
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling the lithium proton exchange of LLZO to enable reproducible processing and performance optimization†
Melanie
Rosen
 ab,
Ruijie
Ye
ac,
Markus
Mann
a,
Sandra
Lobe
a,
Martin
Finsterbusch
*ad,
Olivier
Guillon
ad and
Dina
Fattakhova-Rohlfing
abd
ab,
Ruijie
Ye
ac,
Markus
Mann
a,
Sandra
Lobe
a,
Martin
Finsterbusch
*ad,
Olivier
Guillon
ad and
Dina
Fattakhova-Rohlfing
abd
aInstitute of Energy and Climate Research – Materials Synthesis and Processing (IEK-1), Forschungszentrum Jülich GmbH, 52425 Jülich, Germany. E-mail: m.finsterbusch@fz-juelich.de
bFaculty of Engineering, Center for Nanointegration Duisburg-Essen, Universität Duisburg-Essen, Lotharstr. 1, 47057 Duisburg, Germany
cInstitute for Power Electronics and Electrical Drives (ISEA), RWTH Aachen University, Jägerstr. 17-19, 52066 Aachen, Germany
dHelmholtz Institute Münster: Ionics in Energy Storage (IEK-12), Forschungszentrum Jülich GmbH, Corrensstr. 46, 48149 Münster, Germany
First published on 22nd January 2021
Abstract
Ceramic solid state-electrolytes attract significant attention due to their intrinsic safety and, in the case of the garnet type Li6.45Al0.05La3Zr1.6Ta0.4O12 (LLZO), the possibility to use Li-metal anodes to provide high energy densities on a cell and battery level. However, one of the major obstacles hindering their wide-spread application is the translation and optimization of production processes from laboratory to industrial scale. Even though the plausibility of manufacturing components and cells via wet processing routes like tape casting and screen printing has been shown, the impact of the sensitivity of LLZO to air and protic solvents due to Li+/H+-exchange is not fully understood yet. An uncontrolled alteration of the powder surface results in poorly reproducible processing characteristics and electrochemical performance of the final battery components and full cells. This knowledge gap is the cause of the large performance variations reported across different research labs worldwide and is unacceptable for up-scaling to industrial level. To close this gap, the influence of the Li+/H+-exchange taking place at various steps in the manufacturing process was systematically investigated in this study. For the first time, this allowed a mechanistic understanding of its impact on the processability itself and on the resulting electrochemical performance of a free-standing LLZO separator. The importance of a close control of the pre-treatment and storage conditions of LLZO, as well as contact time with the solvent could be extracted for each step of the manufacturing process. As a result, we were able to optimize the processing of thin, dense, free standing LLZO separators and significantly improve the total Li-ion conductivity to 3.90 × 10−4 S cm−1 and the critical current density to over 300 μA cm−2 without making structural changes to separator or the starting material. These findings do not only enable a deeper understanding and control over the manufacturing process, but also show potential for further improvement of cell concepts already existing in literature.
Introduction
Since its discovery in 2007,1 the garnet-type solid Li-ion conductor LLZO attracts significant attention due to a unique combination of properties that set it apart from other solid electrolytes, namely its relatively high Li-ion conductivity combined with the stability against metallic lithium.2 This combination allows for LLZO to not only be used as the single electrolyte in full cells,3 but also enables its use as protective separator when combining otherwise degradation-prone electrolytes with lithium metal anodes.4 Another attractive feature of LLZO is that the powder and battery components can be processed and handled in air.1 Since the time of its discovery, significant progress has been achieved in increasing the bulk ionic conductivity of LLZO via the introduction of various dopants, resulting in an increase from the original 3 × 10−4 S cm−1 for Al-doped LLZO to 1.84 × 10−3 S cm−1 for Ga-doped LLZO.5 However, to make LLZO an industrial viable alternative to liquid electrolytes, not only the bulk conductivity, but also the total ionic conductivity of polycrystalline ceramic electrolytes needs to be as high as possible. The total conductivity is directly related to the density of the sintered samples, which can be increased e.g. by applying pressure6 or introducing sintering agents,7,8 and can be mainly governed by the conductivity of grain boundaries. Especially these grain boundaries, which are always present in polycrystalline samples, are very sensitive to the morphology of the LLZO powders as well as their surface composition. Besides their strong impact on conductivity, grain boundary properties are also essential for other properties of LLZO-based components such as their mechanical stability1 and the resilience to the growth of metallic Li dendrites that are formed during cycling at high current densities.10 The critical current densities (CCD) that can be achieved in the Li metal/LLZO half-cells are an important performance indicator of functional components made of LLZO.11 Achievable CCD values are mainly affected by interface properties such as the presence of an alloy-forming interphase,12 coatings to increase the LLZO wetting with lithium LLZO,13,14 or a reduction of surface defects.15 However, since dendrite formation takes place predominately at the grain boundaries,12 the chemical and mechanical properties of the grain boundaries in the final separator also have a major impact on the CCD. Therefore, the understanding and control of the factors influencing the grain boundary properties are essential for the reliable development and optimization of LLZO-based components and batteries.In contrast to e.g. sulphide based solid electrolytes, LLZO can be handled in ambient air. However, in the presence of water the surface undergoes a very fast Li+/H+-exchange with formation of a poorly conductive LiOH and subsequently Li2CO3-layer on the surface. This process takes place when LLZO is exposed to the humidity in air16–19 and to all solvents commonly used for wet-processing of ceramic components.20 Although this Li+/H+-exchange on the LLZO surface during storage in air (or any atmosphere with traces of humidity) or exposure to solvents is practically unavoidable, little is known about the influence of this exchange on the sintering behaviour and processability of LLZO powders via wet-processing routes and on the resulting component properties.
Some encouraging studies already exist that show the possibility of large-scale synthesis of LLZO in air21,22 and component manufacturing via solvent based fabrication routes.4,23–27 Thus, for industrial application of LLZO, air and solvent contributions to the Li+/H+-exchange need to be considered. However, the treatment of the material prior to component fabrication, such as storage conditions or pre-treatment, are often not detailed. Also, while stating the introduction of Li-excess during the synthesis to accompany loss during processing, many studies refrain from reporting the actual chemical composition of their samples during or after processing in favour of reporting just the target composition. Furthermore, the contact time between LLZO and the solvent during the wet-processing is scarcely reported and varies greatly between individual works. Thus, a detailed understanding of the impact of air exposure and processing in solvents is hard to generate from the existing work.
To close this knowledge gap, the change of LLZO during air exposure and in the solvent based processing steps was investigated in this study, including the influence on the electrochemical performance of the resulting components. The LLZO powder obtained after lab-scale synthesis typically requires a wet-milling process to acquire a suitable particle size for tape-casting. However, on industrial scale, suitable particle size distributions might be acquired directly after an optimized synthesis route. To investigate to influence of storage conditions, the milled LLZO powder was compared to powders annealed in air and argon after wet-milling. Both annealed powders were also stored in air and solvent for increasing amounts of time to produce a variety of surfaces by the inevitable Li+/H+-exchange. Combined with surface analysis techniques, this enabled us to obtain a mechanistic understanding of the effect of storage and processing of LLZO in air and solvents. This knowledge was used to improve the processing parameters for tape casting of LLZO resulting in a large improvement of both total ionic conductivity and CCD and pushed it closer towards feasibility for industrial application.
