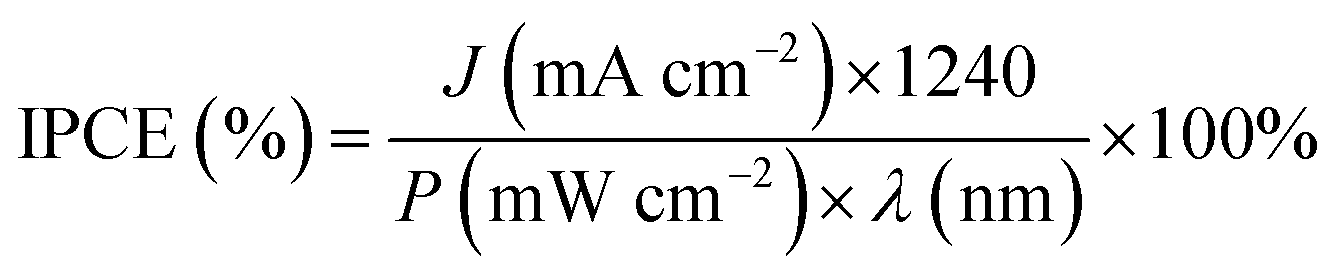DOI:
10.1039/D0TA11494D
(Paper)
J. Mater. Chem. A, 2021,
9, 12214-12224
Revealing the true impact of interstitial and substitutional nitrogen doping in TiO2 on photoelectrochemical applications†
Received
25th November 2020
, Accepted 29th April 2021
First published on 30th April 2021
Abstract
Application of photocatalysts that strongly absorb within the visible range is a common strategy to improve the efficiency of photoelectrochemical (PEC) systems; this may translate to high photocurrents, but it is not always the case. Here, we show that nitrogen doping enhances visible light absorption of TiO2; however, it does not necessarily result in improved PEC performance. Depending on the applied external potential, N-doping can improve, or degrade, PEC performance either under water oxidation conditions or via hole scavenging (Na2S/Na2SO3). In this work, we developed a holistic approach to evaluate the true impact of N doping in TiO2 on PEC performance. Interstitial and substitutional N doping are experimentally explored for the first time through a simple and novel PEC approach which complemented X-ray photoelectron analyses. Using this approach, we show that interstitial N doping of anatase TiO2 dominates up to 400 °C and substitutional doping up to ca. 600 °C, without rutile formation. This reveals that the bottleneck for doping higher N-concentrations in TiO2 is the direct transformation to thermodynamically favorable N-rich phases, such as TiN/Ti2N at 700 °C, inhibiting the formation of rutile phase. Transmission electron microscopy revealed that N doping proceeds mainly from the inner to the outer tube walls via nitridation and follows a preferential pathway from interstitial to substitutional doping. Direct PEC experimental evidence on visible light activation of N doped TiO2, and the location of interband states, showed acceptor levels of 1.0 eV for substitutional and 0.7 eV for interstitial doping above the TiO2 valence band maximum. In addition, due to O vacancies and Ti3+ species, donor levels below the conduction band minimum were also created. These levels act as trapping/recombination centers for charge carriers and, therefore, the gain in the visible range due to N doping does not translate to an improved PEC performance by these structural defects. Ultimately, we show that whilst there is a benefit of visible light absorption through N doping in TiO2, the PEC performance of the samples only surpasses pristine TiO2 at relatively high biasing (>0.3 V vs. Ag/AgCl).
1. Introduction
Over the past few decades, titanium dioxide (TiO2) solar energy harvesting devices have been widely applied in dye sensitized solar cells, sensors, photoelectrochemical (PEC) water splitting and the photocatalytic degradation of pollutants.1–6 However, the large bandgap energy of TiO2 (anatase, E ∼ 3.3 eV) limits its utilization in solar energy devices as it can be only excited in the UV region which accounts for 4% of the solar irradiation that reaches the Earth's surface. Hence, to improve TiO2 photoelectrode performance for solar applications, absorption in the visible light spectrum must be encompassed. This can be achieved by the structural doping and the formation of a heterojunction between TiO2 with smaller bandgap semiconductors (e.g. CdSe).7–10 Metal (cationic) and non-metal (anionic) doping of TiO2 has been shown to improve its electronic and optical properties beyond what is attainable by pure TiO2.11–16 Nevertheless, doping can only be effective for better performance if the obtained electronic structure of the semiconductor is adequate for a particular photocatalytic reaction.
Nitrogen doping of TiO2 is a promising alternative to improve its visible light absorption for photocatalytic applications.7,17,18 The most common approaches to prepare N doped TiO2 are magnetron sputtering,19,20 annealing in a N2 or NH3 flux,21,22 washing with nitrogen precursors23 and ion implantation.24 It has been suggested that the N atoms substitute oxygen atoms in the TiO2 lattice and new N 2p states are introduced that can be either shallow or deep level states.25 Both states can extend the spectral range for light absorption to longer wavelengths.26 For N substitution, N 2p states hybridize with O 2p states causing the valence band (VB) to shift upwards; however, some reports also suggest substitutional N doping results in discrete interband states above the VB rather in the formation of a continuous VB,27,28 Other reports suggest that N doping can also be interstitial.29,30 XPS can be used to determine whether N doping is interstitial or substitutional in the TiO2 structure; however, it cannot infer the exact location of N 2p levels in the bandgap, which is crucial information for photoelectrochemical (PEC) reactions.
N doped TiO2 has been applied in PEC reactions, particularly in oxidation reactions where hole scavenging from the photoanode surface decreases electron–hole recombination and improves photocurrents.31–35 PEC water splitting using N doped TiO2 has been studied in conventional supporting electrolytes (i.e. NaOH, Na2SO4etc.); however, due to the sluggish water oxidation kinetics in these electrolytes, the competition between hole scavenging and hole diffusion to the photoanode makes it difficult to understand its PEC performance.25,36–38 One way to efficiently scavenge the photogenerated holes and suppresses surface recombination is to utilize a sacrificial redox couple in the electrolyte.39–42 This PEC reaction will not be the water oxidation reaction (overall water splitting) but involves the oxidation of the redox species at the photoanode (n-type) surface. Another way to improve PEC performance is to apply higher external biasing. Particularly, for N doped photoelectrodes, high biasing is applied to show an improved performance in J–V curves which hinders the negative effect of doping in many reports.25,37,43 As a result, the relationship between the structural and electronic properties of N doped TiO2 and their photoelectrochemical properties remains unclear leading to poorly interpretated results.
Herein, we determine the mechanism of N doping in TiO2 nanotubes prepared by nitridation, including the limit of N content in the TiO2 NTs structure. We then, developed a new methodology to distinguish interstitial and substitutional doping, via PEC measurements, to fully understand the electronic structure of N doped TiO2 nanotubes in PEC reactions. We present a holistic approach that enabled us to evaluate the true PEC performance of N doped TiO2.
2. Experimental section
2.1 Synthesis of N doped TiO2 NT
Titanium disks (98.6% purity, 30 mm diameter, 1 mm thick) were polished and cleaned ultrasonically in acetone and water for 30 min each. Anodization was performed at room temperature in a custom Teflon cell with a two-electrode configuration (Ti disk anode and a polished Cu disk cathode (30 mm diameter)) separated by 1 cm and immersed in an ultrasonic bath. The electrolyte was composed of ethylene glycol (ETG, 99.5%), ammonium fluoride (0.5 wt%, 98%, Synth) and deionized water (10 wt%).9 An external biasing of 30 V was applied across the electrodes for 1 h with an initial ramping of 10 V s−1 for 30 s. The current vs. time curves were monitored by the Ideial, Dalton Vidor software incorporated with the power supply. The anodized disks were then cleaned with deionized water before being subjected to thermal nitridation at temperatures ranging from 400 °C to 800 °C for 3 h in an ammonia![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :argon (1:9 vol/vol) gas flux. The gas flux rate and heating and cooling rate of the furnace were varied. Unless stated otherwise, nitridation was conducted at 400, 500, 600, 700 or 800 °C (heating ramp of 10 °C min−1) using an ammonia flux of 100 mL min−1. The respective samples were named: TN-400, TN-500, TN-600, TN-700 and TN-800. A control sample of TiO2 NT was heat treated at 400 °C in air and labelled pure TiO2.
:argon (1:9 vol/vol) gas flux. The gas flux rate and heating and cooling rate of the furnace were varied. Unless stated otherwise, nitridation was conducted at 400, 500, 600, 700 or 800 °C (heating ramp of 10 °C min−1) using an ammonia flux of 100 mL min−1. The respective samples were named: TN-400, TN-500, TN-600, TN-700 and TN-800. A control sample of TiO2 NT was heat treated at 400 °C in air and labelled pure TiO2.