Experimental
Synthesis and powder conditioning
Li6.45Al0.05La3Zr1.6Ta0.4O12 starting powder was prepared via solid-state-synthesis. Stoichiometric amounts of LiOH·H2O (Merck, 98%), La2O3 (Merck, 99%, pre-dried at 900 °C for 10 h), ZrO2 (Treibacher, 99.5%), Ta2O5 (Inframat, 99.95%) and Al2O3 (Inframat, 99.9%) were used. 20 wt% excess of LiOH was added to compensate for lithium evaporation during high-temperature treatment. La2O3 was dried at 900 °C for 10 h before weighing in. The mixture was thoroughly mixed (Retsch, 200 M) and pressed into pellets before the first calcination step at 850 °C for 10 h. The obtained pellets were crushed and ground by electrical mortar, pressed to pellets again and calcined a second time at 1000 °C for 10 h. The pellets were again crushed and milled by mortar. The resulting LLZO powder shows cubic crystal phase with few impurities.To reduce the particle size for further processing, the obtained powder was milled in ethanol with ZrO2 jar and balls at 1000 rpm for 15 minutes using a planetary ball mill (Pulverisette 7 premium, Fritsch) and subsequently dried at 70 °C for 8 h. All processing steps up to this point have been carried out in ambient air.
Tape casting
To minimize the dwell time of the powder in the slurry, a solution of the dispersant (BYK 180, Altana), binder (PVB98, Sigma Aldrich) and additives (PEG400, Sigma-Aldrich and Solusolv, Solutia Inc.) in a mixture of ethanol (VWR, 99.6%) and butanone (VWR, 99.6%) was prepared beforehand. For all slurries, the same batch of polymer solution was used and the ratio of solution to ceramic powder was kept constant. To prepare the slurry, the LLZO powder was added to the solution and homogenized in a planetary mixer (Thinky) at 1500 rpm for two minutes. The obtained slurry was cast onto a mylar foil. The gap height was fixed to 350 μm and kept constant for all samples. After drying for 10 h at room temperature, the obtained green tapes were compacted and laminated with 125 MPa at 80 °C for 2 min. After pressing, the green tapes were cut into 12 mm diameter discs, transferred into an Al2O3 crucible, and sintered for 10 h at 1175 °C in air to obtain free standing LLZO separators.Material characterization
The particle size distribution (ESI 1†) of the three starting powders was checked carefully via laser diffraction using a LA950 (Horiba Scientific) with a 650 nm and a 405 nm laser source, data analysed via Mie-theory, to exclude their influence on the viscosity of the slurry. The phase purity was measured using a Bruker D4 Endeavor (ESI 2†) equipped with a 1D detector LYNXEY using monochromatized Cu Kα radiation. To obtain information on the protonation of the powder surface, Raman spectra were collected using a Renishaw InVia Raman Microscope equipped with a solid-state excitation laser (532 nm) and a 2400 lines per mm grating. The measurement was conducted at a laser power of 2.5 mW to avoid laser-induced damage of the material. The as-milled and freshly annealed powders were pressed between two microscopic glass slides and sealed by hot-glue to avoid contamination from ambient air. In order to improve the statistics 1071 spectra over an area of 100 μm × 40 μm with a step size of 2 μm were collected. The exposure time was 1 s per spectrum. The cosmic rays of the raw data were removed by using the function of the Wire 5.2 software (Renishaw). The spectra were normalized to the maximum height between 600 cm−1 and 700 cm−1 and subsequently averaged using an in-house developed Python script. Additionally, X-ray photoelectron spectroscopy (XPS) measurements (Phi5000 VersaProbe II, ULVAC-Phi Inc.) were performed. To avoid air exposure of the Ar-annealed sample, all samples were mounted inside an Ar filled glovebox onto the sample holder and transferred using an air-tight transfer chamber. Survey scans used a pass energy of 187.5 eV and were quantified using Shirley background and empirical sensitivity factors for C 1s, O 1s, La 3d5, Zr 3d and Li 1s. The viscosities of the slurries were measured between 0.01 s−1 and 1000 s−1 with a rotational shear rheometer (Anton Paar, MCR301) using a plate–plate setup. The densities of the sintered, free standing LLZO separators were measured using Archimedes method with water with every value being the mean of measurements on two separate specimen. The microstructure was analysed using a scanning electron microscope (Hitachi, TM3000) on a wet-polished cross-section. Since the obtained separators were too thin for removal of the Li2CO3 by conventional polishing with sandpaper, they were instead heated in argon for 2 h at 750 °C. This ensured a comparable interface for the electrochemical characterization, details can be found here.16 Blocking electrodes were applied by sputtering gold onto the fresh surface (2 min sputter time, Cressington 108cuto Coater). Using a BioLogic VMP-300 multipotentiostat, the room temperature impedance of the pellets was measured, and the resistance and conductivity extracted. The frequency was varied from 7 MHz to 1 Hz with an electrical field perturbation of 10 mV mm−1. To investigate dendrites formation, symmetrical Li|LLZ|Li cells were assembled under argon atmosphere. A thin gold interlayer was sputtered onto the sample surface (∼30 nm, Cressington 108cuto Coater). Freshly calandered metallic lithium was pressed on each side by hand, the sample then placed between two Ni discs and the stack heated to 250 °C to ensure good contact. Critical current density measurements were conducted at 50 °C using the same multipotentiostat mentioned above. All electrochemical measurements were carried out in a Swagelok cell.Results and discussion
Particle pre-treatment
Solid-state synthesis of cubic LLZO phase with a high ionic conductivity typically requires calcination step at high temperatures of around 1000 °C. To compensate Li evaporation, some excess of the lithium source LiOH was added to the synthesis. This excess also needs to be present in the final LLZO powder, to compensate losses during the sintering process. To produce battery components, the received LLZO powders have to be processed further, which typically includes wet-milling to control the particle size, often followed by solvent-based processing steps such as tape casting or extrusion. Finally, the powders often need to be stored after synthesis or between processing steps in an ambient air with traces of humidity. Since LLZO is prone to a Li+/H+-exchange in humid air5 and protic solvents,7 storage and processing steps unavoidably lead to changes in the particle surface composition. In that process, LLZO undergoes a Li+/H+-exchange with ambient water, to form protonated LLZO and LiOH:| Li6.4La3.2Zr1.6Ta0.4Al0.02O12 + xH2O → Li6.4−xHxLa3.2Zr1.6Ta0.4Al0.02O12 + xLiOH | (1) |
| LiOH + 1/2CO2 → 1/2H2O + 1/2Li2CO3 | (2) |
These reactions can be reversed with a suitable heat treatment above 673 K (ref. 28) in both air and inert atmosphere. These phenomena have been investigated in great detail for sintered LLZO components. Nevertheless, a detailed analysis of the behaviour of LLZO powder in presence of excess lithium-sources is needed, to gain a better understanding of the influence on the particle surface and consecutively the grain boundaries.
Similarly to LiOH formed during the protonation of LLZO, the excess LiOH can also react to Li2CO3 in presence of CO2viaeqn (2) and will be labelled as Li2CO3(ex) in the following. To reverse this reaction and re-form the excess LiOH, ambient water needs to be present. However, in inert atmosphere no water is present and only a direct decomposition of Li2CO3 according to
| Li2CO3 →Li2O + CO2 | (3) |
To investigate the impact of different surfaces produced by the Li+/H+-exchange, a sample matrix using the same starting material which is then exposed to different storage conditions in air and solvent was designed. As starting material, calcined and milled LLZO powder (see Experimental part for the details) was divided into three parts. The first part was directly used for fabricating tape cast samples and thus labelled as “milled”. From the other two part, one was annealed in air (assigned further as “air0”) and one in argon atmosphere (assigned further as “argon0”) for 10 h at 750 °C.
Furthermore, to increase the impact of storage in air and solvent, parts of each of the two annealed subsets were stored in air and immersed in ethanol for increasing amounts of time. This experimental matrix, compiled in Fig. 1, allows a systematic assessment of the impact of both ambient air and solvent onto the processability via tape casting and electrochemical performance of the final component.