2.2 Characterization
Field Emission Gun Scanning Electron Microscopy (FEG-SEM) JEOL 7000F FEG-SEM was conducted at 10 kV. A JEOL FEG JEM-2100F transmission electron microscope (TEM), operated at an accelerating voltage of 200 kV and equipped with an energy-dispersive X-ray spectrometer (Noran Seven), was used for classical TEM imaging and High-Resolution Electron Microscopy (HR-TEM). X-Ray Diffraction (XRD) in Bragg Brentano geometry was performed in a D500 Siemens diffractometer (2θ range: 15–60°, step size: 0.05°, 5 s per step, Cu Kα radiation (λ = 1.54 Å)). The crystallite size, calculated from the Scherer equation, and the relative peak intensity ratios (I(101)/I(200)) of anatase TiO2 are displayed in Table S1.† Grazing angle incidence X-ray diffraction (GIXRD) was recorded by Bruker D8 Advanced (2θ range: 15–60°, step size: 0.02°, 1.5 s per step, incident angle: 0.5°, Cu Kα radiation (λ = 1.54 Å)). The UV-Vis absorption spectra were recorded in a CARY 5000 spectrophotometer in diffuse reflectance mode and data was converted to Kubelka–Munk function for Tauc plots. Surface chemical composition was studied by X-ray photoelectron spectroscopy (XPS) performed in a Kratos AXIS ULTRA DLD with Al Kα radiation (1486.6 eV) as the monochromated X-ray source. Survey spectra and regions in high resolution were recorded with pass energies of 80 eV and 20 eV, respectively. Data deconvolution was carried out in CasaXPS and the advantageous C 1s peak at 284.8 eV was chosen for spectra calibration.
2.3 Photoelectrochemical studies
Photoelectrochemical measurements were carried out using an Autolab (PGSTAT 100N) potentiostat. The experiments were performed in a quartz cell using a standard three-electrode configuration employing TiO2 nanotubes as the working electrode, a Pt mesh as the counter electrode and Ag/AgCl as the reference electrode in an aqueous solution of Na2S (0.24 M) and Na2SO3 (0.35 M) and, for comparison, a KOH solution (1 M) was also used. Prior to the measurements the electrolytes were purged with Ar gas for 30 min. The working electrode was irradiated with a 300 W Xenon lamp solar simulator (Oriel) calibrated to AM 1.5G (100 mW cm−2) intensity. The linear sweep voltammetry (LSV) curves were recorded in both dark and 1 Sun illumination at a sweep rate of 10 mV s−1. Nyquist plots were performed at the frequency window of 100 kHz to 100 mHz for signal amplitude of 10 mV under illumination applying open circuit potential. The interband states position was studied in chronoamperometry by keeping the working electrode in the dark and under illumination for 10 s each at varying wavelengths with 20 nm increments starting at 600 nm and decreasing to 280 nm using Cornerstone Oriel monochromator. Long time stability tests were performed by chronoamperometry in two electrodes configuration using TiO2 as the photoanode and Pt as the cathode at 0 V under AM 1.5G illumination. The evolved gases were determined by gas chromatography (Agilent 7890A GC) using a gastight syringe of 100 μL volume. The faradaic efficiency (FE%) was calculated by:
where n = 2 for H2 evolution and n = 4 for O2 evolution and F is the faradaic constant.
Incident photon-to-electron conversion efficiency (IPCE) measurements were performed in Keithley 2400 SourceMeter in two electrodes cell, using a Xenon lamp (300 W) and a cornerstone monochromator (Newport) and calculated using the relation:
where “
P” is the incident light power measured by a photodiode of known responsivity.
3. Results and discussion
3.1 Evolution of thermal nitridation process in TiO2 NTs
The synthesized TiO2 NTs presented smooth walls, open tops and closed bottoms as observed by SEM and TEM images (Fig. S1†). The average tube length, diameter and wall thickness were 2.1 ± 0.05 μm, 130 ± 10 nm and 12 ± 2 nm, respectively. The NTs were subjected to nitridation at different temperatures and analyzed by SEM (Fig. S2†). For TN-400 and TN-500, no significant morphological changes were observed whereas, for TN-600 and TN-700 NTs, cracks and agglomeration were detected. For TN-800, the NTs completely collapsed forming a solid bulk like structure. HR-TEM analyses were performed to gain further insights on the NTs morphology and microstructure along their length (Fig. 1). For TN-400 and TN-500, a d-spacing of 0.24 nm was found which corresponded to the (103) plane of anatase TiO2 (Fig. 1a). The tube walls of TN-600 and TN-700 were visibly rougher than the TN-400, TN500 and pure TiO2 NT samples (Fig. S1c†), and new crystalline structures appeared. TN-600 presented both TiO2 and TiN, identified by the d-spacings of 0.24 and 0.21 nm attributed to (103) of anatase TiO2 and (200) of TiN, respectively (Fig. 1b). In TN-700, the planes related to TiO2 were no longer observed; instead, a mixture of N rich phases were found, characterized by the (111) plane of TiN and the d-spacing of 0.17 nm attributed to (211) plane for Ti2N (Fig. 1c). These results clearly indicated that the nitridation temperature strongly affects the morphology and microstructure of TiO2 NTs and demonstrated that the anatase crystal structure is thermally stable up to 600 °C. Above this temperature, nitridation results in the formation of nitrogen rich phases, i.e. TiN and Ti2N.
 |
| | Fig. 1 TEM and HRTEM images of (a) and (b) TN-400 (c) and (d) TN-500, (e) and (f) TN-600 and (g) and (h) TN-700; the inset high magnification micrographs represent the selected area of each sample. TN-400 and TN-500 are mainly anatase whilst TN-600 and TN-700 presented the formation of N rich phases along with cracks and agglomerations in tubular structure. | |
During nitridation, the N content is abundant and continuously available and its diffusion, particularly near the inner walls of the NTs, can help us understand the nitridation mechanism for N doping in TiO2 NTs. Therefore, HR-TEM was acquired from the top of TN-600 and TN-700 samples (Fig. 2). In TN-600, the inner wall showed a thin layer (ca. 3 nm thick) with a d-spacing of 0.21 nm corresponding to the (200) plane of TiN. Radiating outwards from the inner walls, the nanotubes presented a d-spacing of 0.2 nm relating to the (200) plane of anatase TiO2. For TN-700, the thickness of TiN almost doubles (ca. 5.8 nm thick) when compared to TN-600 and a d-spacing of 0.13 nm attributed to Ti2N was observed. These analyses clearly suggested that N atoms diffused from the inner to the outer NT walls starting from N doping followed by the formation of TiN and provided important evidence towards elucidating the mechanism of N-doping in TiO2 NTs via the nitridation process.
 |
| | Fig. 2 HRTEM images taken from the top of TN-600 (a) and (b) and TN-700 (c) and (d); insets are the high magnification micrographs representing the selected area of each sample. The TN-600 sample exhibited d-spacings associated with a TiN phase from its innermost tube walls changing to a TiO2 phase for the outer walls, this highlighted that the nitridation process was initiated from the inside of the tube and the nitrogen atoms migrate outwards. The TN-700 sample displays the same characteristics with a higher N content for the internal walls relative to the external walls. | |
3.2 Determination of limiting factors in high N doping of TiO2
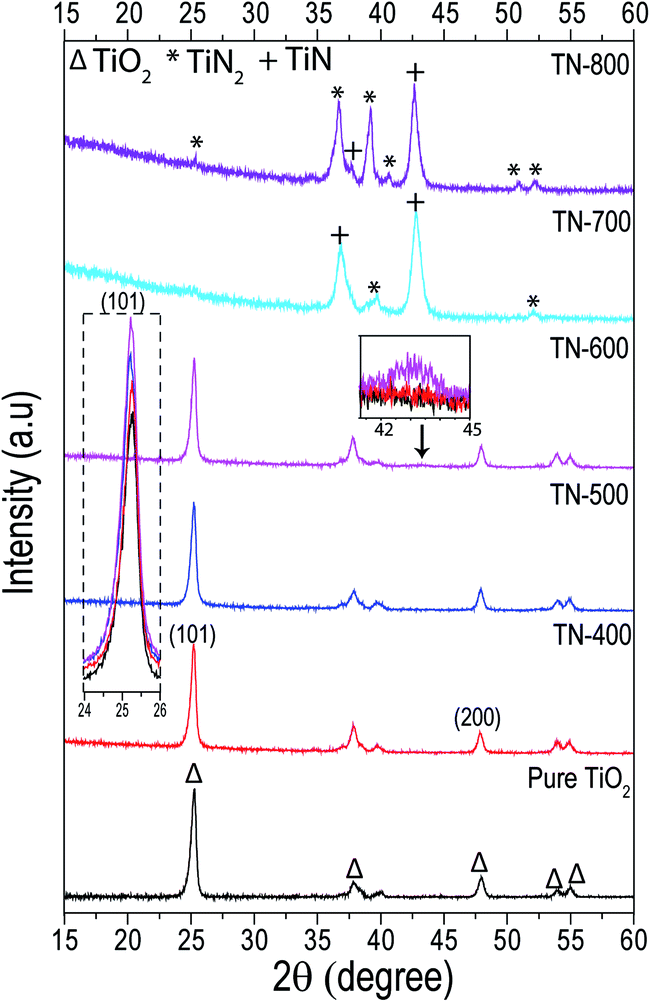
In the PXRD of samples with nitridation temperatures between 400 and 600 °C, the main crystallographic phase was anatase, when compared to pure TiO2, a very small shift in the peak position of the (101) plane towards lower angles was observed (Fig. 3, inset). No significant change in crystallite size was measured between the nitrided samples (ca. 21.5 nm); however, they were all smaller than pure TiO2 (ca. 24 nm). In addition, for fixed nitridation temperature, the ramp rate did not affect the crystal structure (Table S1†). The relative peak intensity ratios of anatase (I(101)/I(200)) in the nitrided samples also showed little deviation and they were all higher compared to the intensity ratio for TiO2 NTs. It is known that N doping increases the dimension of the unit cell of TiO2 as the ionic radii of N is slightly greater than O which can also increase the micro strain. This creates defects at different terminations of the (101) and (200) planes and may contribute to increasing the relative intensity of the anatase peak ratios for the nitrided samples when compared to pure TiO2.19,36,44–46
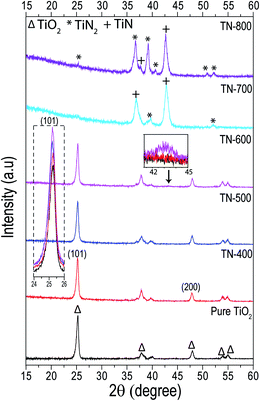 |
| | Fig. 3 Grazing angle XRD patterns of TiO2 nanotubes nitrided at different temperatures for 3 h (flux rate of 100 mL min−1) recorded at a very small grazing angle of 0.5° to remove substrate contribution. The dashed line inset shows the TiO2 (101) plane and is the dominant peak for the pure TiO2 sample as well as the TN-400, TN-500 and TN-600 samples. The solid line inset emphasizes the onset formation of the TiN phase in TN-600 (∼43°). Both TiN and TiN2 feature in the TN-700 and TN-800 samples with a negligible, if any, contribution by TiO2 itself. | |
Furthermore, stacking of the pure TiO2, TN-400, TN-500 and TN-600 diffractograms, shows a small, broad diffraction peak for the TN-600 sample (ca. 2θ ∼ 43°) which corresponded to TiN (inset of Fig. 3 and S3†), corroborating the formation of a thin layer of TiN observed in HRTEM (Fig. 1 and 2). It is well-established that TiO2 NTs undergo a transition from anatase to rutile between 600 and 700 °C in air.47 Interestingly, in this work, the rutile formation is totally inhibited and at 700 °C the anatase phase completely disappears followed by the appearance of TiN and smaller peaks of TiN2 which further intensifies for TN-800. TN-700, which was mainly TiN (semi-metallic in nature), sustained its tubular form. The formation of TiN rather than the solid–solid phase transition to rutile is related to the limited amount of nitrogen that TiO2 can sustain.20 Once N saturates the oxide matrix, the anatase phase transforms directly to N rich phases, such as TiN and/or TiN2. Here, nitridation temperature defined the N doping limit but other techniques, such as magnetron sputtering, follow the same trend.19,20 The Gibbs free energy of formation of TiN is lower than anatase to rutile formation energy and their transformation energy difference is very small.21,48 It is known that anatase cannot support large crystallite sizes and rutile formation requires a contraction of the unit cell, mainly along the c-axis, to make it dense.49 N doping expands the unit cell and harsh nitridation results in a crystallite size that is too large for anatase to support.44 TiN formation which is less negative in Gibbs free energy and is then thermodynamically favorable is the main hurdle to high N concentration doping in TiO2.
3.3 Identifying the pathway to interstitial and substitutional N doping and the effect on PEC properties
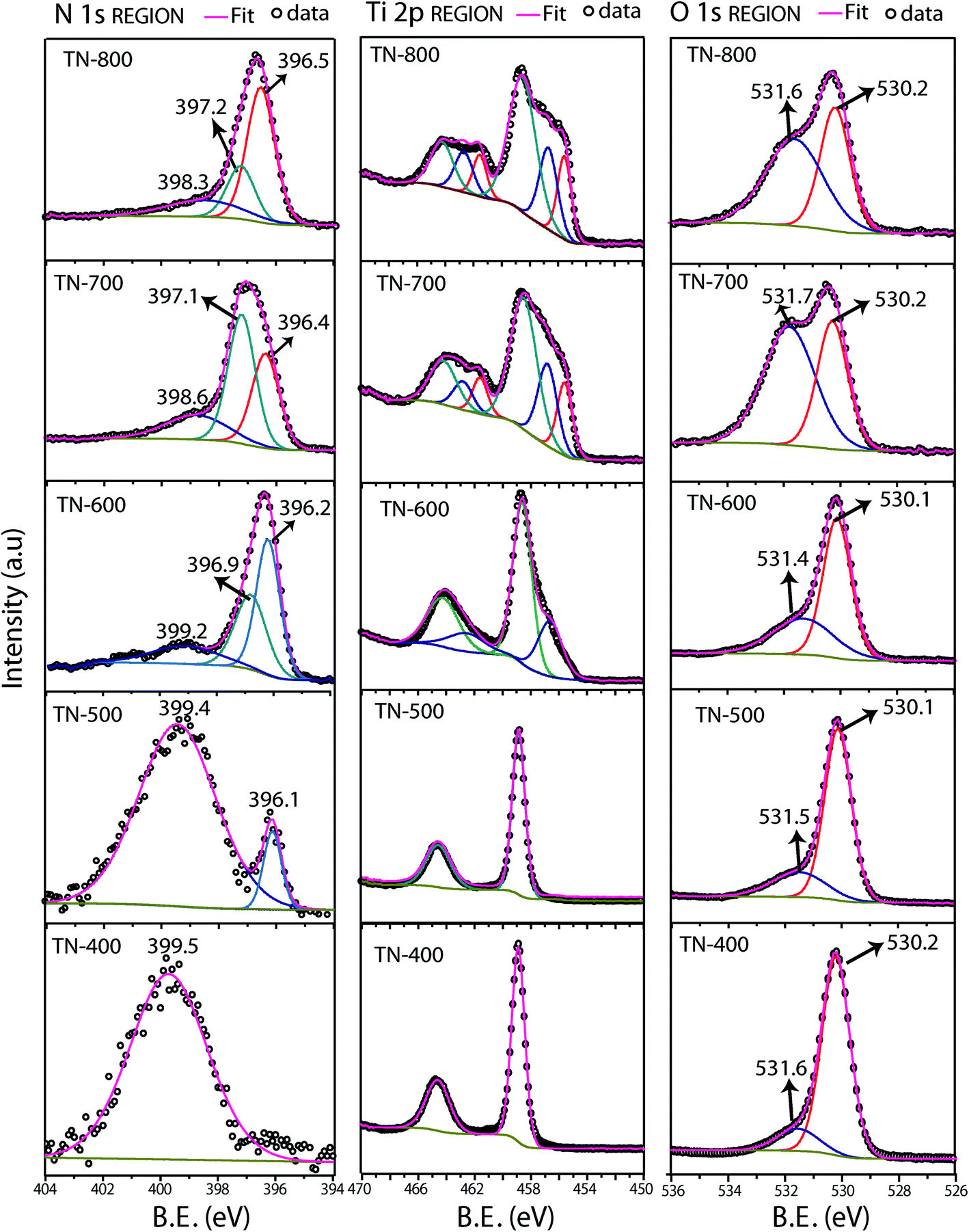
Deconvoluted XPS spectra were performed as a function of different nitridation temperatures to understand the chemical composition of the samples (Fig. 4 and Table S2†). The Ti 2p core level of TN-400 and TN-500 present one doublet corresponding to Ti4+ of TiO2 but, for TN-600, an additional Ti 2p3/2 at 456.5 eV appears corresponding to Ti3+ species.50,51 For TN-700 and TN-800, Ti 2p3/2 peaks at 455.6 eV corresponded to Ti2N.52 Hoang et al. have detected Ti3+ species in N–TiO2via Electron paramagnetic resonance (EPR) spectroscopy.43 These species form donor levels below the CB of TiO2.43,53 TN-400 exhibits a single N 1s peak at the BE of ∼399.5 eV and this peak is also present in TN500 which confirms interstitial doping. The additional N 1s peak at ∼396.1 eV for TN-500 is associated to the substitutional N doping.24,43,51,54 The N 1s spectra of TN-600 was much different than TN-400 and TN-500, as an additional peak at 396.9 eV was present due to the formation of TiN (also seen in XRD and HRTEM). Additionally, the peak related to N substitution increased in intensity, compared to the interstitial peak, demonstrating higher N doping for TN-600 when compared with TN-500. These results also demonstrated that the interstitial doping was favorable at relatively low temperatures, such as 400 °C. Further increasing the temperature results in a mixture of both types of doping followed by a dominant substitutional N-doping in TN-600. For TN-700 and TN-800, the peaks at ca. 396.3 and 397.1 eV correspond to TiN and Ti2N, respectively, with their respective satellite peaks appearing at ca. 398.3 eV.52,55 The N 1s spectra were acquired from multiple areas of the same sample to ensure the reproducibility (Fig. S4†). In the O 1s spectra, the peak at 530.1 eV, from O in the TiO2 anatase lattice, and at 531.5 eV, corresponding to surface adsorbed hydroxide species, were observed for all samples. As the nitridation temperature increased, the hydroxide content also increased which was attributed to larger surface defects. EDS elemental mapping of TN-700 showed that the top of the tubes were shown to be largely populated by Ti atoms, i.e. N/O atoms were removed from the top surface due to harsh reducing ability of NH3 at higher nitridation temperatures (Fig. S5†).