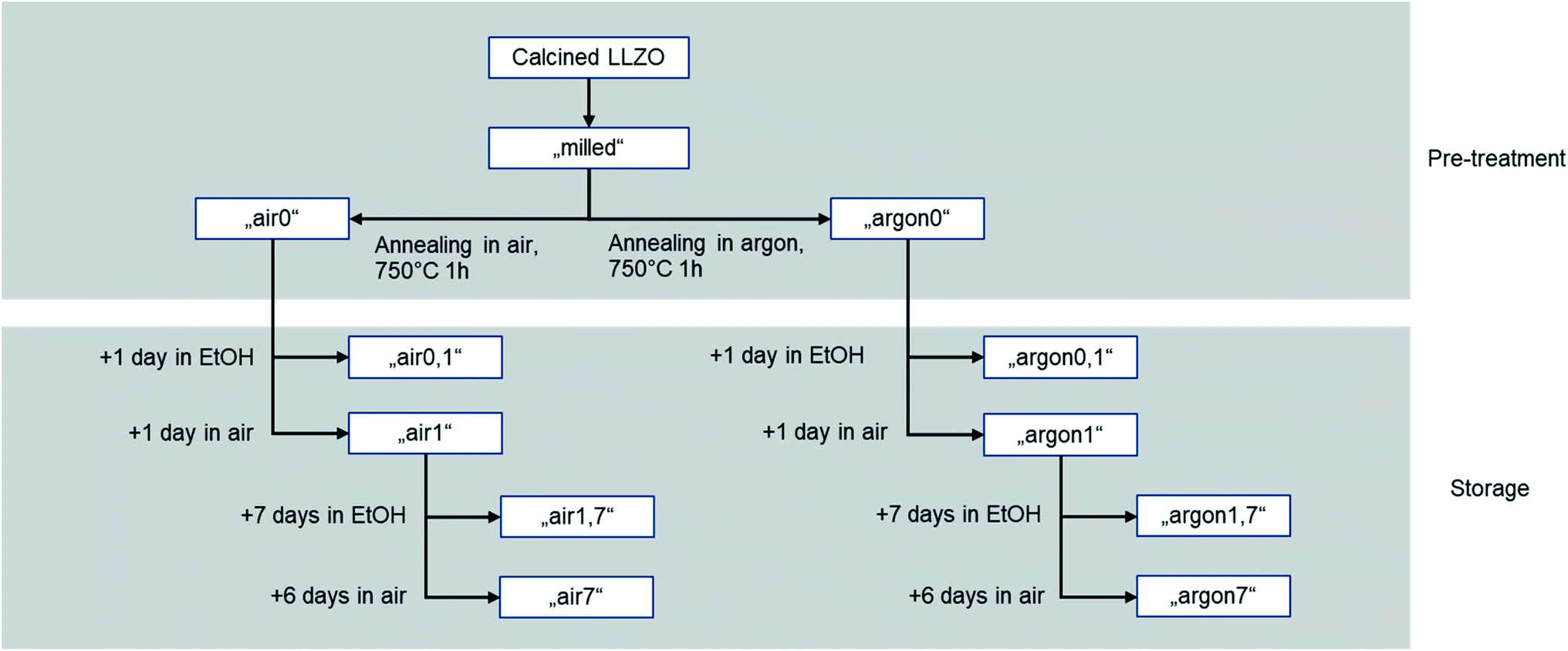 | ||
| Fig. 1 Overview of treatment and storage conditions of all samples. | ||
To analyse the total Li2CO3 content and LLZO surface composition after milling and assess the impact of annealing in Ar and air, we used Raman spectroscopy (Fig. 2). In contrast to X-ray diffraction, which does not show significant difference between as-milled and annealed powder samples, Raman spectroscopy is very sensitive to Li2CO3 and various LLZO crystal phases (cubic vs. tetragonal) and probes only the surface of the particles as the information depth is only several nm. The as-milled LLZO powder (Fig. 2 blue) shows a significant Li2CO3 peak at 1090 cm−1. From previous studies,7 we expect LLZO to undergo a significant proton exchange during high-energy milling in ethanol, the formation of protonated LLZO and lithium hydroxide, as described in (1). Subsequently, during drying in air, lithium carbonate forms from the freshly produced LiOH as well as the excess LiOH, as described in (2). The spectrum of the sample annealed in Ar (Fig. 2 purple) shows good agreement with the high-temperature cubic garnet phase, whereas the milled sample (Fig. 2 blue) and the sample annealed in air (Fig. 2 green) show significant changes of the spectrum between 100 cm−1 and 800 cm−1, that cannot be attributed to the formation of Li2CO3. These spectra indicate the presence of the low-temperature cubic LLZO phase due to the protonation and additional adsorption of CO2 and subsequent incorporation of CO32− into the crystal lattice.29,30,35 Besides a much lower ionic conductivity than the high temperature cubic phase, it is also more ordered and could lead to differences in the interaction of solvents and dispersants in the subsequent wet-processing route.
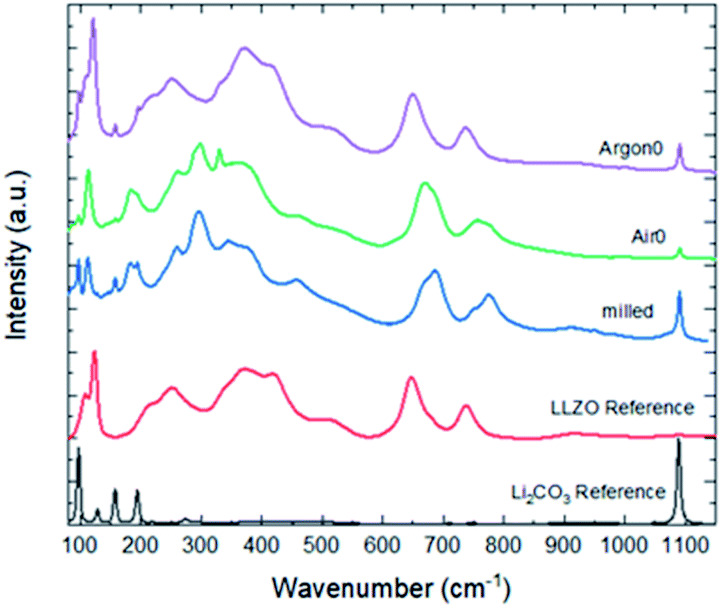 | ||
| Fig. 2 Raman spectra of powders with different pre-treatments in comparison to sintered and polished cubic LLZO34 and commercially available Li2CO3. | ||
During the annealing in air (Fig. 2 green), the overall amount of Li2CO3 is greatly reduced, since Li2CO3(LLZO) can react back to LLZO. Li2CO3(ex) can also react back to LiOH, using atmospheric water as a reaction partner. CO32− is also removed from the LT-cubic phase of LLZO during the annealing. However, since the annealing takes place in a crucible in open but stagnant atmosphere, both the CO32−-stabilized low-temperature cubic phase of LLZO and some Li2CO3(LLZO) and Li2CO3(ex) can be formed again during cooling. However, as the intensities are much smaller compared to the milled powder, they reform to smaller degree as the reaction time is greatly decreased.
During the heating in argon atmosphere (Fig. 2 purple), the partial pressure of water and CO2 is negligible. Thus, Li2CO3(LLZO) can react back according to (2) and (1) to reform LLZO by removing protons from the structure and emitting CO2 and H2O. However, the Li2CO3(ex) cannot easily react back according to (2) to reform LiOH, as no water is available from the oven atmosphere and the direct thermal decomposition of Li2CO3(ex) according to (3) does not occur at the temperatures chosen in this study.