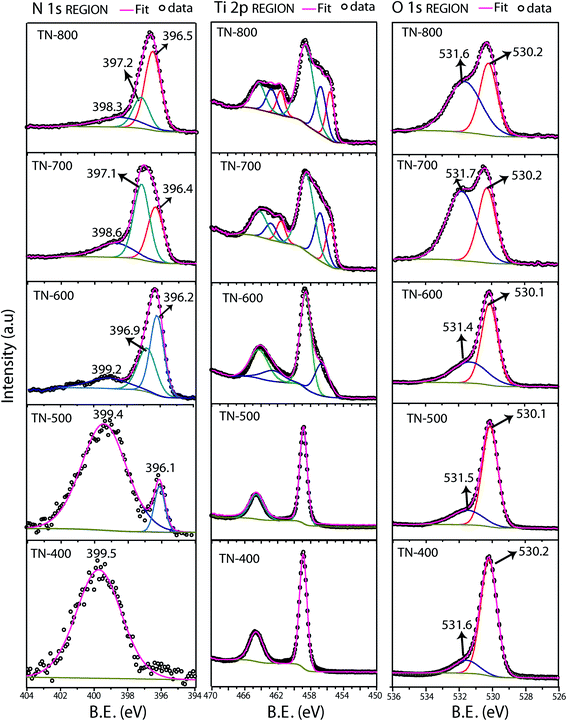 |
| | Fig. 4 Deconvoluted high resolution XPS spectra of N doped TiO2 of N 2p, Ti 2p and O 1s regions. TN-400 exhibited interstitial doping and substitutional doping started from 500 °C and intensified for TN-600. Increase in nitridation temperature (starting from 600 °C) resulted in the formation of N rich phases. | |
UV-Vis diffused reflectance spectroscopy measurements were performed for all the samples to investigate the implications of nitridation on the NTs optical properties (Fig. S6, bandgaps are displayed in the Table S1†). Pure TiO2, TN-400, TN-500 and TN-600 showed similar bandgaps (ca. 3.3 eV), as the anatase structure was the main phase present in these samples. However, lower energy bands were observed for TN-400, TN-500 and TN-600, which were related to N doping of TiO2 and formation of reduced Ti species such as Ti3+.32,38,51 For TN-700 and TN-800 a much larger absorption peak appears at lower energies, which is clearly related to the complete formation of titanium nitride species.56 To compare the optical absorption of the prepared TiO2 nanotubes, commercially available P25 was nitrided at different temperatures. The UV-Vis diffused reflectance spectra of these samples are displayed in Fig. S7.† Compared to as-received P25, the nitrided P25 did not show any significant difference in absorption edge (bandgap: ca. 3.3 eV) and low intensity absorption modes were observed within the visible range, which was the same behavior observed for TiO2 nanotubes (Fig. S6†). In line with the literature, the absorption transitions below the bandgap energy were also observed (inset Fig. S7†) which correspond to Ti3+ species.28 These results suggested the nitridation process has the same influence on the optical properties of TiO2, whether it is a film or powder.
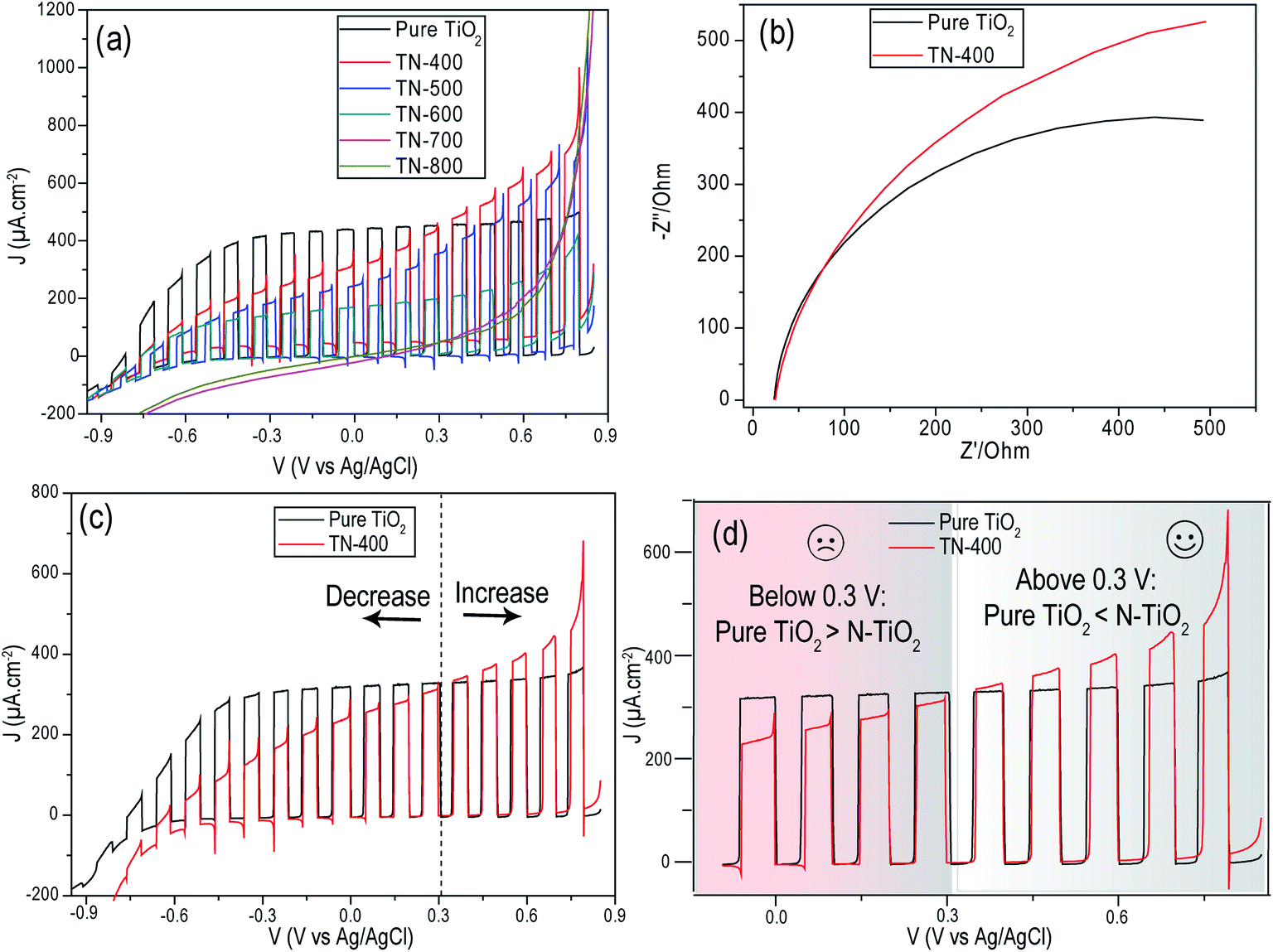
The LSV chopped curves (under AM 1.5G, 1 Sun illumination) were compared and indicated that pure TiO2 presents an improved performance and better fill factor (exhibiting lower resistance) when compared to nitrided samples (Fig. 5 and S8†). Particularly, at lower applied potentials, the decrease in the photocurrent from N doped samples is noteworthy; however, at higher positive potentials, N doping presents improved photocurrents. This can be understood from their better light absorption, as well as the larger band bending at the interface due to a higher electric field that prevents charge recombination and improves hole migration to the electrode surface.39,40 In addition, transient photocurrent overshoots, that are both anodic and cathodic, are observed for N doped samples, indicative of charge recombination.35,39,40 Pure TiO2 does not show these transient overshoots at any applied potential. Upon increasing nitridation temperature, the photocurrent kept decreasing up to 600 °C. Samples TN-400 and TN-500, with larger interstitial doping, were far better than TN-600 which exhibited substitutional doping. Interestingly, the absence of a photoresponse from the TN-700 and TN-800 samples indicated they had lost their semiconducting properties altogether.