Overall, this leads to the much higher signal for Li2CO3 observed in the Raman spectrum of the Ar-annealed sample (Fig. 2 purple). Furthermore, CO32− is again removed from the LT-cubic phase of LLZO, but in contrast to the annealing in air, the partial pressures of CO2 and H2O remain low over the course of the treatment. Thus, during the cooling, CO2 preferably reacts with the excess LiOH, rather than reincorporating into the crystal structure of LLZO preserving the HT-cubic LLZO structure observed in Fig. 2 green. In summary, the Ar-annealed sample shows a higher amount of Li2CO3(ex), but the surface of the particles is very close to the high temperature cubic phase.
To obtain more detailed information about the surface composition of the particles, X-ray photoelectron spectroscopy (XPS) was employed on the same powders and the total amount of surface species extracted (Fig. 3). Verifying the Raman results, the comparison of the air-to the Ar-annealed powder (Fig. 3 green and purple, respectively) shows increasing amounts of C due to the higher amount of Li2CO3. Interestingly, for La and Zr the XPS measurements show a higher value for the milled powder, whereas the Ar and air annealed powders are about the same. Since La and Zr signal are proportional to the bulk LLZO measured via XPS, this indicates that the milled powder has a thinner or incomplete coating with LiOH and/or Li2CO3 compared to the air- or Ar-annealed ones. The most plausible explanation of this effect is, that the excess LiOH and Li2CO3 formed after milling does not homogeneously cover the surface but forms small particles itself (e.g. see schematic representation in Fig. 9, second column). During the high temperature treatment at 750 °C both LiOH (Tm = 426 °C) and Li2CO3 (Tm =720 °C) melt, coat and partially connect the particles (Fig. 9, third column). From the C signal in XPS and the Raman measurements we can now confirm that the composition of these coatings is LiOH-rich for air annealed powders and Li2CO3-rich for the Ar annealing. It can be expected that these coatings on the particle surface and their different compositions will have a significant effect on their behaviour in a subsequent wet-chemical processing step and final component performance.
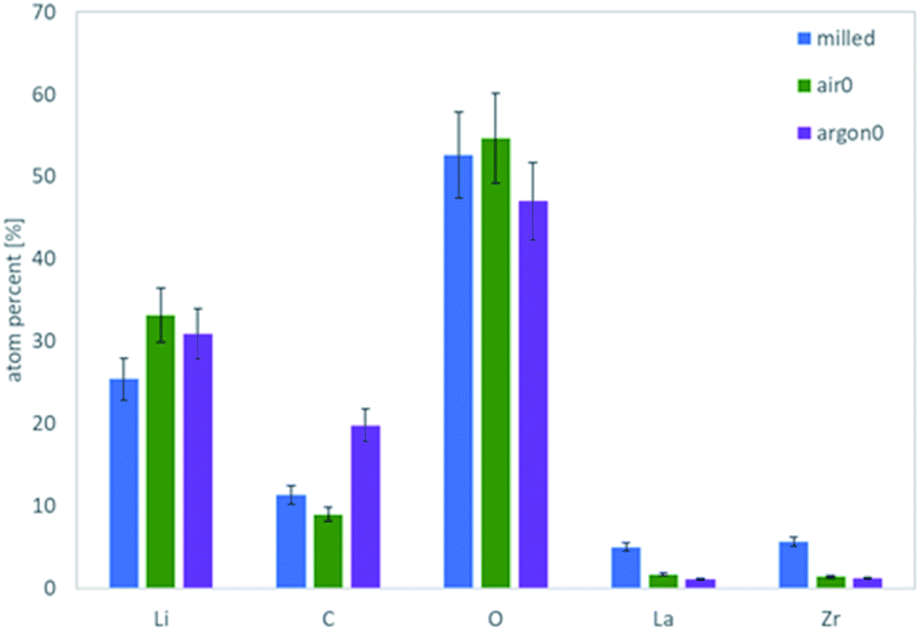 | ||
| Fig. 3 Absolute atomic concentration of the major elements as determined via XPS for the milled powder (blue), the air annealed (green) and the Ar annealed (purple) sample. | ||
Rheological characterization
The processability of tape-casting slurries is determined by a delicate interplay of several properties. To allow an analysis of the influence of the pre-treatment and resulting differences in particle surface of the powders, the choice and amount of all organic parts was kept constant. Thus, all changes in the rheological behaviour of the slurry can be attributed on the influence of the ceramic powder. The recipe was chosen to enable successful casting and drying while tolerating a wide range of rheological behaviour. Suspensions, as well as polymer solutions, are expected to show non-newtonian behaviour. Therefore, the rheological behaviour of the slurry must be investigated over the whole range of shear rates during the tape-casting process, storage, and preparation. Strong shear-thinning is an ideal behaviour for tape-casting slurries, as the low viscosity during casting allows an even distribution of the slurry, while the high viscosity during storage and drying stops the particles from settling. In this study, the changes of the rheological behaviour can be assigned to the ability of the steric dispersant to attach to the particle surface. The steric dispersant used in this study is designed to attach to metal oxide particles in an alkaline environment and is therefore highly sensitive to secondary phases on the particle surface. Low coverage of the particle surface with dispersant leads to higher viscosities and stronger shear thinning, as the particles form weak agglomerates and excess dispersant raises the apparent solvent viscosity. Full coverage of the particle surface prevents agglomeration of the particles and results in a lower viscosity. This behaviour is generally preferred, while the well dispersed ceramic particles will lead to more predictable casting and sintering behaviour and higher reproducibility. Nevertheless, the viscosity profile of the slurry can be varied within certain limits by simply changing the solvent content.The rheological behaviour is shown in Fig. 4. All starting powders (milled (blue), air0 (green) and argon0 (red)) showing shear-thinning behaviour. When getting into contact with the solvent during preparation of the slurry, fast protonation of the surface is expected, as LiOH is formed and consecutively dissolved in the surrounding solvent as proposed by R. Kun et al.20 In our work, the effect of the excess LiOH and Li2CO3 on the particle surface (as determined by Raman and XPS) on the protonation in the solvent and the resulting rheological behaviour needs to be considered. On the one hand, the excess LiOH dissolves very well in ethanol, on the other hand, Li2CO3 has a very low solubility, which means it will remain in its original state and place upon contact with the solvent. From the variation of the viscosity in Fig. 4 in the shear rate range of 0.01 to 1 s−1 we can see there is clear correlation between the Li2CO3 content of the sample, as determined by Raman and XPS measurements, and the viscosity of the slurry. The as milled powder has the highest surface area uncovered by excess material, which has already been protonated by the previous exposure to the solvent and therefore allows fast attachment and full coverage of the dispersant, resulting in the lowest viscosity. The air-annealed sample (air0) is mainly coated by LiOH which can also be dissolved by the solvent. The subsequent protonation of the free particle surface allows for still reasonable coverage of the dispersant and results in medium high viscosities. The Ar-annealed powder (argon0) features a Li2CO3-rich coating of the particles, which has a much lower solubility than LiOH and thus only allows poor coverage with the dispersant (see Fig. 9 fourth column).
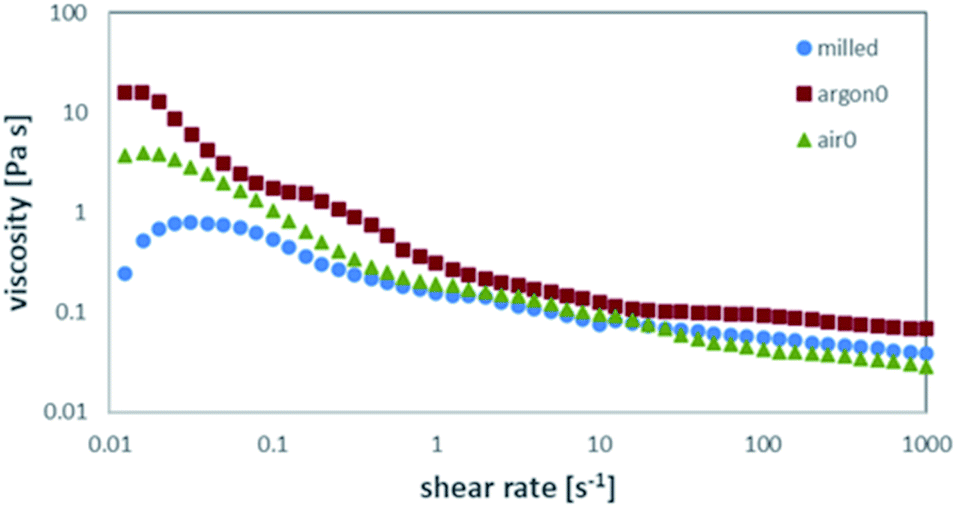 | ||
| Fig. 4 Viscosity over shear-rate measurements for powders after different pre-treatments. | ||
In addition to the strong impact of the surface pre-treatment of starting powders on their rheological behaviour, further significant changes in the rheological behaviour are observed when the LLZO powders are stored for different time both in air and a solvent.
When comparing the behaviour of the freshly annealed powders to the ones stored in air for prolonged times, a clear change can be observed for the air-annealed powders stored in air (Fig. 5a) and the Ar annealed powders stored in air (Fig. 5b). The air-annealed powder shows a decrease in viscosity, caused by the slow protonation of the LLZO surface when in contact with ambient air and the subsequently better coverage with dispersant in the slurry. After one day of exposure to ambient air, the obtained viscosity is close to the viscosity of the as-milled slurry, which means the protonation of the surface is almost complete.
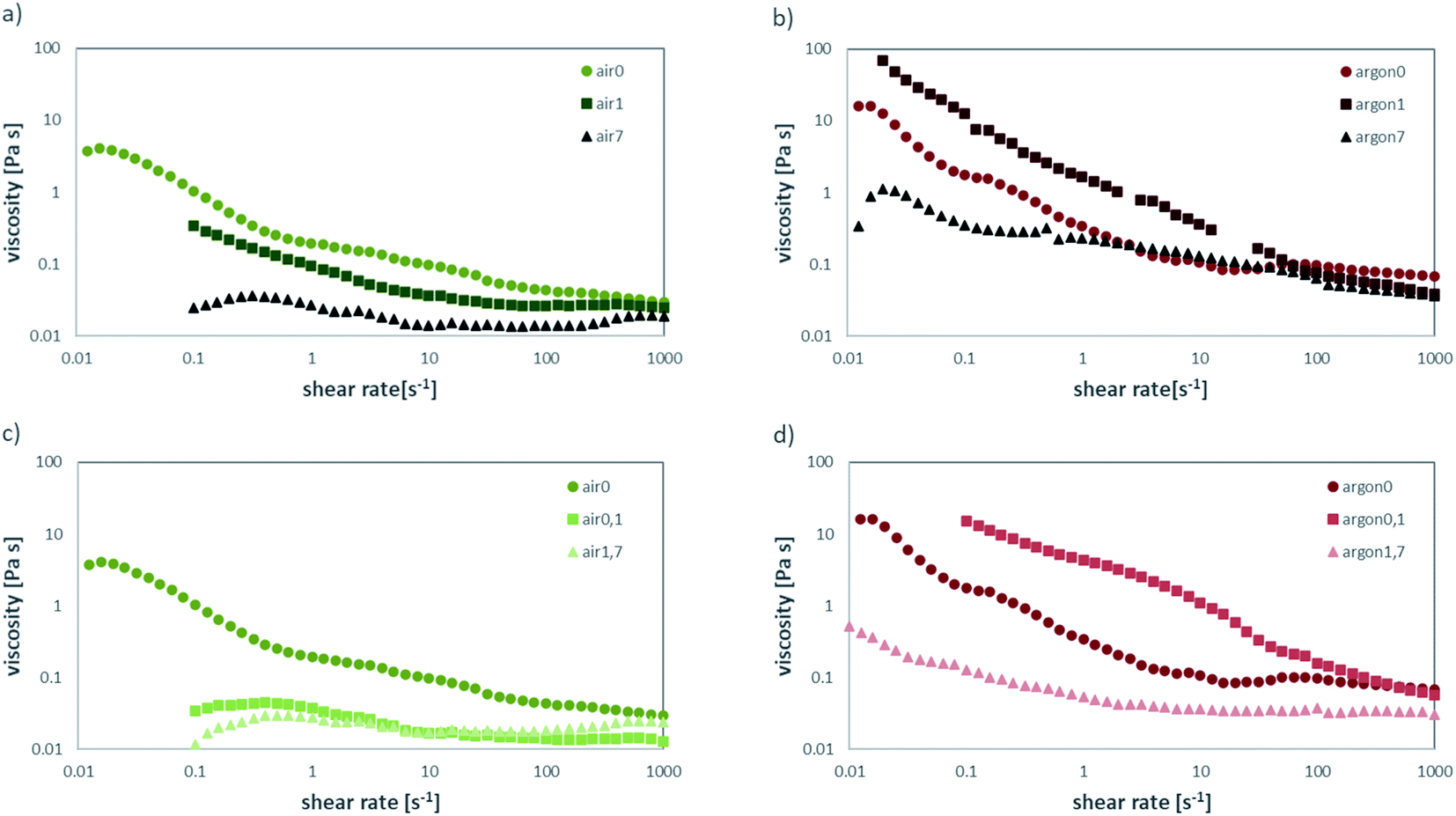 | ||
| Fig. 5 Viscosity over shear-rate measurements for powders for (a) pre-treatment in air, storage in air (b) pre-treatment in argon, storage in air (c) pre-treatment in air, storage in slurry (d) pre-treatment in argon, storage in slurry. | ||
The behaviour or the Ar-annealed powders is less linear. As the partial pressures of water and CO2 are greatly lowered in inert atmosphere, short exposure to air causes the unreacted species obtained after Ar annealing to react with ambient moisture and CO2 to reach equilibrium. As a result, the thicker, insoluble layer of Li2CO3 covering the particles allows for less protonation of LLZO. Therefore, these particles are less covered with dispersant, causing an increase in viscosity of the obtained slurry. With prolonged exposure to ambient air, protonation of the LLZO surface through the Li2CO3 layer can take place, allowing for a better coverage with dispersant in the slurry. Thus, the viscosity of the obtained slurry is vastly lowered (Fig. 5b light red). After 7 days in air, the viscosities of air-annealed and Ar-annealed powders are almost the same.