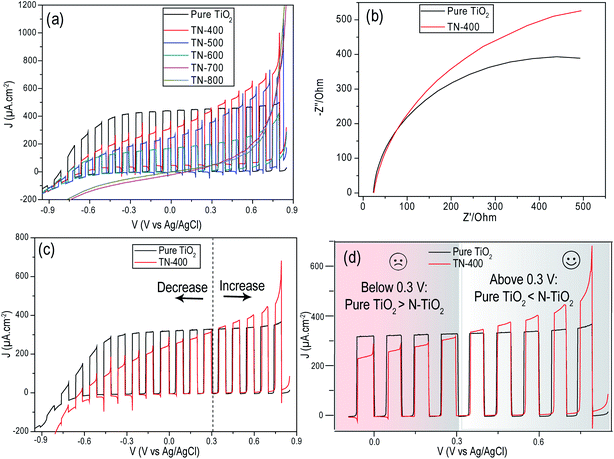 |
| | Fig. 5 (a) Chopped LSV curves of N doped TiO2 prepared at variable nitridation temperatures and (b) Nyquist plots of pure TiO2 and TN-400. The measurements were performed in Na2S (0.24 M) and Na2SO3 (0.35 M). (c) Chopped LSV curves of pure TiO2 and TN-400 in KOH (1 M) with a cutoff voltage of (∼0.3 V) and (d) in the inset selected potential windows from (c) are highlighted demonstrating improved photocurrent from TN-400 at higher positive potentials as compared to pure TiO2 and dominancy of pure TiO2 at lower positive potentials. | |
The purely interstitially doped TN-400 sample was selected to study the interfacial charge transfer resistance as it possessed the best photocurrent of the nitrided samples (Fig. 5). The larger arc diameter of TN-400 in the Nyquist plots (Fig. 5b), relative to pure TiO2, indicated that TN-400 suffers from higher charge transfer resistance, i.e. N-doping negatively influences the charge transportation properties of TiO2. Using an efficient hole scavenging redox couple in the electrolyte should give better hole transfer characteristics at the solid–liquid interface; however, the interfacial resistance was still remarkably higher for TN-400. This suggested that the formed energetic hole trapping states are filled by photogenerated holes before being scavenged via redox species. For instance, in conventional electrolytes for overall water splitting, recombination of charge carriers and hole transportation appears to be two competitive processes and the reaction remains slow. Therefore, one should expect that the presence of Na2S/Na2SO3 in the electrolyte may help in improving hole scavenging and lead to a better photocurrent; however, we observed the opposite, particularly at lower potentials (<0.3 V vs. Ag/AgCl). Thus, in comparison to pure TiO2, the main issue in N doping of TiO2 is the higher charge transfer resistance related to N related crystal defects in TiO2 which increase with nitridation temperature. In addition, nitridation also results in the formation of N rich phases (semi-metallic) that may act as electron capture sites.
For comparison, chopped LSV curves were performed in KOH (1 M) for both pure TiO2 and TN-400 (Fig. 5d). The overall photocurrent clearly decreased compared to the hole scavenging solution experiments (Fig. 5a). An improved photocurrent was obtained from N doped samples at higher potentials (>0.3 V vs. Ag/AgCl) but pure TiO2 was still superior at lower potentials (<0.3 V vs. Ag/AgCl). There are two phenomena that might impede the enhanced performance: (1) interband states that trap the photogenerated holes and require higher biasing potential for improved hole transfer; and, (2) the photogenerated electron capture by the donor states (instead of their migration to the back contact). The improvement at higher potentials for N doped samples was attributed to better visible light absorption as, at higher external biasing, the band bending is energetic enough to decrease hole trapping and aid hole transfer to oxidize the species in the interfacial vicinity for overall water splitting:
| KOH + H2O → K(aq)+ + OH(aq)− |
| TiO2 + 4hν → 4hVB+ + 4eCB− |
| 4OH− + hVB+ → 2H2O + O2 (photoanode surface) |
| 4H2O + 4eCB− → 4OH− + 2H2 (cathode surface) |
Thus, depending on the choice of applied external potential, one can choose whether N doped TiO2 presents improved performance or not. Clearly, for better performance, one needs higher positive potentials; however, their application will compromise solar based energy conversion. Since TN-400 has presented better PEC performance, compared to all other nitrided samples, it was subjected to a stability test and determination of the faradaic efficiency for water oxidation (1 M KOH). The evolved gases from the photoanode compartment were quantified by GC. Within the first 5 min, the photocurrent dropped from its maximum value of ∼180 μA to ∼50 μA, after 3 h it dropped to ∼20 μA and finally at the end, after 24 h, the photocurrent was 10 μA (Fig. S9†). The observed gaseous products during the chronoamperometry were O2 and N2; where the former kept increasing with time and the latter mainly increased for the first 3 h. The experimental O2 evolution was lower than the expected (inset of Fig. S9†); particularly for first 3 h of the photoreaction which was related to the self-oxidation of TN-400 rationalized by the appearance of N2 in GC. The effect of this could be clearly seen in the faradaic efficiency which decreased from ∼62% to ∼30% in the first 3 h. Upon increasing the reaction time, the photocurrent kept decreasing; though with a slower rate as compared to its initial decay; resulting in an increase of the faradaic efficiency, yet far from 100%. These results clearly indicated