For the storage in solvent, similar trends can be observed. The LLZO annealed in air with prolonged storage time in the slurry (Fig. 5c) shows a significant decrease in viscosity already after only 1 day in ethanol (green), while a longer storage time does not change it much further (light green). Again, this can be explained by ethanol dissolving the LiOH predominately covering the particle surface, exposing fresh particle surface. This saturates the solvent with OH−, which is confirmed by the measured pH values of over 13 for all samples. The dispersant can then attach to the fresh particle surface, leading to the observed drop in viscosity. For Ar-annealed powder, the behaviour for prolonged storage in ethanol (Fig. 5d) is much different and resembles the storage in air (Fig. 5b). In contrast to the air annealed sample, the particles are covered by a much larger amount of Li2CO3, which will not dissolve as easily in ethanol. Thus, after the initial increase in viscosity, the dissolution of LiOH and Li2CO3 covering the surface and subsequent drop in viscosity due to improved attachment of the dispersant is much slower. After 7 days, the behaviour resembles the one for air annealed sample as after 1 day storage in air. In summary, the excess LiOH and Li2CO3, covering the surface after annealing, greatly affects the rheological behaviour of the slurry. Common variation in the recipes like homogenizing the slurry on a roller bench for 24–72 h can result in large deviations in castability, depending on the storage and pre-treatment of the original powder. This poses challenges regarding the variation and control of time scales in the manufacturing processes, demonstrated exemplary in Fig. 6. Fig. 6a shows the green tape cast with freshly Ar-annealed powder (argon0) having only minor defect on the edges of the tape due to surface tension based flow during drying. After 1 day of storage in ethanol (argon0,1) the casting results improve, and the green tape shows no defects. After 7 days of storage in ethanol (argon0,7) the green tape shows major defect on the edge due to surface tension based flow and severe coffee-staining. Defects of such severity do not allow for further processing of this tape.
 | ||
| Fig. 6 Casting behaviour resulting from different rheological profiles (a) argon0 (b) argon0,1 (c) argon0,7. | ||
This difference in casting behaviour can of course be compensated (within limits) by changing the composition of the slurry. However, it clearly demonstrates the importance of a detailed understanding of the influence of the pre-processing and storage on the particle surface and its effect on the slurry viscosity to predict the resulting changes in casting behaviour. Additionally, the impact of these changes in particle surface on the properties of the sintered sample needs to be investigated.
Microstructure and phase purity of sintered sample
Not all slurries produce usable samples. Particularly for the powders stored 7 days in ethanol the casting of tapes was not successful. For the other slurries, flat, free-standing, partly transparent separators were obtained having a slightly yellowish colour (Fig. 7, top). The thickness of the laminated samples can be varied between 50 μm and 240 μm (Fig. 7, bottom). All sintered samples show pure cubic phase LLZO [ESI 3†]. Relative densities of the sintered samples, obtained by Archimedes method, strongly decrease with an increase of the Li2CO3 content of the sample (Table 1). In contrast to some publications claiming that lithium carbonate acts as an additive in a liquid phase sintering of LLZO,31 we found that the lower contents of Li2CO3 are more beneficial for the densification. LiOH32 and even Li2O23 have been shown to improve the sintering of LLZO due to their low melting point. A comparative study on the efficiency of these additives has not been shown, but our results strongly indicate that the progression of the reaction of excess LiOH with CO2 during storage, as well as the reaction of LLZO with ambient air or solvents as a negative impact on the density of the sintered ceramics. Highest relative densities were achieved for process routes that produce no or very little Li2CO3 on the particle surface. | ||
| Fig. 7 Top: picture of successfully sintered LLZO components bottom: corresponding cross-section pictures by SEM. | ||
| Sample | Pre-treatment | Storage | Relative density [%] | Conductivity 25 °C [S cm−1] | Critical current density 50 °C [μA cm−2] | Li2CO3 content |
|---|---|---|---|---|---|---|
| Argon0 | Argon | — | 91.2 | 1.89 × 10−4 | 5.1 | + |
| Argon0,1 | Argon | 1 day ethanol | 88.1 | 2.20 × 10−4 | 35.7 | + |
| Argon1 | Argon | 1 day air | 88.2 | 2.66 × 10−4 | <5.1 | ++ |
| Argon7 | Argon | 7 days air | 88.8 | 2.75 × 10−4 | 114.6 | +++ |
| Argon1,7 | Argon | 7 days ethanol | — | — | — | +++ |
| Air0 | Air | — | 92.8 | 3.90 × 10 −4 | 318.5 | 0 |
| Air0,1 | Air | 1 day ethanol | 91.5 | 2.08 × 10−4 | 35.7 | + |
| Air1 | Air | 1 day air | 89.0 | 1.19 × 10−4 | 127.4 | ++ |
| Air7 | Air | 7 days air | 79.5 | 7.87 × 10−6 | — | ++ |
| Air1,7 | Air | 7 days ethanol | — | — | — | +++ |
| Milled | As-milled | — | 89.4 | 1.09 × 10−4 | 127.4 | +++ |
Electrochemical characterization
Ionic conductivity is generally assumed to be strongly dependent on the relative density of the sample. As this is certainly true when assuming no other changes in the system, it has proven to be an over-simplification with more complex production routes, as it disregards changes in the grain boundaries. Computational assessments33 as well as studies on fully densified, hot-pressed samples9 find a lower conductivity of the grain boundary as compared to the bulk LLZO at room temperature. Therefore, the total conductivity of the component is highly dependent on the modification of the grain boundaries. Existing works focus on model systems to gain a fundamental understanding of the grain boundaries. Experimental work on the influence of the processing on the conductivity, especially of the grain boundaries, has so far only been shown once.32 The authors also report a negative impact of a heat-treatment in inert atmosphere prior to processing on the grain boundary conductivity for samples prepared by SPS at reduced temperatures.Due to the low thickness and high conductivity of the samples, the resolution of bulk and grain boundary contributions was not possible at room temperature. Therefore only total ionic conductivities of the components can be reported, taken from the low frequency intercept of the semicircle corresponding to the onset of the blocking electrode behaviour of the sputtered gold electrodes (Fig. 8, left).
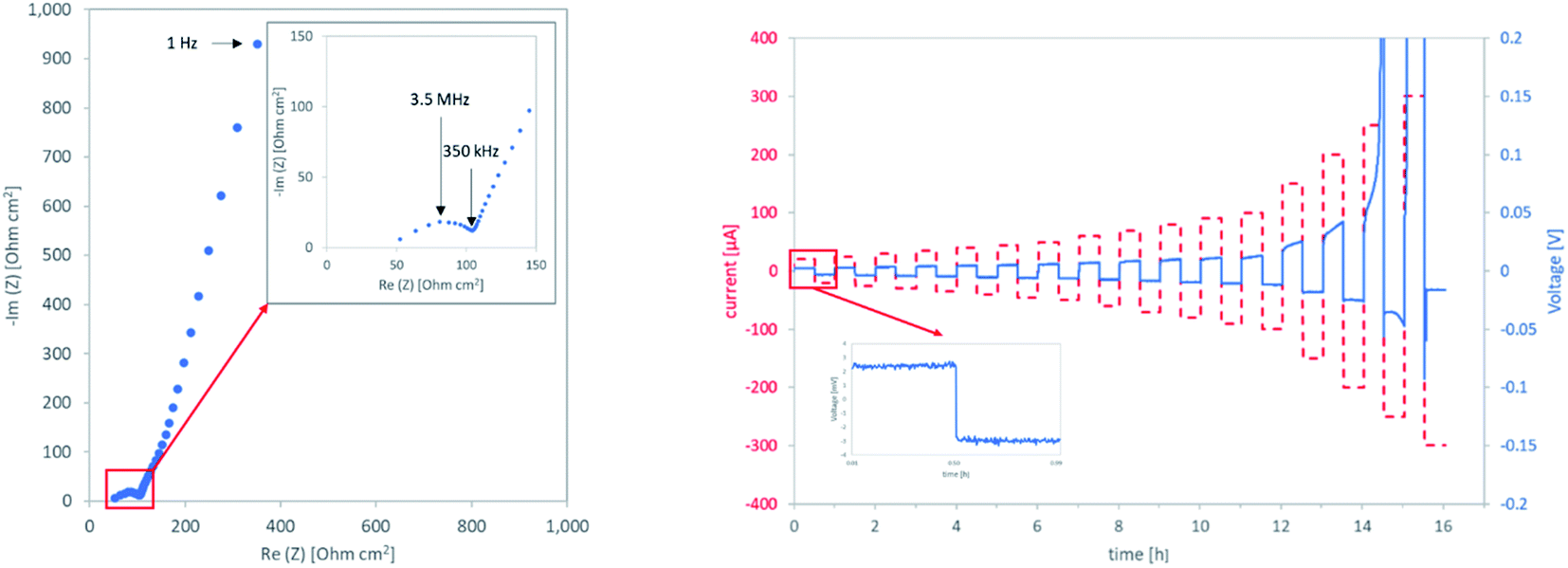 | ||
| Fig. 8 Left: impedance spectrum of air0 at room temperature right: CCD measurement of air0 at 50 °C. | ||
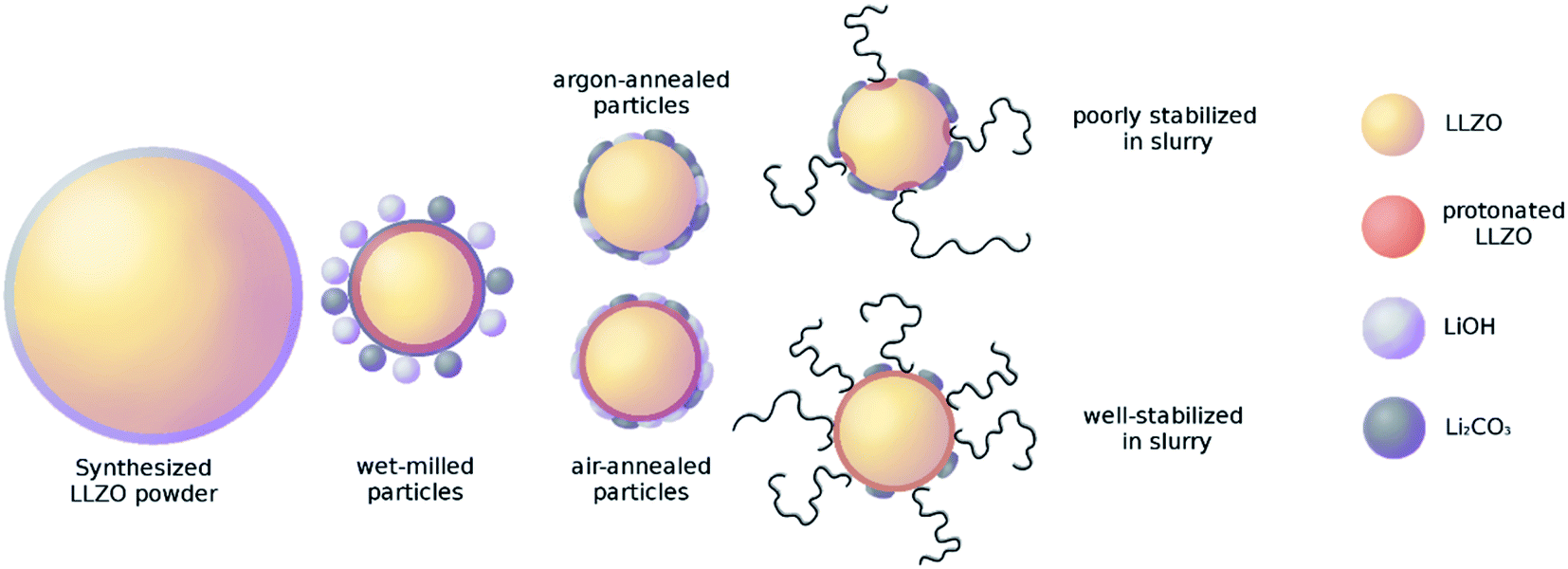 | ||
| Fig. 9 Surface coverage of LLZO particles with different pre-treatments. | ||
No direct correlation between the relative density and conductivity can be drawn from the measurements summarized in (Table 1). Samples obtained from freshly air-annealed LLZO powder show the highest total conductivity of 3.90 × 10−4 S cm−1 at 25 °C. With prolonged storage in air the amount of LiOH decreases in favour of Li2CO3 on the particle surface, resulting in a lower conductivity of the resulting sample. The negative effect of Li2CO3 is even more pronounced for the freshly Ar-annealed sample. Yet, prolonged storage in air improves the conductivity of Ar-annealed samples, as the onset of LLZO-protonation increases the LiOH content. Prolonged storage in the slurry reduces the conductivity of samples obtained from air-annealed powders. Nevertheless, protonation by the solvents seems to have a less disadvantageous influence on the conductivity than the reaction with ambient air, as the subsequent reaction to Li2CO3 is inhibited. The highest ionic conductivity obtained in this work of 3.90 × 10−4 S cm−1 at 25 °C for the components prepared from freshly air-annealed LLZO powder, without the use of sintering aids, is the highest value reported in literature so far.
These findings highlight the importance of a detailed understanding of the influence of the pre-processing and storage on the particle surface and its effect on the ionic conductivity of sintered components.
The critical current densities (CCD) that can be achieved with LLZO separators and Li anodes without failure caused by the development of a lithium dendrite are an important indicator of their performance. To determine CCDs for different LLZO separators prepared in this study, Li plating and stripping experiments were performed on symmetric Li‖LLZO‖Li cells. To improve the wetting of the LLZO with lithium and to minimize the Li/LLZO interfacial resistance,17 a sputtered Au interlayer was introduced to ensure comparable interface resistances amongst all samples. Each sample was characterized via impedance spectroscopy prior to the CCD measurements. Only in the case of argon1, extremely high values for the interface resistance clearly lead to a low CCD. All other samples show similar interfacial resistances (ESI Table 1†), which can be ruled out as the reason for their differences in CCD. Thus, the differences in their CCDs are mainly governed by the differences in the total ionic conductivity of the sample. At low current densities, most samples show flat voltage plateaus, indicating a uniform plating and stripping of the metallic lithium. With higher currents, the growth of a dendrite is marked by an increase in voltage, followed by a sharp decrease, marking the critical failure (Fig. 8, right).
No linear connection of the critical current density to the total ionic conductivity can be derived from our data (Table 1). Since the bulk conductivity should be similar for all samples, a detailed investigation of the dependency of the CCD on the grain boundary structure and composition of LLZO should be undertaken in the future. Still, the overall trends found this far are also valid for the critical current density measurements. Freshly-air annealed powder produces samples with the high CCDs of 0.32 mA cm−2 at 50 °C. Prolonged storage in air causes the protonation of the LLZO surface with subsequent reaction to Li2CO3, causing a decrease in the CCD of the sintered sample. The obtained values match those of the as-milled samples. Sintered samples obtained from freshly argon-annealed powder show very low CCDs, as again the coating of the particles with Li2CO3 negatively influences the grain boundary of the sintered component. Storage of the argon-annealed powder in air for a short time worsens the CCD to <5 μA cm−2, as LiOH remaining on the particle surface after annealing forms additional Li2CO3. Prolonged storage in air allows for the protonation of the LLZO surface, resulting in an improvement of the CCD. The CCD of samples from as-milled powders is similar to the values for both air-annealed and argon-annealed samples after prolonged storage in air.
Prolonged storage in the slurry of the air-annealed powder reduces the CCD of the sintered component vastly, as proton exchange is expected to be faster in the solvent than in air. Yet, prolonged storage in the slurry of the argon-annealed powder improves the CCD, as the protonation is faster in the solvent and the subsequent reaction with CO2 is inhibited.