TN-400 was not stable against photocorrosion for water oxidation. Therefore, we performed gas quantifications and stability tests in the presence of sacrificial reagent (Na2S/Na2SO3) for an irradiation interval of 3 h (Fig. S10a†) and compared against pure TiO2. The photoactivity of pure TiO2 was superior and TN-400 presented an initial photocurrent overshoot (Fig. S10b†) which was not observed for pure TiO2, agreeing with the LSV curves (Fig. 5a and S8†). In the photoanode compartment, no gas evolution was observed (O2/N2) which clearly demonstrated that the photoanode self-oxidation was suppressed and photogenerated holes were efficiently scavenged by S2−/SO32− species according to the following reactions:57
| TiO2 + 2hν → 2hVB+ + 2eCB− |
| SO32− + 2hVB+ + 2OH− → SO42− + H2O |
| SO32− + S2− + 2hVB+ → S2O32− |
In the counter electrode (Pt) compartment H2 gas was detected, however, other reduction reactions may also occur such as:41,57
| S2O32− + 2eCB− → S2− + SO32− |
I–t curves (Fig. S10a†) clearly showed the photoelectrodes were mainly stable. Thus, the presence of Na2S/Na2SO3 in the electrolyte helped suppress the photocorrosion due to efficient hole scavenging. The IPCE spectra of these samples are compared in Fig. S11.† The spectral profiles agree with the literature.36,58,59 TN-400 presented a clear photo-response in the visible region (inset of Fig. S11†); however, the IPCE dropped in the UV-range compared to pure TiO2. This showed that N doping induced interband states that act as recombination centers and can trap photogenerated holes generated by UV light, thus affecting the overall PEC performance of N doped TiO2.59
3.4 Measuring semiconductor bandgap energy and identification of interstitial and substitutional N doping via simple PEC measurement
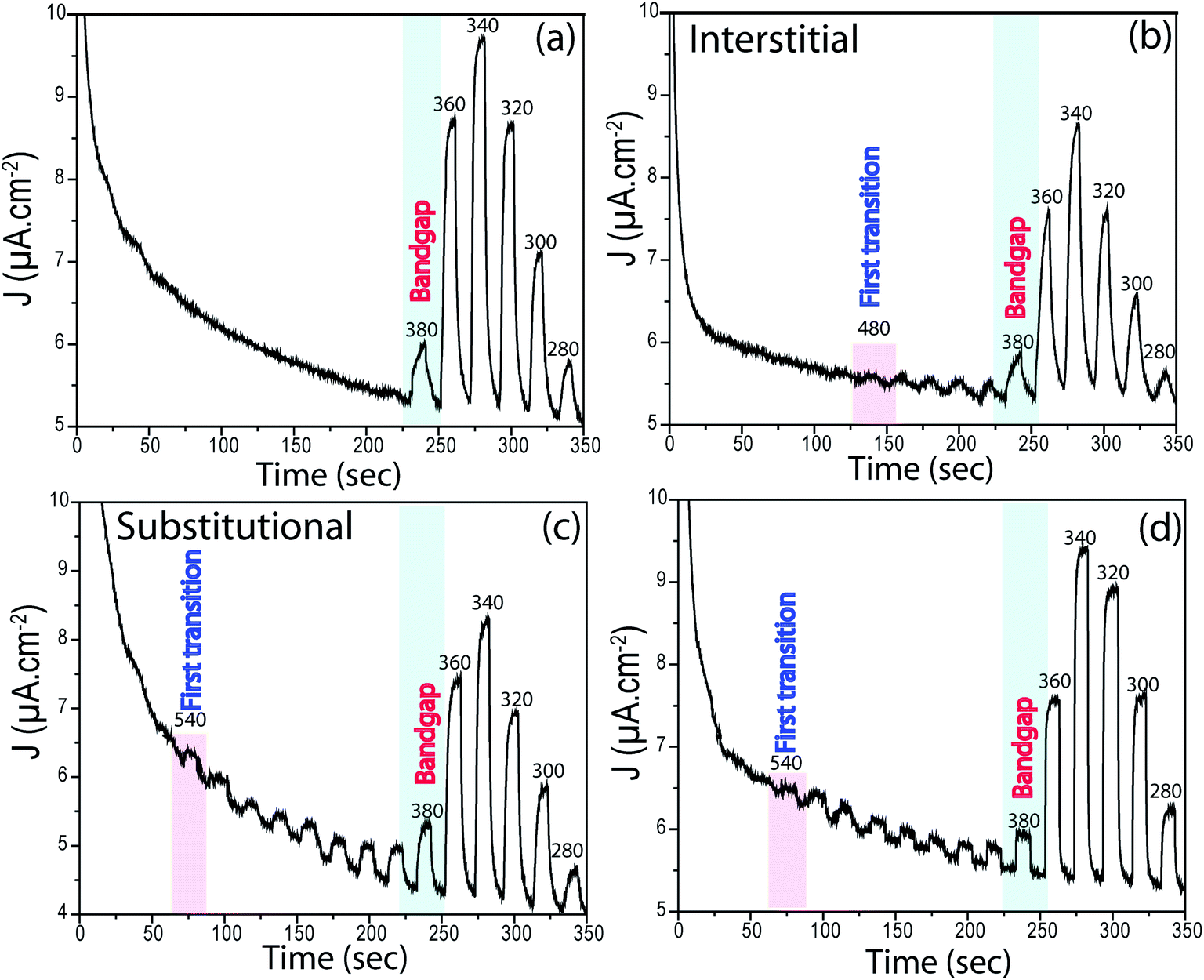
The position of interband states that are created due to N doping was verified by novel PEC measurements performed in chronoamperometry mode. The samples were left in the dark and under illumination for 10 s chopping for each, and the wavelength was decreased from 600 nm to 280 nm in 20 nm increments. As expected, no photocurrent was observed for pure TiO2 in the visible light region. The first photocurrent occurred at ∼380 nm (∼3.3 eV) which shows that these photons are energetic enough to excite electrons from the VB to CB of TiO2, the bandgap energy of TiO2. Indeed, this is a simple, yet novel, approach to identify bandgaps of semiconductors if linear regression in UV-Vis is unfeasible.
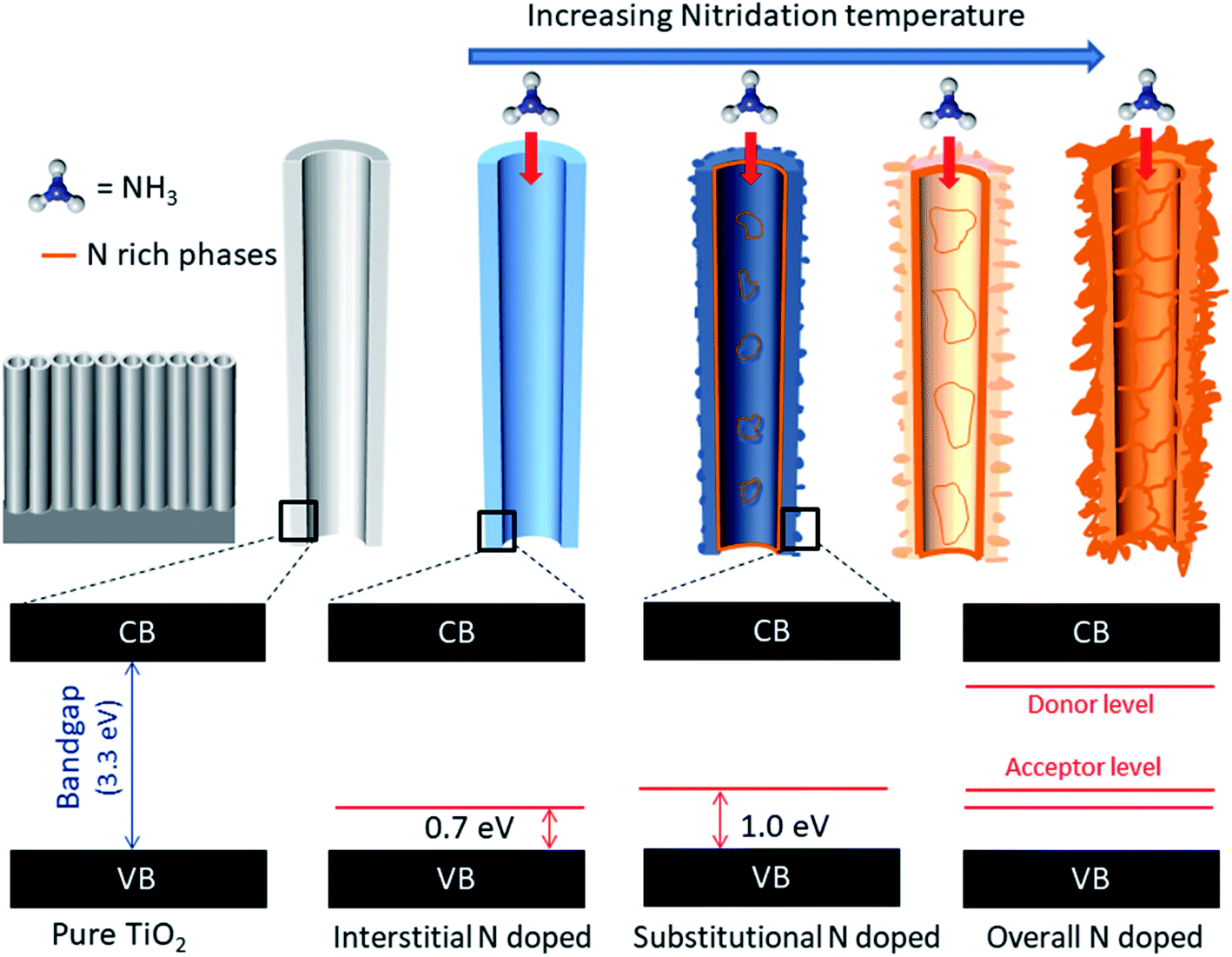
All N-doped TiO2 samples presented photocurrents at higher wavelengths. For TN-400, the first transition occurred at ∼480 nm (∼2.6 eV), whereas for TN-500 it occurred at ∼540 nm (∼2.3 eV). From XPS analyses, TN-400 exhibited interstitial doping, TN-500 had both interstitial and substitutional doping, and TN-600 was substitutional doped (Fig. 4). These results directly confirmed that visible light activation of TiO2 was due to N-doping. In the UV-Vis spectra (Fig. S6†), the bandgap remained mainly unchanged for N-doped TiO2 (Table S1†). This suggests that the visible light activation of TiO2 is based on some localized states in the bandgap. Batzill et al. and Irie et al. suggested that in N doped TiO2 there is a formation of interband states above the VB.27,28 The first transition for interstitial and substitutional doping started from ca. 480 nm (∼2.6 eV) and 540 nm (∼2.3 eV), respectively. These results suggested that the related energy levels for N interstitial and N substitutional doped TiO2 are located at ca. ∼0.7 and ∼1.0 eV above the VB of TiO2 (bandgap energy: 3.3 eV), respectively. Despite absorbing visible light, the photocurrent of N-doped TiO2 nanotubes under polychromatic illumination was less than that of pure TiO2 (at potentials <0.3 V vs. Ag/AgCl). Furthermore, electrochemical impedance spectroscopy showed higher charge transfer resistance for N-doped TiO2 (Fig. 5). N doped TiO2 exhibits N3− 2p character, instead of O2− 2p for pure TiO2, and N 2p is potentially less positive; thereby, slower PEC oxidation kinetics can be expected.36 Furthermore, if N3− replaces O2− the charge balance will necessitate the removal of three O ions from the TiO2 lattice to replace them with two N ions.24,27 Thus, TiO2 will consist of O vacancies and reduced species (e.g. Ti3+ (Fig. 4)) which form donor states below the CB of TiO2.53 Therefore, the overall effect of N doping in the electronic structure of TiO2 is to generate donor levels (due to the O vacancies and Ti3+ species) below CB and acceptor states above the VB (due to N substitutional/interstitial defects). Finally, these levels improve the visible light absorption of TiO2 but they are also responsible for overall photocurrent decreases.