The highest critical current density of 0.32 mA cm−2 was measured for the separator produced from freshly air annealed LLZO powder, which had the shortest exposure times with both air and solvents. Unfortunately, the prevailing majority of publications deal with pressed and sintered LLZO pellets, which are not suitable for the large-scale fabrication of solid state batteries. Most publications on tape-cast LLZO do not report plating-stripping results.22,24 Even so the critical current density obtained for our optimized tape cast membranes is among the highest reported for thin freestanding tape-cast LLZO separators. The only comparable work on tape cast LLZO films by Hitz et al.4 deals with 3D structured separators. Due to a higher contact area of their electrodes the higher total CCD value of the component is achieved. However, the equivalent CCD of their material estimated from the total current and the specific surface area of the porous electrodes is only around 0.25 mA cm−2, which is significantly lower than the value obtained in our work. It can be therefore expected that a combination of 3D structuring that was shown to be beneficial for the cell performance, with the optimized processing route developed in this work would further increase the critical current density of LLZO separators to reach values relevant for industrial application.
Conclusions
For the first time, the impact of the surface degradation of LLZO powder due to Li+/H+-exchange during the pre-treatment, storage and wet-processing has been investigated systematically. Raman and XPS measurements showed the presence of LiOH and Li2CO3 from reaction of LLZO and excess LiOH with ambient air and solvent. While initially they do not fully cover the particle surface, annealing the wet-milled powder in air or argon atmosphere results in a homogenous surface coating. The type of atmosphere determines the exact ratio of LiOH to Li2CO3 in the coating and the surface composition of the underlying LLZO particle. Both heavily impact the particle–dispersant interaction during wet-processing and determine further reactions with ambient air and solvent during storage. These interdependencies pose great difficulties for the reproducibility of wet-processing routes and therefore need to be tightly controlled. Especially, since they also strongly impact the final density, conductivity, and critical current density of the sintered ceramic battery components. Based on these findings, we were able to develop an optimized tape casting route using freshly air annealed powder. Minimization of the storage time in ambient air prior to processing and reduced exposure to the solvent led to significant improvements in conductivity and critical current density. Free standing, thin and dense LLZO separators with thicknesses between 50 μm and 240 μm were prepared. They exhibit the highest reported total Li ion conductivity of 3.90 × 10−4 S cm−1 and the highest critical current density of over 0.3 mA cm−2 without the use of coatings or sintering aids for free-standing LLZO tapes.Author contributions
Writing – original draft: M. R.; writing – review & editing: M. F. and D. F.; investigation: M. R. and S. L.; validation: R. Y. and M. M.; funding acquisition: M. F. and O. G.; project administration: M. F.; conceptualization: M. R., M. F. and D. F.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the German Federal Ministry of Education and Research (BMBF) within of the Festbatt project (FKZ: 13XP0173A) and the LISZUBA project (FKZ: 03XP0115B) and is gratefully acknowledged here. The authors would like to thank Philipp Hecker and Grit Häuschen for the helps in LLZO powder synthesis, Andrea Hilgers for the help with PSD analysis, Volker Bader for the heat treatments, Dr Heinrich Hartmann for the XPS measurements and Hiltrud Moitroux for the photographs. Special thanks to Dr Enkhtsetseg Dashjav for the helpful discussions.Notes and references
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS PubMed
.
- Y. Zhu,
et al.
, Adv. Energy Mater., 2019, 9, 1–11 Search PubMed
- M. Finsterbusch, T. Danner, C. L. Tsai, S. Uhlenbruck, A. Latz and O. Guillon, ACS Appl. Mater. Interfaces, 2018, 10, 22329–22339 CrossRef CAS PubMed
- G. T. Hitz,
et al.
, Mater. Today, 2019, 22, 50–57 CrossRef CAS
- L. Buannic,
et al.
, Chem. Mater., 2017, 29, 1769–1778 CrossRef CAS
- I. N. David, T. Thompson, J. Wolfenstine, J. L. Allen and J. Sakamoto, J. Am. Ceram. Soc., 2015, 98, 1209–1214 CrossRef CAS
- Y. Luo, X. Li, H. Chen and L. Guo, J. Mater. Sci.: Mater. Electron., 2019, 30, 17195–17201 CrossRef CAS
- C. Li, A. Ishii, L. Roy, D. Hitchcock, Y. Meng and K. Brinkman, J. Mater. Sci., 2020, 16470–16481 CrossRef CAS
- A. Sharafi, C. G. Haslam, R. D. Kerns, J. Wolfenstine and J. Sakamoto, J. Mater. Chem. A, 2017, 21491–21504 RSC
- S. Yu and D. J. Siegel, ACS Appl. Mater. Interfaces, 2018, 10, 38151–38158 CrossRef CAS PubMed
- A. Thielmann, R. Isenmann and M. Wietschel, Fraunhofer ISI, 2010, 3–22 Search PubMed
- C. L. Tsai,
et al.
, ACS Appl. Mater. Interfaces, 2016, 10617–10626 CrossRef CAS PubMed
- W. Luo,
et al.
, J. Am. Chem. Soc., 2016, 12258–12262 CrossRef CAS PubMed
- C. Wang,
et al.
, Nano Lett., 2017, 565–571 CrossRef PubMed
- L. Porz,
et al.
, Adv. Energy Mater., 2017, 1701003 CrossRef
- L. Cheng,
et al.
, Phys. Chem. Chem. Phys., 2014, 16, 18294–18300 RSC
- A. Sharafi,
et al.
, Chem. Mater., 2017, 29, 7961–7968 CrossRef CAS
- A. Sharafi,
et al.
, J. Mater. Chem. A, 2017, 5, 13475–13487 RSC
- Y. Jin and P. J. Mcginn, J. Power Sources, 2013, 239, 326–331 CrossRef CAS
- R. Kun,
et al.
, ACS Appl. Mater. Interfaces, 2018, 37188–37197 CrossRef CAS PubMed
- R. Djenadic,
et al.
, Solid State Ionics, 2014, 263, 49–56 CrossRef CAS
- E. Yi, W. Wang, J. Kieffer and R. M. Laine, J. Mater. Chem. A, 2016, 12947–12954 RSC
- K. Gao,
et al.
, J. Alloys Compd., 2019, 791, 923–928 CrossRef CAS
- E. Hanc,
et al.
, J. Solid State Chem., 2017, 51–60 CrossRef CAS
- K. Fu,
et al.
, Energy Environ. Sci., 2017, 1568–1575 RSC
- B. Liu,
et al.
, Energy Storage Mater., 2018, 14, 376–382 CrossRef
- R. Ye,
et al.
, Green Chem., 2020, 22, 4952–4961 RSC
- L. Co, J. Kim and H. Lee, Metall. Mater. Trans. B, 2001, 32, 17–24 CrossRef
- S. Toda,
et al.
, Solid State Ionics, 2013, 233, 102–106 CrossRef CAS
- M. Matsui,
et al.
, R. Soc. Chem., 2014, 1019–1024 CAS
- E. Yi, W. Wang, J. Kieffer and R. M. Laine, J. Power Sources, 2017, 156–164 CrossRef CAS
- R. Hongahally, T. Ito, T. Morimura and R. Bekarevich, J. Power Sources, 2017, 363, 145–152 CrossRef
- S. Yu and D. J. Siegel, Chem. Mater., 2017, 12, 9639–9647 CrossRef
- S. Uhlenbruck,
et al.
, Solid State Ionics, 2018, 320, 259–265 CrossRef CAS
- L. Dhivya,
et al.
, RSC Adv., 2015, 5(116), 96042–96051 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta11096e |
| This journal is © The Royal Society of Chemistry 2021 |