Fig. 7 represents a holistic view of N doped TiO2 formation mechanism and the effect of interstitial/substitutional N doping on the electronic structure of TiO2, by correlating the structural-electronic characterization (Fig. 1–4) with the novel electrochemical characterization proposed in this work (Fig. 5 and 6).
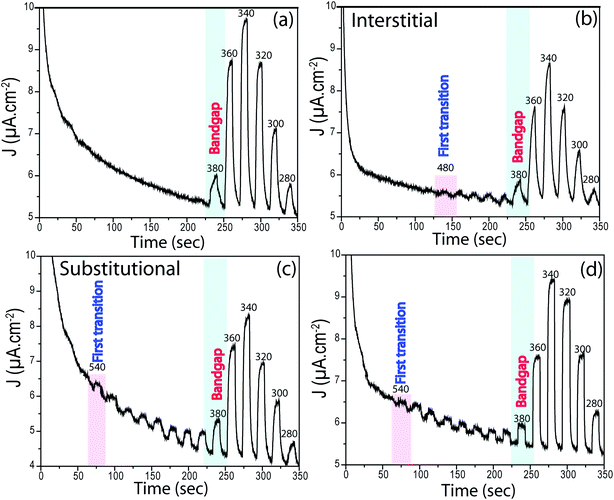 |
| | Fig. 6 Chronoamperometry curves from (a) pure TiO2, (b) TN-400, (c) TN-500 and (d) TN-600 under chopping monochromated light of different wavelengths varied at a regular (light/dark) interval of 10 s at wavelength increment of 20 nm. The first transition, bandgap excitations and some selected wavelengths are given in the inset. The measurements were performed in Na2S (0.24 M) and Na2SO3 (0.35 M). | |
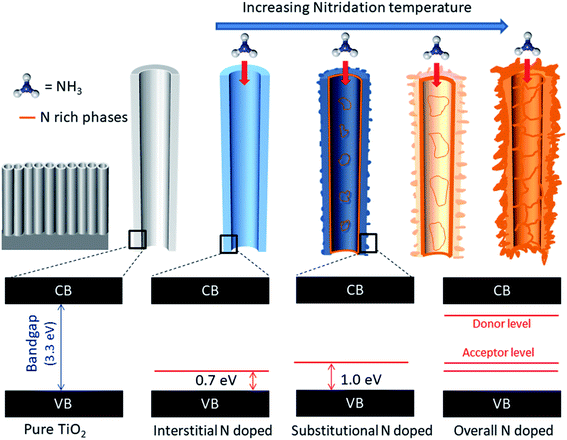 |
| | Fig. 7 Schematic representation of N doping via increasing nitridation temperature and respective bandgaps and the presence of interband states related to interstitial and substitutional doping. | |
4. Conclusion
TiO2 NTs were successfully doped interstitially/substitutionally with N via a nitridation process. N doping into the anatase TiO2 matrix proceeded from interstitial to substitutional doping with increasing nitridation temperature. Once the thermodynamic conditions to form nitrogen rich phases were satisfied, the anatase phase directly transformed to TiN and Ti2N. The lower Gibbs free energy of these phases, and anatase unit cell expansion, prevented higher concentrations of N doping in the TiO2. HR-TEM confirmed that the formation of TiN starts from the inner walls and radiates outward with increasing nitridation temperature. The process generated O vacancies/Ti3+ species and N substitutional/interstitial defects that create donor states below the CB and acceptor states above the VB, respectively. The chronoamperometry measurements, under monochromatic light, presented direct experimental evidence of visible light activation of N doped TiO2. The interband states between 0.7 and 1.0 eV above the VB of TiO2 are responsible for the visible light transitions. This simple and novel measurement can be employed to find the bandgap of semiconductors and can also be used as an evidence to distinguish interstitial and substitutional doping. PEC measurements showed that the photocurrent of N doped TiO2 samples were worse than the pure TiO2 at lower applied potentials (<0.3 V vs. Ag/AgCl) this was attributed to the structural defects that result in increasing charge transfer resistance. Hole scavenging electrolyte (Na2S/Na2SO3) remained ineffective to avoid hole trapping at lower applied potentials. PEC response, which is a characteristic of a semiconductor, was totally lost when the TiN and Ti2N crystal phases were formed as they are semi-metallic. In photostability tests for water oxidation (1 M KOH), N doped TiO2 suffered a quick self-oxidation. However, in Na2S/Na2SO3, the N doped TiO2 nanotubes have shown stable PEC performance. Thus, the sacrificial reagent efficiently scavenged the photogenerated holes which suppressed self-oxidation of the photoelectrode. We conclude that N doping of TiO2 is ineffective for PEC applications as the gain in visible range absorption does not counteract the losses in trapping states and recombination centers.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for financial support from the following Brazilian agencies: CNPq (Process: 424769/2018-2, 309454/2018-2; 307723/2018-6), CAPES-PRINT, (Call: 001/2019-PROPG, UFRGS), FAPERGS (Process: 19/2551-0002287-5) H. Coelho-Júnior and R. L. Sommer acknowledge Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) for financial support. JAF would like to thank the University of Nottingham Beacons of Excellence: Propulsion Futures and EPSRC: LiPPS XPS system, and EP/K005138/1 “University of Nottingham Equipment Account” for providing financial support for this work and the nanoscale and microscale research centre (University of Nottingham, UK) for access to XPS facilities.
References
- J. Bai and B. Zhou, Chem. Rev., 2014, 114, 10131–10176 CrossRef CAS PubMed.
- Q. Guo, C. Zhou, Z. Ma and X. Yang, Adv. Mater., 2019, 31, 1–26 Search PubMed.
- M. Viticoli, A. Curulli, A. Cusma, S. Kaciulis, S. Nunziante, L. Pandolfi, F. Valentini and G. Padeletti, Mater. Sci. Eng., C, 2006, 26, 947–951 CrossRef CAS.
- J. Wang, G. Xu, X. Zhang, J. Lv, X. Zhang, Z. Zheng and Y. Wu, Dalton Trans., 2015, 44, 7662–7672 RSC.
- N. Mintcheva, P. Srinivasan, J. B. B. Rayappan, A. A. Kuchmizhak, S. Gurbatov and S. A. Kulinich, Appl. Surf. Sci., 2020, 507, 145169 CrossRef CAS.
- Z. Zhang, C. Zhao, Y. Duan, C. Wang, Z. Zhao, H. Wang and Y. Gao, Appl. Surf. Sci., 2020, 527, 146693 CrossRef CAS.
- Y. C. Nah, I. Paramasivam and P. Schmuki, ChemPhysChem, 2010, 11, 2698–2713 CrossRef CAS PubMed.
- L. Wei, C. Yu, Q. Zhang, H. Liu and Y. Wang, J. Mater. Chem. A, 2018, 6, 22411–22436 RSC.
- J. A. Fernandes, S. Khan, F. Baum, E. C. Kohlrausch, J. A. Lucena Dos Santos, D. L. Baptista, S. R. Teixeira, J. Dupont and M. J. L. Santos, Dalton Trans., 2016, 45, 9925–9931 RSC.
- J. A. Fernandes, P. Migowski, Z. Fabrim, A. F. Feil, G. Rosa, S. Khan, G. J. Machado, P. F. P. Fichtner, S. R. Teixeira, M. J. L. Santos and J. Dupont, Phys. Chem. Chem. Phys., 2014, 16, 9148–9153 RSC.
- S. K. Gharaei, M. Abbasnejad and R. Maezono, Sci. Rep., 2018, 8, 1–10 Search PubMed.
- D. D. Qin, Q. H. Wang, J. Chen, C. H. He, Y. Li, C. H. Wang, J. J. Quan, C. L. Tao and X. Q. Lu, Sustainable Energy Fuels, 2017, 1, 248–253 RSC.
- I. M. Ulfah, B. M. Bachtiar, A. R. Murnandityas and Slamet, AIP Conf. Proc., 1964, 2018, 020008 Search PubMed.
- C. Das, P. Roy, M. Yang, H. Jha and P. Schmuki, Nanoscale, 2011, 3, 3094–3096 RSC.
- X. Gao, B. Zhou and R. Yuan, Environ. Eng. Res., 2015, 20, 329–335 CrossRef.
- M. Nishida, J. Appl. Phys., 1980, 51, 1669–1675 CrossRef CAS.
- S. A. Ansari, M. M. Khan, M. O. Ansari and M. H. Cho, New J. Chem., 2016, 40, 3000–3009 RSC.
- C. Di Valentin, E. Finazzi, G. Pacchioni, A. Selloni, S. Livraghi, M. C. Paganini and E. Giamello, Chem. Phys., 2007, 339, 44–56 CrossRef CAS.
- M. Kitano, K. Funatsu, M. Matsuoka, M. Ueshima and M. Anpo, J. Phys. Chem. B, 2006, 110, 25266–25272 CrossRef CAS PubMed.
- D. A. Duarte and M. Massi, Mater. Res., 2017, 20, 549–554 CrossRef CAS.
- I. Seki and S. I. Yamaura, Mater. Trans., 2017, 58, 361–366 CrossRef CAS.
- H. Chen, A. Nambu, W. Wen, J. Graciani, Z. Zhong, J. C. Hanson, E. Fujita and J. A. Rodriguez, J. Phys. Chem. C, 2007, 111, 1366–1372 CrossRef CAS.
- E. C. Kohlrausch, M. J. M. Zapata, R. V. Gonçalves, S. Khan, M. D. O. Vaz, J. Dupont, S. R. Teixeira and M. J. Leite Santos, RSC Adv., 2015, 5, 101276–101286 RSC.
- R. Ramos, D. Scoca, R. Borges Merlo, F. Chagas Marques, F. Alvarez and L. F. Zagonel, Appl. Surf. Sci., 2018, 443, 619–627 CrossRef CAS.
- G. R. Torres, T. Lindgren, J. Lu, C. G. Granqvist and S. E. Lindquist, J. Phys. Chem. B, 2004, 108, 5995–6003 CrossRef CAS.
- J. Li and N. Wu, Catal. Sci. Technol., 2015, 5, 1360–1384 RSC.
- M. Batzill, E. H. Morales and U. Diebold, Phys. Rev. Lett., 2006, 96, 1–4 CrossRef PubMed.
- H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483–5486 CrossRef CAS.
- T. H. Kim, G. M. Go, H. B. Cho, Y. Song, C. G. Lee and Y. H. Choa, Front. Chem., 2018, 6, 1–10 CrossRef.
- Q. Xiang, J. Yu, W. Wang and M. Jaroniec, Chem. Commun., 2011, 47, 6906–6908 RSC.
- J. Gomes, J. Lincho, E. Domingues, R. M. Quinta-Ferreira and R. C. Martins, Water, 2019, 11, 373 CrossRef CAS.
- E. Martínez-Ferrero, Y. Sakatani, C. Boissière, D. Grosso, A. Fuertes, J. Fraxedas and C. Sanchez, Adv. Funct. Mater., 2007, 17, 3348–3354 CrossRef.
- Y. K. Lai, J. Y. Huang, H. F. Zhang, V. P. Subramaniam, Y. X. Tang, D. G. Gong, L. Sundar, L. Sun, Z. Chen and C. J. Lin, J. Hazard. Mater., 2010, 184, 855–863 CrossRef CAS PubMed.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, Energy Environ. Sci., 2012, 5, 7626–7636 RSC.
- S. Khan, M. J. L. Santos, C. F. Malfatti, J. Dupont and S. R. Teixeira, Chem.–Eur. J., 2016, 22, 18501–18511 CrossRef CAS PubMed.
- S. Hoang, S. Guo, N. T. Hahn, A. J. Bard and C. B. Mullins, Nano Lett., 2012, 12, 26–32 CrossRef CAS PubMed.
- T. Cottineau, N. Béalu, P. A. Gross, S. N. Pronkin, N. Keller, E. R. Savinova and V. Keller, J. Mater. Chem. A, 2013, 1, 2151–2160 RSC.
- J. Xu, Y. Ao, M. Chen and D. Fu, Appl. Surf. Sci., 2010, 256, 4397–4401 CrossRef CAS.
- D. Klotz, D. A. Grave and A. Rothschild, Phys. Chem. Chem. Phys., 2017, 19, 20383–20392 RSC.
- H. Dotan, K. Sivula, M. Grätzel, A. Rothschild and S. C. Warren, Energy Environ. Sci., 2011, 4, 958–964 RSC.
- P. Gomathisankar, K. Hachisuka, H. Katsumata, T. Suzuki, K. Funasaka and S. Kaneco, Int. J. Hydrogen Energy, 2013, 38, 8625–8630 CrossRef CAS.
- R. P. Lynch, A. Ghicov and P. Schmuki, J. Electrochem. Soc., 2010, 157, G76 CrossRef CAS.
- S. Hoang, S. P. Berglund, N. T. Hahn, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2012, 134, 3659–3662 CrossRef CAS.
- T. Xu, M. Wang and T. Wang, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2019, 34, 55–63 CrossRef CAS.
- L. Huang, K. Zha, S. Namuangruk, A. Junkaew, X. Zhao, H. Li, L. Shi and D. Zhang, Catal. Sci. Technol., 2016, 6, 8516–8524 RSC.
- N. Shibata, A. Goto, S.-Y. Choi, T. Mizoguchi, S. D. Findlay, T. Yamamoto and Y. Ikuhara, Science, 2008, 322, 570–573 CrossRef CAS PubMed.
- J. A. Fernandes, E. C. Kohlrausch, S. Khan, R. C. Brito, G. J. Machado, S. R. Teixeira, J. Dupont and M. J. Leite Santos, J. Solid State Chem., 2017, 251, 217–223 CrossRef CAS.
- S. J. Smith, R. Stevens, S. Liu, G. Li, A. Navrotsky, J. Boerio-Goates and B. F. Woodfield, Am. Mineral., 2009, 94, 236–243 CrossRef CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- B. Tian, W. Yue, C. Wang and J. Liu, Appl. Surf. Sci., 2015, 353, 1156–1163 CrossRef CAS.
- S. Khan, M. J. M. Zapata, D. L. Baptista, R. V. Gonçalves, J. A. Fernandes, J. Dupont, M. J. L. Santos and S. R. Teixeira, J. Phys. Chem. C, 2015, 119, 19906–19914 CrossRef CAS.
- B. Soundiraraju and B. K. George, ACS Nano, 2017, 11, 8892–8900 CrossRef CAS PubMed.
- B. Bharti, S. Kumar, H. N. Lee and R. Kumar, Sci. Rep., 2016, 6, 1–12 CrossRef.
- J. Lynch, C. Giannini, J. K. Cooper, A. Loiudice, I. D. Sharp and R. Buonsanti, J. Phys. Chem. C, 2015, 119, 7443–7452 CrossRef CAS.
- X. Peng, K. Huo, J. Fu, X. Zhang, B. Gao and P. K. Chu, Chem. Commun., 2013, 49, 10172–10174 RSC.
- M. M. Rahman, Z. T. Jiang, P. Munroe, L. S. Chuah, Z. F. Zhou, Z. Xie, C. Y. Yin, K. Ibrahim, A. Amri, H. Kabir, M. M. Haque, N. Mondinos, M. Altarawneh and B. Z. Dlugogorski, RSC Adv., 2016, 6, 36373–36383 RSC.
- C. Ng, J. H. Yun, H. L. Tan, H. Wu, R. Amal and Y. H. Ng, Sci. China Mater., 2018, 61, 895–904 CrossRef CAS.
- H. Sopha, M. Krbal, S. Ng, J. Prikryl, R. Zazpe, F. K. Yam and J. M. Macak, Applied Materials Today, 2017, 9, 104–110 CrossRef.
- S. J. Kim, K. Xu, H. Parala, R. Beranek, M. Bledowski, K. Sliozberg, H. W. Becker, D. Rogalla, D. Barreca, C. Maccato, C. Sada, W. Schuhmann, R. A. Fischer and A. Devi, Chem. Vap. Deposition, 2013, 19, 45–52 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta11494d |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Thais
Lemes Ruwer
b,
Niqab
Khan
a,
Ariadne
Köche
*a,
Thais
Lemes Ruwer
b,
Niqab
Khan
a,
Ariadne
Köche